

Abstract
To improve the properties of poly(butylene succinate) (PBS), a series of poly[(butylene succinate)-co-poly(tetramethylene glycol)]s (PBSTMGs) with different poly(tetramethylene glycol) (PTMG) contents were successfully prepared by the catalyzed melt polycondensation process. The effect of introducing flexible PTMG segments on the properties was investigated, and they were compared to those of PBS. The differential scanning calorimetry results indicated that the melting temperature, crystallization temperature, and crystallinity of PBSTMG copolymers were slightly lower than those of PBS. Furthermore, these thermal parameters decreased gradually with the increase of PTMG content. Dynamic mechanical analysis showed that there was a significant decline of storage modulus (E′) in the overall temperature range of copolymers compared to that of PBS. The incorporation of PTMG did not modify the crystal lattice of PBS according to the wide-angle X-ray diffraction analysis. Because of copolymerization, the size of the spherulites was reduced at high PTMG contents. The soft domain in the copolymers might contribute to the enhanced tear strength of PBSTMG. The elongation at break and impact strength of PBSTMG copolymers were greatly improved as a result of the phase separation structure and lower degree of crystallinity. Especially, when the PTMG content was 10 mol %, the impact strength of the copolymer reached up to 4.5 times that of PBS. In addition, with more soft segments introduced, the biodegradability of the copolymers became much better than that of PBS.
1. Introduction
In recent years, the environmentally friendly and biodegradable polymers have aroused wide concern because of their potential to substitute nonbiodegradable materials. Biodegradable polymers can be degraded into small molecules, water and carbon dioxide, by microorganisms and therefore diminish the problem of environmental pollution caused by the disposal of common plastics.1 On the other hand, with the current increasing demand of global energy and petrochemical products, polymer materials obtained from renewable biomass resources have attracted more and more attention.
A variety of biodegradable polymers,2 such as poly(lactic acid) (PLA),3 polycaprolactone (PCL),4 and polyhydroxybutyrate (PHB),5 have already been found to be widely applicable in the fields of packaging, agriculture, and drug delivery.6 Nevertheless, the applications of aliphatic polyesters are limited7 owing to their poor mechanical properties; thus, chemical and physical modifications are particularly important. Among these polymers, poly(butylene succinate) (PBS) is a typical biodegradable polyester that can be derived from biomass-based resources and exhibits balanced performance in thermal and mechanical properties.8−11 However, its high crystallinity, slow crystallization rate, and low melt strength and tear strength are the main shortcomings during processing and usage. To meet the different requirements of industries, PBS can be copolymerized with other diacids and diols to regulate the properties.12−17
Some prior reports have18,19 revealed that the biodegradability of poly(butylene adipate-co-terephthalate) became worse with the incorporation of aromatic units. Among the factors affecting biodegradability,20−22 the hydrophilic–hydrophobic balance is influenced by the chemical structure and composition of repeating units; thus, the biodegradability of polyesters will be enhanced by increasing the hydrophilicity. One approach is to introduce hydrophilic segments such as polyethers into the molecular backbone to form block copolymers. The mechanical properties of the block copolymers will be affected by unit types, compositional proportions, and soft segment lengths. Kricheldorf et al.23 blended PCL with polyamide and polyethylene so as to improve the mechanical properties. In addition, Albertsson et al.24 have applied poly(ether sulfone)–PTMG copolymers as surgical sutures and drug carriers for their excellent performance.
Copolymerization is an effective modification method to achieve the purpose of reducing the crystallinity, adjusting the degradation rate, and increasing the ductility of PBS. The comonomers can be diacids and diols, including adipic acid, terephthalic acid, ethylene glycol, 1,3-propanediol, 1,6-hexanediol, and so on. The crystallinity of copolymers upon introducing these comonomers is lower than that of PBS, leading to higher ductility.
Djonlagic et al.22 synthesized poly(butylene adipate-co-butylene succinate) by melt polycondensation and studied the impact of copolymerization on the mechanical properties and biodegradability. The toughness and biodegradability of the copolymers enhanced with the increasing content of adipic acid, whereas the melting temperature and crystallinity decreased. Tsutsumi et al.25 investigated poly(butylene succinate-co-butylene terephthalate) by introducing terephthalic acid. The highest degradation rate was observed with 20 mol % terephthalic acid added to succinate acid. A conclusion was drawn in the work that with more terephthalic acid added the glass-transition temperature (Tg) and the mechanical properties of the copolymer were improved and the melting temperature declined when the molar ratio of terephthalic acid to succinate acid was less than 30%.
It has been reported that the biodegradability will be reduced when aliphatic polyesters copolymerized with nonbiodegradable polymers, such as aliphatic–aromatic random copolyesters,26 but will not be significantly reduced with polyethers. Nagata et al.27 synthesized PBS and poly(ethylene glycol) (PEG) copolymers, where PEG molecular weight was in the range from 200 to 2000 g/mol. With an increase in the PEG content, the hydrolysis rate of the copolymer enhanced, whereas the melting temperature, crystallinity, and elongation decreased. Although the biodegradability of the copolymer was improved after the introduction of PEG, the material containing the PEG component was unstable in nature, particularly when exposed to sunlight.
Because of the high degree of crystallinity and the molecular structure of PBS itself, the tear strength and toughness of polyester materials leave much to be desired for films, as well as the low impact strength, seriously affecting the processability and mechanical property.28,29 For expanding the applications, it is very important and necessary to strengthen the tear strength and toughness of PBS, while maintaining the biodegradability. In this work, poly[(butylene succinate)-co-poly(tetramethylene glycol)] (PBSTMG) copolymers were synthesized by melt polycondensation. PBS was used as the hard segment and PTMG as the soft segment to prepare biodegradable polymers with improved properties. By introducing PTMG, the tear resistance and the impact strength of the copolymer were enhanced as a result of the phase separation and lower degree of crystallinity. The relationship between the properties of the copolymers and molar fraction of the soft segments was evaluated, and their behavior was compared to that of PBS.
2. Results and Discussion
2.1. Composition and Structure of PBSTMG Copolymers
The synthesized PBSTMG copolymers with 5, 10, 15, 20, 30, 40, 50, 60, and 70 mol % PTMG in the total diols were denoted PBSTMG-5, PBSTMG-10, PBSTMG-15, PBSTMG-20, PBSTMG-30, PBSTMG-40, PBSTMG-50, PBSTMG-60, and PBSTMG-70, respectively. The structural formula of PBSTMG and the 1H NMR spectrum of PBSTMG-5 are presented in Scheme 1 and Figure 1. Figure 1 shows the proton peaks (c, δ = 1.62–1.71 ppm; b, δ = 4.12 ppm) originating from the methylene group of 1,4-butanediol. The peaks at δ = 2.62 ppm corresponded to the protons (a) in the methylene group of succinic acid (SA), and the methylene protons (d) in PTMG linked to the ether bond showed the peaks at δ = 3.41 ppm. On the basis of these results, the molecular structure of PBSTMG was determined adequately.
Scheme 1. Structural Formula of PBSTMG.

Figure 1.

1H NMR spectrum of the copolymer with 5 mol % PTMG in the total diols (PBSTMG-5).
The composition of the copolymers was calculated from the relative intensities of the characteristic peaks of the SA repeating unit and the methylene groups attached to the ether oxygen of PTMG. Thus, the molar fraction of PTMG units in the synthesized copolymers was calculated and the efficiency of the synthesis reaction could be obtained by comparing with that of the initial feed ratio. The calculation formula is as follows
where Ad and Aa are the integral peak areas of the corresponding peaks. Nd = 52.56 and Na = 4 are the numbers of protons in the corresponding units.
The theoretical and experimental PTMG molar fractions of the synthesized copolymers under different feed ratios are presented in Table 1. As can be seen from Table 1, the molar content of the PTMG unit in the copolymer was approximately the same as that in the initial feed ratio. The ratios of PTMG/1,4-butanediol (BDO) were slightly larger than those in the feed ratio, which was due to the partial volatilization of the butylene glycol and the formation of by-product tetrahydrofuran in the reaction.
Table 1. Composition, Molecular Weight and Polydispersity Index of PBSTMG Copolymers.
|
x(PTMG) (mol %) |
|||||||
|---|---|---|---|---|---|---|---|
| sample | n(BDO):n(SA) | theoretical | experimental | Lna | Mn (×104 g/mol) | Mw (×104 g/mol) | Mw/Mn |
| PBS | 17.64 | 36.27 | 2.06 | ||||
| PBSTMG-5 | 1.14 | 5.00 | 5.16 | 18.38 | 9.49 | 15.30 | 1.61 |
| PBSTMG-10 | 1.08 | 10.00 | 10.24 | 8.77 | 8.66 | 14.70 | 1.70 |
| PBSTMG-15 | 1.02 | 15.00 | 16.95 | 4.90 | 12.23 | 32.55 | 2.66 |
| PBSTMG-20 | 0.96 | 20.00 | 22.18 | 3.51 | 8.89 | 34.24 | 3.85 |
| PBSTMG-30 | 0.84 | 30.00 | 33.68 | 1.97 | 11.99 | 32.97 | 2.75 |
| PBSTMG-40 | 0.72 | 40.00 | 44.74 | 1.24 | 11.97 | 29.82 | 2.49 |
| PBSTMG-50 | 0.60 | 50.00 | 56.31 | 0.78 | 17.74 | 40.85 | 2.30 |
| PBSTMG-60 | 0.48 | 60.00 | 66.47 | 0.50 | 6.62 | 16.74 | 2.53 |
| PBSTMG-70 | 0.36 | 70.00 | 77.18 | 0.30 | 5.07 | 7.93 | 1.56 |
The average sequence lengths of the PBS hard segments (Ln) were calculated from the molar fraction of PBS in the copolymers. Ln = 1/(1 – XPBS) – 1.
The molecular weight information of PBS and PBSTMG copolymers is also listed in Table 1. The number-average molecular weights (Mn) of all investigated copolymers indicated that the copolyesters of high molecular weights were obtained by the synthetic method described in this article.
2.2. Thermal Properties of PBSTMG Copolymers
2.2.1. Melting and Crystallization Behavior Analysis
After the chemical structure of the PBSTMG copolymer was confirmed, the melting and crystallization properties of PBS and PBSTMG were studied by differential scanning calorimetry (DSC). Figure 2 shows the first cooling curves and second heating curves of the copolymers. The thermodynamic parameters are summarized in Table 2. Compared to that of PBS, the melting temperature of PBSTMG copolymers decreased from 114.1 °C for PBS to 78.8 °C for PBSTMG-20. The experimental data indicated that with the increasing PTMG content, the melting temperature, crystallization temperature, and crystallinity of PBSTMG declined gradually.
Figure 2.

DSC thermograms of PBS and PBSTMG copolymers (a) first cooling curves at a cooling rate of 10 °C/min and (b) second heating curves at a heating rate of 10 °C/min.
Table 2. Thermal Properties and Crystallinity of PBS and PBSTMG Copolymers.
| sample | Tm (°C) | Tc (°C) | Tg1 (°C)a | Tg2 (°C)a | ΔHm (J/g) | ΔHc (J/g) | Xc (%)b |
|---|---|---|---|---|---|---|---|
| PBS | 114.1 | 75.7 | –22.9 | 66.9 | 62.3 | 60.5 | |
| PBSTMG-5 | 105.6 | 61.8 | –42.9 | 57.5 | 51.2 | 52.0 | |
| PBSTMG-10 | 101.1 | 58.0 | –56.7 | –26.0 | 50.3 | 49.7 | 45.5 |
| PBSTMG-15 | 82.1 | 47.2 | –63.8 | –26.7 | 24.6 | 27.7 | 25.1 |
| PBSTMG-20 | 78.8 | 47.1 | –67.4 | –31.4 | 20.0 | 16.1 | 14.6 |
The glass-transition temperature (Tg) was adopted from the tan δ peak measured by dynamic thermal analysis.
The degree of crystallinity (Xc) was calculated from the DSC results.
When the molar fraction of the PTMG component reached 20%, the crystallization peak became less obvious, with the corresponding melting peak inconspicuous, and the crystallization ability was greatly weakened, which implied that the copolymerization affected the crystallinity of the polymers to a great degree. As the PTMG content increased, the crystallinity was reduced considerably.
For a typical PBS-based copolymer, the introduction of the third monomer resulted in a significant impact on the melting point and crystallinity. Through copolymerization, the sequence length of crystallizable segments became shorter and the foreign units were excluded from the crystal lattice. The lamella thickness of PBS turned to be thinner with the incorporation of more comonomers. Thus, the melting point and crystallinity of copolymers decreased gradually with the increasing content of the third monomer.
2.2.2. Thermal Stability Analysis
Thermogravimetric analysis (TGA) was used to study the thermal stability, which is very important for manufacture and application. Figure 3 presents the typical TGA curves of PBS and PBSTMG copolymers in a nitrogen atmosphere.
Figure 3.
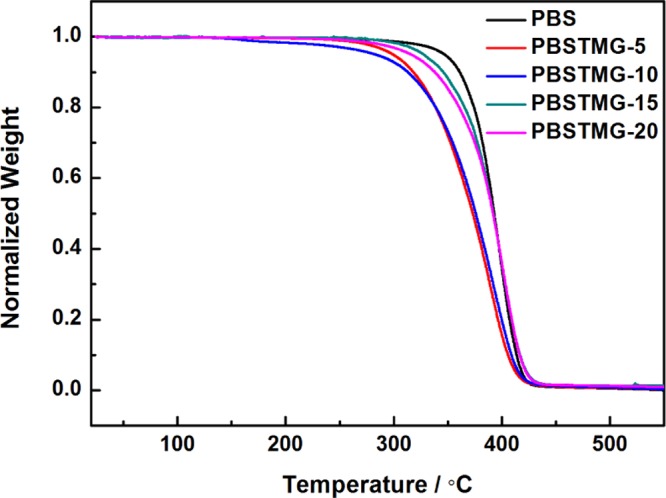
TGA curves of PBS and PBSTMG copolymers in a nitrogen atmosphere at a heating rate of 10 °C/min.
From Figure 3, it could be found that the classic one-step degradation curves were obtained for all samples. The TGA curves of PBSTMG copolymers and PBS were quite similar in shape, indicating that there was no significant difference in the mechanism of thermal degradation. The series of decomposition temperatures of PBSTMG were not much different from those of PBS, which meant that copolymerization had no serious impact on the thermal stability of copolymers.
2.3. Crystalline Structure Analysis
2.3.1. Wide-Angle X-ray Diffraction Analysis (WAXD)
To get further information on the factors affecting the physical properties of PBS and PBSTMG copolymers, WAXD was adopted to confirm the crystalline structure and crystallinity of the samples. It has been reported30 that PBS shows monoclinic α-form crystals with unit cell: a = 0.523 nm, b = 0.908 nm, c = 1.079 nm, β = 123.87°, and the diffraction peaks of crystal planes appear at 19.4° (020), 21.5° (021), 22.5° (110), and 28.8° (111). The X-ray diffraction diagrams of PBS and PBSTMG copolymers showed diffraction peaks at 19.6, 21.8, 22.7, and 29.1°, as revealed in Figure 4. Both PBS and PBSTMG copolymers exhibited almost the same positions of the peaks in the diffractograms. These results confirmed that copolymerization with PTMG did not modify the type of the crystal lattice, characteristic of PBS, and PBSTMG crystallites had a monoclinic crystal structure similar to that of PBS.
Figure 4.
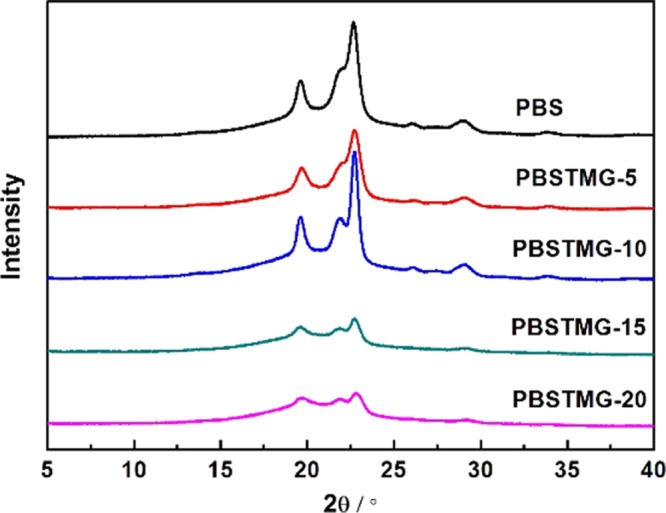
X-ray diffraction diagrams of PBS and PBSTMG copolymers with different contents of PTMG.
As the PTMG content increased, the peaks at each position became less conspicuous. The WAXD analysis indicated that the crystallinity of copolymers decreased gradually with the incorporation of more PTMG, which was in correspondence with the results of DSC.
2.3.2. Polarizing Microscope Analysis
Polarizing optical microscopy (POM) was performed to investigate the spherulite morphology of all copolymers. Figure 5 shows the optical micrographs of the isothermally crystallized PBS and PBSTMG copolymers. There were obvious differences between the spherulite morphologies of PBS and PBSTMG. In addition to the obvious Maltese Cross, POM images of PBS revealed the larger size of spherulites, which consisted of circular bright and gray birefringent bands. Banded spherulites were ubiquitous in polyesters and generally thought to be the result of regular twists of the lamellar crystals.11
Figure 5.

POM images of (a) PBS, (b) PBSTMG-5, (c) PBSTMG-10, (d) PBSTMG-15, and (e) PBSTMG-20 isothermally crystallized at the degree of undercooling of 40 °C.
The phenomenon of banded spherulites was more pronounced in PBSTMG-5 crystals. With the increasing PTMG content, the size of spherulites became increasingly smaller, as presented in Figure 5. Finally, PBS hard segments dispersed in the amorphous phase at 20 mol % PTMG content and the hard segments played a role of physical crosslinking.
2.4. Dynamic Mechanical Analysis (DMA)
DMA was utilized to study the viscoelastic properties of PBS and PBSTMG copolymers. Storage modulus (E′) and damping factor (tan δ) of PBS and PBSTMG copolymers are plotted in Figure 6.
Figure 6.

Temperature dependence of the (a) storage modulus and (b) damping factor of PBS and PBSTMG copolymers.
The change of the PTMG content had a great influence on the storage modulus and glass-transition temperature. In the glassy state, the molecular motion was restricted and hence the values of E′ for all copolymers were higher. The glass-transition temperature could be obtained from Figure 6b. As the PTMG content increased, the loss peaks shifted to the lower temperature and the shape of peaks became narrower. The glass-transition temperature (Tg) of PBSTMG copolymers decreased from −42.9 to −67.4 °C, as shown in Table 2. The curves of PBSTMG-10, PBSTMG-15, and PBETMG-20 all showed two loss peaks in Figure 6, which indicated the existence of phase separation in the copolymerization system.31
2.5. Phase Separation Structure Analysis
The phase separation structure of the copolymer was due to the components being thermodynamically immiscible or the crystallization of a certain component. The atomic force microscopy (AFM) height and phase images in Figure 7 provided convincing evidence of the phase separation structure, which was consistent with the POM and DMA results. The spherulitic morphology observed by POM demonstrated that the crystals still exhibited a continuous phase even at a high PTMG content. When the PTMG content reached 10 mol %, the film formed two different phases in nanoscale. The bright portion represented the crystalline regions, that is, PBS hard segments, and the dark portion represented the amorphous phase, that is, PTMG soft segments. The appearance of phase separation visually revealed that PTMG indeed affected the crystallization kinetics of PBS and the distribution of the particular structure would also influence the mechanical properties to some extent.
Figure 7.

AFM images of (a, b) PBSTMG-5, (c, d) PBSTMG-10, and (e, f) PBSTMG-15. The left and right columns show the height and the phase images, respectively.
2.6. Mechanical Properties
Mechanical property was one of the most important and basic performance of biodegradable polyester materials. Table 3 illustrates the mechanical properties of PBS, PBSTMG copolymers, and PBS-PBSTMG blends. PBS blended with 5, 10, 20, and 30 wt % PBSTMG, which consisted of the same mass fraction of PBSTMG-5, PBSTMG-10, PBSTMG-15, and PBSTMG-20, are denoted PBS-5%PBSTMG, PBS-10%PBSTMG, PBS-20%PBSTMG, and PBS-30%PBSTMG, respectively. The tensile strength of the PBSTMG copolymers was reduced compared to that of PBS. When the content of PTMG was 10 mol % or less, the tensile strength of materials still reached more than 30 MPa. In addition, the elongation at break and impact strength of PBSTMG were greatly improved with an increase in the PTMG content. Especially, when the PTMG content was 10 mol %, the impact strength of the copolymer was about 4.5 times that of PBS.
Table 3. Mechanical Properties of PBS, PBSTMG Copolymers, and PBS-PBSTMG Blends.
| sample | tensile strength (MPa) | elongation at break (%) | impact strength (kJ/m2) |
|---|---|---|---|
| PBS | 37.8 | 323 | 6.7 |
| PBSTMG-5 | 30.5 | 505 | 22.2 |
| PBSTMG-10 | 32.0 | 476 | 30.0 |
| PBSTMG-15 | 23.5 | 473 | a |
| PBSTMG-20 | 12.7 | 573 | a |
| PBS-5%PBSTMG | 34.7 | 317 | 10.4 |
| PBS-10%PBSTMG | 44.1 | 499 | 8.2 |
| PBS-20%PBSTMG | 43.0 | 467 | 8.1 |
| PBS-30%PBSTMG | 34.4 | 379 | 16.7 |
Specimens cannot be ruptured.
There was a significant increase in toughness, elongation, and impact strength of PBSTMG. The results indicated that an appropriate copolymerization ratio could reduce the size of spherulites and crystallinity of polyesters, promote the formation of the phase separation structure, and consequently enhance the toughness and ductility of copolymers, as well as improving the impact strength of materials.
Furthermore, Table 3 also shows the mechanical properties of PBS and PBSTMG blends. By simply blending PBS with PBSTMG, the elongation at break and the impact strength of PBS-PBSTMG blends were all improved to a certain extent compared to those of PBS and the toughness of the blends was much better as well.
PBS films and PBS-PBSTMG blend films were prepared by the film blowing process, the longitudinal direction along the blown film was denoted the MD direction, and the transverse direction perpendicular to the blown film was denoted the TD direction. As shown in Table S1, the mechanical properties of the PBS-PBSTMG blend and PBS films were also studied. Except for the tensile strength of PBS-10%PBSTMG (TD), all PBS-PBSTMG films had higher transverse and longitudinal tensile strengths than those of PBS films, and the elongation was also significantly improved. These results indicated that the blending of PBS and PBSTMG greatly reinforced the mechanical properties of films.
The tear strengths of PBS and PBS-PBSTMG blend films are presented in Table S2. From the data, it was found that the blend films possessed a much better tear strength that PBS films and especially the tear strength of PBS-30%PBSTMG films was about 1.76 times that of PBS films. The films prepared by blending PBS with PBSTMG showed excellent tear resistance property. These results illustrated that the phase separation structure and the lower degree of crystallinity present in copolymers hindered the growth of cracks and thus the tear strength was improved macroscopically.
2.7. Biodegradation Properties
Biodegradability is one of the most attractive and promising properties of PBS. The chemical structure, morphology, and degradation conditions of PBS and its copolymers had a great influence on the biodegradation rate. To investigate the effect of the chemical structure of aliphatic copolyesters on biodegradability, enzymatic degradation experiments were performed with PBS and PBSTMG films (5 mm × 5 mm × 0.15 mm). The biodegradability of the copolymers was characterized by the weight loss of the films in a lipase solution, using the pure phosphate buffer for the blank control tests.
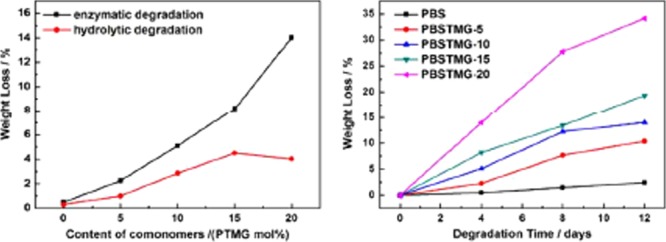
The relationship between the content of PTMG segments and the weight losses of PBS and PBSTMG films after both enzymatic degradation and hydrolytic degradation (without lipase) for 4 days is presented in Figure 8a. During the test time, the weight losses of PBS films in the lipase solution and phosphate buffer were almost the same and the presence of lipase only slightly promoted the degradation of PBS. It could be observed that all PBSTMG films containing hydrophilic soft segments showed a higher weight loss in both types of degradation experiments and the biodegradation rates were significantly improved compared with those of PBS. The introduction of PTMG resulted in the increase of the hydrolysis rate of the copolymers, and the degradation rate accelerated steadily with the addition of more PTMG. In the case of enzymatic degradation, the weight loss was increased from 2.26% of PBSTMG-5 to 14.00% of PBSTMG-20. Meanwhile, the degradation rates of the copolymers in the lipase solution were higher than those in the pure phosphate buffer. The above results could be attributed to the introduction of soft segment PTMG, and the copolymers became more flexible, which led to decreased crystallinity and improved hydrophilicity. Therefore, the degradation rate of PBSTMG turned to be faster than that of PBS. Among the comprehensive impacts on the biodegradability of copolymers, the crystallinity was the main factor affecting the degradation rate.
Figure 8.

Weight losses of (a) PBS and PBSTMG films with different contents of PTMG after enzymatic degradation and hydrolytic degradation (without lipase) for 4 days at 35 °C and (b) PBS and PBSTMG films in enzymatic degradation tests for different days at 35 °C.
To gain more insight into the degradation behavior of PBSTMG, the enzymatic degradation experiments of PBS and PBSTMG films were carried out for different days. It could be observed from Figure 8b that the weight losses of the copolymers were all larger than those of PBS at different degradation periods. On the one hand, for each sample, there was a distinct increase in the weight loss when the degradation time was prolonged and later the curves in Figure 8b became relatively flatter, which illustrated the decreased degradation rate. On the other hand, when more content of PTMG was copolymerized, the weight loss of PBSTMG was increased obviously and the enzymatic degradation rate was improved. The results showed that with the introduction of more soft segments, the biodegradability of the copolymers became better. After 8 days of enzymatic degradation, the PBSTMG-20 films had been partially decomposed into small pieces. The biodegradability of the copolyesters was influenced by the hydrophilic–hydrophobic equilibrium, and the degradation usually began in the amorphous region. Because of the increase of the PTMG content, the flexibility and hydrophilicity of the copolymer segments were gradually enhanced.
3. Conclusions
In this work, PBSTMG copolymers with different PTMG contents were successfully synthesized via introducing PTMG segments in PBS by melt polycondensation. The chemical structure and composition of PBSTMG obtained were confirmed by 1H NMR. The incorporation of PTMG slightly reduced the melting temperature, crystallization temperature, glass-transition temperature, and storage modulus of the copolymers compared with those of PBS. Moreover, crystallinity of PBSTMG decreased gradually with the increase of PTMG content. Phase separation of hard segments and soft segments visually revealed that the introduction of PTMG reduced the size of spherulites and crystallinity of polyesters and indeed affected the mechanical properties of polymers. Regulated by PTMG content, the elongation at break, impact strength, and flexibility of the copolymers were significantly improved. Especially as films, the tear strength of PBSTMG was greatly enhanced, which might be attributed to the lower degree of crystallinity and the phase separation structure that hinder the further growth of cracks. The increased hydrophilicity and decreased crystallinity of the copolymers also resulted in the accelerated biodegradation rate compared to that of PBS. To summarize, all of the analyses on the thermal and mechanical properties and biodegradability demonstrated that the PBSTMG copolymers could be used as biodegradable polyesters for various applications.
4. Experimental Section
4.1. Materials
SA was supplied by Anqing Hexing Chemical Co., Ltd., China, and BDO was supplied by Xilong Chemical Co., Ltd., China. Poly(tetramethylene glycol) (PTMG) with a molecular weight of 1000 g/mol and tetrabutyl titanate (Ti(OBu)4) were supplied by Aladdin Chemistry Co., Ltd., China. Amano lipase from Pseudomonas fluorescens was purchased from Sigma-Aldrich Co., Ltd., China. All materials were used as received.
4.2. Synthesis of PBSTMG Copolymers
The PBSTMG copolymers were prepared by melt polymerization in the bulk, including esterification and polycondensation. The diol component was used in a 20 mol % excess over the diacid component. As a result, the polyester chains after esterification were terminated with diol groups, and in the following polycondensation step, the terminal butanediols would be removed by vacuum to form high-molecular-weight copolymers. In addition, some butanediols would be partially volatilized and form by-product tetrahydrofuran during the reaction. As an example, the synthesis of PBSTMG with 10 mol % PTMG in the total diols was described. A three-necked flask equipped with a nitrogen inlet tube, a condenser, and a mechanical stirrer was charged with 11.8 g (0.1 mol) of SA, 9.72 g (0.108 mol) of 1,4-butanediol, and 12.0 g (0.012 mol) of PTMG.
4.2.1. Esterification Step
The whole reaction system was placed in a well-controlled-temperature oil bath, and the system was purged with nitrogen. The reaction was carried out at 135 °C for 30 min to ensure that the reactants were fully dispersed and melted and then the temperature was increased gradually to 215 °C (20 °C per 30 min). Finally, the system was maintained at 225 °C for 4 h until there was no liquid distillated.
4.2.2. Polycondensation Step
At the second stage, the system was cooled to 150 °C. The water separator and the condenser were removed, 0.5 wt % Ti(OBu)4 was added as a catalyst to the system, and the temperature was increased to 225 °C with continuous stirring. The system was then evacuated to a pressure below 100 Pa for 4 h. The amount of PTMG soft segments in the poly(ether–ester) chains varied between 5 and 70 mol %.
4.3. Preparation of Blend Samples and Blowing Films
The samples were removed in a drying oven at 70 °C for 24 h before mixing. The blending mixing module of a Haake torque rheometer was used, and the temperatures of three mixing stages were set at 150, 180, and 140 °C. The rotation speed was set as 20 min–1, and the mixing time was 20 min. After mixing, the blends were stored at room temperature.
The film blowing process was operated with the extrusion module of the Haake torque rheometer, connected with a flat film extrusion head (die lip size: 0.75 mm × 50 mm) and drafting roll device. The temperatures of the feed zone, screw zone, die, and winding roll were 150, 180, 140, and 30 °C, respectively. The rotation speed was 20 min–1, and the speed of winding roll was about 150 cm/min. The blowing films had an average width of 50 mm, and the thickness was in the range of 0.035–0.075 mm.
4.4. Characterizations
1H NMR (400 MHz) spectra were recorded in CDCl3 solution by a Varian-Gemini-200 instrument with tetramethylsilane as the reference standard. The concentration of each sample was about 5 wt % in the solvent. Chemical shifts were presented in parts per million.
Thermal properties of PBSTMG copolymers were analyzed by Shimadzu DSC-60 (Japan). Samples (3–5 mg) were placed in sealed aluminum crucibles for the DSC tests. Each sample was first heated from room temperature to 150 °C at a heating rate of 10 °C/min, kept at 150 °C for 3 min to eliminate thermal history, then cooled to 30 °C at a cooling rate of 10 °C/min, held at 30 °C for 3 min, and then reheated at the same rate to 150 °C. The crystallinity of PBS and copolymers was calculated by the following function: Xc = (ΔHm/ΔHθm) × 100%, where ΔHm is the melting enthalpy of the sample and ΔHθm is the melting enthalpy of 100% crystalline PBS (ΔHθm = 110.5 J/g).32
TGA was performed on a Shimadzu DTG-60 thermogravimetric analyzer (Japan) in a N2 atmosphere with the flow rate of 40 mL/min. Each sample (3–5 mg) was placed in a sealed aluminum crucible and heated from 30 to 550 °C at a heating rate of 10 °C/min, using an empty aluminum crucible as reference.
WAXD analysis was carried out at room temperature on a Rigaku SmartLab X-ray diffractometer (Japan), using Cu Kα radiation (λ = 0.154 nm). Scanning was operated with 2θ from 5 to 40° at a rate of 4°/min and a step interval of 0.02°.
The spherulitic morphology and nucleation behavior of samples were observed under an Olympus BX41P POM (Japan). The samples were pressed between two glass slides and melted at 160 °C for 5 min to erase any thermal history and then quickly cooled to the crystallization temperature for isothermal crystallization.
DMA was conducted by the Anton Paar MCR301 stress/strain-controlled rheometer (Austria) from −80 to 70 °C at a heating rate of 4 °C/min, with the strain of 0.02% and a frequency of 1 Hz in the tensile configuration.
AFM (Shimadzu SPM-9700) was utilized to characterize the phase separation and micromorphology of PBSTMG copolymers under the tapping mode. The AFM specimens were prepared by casting 50 μL solution of the centrifuged mixture on silicon and dried in a vacuum oven for 12 h to remove the residual solvent.
The tensile properties of PBS and PBSTMG copolymers were tested by a TS-2000 Gotech tester (Taiwan) at room temperature and at a crosshead speed of 100 mm/min. The impact strength of the samples was measured by a Jinjian XJUD-5.5 izod impact machine (China). All specimens were stored at room temperature for 12 h before measurement.
The tear strength is one of the important properties of films. The GB/T 16578-1996 test method was applied in the experiment. The film samples (Figure 9) prepared were of 150 mm × 50 mm with a 75 mm incision in the middle, and the two edges of the trouser-like sample were clamped in the fixture and then tear along the direction of the incision at a speed of 200 mm/min.
Figure 9.

Schematic diagram of the tear strength test.
PBS and PBSTMG films for the enzymatic degradation test were prepared by hot pressing and stored at room temperature. The amano lipase from Pseudomonas fluorescens was dissolved in the phosphate buffer (pH 7.2–7.4) to obtain a 1 mg/mL enzyme solution, and the enzyme-free phosphate buffer solution was used for blank control experiments. The films (5 mm × 5 mm × 0.15 mm) were immersed in degradation solutions and placed in a 35 °C shaking incubator. The samples were removed from the degradation solution every 4 days, rinsed with deionized water, and dried in a vacuum oven for 12 h at room temperature. The degradation test was quantified as the weight loss, and the degradation solution was changed with a freshly prepared one after each measurement.
Acknowledgments
The authors thank the National Basic Research Program of China (973 Program) (grant no. 2014CB932202) and the National Natural Science Foundation of China (grant nos. 51673110 and 51473085) for financial support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00517.
Table S1, mechanical properties of PBS and PBS-PBSTMG blend films; Table S2, tear strengths of PBS and PBS-PBSTMG blend films (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nair L. S.; Laurencin C. T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. 10.1016/j.progpolymsci.2007.05.017. [DOI] [Google Scholar]
- Jun-Hui J. I. Developments and Applications of Fully Biodegradable Plastics. Plastics 2007, 36, 37. [Google Scholar]
- Datta R.; Tsai S. P.; Bonsignore P.; Moon S. H.; Frank J. R. The technological and economic potential of poly(lactic acid) and its derivatives. FEMS Microbiol. Rev. 1995, 16, 221–231. 10.1111/j.1574-6976.1995.tb00168.x. [DOI] [Google Scholar]
- Hutmacher D. W.; Schantz T.; Zein I.; Ng K. W.; Teoh S. H.; Tan K. C. Mechanical properties and cell cultural response of polycaprolactone scaffolds designed and fabricated via fused deposition modeling. J. Biomed. Mater. Res. 2001, 55, 203–216. . [DOI] [PubMed] [Google Scholar]
- Poirier Y.; Dennis D. E.; Klomparens K.; Somerville C. Polyhydroxybutyrate, a biodegradable thermoplastic, produced in transgenic plants. Science 1992, 256, 520–523. 10.1126/science.256.5056.520. [DOI] [PubMed] [Google Scholar]
- Ikada Y.; Tsuji H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. . [DOI] [Google Scholar]
- Zhang S. P.; Gong M.; Dang Y.; Shi S. Q.; Gong Y. K. Research Progress of Poly(butylene succinate). Polym. Bull. 2011, 34, 86–93. [Google Scholar]
- Xu J.; Guo B. H. Poly(butylene succinate) and its copolymers: research, development and industrialization. Biotechnol. J. 2010, 5, 1149–1163. 10.1002/biot.201000136. [DOI] [PubMed] [Google Scholar]
- Zheng L. C.; Chun cheng L. I.; Shao hua W. U.; Zhang D.; Xiao Y. N.; Zhang B.; Zhu W. X. Recent Progress on Poly(butylene succinate):Synthesis,Copolymerization and Functionalization. Polym. Bull. 2016, 9, 115. [Google Scholar]
- Tang Y. R. X. J.; Guo B. H. Tuning The Properties of Biodegradable Poly(Butylene Succinate) Via Random Copolymerization. J. Chem. Proc. Eng. 2014, 1, 205. [Google Scholar]
- Xu J.; Guo B. H.. Microbial Succinic Acid, Its Polymer Poly(butylene succinate), and Applications. In Plastics from Bacteria: Natural Functions and Applications, Microbiology Monographs; Chen G. Q., Ed.; Springer-Verlag: Berlin, 2010; pp 347–388. [Google Scholar]
- Yang L.; Chen M. J.; Zhao Z. Y.; Chen L.; Wang Y. Z. Halogen-free flame retardation of poly(butylene succinate). Chem. Res. Appl. 2012, 8, 116–118. [Google Scholar]
- Zhang S.; Yang J.; Liu X.; Chang J.; Cao A. Synthesis and characterization of poly(butylene succinate-co-butylene malate): a new biodegradable copolyester bearing hydroxyl pendant groups. Biomacromolecules 2003, 4, 437–445. 10.1021/bm0201183. [DOI] [PubMed] [Google Scholar]
- Zeng J. B.; Li Y. D.; Li W. D.; Zhu Q. Y.; Xiong Z.; Yang K. K.; Wang Y. Z. Poly(Ester Urethane) Synthesized Via Chain Extension of Plla and Pbs Prepolymers Using Hdi. Acta Polym. Sin. 2009, 009, 1018–1024. 10.3724/SP.J.1105.2009.01018. [DOI] [Google Scholar]
- Sun Y. B.; Xu J.; Xu Y. X.; Guo B. H. Synthesis and crystallization behavior of biodegradable poly(butylene succinate-co-butylene phenylsuccinate). Acta Polym. Sin. 2006, 006, 745–749. 10.3724/SP.J.1105.2006.00745. [DOI] [Google Scholar]
- Wang G. L.; Guo B. H.; Li R. Synthesis, characterization, and properties of long-chain branched poly(butylene succinate). J. Appl. Polym. Sci. 2012, 124, 1271–1280. 10.1002/app.34034. [DOI] [Google Scholar]
- Wang G. Y.; Qiu Z. B. Crystalline morphology and crystallization kinetics of melt-miscible crystalline/crystalline polymer blends of poly(vinylidene fluoride) and poly(butylene succinate-co-24 mol% hexamethylene succinate). Chin. J. Polym. Sci. 2014, 32, 1139–1148. 10.1007/s10118-014-1499-5. [DOI] [Google Scholar]
- Marten E.; Müller R. J.; Deckwer W. D. Studies on the enzymatic hydrolysis of polyesters. II. Aliphatic–aromatic copolyesters. Polym. Degrad. Stab. 2005, 88, 371–381. 10.1016/j.polymdegradstab.2004.12.001. [DOI] [Google Scholar]
- Müller R. J.; Kleeberg I.; Deckwer W. D. Biodegradation of polyesters containing aromatic constituents. J. Biotechnol. 2001, 86, 87–95. 10.1016/S0168-1656(00)00407-7. [DOI] [PubMed] [Google Scholar]
- Montaudo G.; Rizzarelli P. Synthesis and enzymatic degradation of aliphatic copolyesters. Polym. Degrad. Stab. 2000, 70, 305–314. 10.1016/S0141-3910(00)00139-7. [DOI] [Google Scholar]
- Gan Z.; Abe H.; Kurokawa H.; Doi Y. Solid-state microstructures, thermal properties, and crystallization of biodegradable poly(butylene succinate) (PBS) and its copolyesters. Biomacromolecules 2001, 2, 605–613. 10.1021/bm015535e. [DOI] [PubMed] [Google Scholar]
- Nikolic M. S.; Djonlagic J. Synthesis and characterization of biodegradable poly(butylene succinate-co-butylene adipate)s. Polym Degrad Stab. Polym. Degrad. Stab. 2001, 74, 263–270. 10.1016/S0141-3910(01)00156-2. [DOI] [Google Scholar]
- Kricheldorf H. R.; Mang T.; Jonte J. M. Polylactones. 1. Copolymerizations of glycolide and iε-caprolactone. Macromolecules 1984, 17, 2173–2181. 10.1021/ma00140a051. [DOI] [Google Scholar]
- Albertsson A.; Ljungquist O. Degradable Polymers. II. Synthesis, Characterization, and Degradation of an Aliphatic Thermoplastic Block Copolyester. J. Macromol. Sci., Chem. 1986, 23, 546–548. 10.1080/00222338608063403. [DOI] [Google Scholar]
- Nagata M.; Goto H.; Sakai W.; Tsutsumi N. Synthesis and enzymatic degradation of poly(tetramethylene succinate) copolymers with terephthalic acid. Polymer 2000, 41, 4373–4376. 10.1016/S0032-3861(99)00727-2. [DOI] [Google Scholar]
- Ki H. C.; Park O. O. Synthesis, characterization and biodegradability of the biodegradable aliphatic–aromatic random copolyesters. Polymer 2001, 42, 1849–1861. 10.1016/S0032-3861(00)00466-3. [DOI] [Google Scholar]
- Nagata M.; Kiyotsukuri T.; Takeuchi S.; Tsutsumi N.; Sakai W. Hydrolytic degradation of aliphatic polyesters copolymerized with poly(ethylene glycol)s. Polym. Int. 1997, 42, 33–38. . [DOI] [Google Scholar]
- Pepic D.; Nikolic M. S.; Djonlagic J. Synthesis and characterization of biodegradable aliphatic copolyesters with poly(ethylene oxide) soft segments. J. Appl. Polym. Sci. 2007, 106, 1777–1786. 10.1002/app.26860. [DOI] [Google Scholar]
- Lee H. S.; Park H. D.; Cho C. K. Domain and segment orientation behavior of PBS–PTMG segmented block copolymers. J. Appl. Polym. Sci. 2000, 77, 699–709. . [DOI] [Google Scholar]
- Ihn K. J.; Yoo E. S.; Im S. S. Structure and Morphology of Poly(Tetramethylene Succinate) Crystals. Macromolecules 1995, 28, 2460–2464. 10.1021/ma00111a045. [DOI] [Google Scholar]
- Ojijo V.; Cele H.; Ray S. S. Morphology and Properties of Polymer Composites Based on Biodegradable Polylactide/Poly[(butylene succinate)-co-adipate] Blend and Nanoclay. Macromol. Mater. Eng. 2011, 296, 865–877. 10.1002/mame.201100042. [DOI] [Google Scholar]
- Van Krevelen D. W.Properties of Polymers: Their Correlation with Chemical Structures; Elsevier Science: New York, 1990; pp 189–225. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.