

Abstract
Loc1 and Puf6, which are localized predominantly to the nucleus, are required for the localization and translational repression of the ASH1 mRNA in the yeast, Saccharomyces cerevisiae. During its transport to the daughter cell, the ASH1 mRNA is translationally repressed via associations with She2, Loc1, and Puf6. Here, we investigated the roles of Loc1 and Puf6 in the translation of mRNAs other than that encoding ASH1. In loc1 or puf6 deletion strains, expression of the mating-specific transcription factor, Ste12, was significantly increased at the post-transcriptional level. These phenotypes required the 5’ untranslated region (UTR) of STE12, which carries the putative Puf6-binding sequences. The RNA helicase, Dhh1, which is a known positive regulator for the translation of STE12 mRNA, was found to be functionally connected with Loc1 and Puf6 in the context of Ste12 expression. Our results collectively show that the phosphorylation of the N-terminal Thr16 residue of Dhh1 affects the protein interactions of Dhh1 with Loc1 or Puf6, and consequently regulates Ste12 expression.
Introduction
mRNAs are transcribed in the nucleus and transported to the cytoplasm, where they direct protein synthesis. The key regulatory steps for the eukaryotic gene expression include the nuclear assembly of pre-messenger ribonucleoprotein particles (mRNPs), the proper localization of the mRNA, and the initiation of translation. In the budding yeast, Saccharomyces cerevisiae, the Ash1 protein has been reported to be asymmetrically localized in daughter cells to repress the transcription of HO endonuclease, which is crucial for the mating type switch [1–3]. The ASH1 mRNA is transcribed in the mother cell and transported to the distal tip of the daughter cell, where the protein is translated. During this transport, the translation of the ASH1 mRNA is repressed by its associations with RNA-binding proteins such as She2, Puf6, Loc1, and Khd1 [4–8].
Puf6 belongs to the pumilio/fem-3 domain family whose members are characterized by a conserved RNA-binding domain with eight PUM (pumilio) repeats of ~ 36 amino acids [9,10]. Puf6 represses translation of the ASH1 mRNA by binding primarily to its 3’-UTR which contains the conserved UUGU elements [5]. Loc1 has been implicated in the assembly of nuclear mRNPs [11,12]. Both Puf6 and Loc1 are nuclear proteins that are enriched in the nucleolus. The ASH1 mRNA is exported to the cytoplasm along with Puf6, whereas Loc1 is removed from the ASH1 mRNA complex prior to or during nuclear export [11]. Deletion of LOC1 or PUF6 decreases the efficiency of ASH1 mRNA localization and up-regulates the cytoplasmic translation of the ASH1 mRNA [5,11].
The Ste12 protein is the primary transcriptional activator responsible for initiating the transcription of about 200 mating-specific genes in S. cerevisiae [13,14]. Upon α-factor stimulation, Ste12 dissociates from its inhibitors, Dig1 and Dig2, and binds to promoters containing pheromone-responsive elements (PREs). Additionally, through its binding with the transcription factor Tec1, Ste12 functions as a key transcriptional regulator during the filamentous response [15,16]. Transcription of the STE12 gene itself is activated by α-factor through four PREs located in its promoter [17]. In addition, STE12 expression is reportedly regulated at the translational level under both filamentous growth and mating conditions [18–21].
The Dhh1 protein, which is a member of the DEAD-box RNA helicase family, functions as a mRNA decapping activator in the mRNA decay pathway and is a major component of the cytoplasmic mRNA granules that are known as P-bodies (processing bodies) [22,23]. Dhh1 has been widely studied as a translational repressor, but accumulating evidence shows that it also participates in translational regulation as a positive and gene-specific activator [18,19,24]. The dhh1 deletion mutation significantly decreased the Ste12 protein level without altering the STE12 transcript level during both the mating process and hyphal growth. High-throughput analysis using both ribosome profiling and RNA-seq experiments in dhh1 mutant cells revealed that a significant number of selected mRNAs are positively regulated by Dhh1 [24].
In the present study, we investigated the potential involvement of Loc1 and Puf6 in the translation of mRNAs other than the ASH1 mRNA. We found that Loc1 and Puf6 appear to translationally repress the STE12 mRNA. The loc1 or puf6 deletion mutations increased STE12 expression at the post-transcriptional level. Genetic and co-immunoprecipitation analyses revealed that Loc1 and Puf6 are functionally connected with the RNA helicase, Dhh1, in regulating Ste12 expression. The N-terminal phosphorylation sites of Dhh1 were found to regulate the association of Dhh1 with Loc1 or Puf6.
Results
The translational repressors Loc1 and Puf6 are functionally connected to Dhh1 in regulating Ste12 protein expression
Loc1 and Puf6 are localized predominantly to the nucleus, and are required for the localization and translational repression of the ASH1 mRNA [3,12,25]. We questioned whether Loc1 and/or Puf6 could translationally repress other mRNAs. Previous reports showed that the transcription factor, Ste12, is post-transcriptionally regulated under conditions that promote filamentous growth and mating [18–20]. Here, we analyzed the expression of Ste12 in loc1 or puf6 deletion strain (Fig 1A and 1B). Ste12-HA protein levels were found to be higher in loc1 or puf6 deletion mutant as compared with wild-type cells. Quantitation of STE12 transcripts revealed that the loc1 mutation caused a slight increase in the STE12 mRNA level, and the puf6 mutation did not significantly alter this level compared with the wild-type strain (Fig 1C and 1D). These results suggest that Loc1 and Puf6 repressed the expression of STE12 at the post-transcriptional level.
Fig 1. Ste12 protein levels were increased in loc1 or puf6 deletion strains.

(A) Ste12-HA protein levels were measured by Western blot in wild-type cells, loc1 cells, and loc1 cells carrying the LOC1 plasmid. Tubulin was detected as a loading control. A representative Western blot is shown. Graphs represent quantification of Ste12-HA to tubulin ratio (n = 3 independent replicates). Values are mean ± SD. *p<0.05 (B) Ste12-HA protein levels were assessed in wild-type cells, puf6 cells, and, puf6 cells carrying the PUF6 plasmid. A representative Western blot is shown. Graphs represent quantification of Ste12-HA to tubulin ratio (n = 3 independent replicates). Values are mean ± SD. * p < 0.05. *** p < 0.005 (C) RNA prepared from the cultures listed in (A) was analyzed by quantitative RT-PCR. STE12 mRNA expression was normalized against ACT1 mRNA expression (error bars, mean + S.D.). (D) RNA prepared from the cultures listed in (B) was analyzed by quantitative RT-PCR. STE12 mRNA expression was normalized against ACT1 mRNA expression (error bars, mean + S.D.).
We next examined the genetic interactions of Loc1 and Puf6 with the DEAD-box RNA helicase, Dhh1, because Ste12 protein expression is known to be decreased in dhh1 deletion or domain-specific mutant strains [18–20]. The STE12 transcript is not affected by dhh1 mutations [18, 19]. We deleted DHH1 from the loc1 or puf6 strains and examined Ste12 expression (Fig 2A and 2B). The dhh1loc1 and dhh1puf6 double deletion strains showed Ste12 levels similar to that of the dhh1 strain, indicating that the dhh1 deletion abolished the de-repressed state of Ste12 expression in loc1 or puf6 cells. We hypothesize that Dhh1 regulates Ste12 translation in functional association with Loc1 and Puf6, possibly downstream of these factors (Fig 2D).
Fig 2. Genetic interactions of dhh1, loc1, and puf6 mutations revealed by double-mutant analysis.
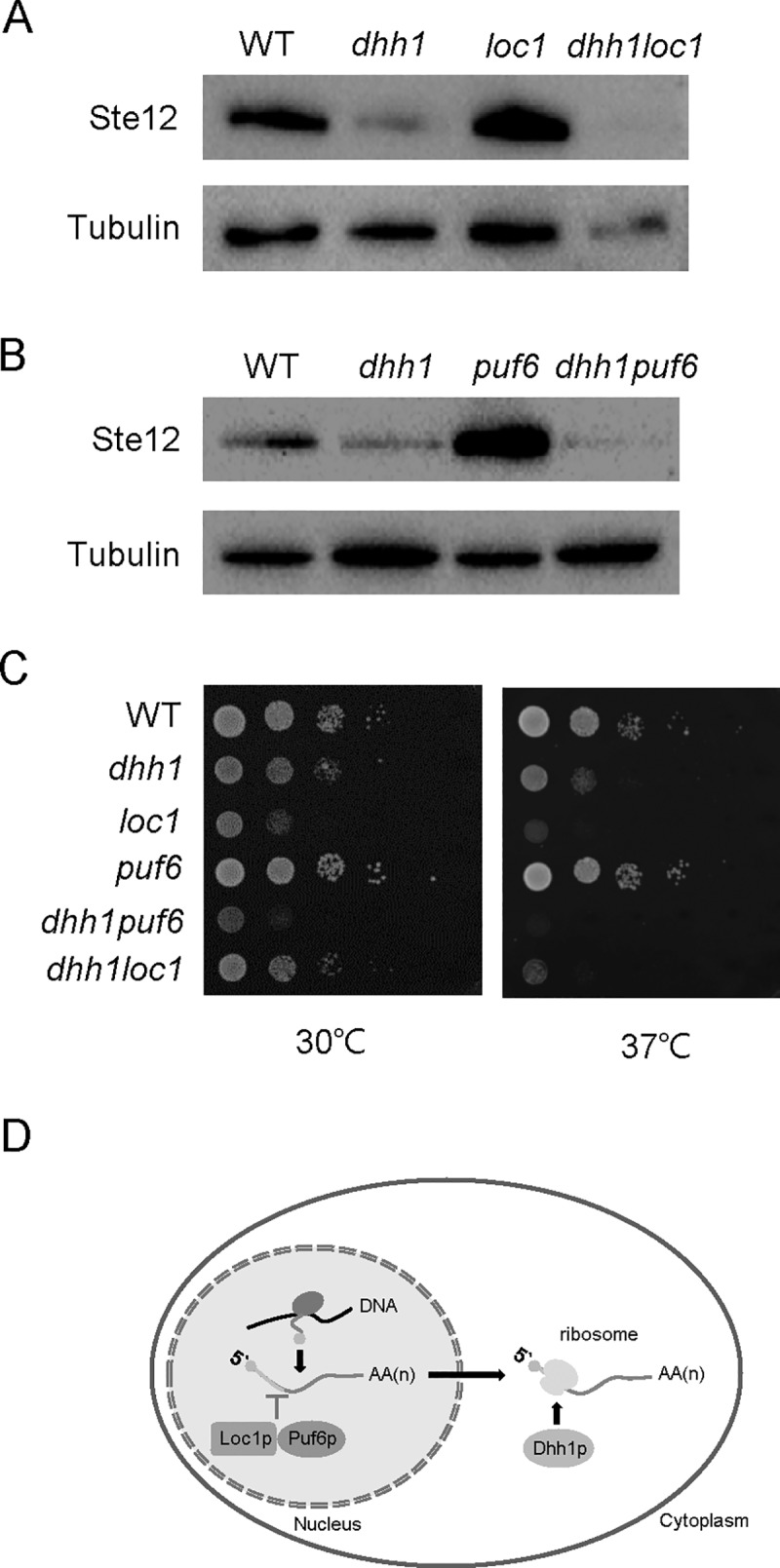
(A) Ste12-HA protein levels were measured in the wild-type, dhh1, loc1, and dhh1loc1 cells by Western blot analysis. Tubulin was used as a loading control. (B) Ste12-HA protein levels were measured in wild-type, dhh1, puf6, and dhh1puf6 cells. (C) Growth defects of the dhh1loc1 and dhh1puf6 double-mutant strains. Ten-fold serial dilutions of overnight cultures of wild-type, dhh1, loc1, puf6, dhh1loc1 and dhh1puf6 cells were spotted on YEPD plates, which were incubated at 30°C and 37°C for 2 days and then photographed. (D) Model of the regulation of STE12 mRNA by Loc1, Puf6, and Dhh1. Loc1 and Puf6 are localized predominantly in the nucleus and repress the expression of STE12 at the post-transcriptional level. Dhh1 positively regulates the translation of the STE12 mRNA, possibly downstream of Loc1 and Puf6.
We analyzed the growth phenotypes of the dhh1loc1 and dhh1puf6 double-deletion strains (Fig 2C). Previous reports indicated that the dhh1 deletion mutant has a growth defect at 37°C and the loc1 deletion mutant shows a severely slow growth phenotype, whereas deletion of PUF6 does not cause any significant growth defect [12, 23, 26]. The dhh1loc1 mutant strain showed a slow growth phenotype at 30°C and a growth defect at 37°C, and thus resembled the dhh1 mutant. The dhh1puf6 mutant strain showed a growth phenotype, almost similar to that of dhh1 cells. These results suggest that Dhh1 interacts genetically with Loc1 and Puf6.
Loc1- and Puf6-mediated repressions are STE12 5’-UTR-dependent
Puf6 represses the ASH1 mRNA by binding to its 3’-UTR, which carries multiple copies of the conserved UUGU element [5,7]. Our sequence analysis of the STE12 mRNA identified UUGU sequences located at nine, 15, and 124 nucleotides upstream of the STE12 start codon (Fig 3A). The length of STE12 5’-UTR has been reported to be 323 nucleotides [27, 28]. Three UUGU sequences were in this 5’-UTR. Four PREs (indicated as -387, 429, -438, and -469) were located in the STE12 promoter region [17].
Fig 3. 5’UTR-dependent repression of the STE12 mRNA.

(A) Schematic representation of the UUGU sequences in the 5’-UTR of STE12 mRNA. The AUG start codon is indicated as +1 in the STE12 ORF. UUGU sequences were found at the -124, -15, and -9 positions. The 5’-UTR starts at -323 position as indicated with a right angle arrow. Sequences indicated with arrows represent the pheromone responsive elements (PREs). (B) Diagrams showing the wild-type STE12::HA and pGAL-STE12::HA constructs. The pGAL-STE12::HA plasmid carried a 800-bp fragment containing the GAL1 promoter region. (C) Western blot analysis of Ste12-HA proteins prepared from wild-type and loc1 cells carrying the STE12::HA or pGAL-STE12::HA plasmids. For STE12::HA (lanes 1 and 2), cells grown to the early log phase (OD600 = 1.0) in 2% glucose medium were used as a control. For galactose induction, cells were grown in SC medium containing 2% raffinose to the early log phase (OD600 = 1.0), and induced with 2% galactose or 2% galactose plus 0.2% glucose for 2 hours. Lanes 3 and 4, 2% raffinose as a sole carbon source; lanes 5 and 6, 2% galactose was added for GAL1 promoter induction; lanes 7 and 8, 2% galactose plus 0.2% glucose. Tubulin was detected as a loading control. (D) Western blot analysis of Ste12-HA proteins prepared from wild-type and puf6 cells carrying STE12::HA or pGAL-STE12::HA plasmids. Cultures and protein analysis were performed essentially as described in (C). (E) Ste12-HA protein levels in cells carrying the wild-type UUGU-STE12-HA or mutant UACU-STE12-HA construct, as revealed by Western blot analysis. Graphs represent quantification of Ste12-HA to GAPDH ratio (n = 2 independent replicates). Values are mean ± SD. *p < 0.05. (F) RNA prepared from the cultures listed in (E) was analyzed by quantitative RT-PCR. STE12 mRNA expression was normalized against ACT1 mRNA expression (error bars, mean + S.D.). (G) RNA immunoprecipitation of Loc1-TAP and Puf6-TAP from the cell extracts of the wild-type or TAP-tagged strain, followed by RNA purification and RT-PCR. STE12-specific primers were used for PCR. ASH1 mRNAs were detected as a binding control.–RT indicates RT-PCR without reverse transcriptase.
To assess whether the abilities of Puf6 and Loc1 to regulate Ste12 protein expression are mediated by the STE12 5’-UTR, we employed a pGAL1-STE12 construct in which the promoter and upstream sequences of the STE12 ORF were replaced with a heterologous GAL1 promoter region (Fig 3B). The plasmid pGAL1-STE12-HA carries about 800 bp fragment of the yeast GAL1 promoter inserted at position -1 of the STE12 start codon. This construct contains the endogenous 3-’UTR of STE12 mRNA. Expression of pGAL1-STE12-HA was induced with 2% galactose in wild-type, puf6 mutant, and loc1 mutant strains. In wild-type STE12-HA cells, the de-repression phenotypes caused by puf6 or loc1 mutation were evident in 2% glucose media (Fig 3C and 3D, lane 1, 2). The levels of pGAL1-STE12-HA expression were similar in wild-type and puf6 mutant cells (lane 5, 6). To avoid artifacts caused by over-expression, the level of galactose induction was modulated by adding 0.2% of glucose to the media (lane 7, 8). We obtained similar results in the wild-type and loc1 mutant cells (Fig 3D). These results suggest that Puf6 and Loc1 modulate STE12 translation in a 5’-UTR-dependent manner. We further questioned whether the putative Puf-binding sites in the 5’UTR of the STE12 mRNA are important for the translational repression of this transcript. The UUGU element at 124 nucleotides upstream of the start codon was mutated to UACU and the resulting plasmid, 5’UACU-STE12-HA, was introduced into the wild-type and puf6 deletion strains. As shown in Fig 3E, the Ste12-HA protein level was significantly higher in a wild-type strain carrying the mutant 5’UACU-STE12-HA mutant compared to a wild-type strain carrying the wild-type 5’UUGU-STE12-HA (lanes 1 and 2). In a puf6 deletion strain, de-repression of Ste12 expression was similarly observed for the wild-type 5’UUGU-STE12-HA and the mutant 5’UACU-STE12-HA (lanes 3 and 4). These results suggest that the UUGU elements in the 5’UTR of the STE12 mRNA are critical for the regulation of STE12 translation. The interaction between the STE12 mRNA and the Loc1 or Puf6 protein in vivo was determined by RNA immunoprecipitation. In this assay, the Loc1-TAP or Puf6-TAP proteins were precipitated from the cell extracts and STE12 mRNAs were detected by RT-PCR analysis. Both Loc1-TAP and Puf6-TAP interacted with the STE12 mRNA, also with the ASH1 mRNA (Fig 3G). It is highly likely that Loc1 and Puf6 bind to the STE12 mRNA to regulate its translation.
Dhh1 and Puf6 regulate the protein expression levels of Cln1 and Gpa2
In a screen performed using polyribosome fractionation, STE12, GPA2, and CLN1 were identified as being translationally regulated during filamentous growth [19]. Cln1 is an mRNA target of 4E-BP-mediated translational repression during growth on rich media [26]. Here, we analyzed the protein expression levels of Cln1 and Gpa2 in dhh1, loc1, and puf6 deletion strains. In the dhh1 mutant strain, the protein levels of Gpa2 and Cln1 were decreased compared to those in wild-type cells, and thus paralleled Ste12 protein expression (Fig 4A). Interestingly, the protein levels of Gpa2 and Cln1 were decreased in the puf6 and loc1 mutant strains compared with wild-type cells, and thus contrasted with the Ste12 protein expression pattern (Fig 4B and 4C). The Gpa2 protein level was only slightly decreased in the loc1 mutant strain. Together, these results suggest that puf6 or loc1 deletion had gene-specific effects on the expression levels of Ste12, Gpa2, and Cln1.
Fig 4. Effects of dhh1, loc1, and puf6 mutations on the protein expression levels of Cln1 and Gpa2.

(A) Western blots of Ste12-HA, Cln1-HA and Gpa2-HA proteins in the dhh1 mutant strain. GAPDH was detected as a loading control. (B) Western blots of Cln1-HA and Gpa2-HA proteins in the puf6 mutant strain. Tubulin was detected as a loading control. (C) Western blots of Cln1-HA and Gpa2-HA proteins in the loc1 mutant strain. Tubulin was detected as a loading control.
Phosphorylation site mutations of DHH1 affect Ste12 expression and the Puf6-Dhh1 interaction
Phosphoproteomic analysis previously identified three phosphorylation sites of Dhh1: Thr10, Ser14 and Thr16 [29–31]. To determine whether the phosphorylation of these amino acid residues are crucial for the functions of Dhh1, we introduced either phospho-deficient (replacement with an alanine) or phospho-mimetic (replacement with a glutamate) mutations at Thr10, Ser14, and Thr16 (Fig 5A). We then examined whether these alterations of each Dhh1 phosphorylation site affected the expression level of Ste12. Phospho-deficient mutants at Thr10, Ser14, and Thr16 by alanine (T10A, S14A, and T16A) did not exhibit substantial defects in Ste12 expression, but the substitution of Thr16 by phospho-mimetic glutamate (T16E) dramatically decreased Ste12 expression (Fig 5B).
Fig 5. Effects of phosphorylation mutations of Dhh1 on Ste12 translation and Dhh1-Puf6 interactions.

(A) Six mutations were introduced at the putative phosphorylation sites in the N-terminus of Dhh1. Mutated amino acids are indicated by gray boxes. (B) Ste12-HA protein levels in cells expressing various phosphorylation-mutant versions of Dhh1, as revealed by Western blot analysis. Protein extracts were prepared from the dhh1 deletion strain (JK400) carrying a DHH1 plasmid (pJI323), vector (pRS316), or mutant plasmid (pJI324-329). GAPDH was detected as a loading control. Graphs represent quantification of Ste12-HA to GAPDH ratio (n = 3 independent replicates). Values are mean ± SD. **p < 0.01. (C) Dhh1 phosphorylation was analyzed using an anti-phosphothreonine antibody. A wild-type BY4741 strain was transformed with DHH1-FLAG (pJI323), vector (pRS316), DHH1-T10A (pJI324), or DHH1-T16A (JI326) plasmids. Cell lysates were immunoprecipitated with anti-FLAG-conjugated agarose and probed with an anti-phosphor-threonine antibody. To detect Dhh1-FLAG, the membrane was re-probed with an anti-FLAG antibody. Graphs represent quantification of phosphorylated Dhh1 to Dhh1-FLAG ratio (n = 3 independent replicates). Values are mean ± SD. *p < 0.05. (D) Dhh1-Puf6 protein interactions, as revealed by co-immunoprecipitation analysis. Wild-type and PUF6-TAP were transformed with DHH1-FLAG (pJI330), DHH1-T16A (pJI331), or DHH1-T16E (pJI332). Cell lysates were immunoprecipitated with anti-FLAG-conjugated agarose in the absence (-) or presence (+) of RNase, and further probed with anti-PAP or anti-FLAG antibodies, respectively. (E) Dhh1-Loc1 protein interactions, as revealed by co-immunoprecipitation analysis. Wild-type and LOC1-TAP were transformed with DHH1-FLAG (pJI330), DHH1-T16A (pJI331), or DHH1-T16E (pJI332). Cell lysates were immunoprecipitated with anti-PAP-conjugated agarose in the absence (-) or presence (+) of RNase, and further probed with anti-PAP or anti-FLAG antibodies, respectively.
To directly analyze the in vivo phosphorylation status of Dhh1, we expressed wild-type and phospho-deficient (T10A and T16A) Dhh1-FLAG, collected these proteins using immunoprecipitation, and performed Western blot analysis with an anti-phosphothreonine antibody. We detected phosphorylation signals in the wild-type DHH1 and DHH1-T10A cells, but this signal was significantly decreased in DHH1-T16A cells (Fig 5C). These results suggest that Dhh1 is phosphorylated in vivo, potentially at Thr16.
One possible mechanism through which Dhh1 phosphorylation may affect Ste12 expression is by altering a protein-protein interaction between Dhh1 and Puf6. To analyze whether there is a direct association between Puf6 and Dhh1, we generated cells carrying chromosomal TAP-tagged PUF6 and plasmid-carried DHH1-FLAG, and compared the abilities of wild-type DHH1, DHH1-T16A, and DHH1-T16E to co-immunoprecipitate with Puf6 (Fig 5D). We observed that there was a protein-protein interaction between Dhh1 and Puf6, and that this interaction was decreased significantly by the DHH1-T16E mutation. When yeast extracts were treated with RNase, Dhh1 was not able to co-precipitate with Puf6, indicating that association of the two proteins was RNA-dependent. A protein interaction between Dhh1 and Loc1 was also observed, but it was not altered by the DHH1-T16E mutation (Fig 5E). These results demonstrate that the phosphorylation of Dhh1 at Thr16 may affect the Dhh1-Puf6 interaction.
Phosphorylation site mutations of DHH1 affect P-body accumulation
Dhh1 is localized throughout the cytoplasm and can be redistributed into discrete foci, called P-bodies, upon various stresses [22,32]. P-bodies contain translationally repressed mRNAs and modulate the storage, decay, and translation of these mRNAs. In such granules, Dhh1 interacts with mRNA decapping enzymes (Dcp1/Dcp2), Xrn1 exoribonuclease, and decapping activators (e.g., Edc3, Scd6, and Pat1) [33,34]. The dhh1 deletion mutation reportedly causes failure of P-body accumulation under glucose depletion or pheromone treatment [18,34]. To investigate whether Dhh1 phosphorylation alters the aggregation of P-body components, we used Dcp2-GFP as a representative P-body marker and examined P-body accumulation in our phosphorylation-site mutant strains (Fig 6). When treated with α-factor, the wild-type cells showed an increase in the number of P-bodies. Interestingly, the phospho-mimetic mutations (T10E, S14E, and T16E) and the phospho-deficient mutation, T10A, were associated with defects in P-body formation.The phospho-deficient mutant cells (S14A and T16A) showed increased levels of P-body accumulation under logarithmic-growth, compared with the wild-type cells. The effects of the phosphorylation site mutations of DHH1 appear to be stress-specific, because the phosphorylation site mutants did not show any significant change in P-body accumulation under glucose deprivation (Fig 6B). These results suggest that the phosphorylation of Dhh1 affects its functions in P-body formation during α-factor treatment.
Fig 6. P-body accumulation in the Dhh1 phosphorylation mutants upon glucose deprivation and α-factor treatment.

The dhh1 deletion mutant strain (JK400) carrying DCP2::GFP (pRP1316) was transformed with the DHH1-FLAG plasmid (pJI323), vector (pRS316), or the various mutant plasmids (pJI324-329). (A) Cultures were grown in the absence (- α) or presence (+ α) of α-factor. (B) Cultures were grown in the presence or absence of glucose. Observations were made under an Olympus BX51 fluorescence microscope using a 100 X objective, and images were processed using the MetaMorph Program software. (C) Graphs represent the quantification of P-body containing cells (%), as conducted in (A) and (B) (n = 2 replicates, > 200 cells). Values are mean ± SD. *** p < 0.005.
Discussion
In this report, we show that Puf6 and Loc1 mediate the translational repression of the STE12 mRNA, and that these roles are functionally connected with Dhh1, which acts as a positive regulator for STE12 mRNA translation. Based on our results obtained from dhh1loc1 and dhh1puf6 double mutant strains and protein-protein interaction assays involving Dhh1, Loc1, and Puf6, we propose that the Puf6-Loc1 mediated repression of the STE12 mRNA precedes the involvement of Dhh1 in this process (Fig 2D). Moreover, the phosphorylation of Dhh1 at Thr16 appears to be important for the physical and functional interactions of Dhh1 with Puf6 and Loc1.
The identification of three UUGU sequences (putative Puf6-binding target) upstream of the STE12 start codon and the mutational analysis of these sequences support our contention that Puf6 and Loc1 regulate the translation of the STE12 mRNA via its 5’-UTR. Puf6 and Loc1 bind directly the 3-’UTR of the ASH1 mRNA in the presence of the She2 protein, and the formed complex is important for the bud-localization and translational control of this transcript [5,7]. The STE12 mRNA has not been reported as a bud-localized transcript, and the Ste12 transcription factor is primarily localized in the nucleus [35,36]. Therefore, it seems likely that the mechanisms underlying Puf6- or Loc1-dependent repression differ depending on the target mRNA(s) [7,37]. In addition, the regulatory factors that associate with Puf6 or Loc1 in the target mRNA-protein complexes are likely to affect this repression mechanism.
A recent report showed that Puf6 and Dhh1 act together in the translational repression of ASH1 mRNA with Dhh1 binding to the 5’-UTR of the ASH1 mRNA and interacting with Puf6 [38]. Here, we show that Dhh1 and Puf6 interact on both genetic and protein levels to mediate the translation of the STE12 mRNA. In particular, our phospho-mimetic substitution of the phosphorylation site residue, Thr16, in the N-terminus of Dhh1 (DHH1-T16E) dramatically decreased Ste12 expression and the Dhh1-Puf6 protein interaction. We observed that this Dhh1-Puf6 interaction appeared to be RNA-dependent. We hypothesize that the binding of Puf6 to the 5’-UTR of the STE12 mRNA suppresses its translation. The interaction of Dhh1 with mRNA-bound Puf6 may be critical for the release of the Puf6-mediated repression.
Dhh1 directly interacts with both decapping and deadenylase complexes [22, 34]. Our observation that the phosphorylation site mutations of DHH1 showed defects in P-body accumulation upon mating factor treatment but not under glucose deprivation prompted us to further question whether the N-terminal phosphorylation regulates the interaction of Dhh1 with P-body components, such as the decapping and deadenylase complexes.
The Dhh1 N-terminal region was not included in previous crystal structure or functional domain analyses of Dhh1, and we know little about the roles of the putative phosphorylation sites in this region [20,30,31,39,40]. Our present observation that the N-terminal phosphorylation sites of Dhh1 regulate its association with Puf6 might improve our understanding of the repression mechanisms that are mediated by Puf6, Loc1, and Dhh1.
Materials and methods
Strains and growth conditions
The Saccharomyces cerevisiae strains and plasmids used in this study are listed in Table 1. Double-deletion mutants JK460 and JK461 were derived from JK443 and JK444, respectively, using a PCR-based gene disruption method. Briefly, the dhh1 disruption cassette containing the LEU2 marker was amplified, and the obtained PCR products were transformed into the puf6 or loc1 deletion strains [18].
Table 1. Strains and plasmids used in this study.
| Strains | Genotype | Reference |
| JK147 | MAT a ura3-52 leu2-3, 112 trp1-1, ade2, cyh r | [18] |
| JK400 | MAT a dhh1::LEU2 ura3-52 leu2-3, 112 trp1-1, ade2, cyhr | [18] |
| BY4741 | MAT a his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | Euroscarfa |
| JK433 | MAT a his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 dhh1::kanMX4 | Euroscarf |
| JK443 | MAT a his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 loc1::kanMX4 | Euroscarf |
| JK444 | MAT a his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 puf6::kanMX4 | Euroscarf |
| JK460 | MAT a ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 ura3Δ0 loc1::kanMX4 dhh1::LEU2 | This work |
| JK461 | MAT a ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 ura3Δ0 puf6::kanMX4 dhh1::LEU2 | This work |
| JK445 | MAT a his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 LOC1::TAP | Open Biosystemsb |
| JK446 | MAT a his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 PUF6::TAP | Open Biosystems |
| Plasmid | Genotype | Reference |
| pJI255 | STE12::HA URA3 CEN | [19] |
| pJI256 | CLN1::HA URA3 CEN | [19] |
| pJI275 | pGAL-STE12::HA URA3 CEN | This laboratory |
| pJI277 | DHH1 URA3 CEN | [19] |
| pJI323 | DHH1::FLAG URA3 CEN | This work |
| pJI324 | DHH1-T10A::FLAG URA3 CEN | This work |
| pJI325 | DHH1-S14A::FLAG URA3 CEN | This work |
| pJI326 | DHH1-T16A::FLAG URA3 CEN | This work |
| pJI327 | DHH1-T10E::FLAG URA3 CEN | This work |
| pJI328 | DHH1-S14E::FLAG URA3 CEN | This work |
| pJI329 | DHH1-T16E::FLAG URA3 CEN | This work |
| pJI330 | DHH1::FLAG URA3 2μ | This work |
| pJI331 | DHH1-T16A::FLAG URA3 2μ | This work |
| pJI332 | DHH1-T16E::FLAG URA3 2μ | This work |
| pJI333 | STE12::HA TRP1 CEN | [18] |
| pJI352 | GPA2::HA URA3 CEN | This work |
| pJI354 | CLN1::HA TRP1 CEN | This work |
| pJI355 | GPA2::HA TRP1 CEN | This work |
| pJI356 | LOC1 LEU2 CEN | This work |
| pJI357 | PUF6 LEU2 CEN | This work |
| pJI377 | UACU124 -STE12::HA URA3 CEN | This work |
| pRS426 | URA3 2μ | [41] |
| pRS314 | TRP1 CEN | [42] |
| pRS316 | URA3 CEN | [42] |
| pRP1316 | DCP2::GFP TRP1 CEN | [43] |
a Euroscarf Collection Center (Frankfurt, Germany)
b Open Biosystems (Colorado, USA)
Yeast strains were grown in YEPD (1% yeast extract, 2% peptone, 2% glucose) or SC (synthetic complete; 0.67% yeast nitrogen base w/o amino acid, 2% glucose, all required amino acids) medium. For galactose induction, cells were grown in SC medium containing 2% raffinose to the early log phase (OD600 = 1.0) and induced with 2% galactose or 2% galactose plus 0.2% glucose for 2 hrs. For α-factor induction, cells were grown in SC medium to the early log phase (OD600 = 0.5), and treated with α-factor (5 μM, 1:100 dilution of a 0.5 mM solution in methanol; Sigma-Aldrich, St. Louis, MO, USA) for 30–90 min. For glucose deprivation, cells were centrifuged and washed with SC lacking glucose. Pellets were resuspended in SC lacking glucose and incubated for 10 min.
Plasmid construction and site-directed mutagenesis
The DHH1-FLAG plasmid (pJI323) was constructed using overlap-extension PCR method. Two PCR-amplified products sharing the FLAG sequence were assembled by PCR and subsequently cloned into the pRS316 vector. Phosphorylation site mutations for Dhh1 were generated in pJI323 by using mutagenic oligonucleotide primers (Table 2). After PCR amplification, the template DNA was digested with DpnI. All mutant constructs were confirmed by sequence analysis. pJI377 plasmid was constructed by using mutagenic oligonucleotide primers and pJI255 template.
Table 2. Primers used in this study.
| Primer name | Sequence |
|---|---|
| Dhh1-F | GGGCTGCAGGACCAACAAACCAATA |
| Dhh1-R | GGGACTAGTGTGATAGTAATAAAA |
| Dhh1-Flag-F2 | GTATCGATGGATTACAAGGATGACGACGATAAGATCTAAAGAATATCTAAGAAA |
| Dhh1-Flag-R2 | GATCTTATCGTCGTCATCCTTGTAATCCATCGATACATACTGGGGTTGTGACTG |
| Dhh1-T10A F | CCATCAATAATAACTTCAACGCTAATAATAACAGTAATACGG |
| Dhh1-T10A R | CCGTATTACTGTTATTATTAGCGTTGAAGTTATTATTGATGG |
| Dhh1-S14A F | CTTCAACACTAATAATAACGCTAATACGGATCTCGATCGG |
| Dhh1-S14A R | CCGATCGAGATCCGTATTAGCGTTATTATTAGTGTTGAAG |
| Dhh1-T16A F | CTAATAATAACAGTAATGCGGATCTCGATCGGGAC |
| Dhh1-T16A R | GTCCCGATCGAGATCCGCATTACTGTTATTATTAG |
| Dhh1-T10E F | CCATCAATAATAACTTCAACGAAAATAATAACAGTAATACGG |
| Dhh1-T10E R | CCGTATTACTGTTATTATTTTCGTTGAAGTTATTATTGATGG |
| Dhh1-S14E F | CTTCAACACTAATAATAACGAAAATACGGATCTCGATCGGG |
| Dhh1-S14E R | CCCGATCGAGATCCGTATTTTCGTTATTATTAGTGTTGAAG |
| Dhh1-T16E F | CTAATAATAACAGTAATGAGGATCTCGATCGGGAC |
| Dhh1-T16E R | GTCCCGATCGAGATCCTCATTACTGTTATTATTAG |
| UACU124-Ste12 F | CAGGTAAGCACTGAAGACTACTTTTATAAGTGTCCCAAGCG |
| UACU124-Ste12 R | CGCTTGGGACACTTATAAAAGTAGTCTTCAGTGCTTACCTG |
| STE12-qRT F | GTATCTCCTAGCGACCCTAC |
| STE12-qRT R | AGTTTGCTGGCCAGAGTTGT |
| ACT1-qRT F | CTGCCGGTATTGACCAAACT |
| ACT1-qRT R | CGGACATAACGATGTTACCG |
| ASH1-RIP F | ACGACGCTTAGAGGAGTAGA |
| ASH1-RIP R | GTTGTCGAGTTTCATCACCA |
| STE12-RIP F | CAGTAAAGCTACTCCGGGCGA |
| STE12-RIP R | AGGCGGGCTCATTAACGGGC |
Western blot analysis
Total protein preparation and Western blotting were performed as previously described [18]. HA-tagged proteins were detected with an anti-HA monoclonal antibody 12CA5 (Roche, Manheim, Germany). Horseradish peroxidase-conjugated anti-mouse antibody (Santa Cruz Biotech, Santa Cruz, CA, USA) was utilized as the secondary antibody. Tubulin protein was detected using a monoclonal anti-α-tubulin antibody (Sigma-Aldrich). Protein bands were detected using the EPDTM Western reagent (Elpis-Biotech, Daejeon, KR).
Total RNA extraction and qRT-PCR
Total RNA was prepared as described previously [18]. cDNA was synthesized from total RNA by reverse transcriptase (RT) using an M-MLV cDNA synthesis kit (Enzynomics, Daejeon, Kr). STE12 mRNA levels were measured by quantitative real-time PCR (qRT-PCR) using SYBR green I (GenetBio, Daejeon, Kr). All values were normalized to the level of ACT1 (actin) mRNA. STE12-specific primers and ACT1-specific primers (Table 2) were used to amplify 100-bp fragments of each coding sequence.
Fluorescence microscopy
Cells were treated with α-factor or grown under glucose-deprived conditions and observed under an Olympus BX51 Fluorescence Microscope using a 100 X objective. The images were processed using the MetaMorph Program software.
Immunoprecipitation and phosphorylation assay
Cells were grown in SC medium to logarithmic phase (OD600 = 1.0), harvested, and lysed with glass beads in lysis buffer (50 mM HEPES/KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, 10 mM NaF, 1 mM Na3VO4). Immunoprecipitation was carried out using an anti-Flag antibody (Sigma-Aldrich, St. Louis, MO, USA). Briefly, 20 μl of protein A/G beads (Santa Cruz Biotech) were added to 1 ml of freshly prepared lysate and incubated for 2 hours on a turning wheel at 4°C. Immunopellets were washed seven times in lysis buffer and recovered by boiling in 2X SDS buffer for 10 min at 100°C. For co-immunoprecipitation experiments, Dhh1 was detected using a monoclonal anti-FLAG antibody (Sigma-Aldrich). Puf6 and Loc1 were detected using a monoclonal anti-PAP antibody (Sigma-Aldrich, St. Louis, MO, USA). HRP-conjugated anti-mouse antibody (Santa Cruz Biotech) was utilized as a secondary antibody.
RNA immunoprecipitation (RIP)
Cells were grown to logarithmic phase (OD600 = 1.0), harvested, and lysed with glass beads in 200 μl FA lysis buffer (50 mM HEPES/KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium deoxycholate, 0.4 U/μl RNasin, 1X protease inhibitor cocktail). Immunoprecipitation was carried out using an anti-PAP antibody (Sigma-Aldrich, St. Louis, MO, USA) and protein G-sepharose bead (GE healthcare, Chicago, IL, USA). Immunopellets were washed three times and suspended in ChIP elution buffer (100 mM Tris-Cl, pH 8.0, 10 mM EDTA, 1% SDS, 0.4 U/μl RNasin). The 5M NaCl and 0.1 μg/μl proteinase were added to 150μl of supernatant and the mixture was incubated at 42°C for 1 hr, and then at 65°C for 1 hr. RNA preparation was carried out as described above and RT-PCR were carried out using STE12-specific or ASH1-specific primers (Table 2).
Acknowledgments
We thank R Parker for kindly providing the Dcp2-GFP plasmid. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2015R1C1A2A01051577).
Data Availability
All relevant data are within the paper.
Funding Statement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2015R1C1A2A01051577) to J. Kim. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Sil A, Herskowitz I. Identification of asymmetrically localized determinant, Ash1p, required for lineage-specific transcription of the yeast HO gene. Cell. 1996; 84: 711–722. 10.1016/s0092-8674(00)81049-1 [DOI] [PubMed] [Google Scholar]
- 2.Long RM, Singer RH, Meng X, Gonzalez I, Nasmyth K, Jansen RP. Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science. 1997; 277: 383–387. 10.1126/science.277.5324.383 [DOI] [PubMed] [Google Scholar]
- 3.Heym RG, Niessing D. Principles of mRNA transport in yeast. Cell Mol Life Sci. 2012; 69: 1843–1853. 10.1007/s00018-011-0902-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besse F, Ephrussi A. Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat Rev Mol Cell Biol. 2008; 9: 971–980. 10.1038/nrm2548 [DOI] [PubMed] [Google Scholar]
- 5.Gu W, Deng Y, Zenklusen D, Singer RH. A new yeast PUF family protein, Puf6p, represses ASH1 mRNA translation and is required for its localization. Genes Dev. 2004; 18: 1452–1465. 10.1101/gad.1189004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irie K, Tadauchi T, Takizawa PA, Vale RD, Matsumoto K, Herskowitz I. The Khd1 protein, which has three KH RNA-binding motifs, is required for proper localization of ASH1 mRNA in yeast. EMBO J. 2002; 21: 1158–1167. 10.1093/emboj/21.5.1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shahbabian K, Jeronimo C, Forget A, Robert F, Chartrand P. Co-transcriptional recruitment of Puf6 by She2 couples translational repression to mRNA localization. Nucleic Acids Res. 2014; 42: 8692–8704. 10.1093/nar/gku597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chartrand P, Meng XH, Huttelmaier S, Donato D, Singer RH. Asymmetric sorting of ash1p in yeast results from inhibition of translation by localization elements in the mRNA. Mol Cell. 2002; 10: 1319–1330. [DOI] [PubMed] [Google Scholar]
- 9.Qiu C, McCann KL, Wine RN, Baserga SJ, Hall TM. A divergent Pumilio repeat protein family for pre-rRNA processing and mRNA localization. Proc Natl Acad Sci USA. 2014; 111: 18554–18559. 10.1073/pnas.1407634112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russo J, Olivas WM. Conditional regulation of Puf1p, Puf4p, and Puf5p activity alters YHB1 mRNA stability for a rapid response to toxic nitric oxide stress in yeast. Mol Biol Cell. 2015; 26:1015–1029. 10.1091/mbc.E14-10-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niedner A, Muller M, Moorthy BT, Jansen RP, Niessing D. Role of Loc1p in assembly and reorganization of nuclear ASH1 messenger ribonucleoprotein particles in yeast. Proc Natl Acad Sci USA. 2013; 110: E5049–5058. 10.1073/pnas.1315289111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang YT, Ting YH, Liang KJ, Lo KY. The Roles of Puf6 and Loc1 in 60S Biogenesis are interdependent, and both are required for efficient accommodation of Rpl43. J Biol Chem. 2016; 291: 19312–19323. 10.1074/jbc.M116.732800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2004; 25: 1465–1476. 10.1016/j.peptides.2003.10.022 [DOI] [PubMed] [Google Scholar]
- 14.Roberts CJ, Nelson B, Marton MJ, Stoughton R, Meyer MR, Bennett HA, He YD, Dai H, Walker WL, Hughes TR et al. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science. 2000; 287: 873–880. 10.1126/science.287.5454.873 [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Styles CA, Fink GR. Elements of the yeast pheromone response pathway required for filamentous growth of diploids. Science. 1993; 262: 1741–1744. 10.1126/science.8259520 [DOI] [PubMed] [Google Scholar]
- 16.Madhani HD, Fink GR. Combinatorial control required for the specificity of yeast MAPK signaling. Science. 1997; 275: 1314–1317. 10.1126/science.275.5304.1314 [DOI] [PubMed] [Google Scholar]
- 17.Houser JR, Ford E, Nagiec MJ, Errede B, Elston TC. Positive roles for negative regulators in the mating response of yeast. Mol Syst Biol. 2012; 8: 586 10.1038/msb.2012.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ka M, Park YU, Kim J. The DEAD-box RNA helicase, Dhh1, functions in mating by regulating Ste12 translation in Saccharomyces cerevisiae. Biochem Biophys Res Comm. 2008; 367: 680–686. 10.1016/j.bbrc.2007.12.169 [DOI] [PubMed] [Google Scholar]
- 19.Park YU, Hur H, Ka M, Kim J. Identification of translational regulation target genes during filamentous growth in Saccharomyces cerevisiae: regulatory role of Caf20 and Dhh1. Eukaryot Cell. 2006; 5: 2120–2127. 10.1128/EC.00121-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung D, Ahn J, Rhee B, Kim J. Mutational analysis of the RNA helicase Dhh1 in Ste12 expression and yeast mating. J Microbiol. 2017; 55: 373–378. 10.1007/s12275-017-7020-4 [DOI] [PubMed] [Google Scholar]
- 21.Park K, Lee YS, Jung D, Kim J. Roles of eIF4E-binding protein Caf20 in Ste12 translation and P-body formation in yeast. J Microbiol. 2018; 56: 744–747. 10.1007/s12275-018-8230-0 [DOI] [PubMed] [Google Scholar]
- 22.Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005; 122: 875–886. 10.1016/j.cell.2005.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng-Rogenski SS, Chong JL, Thomas CB, Enomoto S, Berman J, Chang TH. Functional conservation of Dhh1p, a cytoplasmic DExD/H-box protein present in large complexes. Nucleic Acids Res. 2003; 31: 4995–5002. 10.1093/nar/gkg712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jungfleisch J, Nedialkova DD, Dotu I, Sloan KE, Martinez-Bosch N, Bruning L, Raineri E, Navarro P, Bohnsack MT, Leidel SA et al. A novel translational control mechanism involving RNA structures within coding sequences. Genome Res. 2017; 27: 95–106. 10.1101/gr.209015.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen Z, Paquin N, Forget A, Chartrand P. Nuclear shuttling of She2p couples ASH1 mRNA localization to its translational repression by recruiting Loc1p and Puf6p. Mol Biol Cell. 2009; 20: 2265–2275. 10.1091/mbc.E08-11-1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cridge AG, Castelli LM, Smirnova JB, Selley JN, Rowe W, Hubbard SJ, McCarthy JE, Ashe MP, Grant CM, Pavitt GD. Identifying eIF4E-binding protein translationally-controlled transcripts reveals links to mRNAs bound by specific PUF proteins. Nucleic Acids Res. 2010; 38: 8039–8050. 10.1093/nar/gkq686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.David L, Huber W, Granovskaia M, Toedling J, Palm CJ, Bofkin L, Jones T, Davis RW, Steinmetz LM. A high-resolution map of transcription in the yeast genome. Proc Natl Acad Sci USA. 2006; 103: 5320–5325. 10.1073/pnas.0601091103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rachfall N, Schmitt K, Bandau S, Smolinski N, Ehrenreich A, Valerius O, Braus GH. RACK1/Asc1p, a ribosomal node in cellular signaling. Mol Cell Proteomics. 2013; 12:87–105. 10.1074/mcp.M112.017277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics. 2008; 7: 1389–1396. 10.1074/mcp.M700468-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Braun KA, Vaga S, Dombek KM, Fang F, Palmisano S, Aebersold R, Young ET. Phosphoproteomic analysis identifies proteins involved in transcription-coupled mRNA decay as targets of Snf1 signaling. Sci Signal. 2014; 7: ra64 10.1126/scisignal.2005000 [DOI] [PubMed] [Google Scholar]
- 31.Shively CA, Kweon HK, Norman KL, Mellacheruvu D, Xu T, Sheidy DT, Dobry CJ, Sabath I, Cosky EE, Tran EJ et al. Large-scale analysis of kinase signaling in yeast pseudohyphal development identifies regulation of ribonucleoprotein granules. PLoS Genet. 2015; 11: e1005564 10.1371/journal.pgen.1005564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutta A, Zheng S, Jain D, Cameron CE, Reese JC. Intermolecular interactions within the abundant DEAD-box protein Dhh1 regulate its activity in vivo. J Biol Chem. 2011; 286: 27454–27470. 10.1074/jbc.M111.220251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brengues M, Teixeira D, Parker R. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science. 2005; 310: 486–489. 10.1126/science.1115791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teixeira D and Parker R. Analysis of P-body assembly in Saccharomyces cerevisiae. Mol Biol Cell. 2007; 18:2274–2287. 10.1091/mbc.E07-03-0199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shepard KA, Gerber AP, Jambhekar A, Takizawa PA, Brown PO, Herschlag D, DeRisi JL, Vale RD. Widespread cytoplasmic mRNA transport in yeast: identification of 22 bud-localized transcripts using DNA microarray analysis. Proc Natl Acad Sci USA. 2003; 100: 11429–11434. 10.1073/pnas.2033246100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003; 425: 686–691. 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- 37.Quenault T, Lithgow T, Traven A. PUF proteins: repression, activation and mRNA localization. Trends Cell Biol. 2011; 21: 104–112. 10.1016/j.tcb.2010.09.013 [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Meng X, Li D, Chen S, Luo J, Zhu L, Singer RH, Gu W. Binding of DEAD-box helicase Dhh1 to the 5'-untranslated region of ASH1 mRNA represses localized translation of ASH1 in yeast cells. J Biol Chem. 2017; 292: 9787–9800. 10.1074/jbc.M117.776492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng Z, Coller J, Parker R, Song H. Crystal structure and functional analysis of DEAD-box protein Dhh1p. RNA. 2005; 11: 1258–1270. 10.1261/rna.2920905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mugler CF, Hondele M, Heinrich S, Sachdev R, Vallotton P, Koek AY, Chan LY, Weis K. ATPase activity of the DEAD-box protein Dhh1 controls processing body formation. Elife. 2016; 5: e18746 10.7554/eLife.18746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992; 110: 119–122. 10.1016/0378-1119(92)90454-w [DOI] [PubMed] [Google Scholar]
- 42.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989; 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segal SP, Dunckley T, Parker R. Sbp1p affects translational repression and decapping in Saccharomyces cerevisiae. Mol Cell Biol. 2006; 26: 5120–5130. 10.1128/MCB.01913-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.