

Abstract
The addition of highly polar and aprotic cosolvents to ionic liquids has proven to considerably decrease the viscosity of the solution and improve mass transfer in many chemical reactions. In this work, the interactions between a representative pyridinium-based ionic liquid, N-butylpyridinium dicyanamide ([Bpy][DCA]), and a cosolvent, dimethylsulfoxide (DMSO), were studied in detail by the combined use of attenuated total reflection Fourier transform infrared spectroscopy, hydrogen nuclear magnetic resonance (1H NMR), and density functional theory calculations. Several species in the [Bpy][DCA]–DMSO mixtures have been identified, that is, ion clusters can translate into ion pairs during the dilution process. DMSO formed hydrogen bonds (H bonds) simultaneously with [Bpy]+ cations and [DCA]− anions but stronger hydrogen-bonding interactions with the [Bpy]+ cations than the [DCA]− anions, and the intrinsic hydrogen-bond networks of IL were difficult to interrupt at low DMSO concentrations. Interestingly, hydrogen-bonding interactions reach the strongest when the molar fraction of DMSO is 0.4–0.5. Hydrogen-bonding interactions are prominent in the chemical shifts of hydrogen atoms in [Bpy]+ cations, and anisotropy is the main reason for the upfield shifts of DMSO in the presence of [Bpy][DCA]. The theoretical calculations offer in-depth studies of the structural evolution and NMR calculation.
1. Introduction
Ionic liquids (ILs) are low melting salts, thus forming liquids that are composed entirely of cations and anions.1−4 Because of the development of green chemistry, as well as their unique properties, and safety, environmentally benign ILs have become the new paradigm as solvents for eliminating the use of toxic and hazardous chemicals and also offering applications in catalysis,5 gas separation,6−8 and other promising fields.9−12 The properties of ILs have been investigated with particular efforts to make a better use of ILs, including their viscosity, density, solubility, and thermal and chemical properties.13−16 It has been well-established that hydrogen-bonding interaction17 between cations and anions of ILs is one of the key features, which has a significant influence on their properties.14,16,18−23 What is more, the presence of water,24−26 alcohol,27−29 dimethylsulfoxide (DMSO),30,31 or other cosolvents32,33 is believed to make structural changes in the hydrogen-bonding networks of ILs, as well as their physical and chemical properties, such as viscosity.22,32 These properties are closely related to the applications of ILs and their binary solutions.
Among these, DMSO is a widely used cosolvent in chemical engineering processes, completely miscible with water and a very wide range of organic and inorganic substances.34 For instance, adding DMSO into ILs is beneficial to the utilization of cellulose.35 Gomes et al. reported a much faster dissolution of cellulose when adding DMSO as a cosolvent in [Bmim][OAc].36 As expected, DMSO decreased the viscosity and increased the conductivity of the mixtures, thus facilitating mass transport. The one-pot conversion of cellulose into 5-hydroxymethylfural (5-HMF) achieved a high yield in DMSO–[Bmim][AlCl4],37 and the catalytic system also showed a promising potential in the large-scale production of 5-HMF. Besides, Hansmeier reported that imidazolium- and pyridinium-based ILs outperform the ILs reported so far for the removal of sulfur- and nitrogen-containing heteroaromatics from petrochemical streams.38 It is worth noticing that aromatic cations bearing a low viscosity and an effective mass transfer contribute to short extraction times.39 Zhang’s work proved that pyridinium-based ILs with [DCA]− anions have a better extractive selectivity of sulfur compounds than those with [SCN]− and [Tf2N]−.40 Also, pyridinium- or dicyanamide-based ILs are scrutinized and applied in CO2 capture,41−43 while the relatively high viscosities of task-specific ILs might limit their eventual applications in large-scale gas scrubbing.44 In this case, this problem is likely to be solved by adding a cosolvent. Because of their unique set of properties (e.g., high oxygen solubility, good conductivity, large potential windows, and so forth), ILs are also considered as electrolytes for Li–O2 batteries.45−48 Bruce et al. have shown that Li–O2 batteries based on DMSO have a high reversibility and cell capacity,49 with only a slight decrease in the cell performance observed after 100 cycles. Zhao improved the solubility of O2 and the reversibility of oxygen reduction reaction and oxygen evolution reaction cell performance using a mixed electrolyte based on DMSO/[BMP][Tf2N].30 Furthermore, many [DCA]− anion-based ILs are shown to be suitable for several different electrochemical applications.50−53
The works cited above have greatly enriched our knowledge of the superiority of IL–DMSO binary systems, and most of these research studies have focused on their practical performance. However, limited work has been devoted to microstructural properties and hydrogen-bonding interactions between ILs and DMSO. Recently, the combination use of attenuated total reflection Fourier transform infrared spectroscopy (ATR–FTIR) and density functional theory (DFT) calculations has been an effective method for shedding light on microstructural interactions. Zheng’s work demonstrated that alkyl Calkyl–H-involved H bonds were weakened, whereas the aromatic Cring–H-involved H bonds were strengthened during the dilution process54 and that the different behaviors of [Bmim][BF4]–DMSO complexes may guide us to explore their applications in cellulose dissolution and other practices.55 Wang reported the strong anion···HOH···anion H bonds accompanied by the H bonds between water and aromatic C–H of [Bpy][BF4],56 which is the first detailed spectroscopic investigation of water and a pyridinium-based IL. The aromatic C–H group was proven to be superior to the alkyl chain C–H group in forming H bonds both experimentally and theoretically. To the best of our knowledge, He et al. also did much interesting work in the field of ILs and small molecular solvents and found that H bonds are universally involved and play an important role for the miscibility of DMSO and [C4Py][SCN].57 Their work has enhanced our perspective on the interactions between ILs and cosolvents. However, few works explain why binary solutions of ILs with DMSO have superiority in application than pure ILs or pure solvents at the molecular level.
In this work, the hydrogen-bonding interaction between N-butylpyridinium dicyanamide [Bpy][DCA] and DMSO (Figure 1) was investigated. The blends of [Bpy][DCA]/DMSO across the full concentration range from pure ILs to pure DMSO were tested, and deuterated DMSO (DMSO-d6) was used to avoid the absorption overlap of C–H stretching bands of DMSO with that of [Bpy][DCA]. Furthermore, the molecular interactions in these mixtures were discussed from the results of ATR–FTIR spectroscopy, two-dimensional (2D) correlation spectra, 1H NMR, and DFT calculations.
Figure 1.

Structures of [Bpy][DCA] IL and DMSO.
2. Experimental and Computational Methods
2.1. Chemicals and Sample Preparation
The pyridinium-based IL [Bpy][DCA] was purchased from Linzhou Keneng Material Technology Co, Ltd with a purity of 99 wt %. [Bpy][DCA] is a dark yellow liquid at room temperature, and it was further dried in the vacuum oven at 348.15 K for 96 hours. Water content was measured using the Karl Fischer coulometric titration, and the final water content was below 0.1 wt %. The purity of [Bpy][DCA] was confirmed by 1H NMR using a JEOL JNMECA 600 NMR spectrometer: δH (600 MHz, DMSO, TMS/CCl4), 8.7 (2H, d, C2,6–H), 8.09 (1H, t, C4–H), 7.63 (2H, t, C3,5–H), 4.2 (2H, t, C7–H), 1.38 (2H, m, C8–H), 0.69 (2H, m, C9–H), 0.21 (3H, t, C10–H). Also, DMSO (>99%) and DMSO-d6 (D, 99.8%) were purchased from Cambridge Isotope Laboratories without further purification.
The binary mixtures of [Bpy][DCA]–DMSO and [Bpy][DCA]–DMSO-d6 were prepared according to a series of molar concentrations. The mole fractions of DMSO-d6 in the mixtures of [Bpy][DCA]–DMSO-d6 were 0.1001, 0.1960, 0.3000, 0.3982, 0.4977, 0.5979, 0.7008, 0.7988, and 0.9004. The mole fractions of DMSO in the binary solutions were 0.0979, 0.2030, 0.3036, 0.4001, 0.5009, 0.5999, 0.7001, 0.7994, and 0.8998. Also, one set of experiments included two samples of pure ILs and pure small molecular solvents (DMSO or DMSO-d6).
2.2. ATR–FTIR Spectra
To obtain the excess absorption spectra of the binary mixtures, ATR–FTIR spectra were collected within the range of 4000–650 cm–1 at 298.35 K using the Nicolet 5700 FTIR spectrometer equipped with a mercury–cadmium–telluride detector. Two horizontal ATR cells were employed in the experiments to hold the samples. A ZnSe crystal with incident angles of 45° and 12 reflections was applied to examine the absorbance of C–H stretching vibration, and a Ge crystal with an incident angle of 60° and seven reflections was applied to examine the strong absorbance of C≡N vibration. The spectra were recorded with a resolution of 2 cm–1, a zero filling factor of 2, and 32 parallel scans. For each sample, three parallel measurements were carried out. The refractive indexes of the binary solutions and the pure compound were measured with a refractometer at 298.35 K. The formula suggested by Hansen was used to do the ATR corrections.58
2.3. Excess Infrared and 2D Correlation Spectroscopy
The theory of excess infrared (IR) spectroscopy had been developed by Yu’s group,59,60 and its applications were developed to examine the microstructures of different binary systems.28,29,55,61 The definition of excess spectrum was proposed as the difference between the spectrums of the real solutions and those of the ideal solutions under identical conditions. The equation of excess molar absorbance used for the calculations was derived from the Beer–Lambert law (eq 1)
| 1 |
where A is the absorbance of the binary mixtures from ATR–FTIR, d is the light path length through the ATR crystal, x1 and x2 are the molar fractions of the two components, C1 and C2 are the molar concentrations of the two components, and ε1* and ε2 are the ideal molar absorbance coefficients of the two components at their pure states, respectively. εE is used to resemble the integral value of excess spectrum within a certain range of the absorbance band. The calculations of excess IR spectra were performed with the original data using the MATLAB R2015b software, that is, truncation, subtraction, and baseline correction.
2D correlation spectroscopy was proposed to investigate the sequence of two functional groups (groups 1 and 2) interacting with group 3.62 2D correlation spectra were performed with 2D shige.63 In the 2D correlation map, correlation peaks on the diagonal are called automatic peaks, and the peaks outside the diagonal are called cross-peaks. The red and blue colors represent the positive and negative signs of these cross-peaks, respectively [φ (v1, v2) in synchronous spectra and ψ (v1, v2) in asynchronous spectra]. If φ (v1, v2) > 0 and ψ (v1, v2) > 0, functional group 1 prefers over group 2 to interact with group 3; if φ (v1, v2) > 0 and ψ (v1, v2) < 0, group 2 prefers over group 1 to interact with group 3 and vice versa.64 The synchronous and asynchronous spectroscopies are helpful to determine the sequence of molecular interactions between the functional groups.
2.4. 1H NMR Characterizations
The 1H NMR characterizations for pure DMSO, [Bpy][DCA], and the mixtures of [Bpy][DCA]–DMSO were conducted on a JEOL JNMECA 600 NMR spectrometer (600 MHz) at 298.15 K. To avoid the influence of possible interactions between the standard chemical tetramethylsilane (TMS) and the samples, the solution of TMS in carbon tetrachloride (CCl4) was chosen as the external standard.
2.5. DFT Calculations
DFT was performed to investigate the molecular structures of [Bpy][DCA] and DMSO and the interactions between these two components using the Gaussian 09 package.65 All geometric optimizations were carried out at the B3LYP/6-31++g(d,p) level without an imaginary vibrational frequency, and IR frequencies were calculated at the B3LYP/6-311++g(d,p) level. The stable structures, interaction energies, and frequency analyses had been successfully obtained for the cation [Bpy]+, anion [DCA]−, and ion pairs [Bpy][DCA], as well as the complexes of [Bpy][DCA]–DMSO and ion clusters–DMSO. The interaction energy was calculated according to eq 2
| 2 |
3. Results and Discussion
3.1. FTIR Spectroscopy of Pure [Bpy][DCA], DMSO, and DMSO-d6
The ATR–FTIR spectra of pure [Bpy][DCA], DMSO, and DMSO-d6 are shown in Figure 2. The absorbance above 3000 cm–1 (3131 cm–1 and 3058 cm–1) is attributed to the stretching vibrations of ν(Cring–H) of the aromatic pyridinium ring; absorbances at 2962, 2936, and 2875 cm–1 are attributed to the stretching vibrations of ν(Calkyl–H) of the butyl substituent of the pyridinium ring, including (−CH2−) and (−CH3) vibrations. Apart from the main C–H vibrations of cations, the signals at 2100–2300 cm–1 show the existence of the triple bond between the terminal N atoms and the C atom of the [DCA]− anion. According to Jürgens,66 three remarkable vibration bands at 2228, 2190, and 2128 cm–1 may be attributed to the absorbance peaks of νs(C≡N), ν(C–N), and νas(C≡N), respectively. As demonstrated by the literature,21 H bonds and electrostatic interactions are prevalent in ILs. It is suggested that cations and anions in [Bpy][DCA] would have hydrogen-bonding networks appearing as Cring–H···C≡N and Calkyl–H···C≡N in pure [Bpy][DCA].
Figure 2.
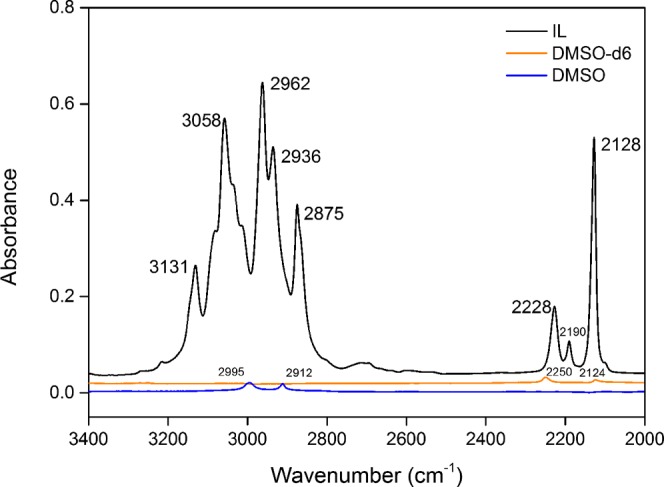
ATR–FTIR spectra of [Bpy][DCA], DMSO, and DMSO-d6 in the range of 3400–2000 cm–1.
The absorbance spectrum of pure DMSO is relatively smoother than that of [Bpy][DCA]. Two absorption peaks at 2995 and 2912 cm–1 are assigned to νas(C–H) and νs(C–H), respectively. However, the two absorption bands overlap with the ν(C–H) group of [Bpy]+ cations in the range of 3200–2800 cm–1; thus, the binary system of [Bpy][DCA]–DMSO-d6 was chosen for the analysis of ν(Calkyl–H) and ν(Cring–H). The absorbance spectrum of pure DMSO-d6 shows that two significant peaks appear at 2250 and 2124 cm–1. The two bands may overlap with ν(C≡N) of [Bpy][DCA] in the binary solutions; thus, [Bpy][DCA]–DMSO was chosen to distinguish ν(C≡N) from ν(C–D).
3.2. ATR–FTIR and Excess IR Spectra Analyses
The results of ATR–FTIR and excess IR spectra are shown in Figure 3, and the mole fractions of IL [Bpy][DCA] are marked from 1 to 0 (top to bottom) in A, B, and C. To avoid the intersection of excess peaks around 2130 cm–1, mole fractions of IL are marked reversely from 0 to 1 (top to bottom). The spectra of pure [Bpy][DCA] and DMSO/DMSO-d6 are indicated by dashed lines. The partial absorbance of ν(C–H) of the [Bpy]+ cation in the binary solutions of [Bpy][DCA]–DMSO-d6 is depicted in Figure 3A, whereas the absorbance of ν(C≡N) of the [DCA]− anion in the mixtures of [Bpy][DCA]–DMSO is shown in Figure 3C. Within the range of 3200–2800 cm–1, the absorption bands at 3131 and 3058 cm–1 may be attributed to the vibrations of pyridinium ring C–H stretches, whereas the three peaks with wavenumbers lower than 3000 cm–1 are attributed to the alkyl chain C–H stretches.56,57 Because of the coupling vibrations, the two bands (3131 and 3058 cm–1) cannot correspond to the specific C–H group of the pyridinium ring. (Notice that the intensity of the absorption bands decreases monotonically with the increase in the molar fractions of DMSO-d6 in the systems.) Meanwhile, when it comes to the pure DMSO-d6, almost no absorption peaks appear in this region. As we know, H bonds are prevalent between cations and anions in ILs,21 and ILs can easily form H bonds with other organic compounds. With increasing DMSO-d6 concentration, the interionic H bonds become successively weaker because of the eventual formation of C–H···O=S between the [Bpy]+ cation and DMSO or C–D···C≡N between DMSO-d6 and the [DCA]− anion. Consequently, the intrinsic H bonds between the cations and the anions are weakened, resulting in the reduced strength of ν(C–H). In general, the reduced strength of ν(C–H) is accompanied with an elongation of C–H bond and the vibration frequency will experience a red shift.67,68 To make a clear understanding of the wavenumber shifts of Cring–H and Calkyl–H in the presence of DMSO-d6, the results in Figure 3E show the wavenumber shifts of ν(Cring–H) and ν(Calkyl–H), respectively. Besides the absorption band at 3058 cm–1 (4.5 cm–1 red shifts), other peaks do not exhibit apparent frequency changes during the dilution process, with less than 1 cm–1 blue shifts, which is in agreement with the literature that H bonds involving Cring–H are proper red-shift H bonds, whereas those related to Calkyl–H are improper red-shift H bonds.67,68 The red shift of the band at 3058 cm–1 may be attributed to the replacement of strong interionic H bonds between [Bpy]+ and [DCA]− by weak long-range interactions between DMSO-d6 and [Bpy][DCA], whereas the weaker H bonds Cring–H···O=S appear at lower vibrational frequencies. The significant red shift implies that ν(Cring–H) 3058 cm–1 prefers to interact with DMSO-d6. It is worthy to notice that the slight blue shifts of Calkyl–H can be attributed to two aspects. One is the formation of blue-shift H bond by the Calkyl–H moiety itself. The other one is that the arrangement of DMSO-d6 molecules at the top or the bottom of the pyridinium ring causes the probable C–H···π interactions, through which the lone-pair electrons of DMSO-d6 oxygen will transfer to the pyridinium ring. The increased electron density will rearrange in the pyridinium cation, leading to charge-assisted H bonds involving Calkyl–H.69 The newly emerged H-bonding interactions between [Bpy][DCA] and DMSO-d6 can be identified by a clear deviation from the ideal state in the excess IR spectra. The excess IR spectra of all C–H groups are shown in Figure 3B (the dashed lines indicate the ideal state with εE = 0), where the most obvious feature is the positive bands from both Cring–H and Calkyl–H. It can be seen that there are seven peaks (which can also be found in ATR–FTIR, Figure 3A) at 3131, 3081, 3058, 3015, 2962, 2936, and 2875 cm–1, with 3058 cm–1 having the greatest deviation from the ideal state. These five positive peaks are attributed to the stretching vibrations of Cring–H and Calkyl–H, which indicate that the IR activities of the Cring–H and Calkyl–H vibrations are enhanced.70 It is clear to see that one positive peak at around 3010 cm–1 gradually becomes stronger with the decrease of mole fractions of IL from 1 to 0, which is attributed to the newly arisen H-bond Cring–H···O=S. The newly arisen H bond at a lower wavenumber is thought to lead to the obvious red shift of 3058 cm–1, which also proves that the peak at 3058 cm–1 plays a key role in hydrogen-bonding interactions when adding DMSO to [Bpy][DCA]. Compared with Zheng’s work,55 the positive excess peaks indicate that both pyridinium Cring–H- and Calkyl–H-involved H bonds are enhanced when interacting with DMSO-d6, whereas the excess IR spectra show both positive and negative peaks for Cring–H of imidazolium-based ILs. According to the study reported previously,21 the presence of two electron-withdrawing N atoms in the imidazolium ring makes C2–H much more acidic than C4,5–H, and C2–H will act as the key site of the H-bond donor. Although the charge difference in the pyridinium Cring–H is not as significant as that in the imidazolium ring, the acidity of all pyridinium H atoms is small and all Cring–H can act as the H-bond donors to interact with DMSO-d6. The charge difference between [Bmim]+ and [Bpy]+ performed by a natural population analysis can be found in Figure S8.71 In addition, the two alkyl chains of the imidazolium cation make DMSO-d6 spatially infeasible to approach C2–H, whereas the pyridinium cation contains only one alkyl chain, resulting in DMSO-d6 preferring to interact with the anions of imidazolium ILs but with both cations and anions in the case of pyridinium ILs.56 Therefore, adding DMSO-d6 evidently weakened interionic H bonds in imidazolium-based ILs but did not significantly affect the H bonds involving pyridinium Cring–H. Meanwhile, the pyridinium ring can form more H bonds with DMSO-d6 because of more H-bond donors. Consequently, an apparent decrease in interionic H bonds and an increase in the ion pair–DMSO interactions in imidazolium-based ILs can occur, which is related to the negative and positive bands in Zheng’s work. However, in this study, the electrons of cations distribute at a uniform density, and the addition of DMSO will lead to the enhanced IR activity of almost all C–H bonds of [Bpy][DCA], as well as increase the number of H bonds involving the pyridinium ring. The results can be shown by the observable positive excess peaks in the respective C–H vibrational modes.
Figure 3.

ATR–FTIR (A) and excess IR (B), indicating the spectra of C–H stretching vibrations in [Bpy][DCA]/DMSO-d6 mixtures from 3200 to 2800 cm–1, and ATR–FTIR (C) and excess IR (D), indicating the spectra of C≡N in [Bpy][DCA]/DMSO mixtures from 2300 to 2050 cm–1. E and F showing the wavenumber shifts of C–H and C≡N vibrations during the dilution, respectively.
In the 2300–2050 cm–1 region, DMSO does not absorb, and strong absorption bands are observed from the [DCA]− anion of [Bpy][DCA]. Three stretching vibrations at 2228, 2190, and 2128 cm–1 may be attributed to the typical signals of νas(C≡N), ν(C–N), and νs(C≡N), respectively (Figure 3C).66,72 The most obvious feature is that the absorption intensity of the three bands decreases with the addition of DMSO. Because the intensity of the positive bands near 2130 cm–1 gradually increases with decreasing mole fractions of [Bpy][DCA], sequences of excess IR spectra in Figure 3D are reversed in comparison with that in Figure 3C for avoiding the intersection of these bands. The wavenumber shifts of ν(C≡N) are shown in Figure 3F, and all of the C≡N group experiences a monotonic red shift. It has been suggested that [DCA]− can form H bonds with the C–H groups of DMSO that are weaker than those of the intrinsic H-bond networks of IL;57,73,74 the positive and negative peaks in excess IR Figure 3D at the fixed positions (nearly fixed zero point) are attributed to Cring/Calkyl–H···C≡N and CDMSO–H···C≡N. The positive peak near 2225 cm–1 indicating CDMSO–H···C≡N appears at a lower wavenumber than the negative peaks indicating H bonds between the cations and the anions. These results also suggest that the formation of CDMSO–H···C≡N gives rise to the red shift of C≡N vibrations (shown in Figure 3F). The addition of DMSO would have a positive effect on the formation of H bonds, especially when the mole fraction of DMSO is around 0.4–0.5. With the help of excess IR spectra, the newly emerged H-bond species can be identified, which strongly support the wavenumber shifts in the ATR–IR spectra.
3.3. Analysis of 2D Correlation Spectra
The sequential order of the interactions of Cring–H and Calkyl–H with DMSO can be evaluated by the 2D correlation spectra. The presence of DMSO-d6 results in a different chemical environment for the C–H groups in the [Bpy][DCA]–DMSO-d6 system. The synchronous and asynchronous spectra are shown in Figure 4. There are more correlation bands in the asynchronous spectrum (Figure 4B) than in the synchronous spectrum (Figure 4A) because the asynchronous cross-peaks result from the relative dissimilarity of the intensity variation behavior. Specifically, two main cross-correlation peaks are observed in Figure 4B, located at (3058 cm–1 and 2945 cm–1) and (3031 cm–1 and 2950 cm–1). The two central peaks are taken as the representative absorptions for Cring–H and Calkyl–H groups. Positive cross-peaks at (3058 cm–1 and 2945 cm–1) and (3031 cm–1 and 2950 cm–1) are seen in the synchronous spectrum because of the same changing direction of the absorption coefficients of Cring–H and Calkyl–H. In the asynchronous spectrum, the cross-peaks at (3058 cm–1 and 2945 cm–1) and (3031 cm–1 and 2950 cm–1) are also positive. Thus, the signs of both synchronous and asynchronous cross-peaks are the same. According to Noda’s rule,64,75 DMSO prefers over the alkyl chain for interacting with the pyridinium ring. The results are consistent with the wavenumber shifts, and Cring–Hs are more susceptible to the dilution process.
Figure 4.

2D synchronous (A, −0.02–3.92) and asynchronous (B, −0.20–0.20) correlation spectra contour maps of [Bpy][DCA]/DMSO-d6 mixtures in the 3100–2850 cm–1 region. The red and blue regions represent the positive and negative response signals, respectively.
3.4. 1H NMR Analysis
Assignment of the 1H NMR signals to the hydrogen atoms of pure [Bpy][DCA] is displayed in Figure 5A (black line), and the chemical shift changes of individual hydrogen atoms during the dilution process are evaluated in Figure 5B (Cring–H), 5C (Calkyl–H), and 5D (DMSO). The chemical shift of all hydrogen atoms in [Bpy]+ cations gradually moves downfield with increasing mole concentrations of DMSO (from top to bottom), whereas the hydrogen atoms in DMSO move upfield with increasing mole concentrations of IL [Bpy][DCA] (from bottom to top) in Figure 5A. The chemical shift variation (Δδ) is expressed by the differences between the chemical shifts of the mixtures and those of the pure chemicals. The chemical shift variations are C2,6–H (0.28 ppm), C4–H (0.34 ppm), C3,5–H (0.35 ppm), C7–H (0.27 ppm), CDMSO–H (−0.31 ppm), C8–H (0.35 ppm), C9–H (0.42 ppm), and C10–H (0.49 ppm). As can be seen, both Cring–H and Calkyl–H have positive Δδ values of hydrogen atoms in Figure 5B, C, in contrast to the negative Δδ values of hydrogen atoms on the methyl groups of DMSO in Figure 5D. This proves that hydrogen atoms of the [Bpy]+ cation show downfield shifts with the addition of DMSO to [Bpy][DCA], whereas the hydrogen atoms of DMSO experience upfield shifts with increasing mole concentrations of [Bpy][DCA]. It is well-known that the hydrogen atoms show positive chemical shifts and move downfield when they are attracted by electron-withdrawing groups to form H bonds;76−78 thus, the 1H NMR data undoubtedly demonstrate that the addition of DMSO significantly influences hydrogen-bonding interactions between the cations and the anions of [Bpy][DCA], and both Calkyl–H and Cring–H can form H bonds with DMSO. With the changes in proportions of [Bpy][DCA]/DMSO, the electrostatic environment around the ionic cations and anions is changed. The interactions between the cations and DMSO, anions and DMSO, and cations and anions slightly change the environment around the C–H groups, resulting in the changes in the chemical shifts of hydrogen atoms in 1H NMR. This observation supports our previous determination that the excess IR of H bonds involving the hydrogen atoms of [Bpy]+ is strengthened with the newly emerged Cring–H···O=S and Calkyl–H···O=S H bonds, and the presence of ILs weakens the self-associated H-bond networks of DMSO. However, hydrogen-bonding interactions are not the main reason for the upfield shift of hydrogen atoms of DMSO, which can be explained by the shielding effects when the methyl group of DMSO is located above the aromatic pyridinium ring.55
Figure 5.

1H NMR assignment of the pure [Bpy][DCA] (A, black line), chemical shift changes of the Cring–H of the [Bpy]+ cation (B), Calkyl–H of the [Bpy]+ cation (C), and H atoms of the methyl group of DMSO (D) in [Bpy][DCA]–DMSO binary systems.
3.5. DFT Calculations of Interactions between [Bpy][DCA] and DMSO
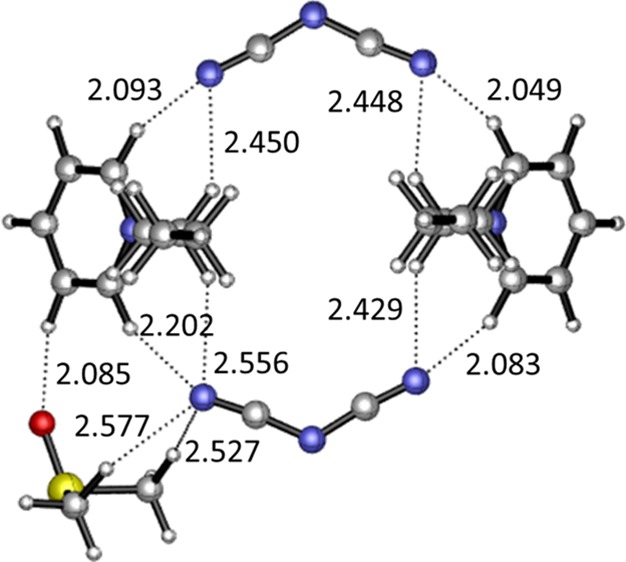
All of the structures were optimized using the DFT theory at the B3LYP/6-31++g(d,p) level. The angles of C–H···N and C–H···O are all between 90° and 180°, and the sums of the van der Waals radii of H and N atoms(2.75 Å) and H and O atoms(2.72 Å)79 are applied as criteria for the formation of H bonds between the donors and the acceptors.
The electrostatic potential surfaces of the individual cation, anion, and DMSO are shown in Figure 6, where the blue-colored surfaces represent the positive electrostatic potential and the red-colored surfaces represent the negative electrostatic potential. As can be seen in this figure, all regions around the [Bpy]+ cation are positive electrostatic potential, and because of its darker blue-colored surface, the positive charges are more concentrated surrounding the pyridinium ring than the alkyl chain. Obviously, negative charges are more uniformly dispersed around the [DCA]− anion. In the structure of DMSO, the negative electrostatic potential is distributed mainly around the oxygen atoms, with a less positive charge around the two methyl groups. The positive and negative electrostatic potential plays an important role in the formation of H bonds.
Figure 6.

Electrostatic potential surfaces of (A) [Bpy]+, (B) [DCA]−, and (C) DMSO (Isov = 0.0004).
A series of possible geometrics of [Bpy][DCA] ion pairs are optimized using the DFT theory (Figure S1A–C). The anions formed H bonds with the hydrogen atoms of the pyridinium ring and the butyl chain simultaneously, and the [DCA]− anion would appear directly above the pyridinium ring (Figure S1C). In addition, the hydrogen atoms of the pyridinium ring have stronger hydrogen-bonding abilities than Calkyl–H because of their larger interaction energy (317 kJ/mol > 305.89 kJ/mol). In the complexes of the [Bpy]+ cation and DMSO (Figure S1D–F), it is found that the interaction energies of structures D (74.25 kJ/mol) and E (73.20 kJ/mol) are larger in absolute values than the interaction in structure F (59.13 kJ/mol), indicating that complexes D and E are more stable than F. The difference between the interaction energy is explained by the electron-withdrawing effect of the adjacent nitrogen atom. The results of the interaction between the [DCA]− anion and DMSO (Figure S1G, H) are distinct from the cation–DMSO, in which the methyl group of DMSO is most favorable for forming H bonds with the [DCA]− anion because of the electron deficit in the methyl group. Comparing the interaction energies of [Bpy]+–DMSO and [DCA]−–DMSO, all of the [Bpy]+–DMSO complexes have larger absolute values of interaction energies than those of [DCA]−–DMSO, indicating that the interaction between DMSO and the [Bpy]+ cation is stronger than that between the [DCA]− anion and DMSO. What is more, owing to the strong electrostatic attraction, the interaction energy of [Bpy][DCA] is much larger than those of [Bpy]+–DMSO and [DCA]−–DMSO complexes.
The complexes of [Bpy][DCA]–DMSO are shown in Figure S2. It has been found that the electrostatic interaction of the [Bpy]+ cation and the [DCA]− anion is always dominant in the presence of a low mole fraction of DMSO, and the ion pair structures are not interrupted by the DMSO molecules. Both the S=O and methyl groups participate in the hydrogen-bonding interaction, wherein S=O prefers to interact with the H atoms of the [Bpy]+ cation, and the methyl group preferentially forms H bonds with the N atom of the [DCA]− anion. It is obvious that the H bonds between Cring–H and DMSO lead to a larger interaction energy (371.96 kJ/mol in Figure S2C) than that between alkyl C–H and DMSO (348.05 kJ/mol in Figure S2A), which in turn implies that the favorable interaction sites for [Bpy][DCA]–DMSO complexes are the Cring–H and [DCA]− anions. The interaction energies in Figure S2B and D are relatively high mainly because of the anion−π interaction between [Bpy]+ and [DCA]−.
To validate the dilution process, the larger complex consisting of two [Bpy]+ cations and two [DCA]− anions was selected as the ion cluster structure in high IL concentrations.78,80−82 The geometrics of the ion cluster are shown in Figure S3. The cations and anions are uniformly cross-arranged, and both terminal nitrogen atoms of the [DCA]− anions form double H bonds with the adjacent [Bpy]+ cations. The interaction between the ion cluster and DMSO is considered in Figure S3B, where DMSO can form H bonds with [Bpy][DCA]. However, a small amount of DMSO only attacks the structure on the side of the cluster and is not strong enough to deconstruct the strong interaction between the cations and the anions, which makes the cluster complex stable and a stronger hydrogen-bonding interaction at xDMSO = 0.4–0.5. The topology analysis was employed for the two clusters in Figure S3, and the results are shown in Table S1 and Figure S4. The topology structures show the existence of bond critical points between the two atoms. To some extent, the lengths of the H bonds (C5–H11···N57, C12–H14···N57, C30–H37···N58, C41–H42···N58, C34–H40···N28, C41–H43···N28, C1–H8···N29, and C12–H13···N29) in the cluster 2[Bpy]+–2[DCA]− were elongated after adding DMSO. Meanwhile, the electron density (ρBCP) at these H-bond critical points decreased, and ρBCP increased at C60–H63···N58, C64–H66···N58, and C31–H38···O68, which resulted from the interaction between DMSO and the IL cluster. The higher ρBCP (0.281 au) and negative ∇2ρBCP (−1.017 au) also indicate the covalent characteristic of the H-bond C64–H66···N58. With the increased mole fractions of DMSO in the [Bpy][DCA]–DMSO binary system, more DMSO is involved in the formation of H bonds with ILs, and the big clusters are dissociated to ion pairs. The completed destruction of ion pairs occurs at very high concentrations of DMSO.
Apart from the structure analysis, a comparison between the experimental IR spectrum and the vibration modes of the two clusters is shown in Figure 7. To represent the binary solutions, [Bpy][DCA]/DMSO was selected with the mole fraction of xIL = 0.9, denoted by the solid line. The calculated clusters [Bpy]+–[DCA]−–DMSO (Figure S2B) and 2[Bpy]+–2[DCA]−–DMSO (Figure S3B) may become microscopic models to describe the structures in their real solutions, denoted by the dashed lines. It should be noted that the frequencies were scaled by dividing the maximum value of each set of data to make the intensity comparable.17,83 It is clearly seen that the main features of the measured spectra of [Bpy][DCA]/DMSO are reproduced by the DFT-calculated vibrational frequencies. Because of some uncertainties of theoretical methods, the peaks of the main C–H vibrational bands (3131, 3081, 2962, 2936, and 2875 cm–1) and C≡N vibrational bands (2228 cm–1 and 2128 cm–1) are sharper than the experimental results,84 especially for DMSO interacting at a higher IL concentration (2[Bpy]+–2[DCA]−–DMSO). The strong agreement with the major bands provided by DFT calculations and experimental measurements can also be assigned rationally by the structures of these theoretical clusters.17
Figure 7.
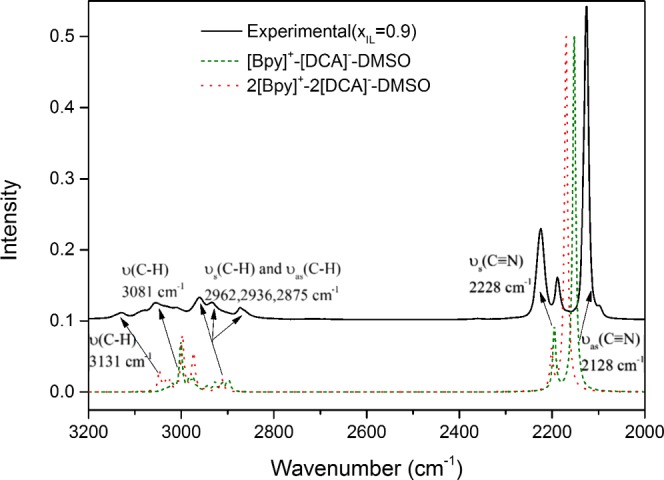
Measured ATR–FTIR spectra of [Bpy][DCA]/DMSO (xIL = 0.9) at 298.35 K compared to the vibrational frequencies of the corresponding clusters [Bpy]+–[DCA]−–DMSO and 2[Bpy]+–2[DCA]−–DMSO calculated at the B3LYP/6-311++g(d,p) level of the theory (all of the calculated bands were corrected by the factor 0.96385).
The chemical shift of the hydrogen atoms in the binary system was also investigated using DFT calculations. Because the structure in Figure S2B was optimized from Figure S1A, the two complexes were selected to perform the NMR calculation, with the results of the chemical shifts in the experimental and theoretical calculations compared in Table 1. As summarized in this table, all of the hydrogen atoms of the [Bpy]+ cation exhibit a downfield shift in the presence of DMSO, and the hydrogen atoms of DMSO exhibit an upfield shift in the presence of [Bpy][DCA]. The theoretical data are in good agreement with the experimental results, indicating that the newly emerged hydrogen-bonding interactions between [Bpy][DCA] and DMSO are the foremost reason for the positive chemical shift variations of the cationic hydrogen atoms and that the anisotropy effect of the pyridinium ring serves prominently to explain the negative chemical shift of DMSO. Another thing worth mentioning is that the calculated chemical shifts have the same trend with the experimental data but differ in specific values, which may be explained by the difference between the bulk property and the local quality at the molecular level.
Table 1. Comparison of Chemical Shifts of 1H NMR Data in Experiments and Theoretical Calculation.
| experimental data (xDMSO = 0.4) |
theoretical
data |
|||||
|---|---|---|---|---|---|---|
| [Bpy][DCA] | [Bpy][DCA]–DMSO | DMSO | [Bpy][DCA] | [Bpy][DCA]–DMSO | DMSO | |
| C2,6–H | 8.60 | 8.69 | 8.22 | 9.05 | ||
| C4–H | 8.09 | 8.17 | 6.57 | 7.27 | ||
| C3,5–H | 7.62 | 7.71 | 8.08 | 5.92 | ||
| C7–H | 4.17 | 4.24 | 2.60 | 2.76 | ||
| C8–H | 1.43 | 1.51 | 1.53 | 1.89 | ||
| C9–H | 0.74 | 0.85 | 0.74 | 0.83 | ||
| C10–H | 0.28 | 0.40 | 0.43 | 0.46 | ||
| DMSO | 2.18 | 2.51 | 0.82 | 1.8 | ||
4. Conclusions
In this work, the hydrogen-bonding interactions between the components in the binary solutions consisting of [Bpy][DCA] and DMSO were studied using ATR–FTIR spectroscopy, 1H NMR, and DFT calculation. It may be concluded that the [Bpy]+ cation-involved hydrogen-bonding interactions are enhanced during the dilution process. With the addition of DMSO, the number of C–H···N≡C H bonds between the cations and the anions is reduced rather than the newly formed H bonds C–H···O=S between the cations and DMSO and C–H···C≡N between both the methyl group of DMSO and anions. DMSO molecules preferentially have a stronger interaction with the [Bpy]+ cations than the [DCA]− anions, and the ring C–Hs are stronger hydrogen donors than the alkyl C–Hs. Consequently, quantum chemical calculations suggest that intrinsic hydrogen-bond networks are difficult to interrupt by a low concentration of DMSO but can be dissociated from big ion clusters to small ion pairs and even to separated ions at extremely high concentrations of DMSO. In addition, the theoretical data of 1H NMR are in agreement with the experimental results, showing that hydrogen-bonding interactions are prominent in determining the downfield shift of hydrogen atoms of the [Bpy]+ cation and that the location of DMSO in the shielding cone of the pyridinium ring is responsible for the upfield shift of the methyl group in DMSO. The in-depth study of the hydrogen-bonding interaction between [Bpy][DCA] and DMSO deepens our understanding of the properties of binary systems, which may further inspire chemists to explore the applications of ILs.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21406230 and 91434203), the Innovation International Team CAS (20140491518), the Youth Innovation Promotion Association CAS (2017066), the Beijing Natural Science Foundation (2182068), and the Key Research Program of Frontier Sciences, CAS (QYZDB-SSW-SLH022). We are grateful to Dr. Jing Xu and Prof. Zhiwu Yu, Tsinghua University for their help in ATR–FTIR and NMR analyses and helpful discussions. We are also grateful to Prof. Jun Yang, the Institute of Process Engineering, Chinese Academy of Sciences, for his great help in the whole process.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01805.
Structures of ion pairs, cation–DMSO, and anion–DMSO ; possible geometrics and interaction energies of [Bpy][DCA]–DMSO; optimized geometrics for the ion cluster (2[Bpy]+-2[DCA]–) and one DMSO molecule interacting with the ion cluster (2[Bpy]+-2[DCA]–-DMSO); critical points for ion clusters; electron density difference around the H-bond C31-H38···O68, H-bond C64-H66···N58, and H-bond C60-H63···N58; natural population analysis images; electron density at bond critical point; chemical shifts of hydrogen atoms in the binary system when x(DMSO) = 0.4; and chemical shift changes of hydrogen atoms (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- He H.; Zhang S.; Liu X.; Wang J.; Yao X.; Zhang X. Structures and hydrogen bonds of biodegradable naphthenate ionic liquids. Fluid Phase Equilib. 2013, 360, 169–179. 10.1016/j.fluid.2013.09.007. [DOI] [Google Scholar]
- Zhang S.; Sun N.; He X.; Lu X.; Zhang X. Physical properties of ionic liquids: Database and evaluation. J. Phys. Chem. Ref. Data 2006, 35, 1475–1517. 10.1063/1.2204959. [DOI] [Google Scholar]
- Welton T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. 10.1021/cr980032t. [DOI] [PubMed] [Google Scholar]
- Dong K.; Zhang S.; Wang Q. A new class of ion-ion interaction: Z-bond. Sci. China: Chem. 2015, 58, 495–500. 10.1007/s11426-014-5147-2. [DOI] [Google Scholar]
- Boroujeni K. P.; Shirazi E. R.; Doroodmand M. M. Synthesis of α-aminophosphonates using carbon nanotube supported imidazolium salt-based ionic liquid as a novel and environmentally benign catalyst. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 191, 683–688. 10.1080/10426507.2015.1072182. [DOI] [Google Scholar]
- Blanchard L. A.; Gu Z.; Brennecke J. F. High-pressure phase behavior of ionic liquid/CO2 systems. J. Phys. Chem. B 2001, 105, 2437–2444. 10.1021/jp003309d. [DOI] [Google Scholar]
- Cadena C.; Anthony J. L.; Shah J. K.; Morrow T. I.; Brennecke J. F.; Maginn E. J. Why is CO2 so soluble in imidazolium-based ionic liquids?. J. Am. Chem. Soc. 2004, 126, 5300–5308. 10.1021/ja039615x. [DOI] [PubMed] [Google Scholar]
- Bates E. D.; Mayton R. D.; Ntai I.; Davis J. H. CO2 capture by a task-specific ionic liquid. J. Am. Chem. Soc. 2002, 124, 926–927. 10.1021/ja017593d. [DOI] [PubMed] [Google Scholar]
- Wilkes J. S.; Levisky J. A.; Wilson R. A.; Hussey C. L. Dialkylimidazolium chloroaluminate melts: A new class of room-temperature ionic liquids for electrochemistry, spectroscopy and synthesis. Inorg. Chem. 1982, 21, 1263–1264. 10.1021/ic00133a078. [DOI] [Google Scholar]
- Mahajan S.; Sharma R.; Mahajan R. K. An investigation of drug binding ability of a surface active ionic liquid: Micellization, electrochemical, and spectroscopic studies. Langmuir 2012, 28, 17238–17246. 10.1021/la303193n. [DOI] [PubMed] [Google Scholar]
- Smiglak M.; Pringle J. M.; Lu X.; Han L.; Zhang S.; Gao H.; MacFarlane D. R.; Rogers R. D. Ionic liquids for energy, materials, and medicine. Chem. Commun. 2014, 50, 9228–9250. 10.1039/c4cc02021a. [DOI] [PubMed] [Google Scholar]
- Swatloski R. P.; Spear S. K.; Holbrey J. D.; Rogers R. D. Dissolution of cellose with ionic liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. 10.1021/ja025790m. [DOI] [PubMed] [Google Scholar]
- Fannin A. A. Jr.; Floreani D. A.; King L. A.; Landers J. S.; Piersma B. J.; Stech D. J.; Vaughn R. L.; Wilkes J. S.; Williams J. L. Properties of 1, 3-dialkylimidazolium chloride-aluminum chloride ionic liquids. 2. Phase transitions, densities, electrical conductivities, and viscosities. J. Phys. Chem. 1984, 88, 2614–2621. 10.1021/j150656a038. [DOI] [Google Scholar]
- Ghani N. A.; Sairi N. A.; Aroua M. K.; Alias Y.; Yusoff R. Density, surface tension, and viscosity of ionic liquids (1-ethyl-3-methylimidazolium diethylphosphate and 1,3-dimethylimidazolium dimethylphosphate) aqueous ternary mixtures with MDEA. J. Chem. Eng. Data 2014, 59, 1737–1746. 10.1021/je400562z. [DOI] [Google Scholar]
- Mokhtarani B.; Sharifi A.; Mortaheb H. R.; Mirzaei M.; Mafi M.; Sadeghian F. Density and viscosity of pyridinium-based ionic liquids and their binary mixtures with water at several temperatures. J. Chem. Thermodyn. 2009, 41, 323–329. 10.1016/j.jct.2008.09.001. [DOI] [Google Scholar]
- Singh G.; Kumar A. Ionic liquids: Physico-chemical, solvent properties and their applications in chemical processes. Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem. 2008, 47, 495–503. [Google Scholar]
- Dong K.; Song Y.; Liu X.; Cheng W.; Yao X.; Zhang S. Understanding structures and hydrogen bonds of ionic liquids at the electronic level. J. Phys. Chem. B 2012, 116, 1007–1017. 10.1021/jp205435u. [DOI] [PubMed] [Google Scholar]
- Marciniak A. The solubility parameters of ionic liquids. Int. J. Mol. Sci. 2010, 11, 1973–1990. 10.3390/ijms11051973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornellas A.; Perez L.; Comelles F.; Ribosa I.; Manresa A.; Garcia M. T. Self-aggregation and antimicrobial activity of imidazolium and pyridinium based ionic liquids in aqueous solution. J. Colloid Interface Sci. 2011, 355, 164–171. 10.1016/j.jcis.2010.11.063. [DOI] [PubMed] [Google Scholar]
- De la Parra C. J.; Zambrano J. R.; Bermejo M. D.; Martín Á.; Segovia J. J.; Cocero M. J. Influence of water concentration in the viscosities and densities of cellulose dissolving ionic liquids. Correlation of viscosity data. J. Chem. Thermodyn. 2015, 91, 8–16. 10.1016/j.jct.2015.07.015. [DOI] [Google Scholar]
- Hunt P. A.; Ashworth C. R.; Matthews R. P. Hydrogen bonding in ionic liquids. Chem. Soc. Rev. 2015, 44, 1257–1288. 10.1039/c4cs00278d. [DOI] [PubMed] [Google Scholar]
- Ciocirlan O.; Croitoru O.; Iulian O. Viscosity of binary mixtures of 1-ethyl-3-methylimidazolium tetrafluoroborate ionic liquid with four organic solvents. J. Chem. Thermodyn. 2016, 101, 285–292. 10.1016/j.jct.2016.06.015. [DOI] [Google Scholar]
- Zhang S.; Huo F. Angstrom science: Exploring aggregates from a new viewpoint. Green Energy Environ. 2016, 1, 75–78. 10.1016/j.gee.2016.04.008. [DOI] [Google Scholar]
- Méndez-Morales T.; Carrete J.; Cabeza Ó.; Gallego L. J.; Varela L. M. Molecular dynamics simulation of the structure and dynamics of water-1-alkyl-3-methylimidazolium ionic liquid mixtures. J. Phys. Chem. B 2011, 115, 6995–7008. 10.1021/jp202692g. [DOI] [PubMed] [Google Scholar]
- Porter A. R.; Liem S. Y.; Popelier P. L. A. Room temperature ionic liquids containing low water concentrations—A molecular dynamics study. Phys. Chem. Chem. Phys. 2008, 10, 4240–4248. 10.1039/b718011j. [DOI] [PubMed] [Google Scholar]
- Anthony J. L.; Maginn E. J.; Brennecke J. F. Solution thermodynamics of imidazolium-based ionic liquids and water. J. Phys. Chem. B 2001, 105, 10942–10949. 10.1021/jp0112368. [DOI] [Google Scholar]
- He H.; Chen H.; Zheng Y.; Zhang X.; Yao X.; Yu Z.; Zhang S. The hydrogen-bonding interactions between 1-ethyl-3-methylimidazolium lactate ionic liquid and methanol. Aust. J. Chem. 2013, 66, 50–59. 10.1071/ch12308. [DOI] [Google Scholar]
- Zhang Q.-G.; Wang N.-N.; Wang S.-L.; Yu Z.-W. Hydrogen bonding behaviors of binary systems containing the ionic liquid 1-butyl-3-methylimidazolium trifluoroacetate and water/methanol. J. Phys. Chem. B 2011, 115, 11127–11136. 10.1021/jp204305g. [DOI] [PubMed] [Google Scholar]
- Li Q.; Wang N.; Zhou Q.; Sun S.; Yu Z. Excess infrared absorption spectroscopy and its applications in the studies of hydrogen bonds in alcohol-containing binary mixtures. Appl. Spectrosc. 2008, 62, 166–170. 10.1366/000370208783575663. [DOI] [PubMed] [Google Scholar]
- Khan A.; Zhao C. Enhanced performance in mixture DMSO/ionic liquid electrolytes: Toward rechargeable Li-O2 batteries. Electrochem. Commun. 2014, 49, 1–4. 10.1016/j.elecom.2014.09.014. [DOI] [Google Scholar]
- Huo F.; Liu Z.; Wang W. Cosolvent or antisolvent? A molecular view of the interface between ionic liquids and cellulose upon addition of another molecular solvent. J. Phys. Chem. B 2013, 117, 11780–11792. 10.1021/jp407480b. [DOI] [PubMed] [Google Scholar]
- Marsh K. N.; Boxall J. A.; Lichtenthaler R. Room temperature ionic liquids and their mixtures—A review. Fluid Phase Equilib. 2004, 219, 93–98. 10.1016/j.fluid.2004.02.003. [DOI] [Google Scholar]
- Sarkar S.; Mandal S.; Ghatak C.; Rao V. G.; Ghosh S.; Sarkar N. Photoinduced electron transfer in an imidazolium ionic liquid and in its binary mixtures with water, methanol, and 2-propanol: Appearance of marcus-type of inversion. J. Phys. Chem. B 2012, 116, 1335–1344. 10.1021/jp2075995. [DOI] [PubMed] [Google Scholar]
- Jacob S. W.; Jack C.. Dimethyl sulfoxide (DMSO) in trauma and disease; CRC Press, 2015. [Google Scholar]
- Zhao Y.; Liu X.; Wang J.; Zhang S. Insight into the cosolvent effect of cellulose dissolution in imidazolium-based ionic liquid systems. J. Phys. Chem. B 2013, 117, 9042–9049. 10.1021/jp4038039. [DOI] [PubMed] [Google Scholar]
- Andanson J.-M.; Bordes E.; Devémy J.; Leroux F.; Pádua A. A. H.; Gomes M. F. C. Understanding the role of co-solvents in the dissolution of cellulose in ionic liquids. Green Chem. 2014, 16, 2528–2538. 10.1039/c3gc42244e. [DOI] [Google Scholar]
- Xiao S.; Liu B.; Wang Y.; Fang Z.; Zhang Z. Efficient conversion of cellulose into biofuel precursor 5-hydroxymethylfurfural in dimethyl sulfoxide-ionic liquid mixtures. Bioresour. Technol. 2014, 151, 361–366. 10.1016/j.biortech.2013.10.095. [DOI] [PubMed] [Google Scholar]
- Hansmeier A. R.; Meindersma G. W.; de Haan A. B. Desulfurization and denitrogenation of gasoline and diesel fuels by means of ionic liquids. Green Chem. 2011, 13, 1907–1913. 10.1039/c1gc15196g. [DOI] [Google Scholar]
- Asumana C.; Yu G.; Li X.; Zhao J.; Liu G.; Chen X. Extractive desulfurization of fuel oils with low-viscosity dicyanamide-based ionic liquids. Green Chem. 2010, 12, 2030–2037. 10.1039/c0gc00118j. [DOI] [Google Scholar]
- Gao H.; Zeng S.; Liu X.; Nie Y.; Zhang X.; Zhang S. Extractive desulfurization of fuel using N-butylpyridinium-based ionic liquids. RSC Adv. 2015, 5, 30234–30238. 10.1039/c5ra03762j. [DOI] [Google Scholar]
- Hou Y.; Baltus R. E. Experimental measurement of the solubility and diffusivity of CO2 in room-temperature ionic liquids using a transient thin-liquid-film method. Ind. Eng. Chem. Res. 2007, 46, 8166–8175. 10.1021/ie070501u. [DOI] [Google Scholar]
- Muldoon M. J.; Aki S. N. V. K.; Anderson J. L.; Dixon J. K.; Brennecke J. F. Improving carbon dioxide solubility in ionic liquids. J. Phys. Chem. B 2007, 111, 9001–9009. 10.1021/jp071897q. [DOI] [PubMed] [Google Scholar]
- Yunus N. M.; Mutalib M. I. A.; Man Z.; Bustam M. A.; Murugesan T. Solubility of CO2 in pyridinium based ionic liquids. Chem. Eng. J. 2012, 189, 94–100. 10.1016/j.cej.2012.02.033. [DOI] [Google Scholar]
- Gutowski K. E.; Maginn E. J. Amine-functionalized task-specific ionic liquids: A mechanistic explanation for the dramatic increase in viscosity upon complexation with CO2 from molecular simulation. J. Am. Chem. Soc. 2008, 130, 14690–14704. 10.1021/ja804654b. [DOI] [PubMed] [Google Scholar]
- Cecchetto L.; Salomon M.; Scrosati B.; Croce F. Study of a Li-air battery having an electrolyte solution formed by a mixture of an ether-based aprotic solvent and an ionic liquid. J. Power Sources 2012, 213, 233–238. 10.1016/j.jpowsour.2012.04.038. [DOI] [Google Scholar]
- De Giorgio F.; Soavi F.; Mastragostino M. Effect of lithium ions on oxygen reduction in ionic liquid-based electrolytes. Electrochem. Commun. 2011, 13, 1090–1093. 10.1016/j.elecom.2011.07.004. [DOI] [Google Scholar]
- Monaco S.; Soavi F.; Mastragostino M. Role of oxygen mass transport in rechargeable Li/O2 batteries operating with ionic liquids. J. Phys. Chem. Lett. 2013, 4, 1379–1382. 10.1021/jz4006256. [DOI] [PubMed] [Google Scholar]
- Li Q.; Chen J.; Fan L.; Kong X.; Lu Y. Progress in electrolytes for rechargeable Li-based batteries and beyond. Green Energy Environ. 2016, 1, 18–42. 10.1016/j.gee.2016.04.006. [DOI] [Google Scholar]
- Peng Z.; Freunberger S. A.; Chen Y.; Bruce P. G. A reversible and higher-rate Li-O2 battery. Science 2012, 337, 563–566. 10.1126/science.1223985. [DOI] [PubMed] [Google Scholar]
- Deng M.-J.; Chen P.-Y.; Leong T.-I.; Sun I.-W.; Chang J.-K.; Tsai W.-T. Dicyanamide anion based ionic liquids for electrodeposition of metals. Electrochem. Commun. 2008, 10, 213–216. 10.1016/j.elecom.2007.11.026. [DOI] [Google Scholar]
- MacFarlane D. R.; Golding J.; Forsyth S.; Forsyth M.; Deacon G. B. Low viscosity ionic liquids based on organic salts of the dicyanamide anion. Chem. Commun. 2001, 1430–1431. 10.1039/b103064g. [DOI] [Google Scholar]
- Yoon H.; Lane G. H.; Shekibi Y.; Howlett P. C.; Forsyth M.; Best A. S.; MacFarlane D. R. Lithium electrochemistry and cycling behaviour of ionic liquids using cyano based anions. Energy Environ. Sci. 2013, 6, 979–986. 10.1039/c3ee23753b. [DOI] [Google Scholar]
- Blahut A.; Dohnal V. Interactions of volatile organic compounds with the ionic liquid 1-butyl-1-methylpyrrolidinium dicyanamide. J. Chem. Eng. Data 2011, 56, 4909–4918. 10.1021/je200822w. [DOI] [Google Scholar]
- Zheng Y.-Z.; He H.-Y.; Zhou Y.; Yu Z.-W. Hydrogen-bonding interactions between [BMIM][BF4] and dimethyl sulfoxide. J. Mol. Struct. 2014, 1069, 140–146. 10.1016/j.molstruc.2014.01.013. [DOI] [Google Scholar]
- Zheng Y.-Z.; Zhou Y.; Deng G.; Yu Z.-W. Hydrogen-bonding interactions between a nitrile-based functional ionic liquid and DMSO. J. Mol. Struct. 2016, 1124, 207–215. 10.1016/j.molstruc.2016.01.071. [DOI] [Google Scholar]
- Wang N.-N.; Zhang Q.-G.; Wu F.-G.; Li Q.-Z.; Yu Z.-W. Hydrogen bonding interactions between a representative pyridinium-based ionic liquid [BuPy][BF4] and water/dimethyl sulfoxide. J. Phys. Chem. B 2010, 114, 8689–8700. 10.1021/jp103438q. [DOI] [PubMed] [Google Scholar]
- He H.; Chen H.; Zheng Y.; Zhang S.; Yu Z. Hydrogen-bonding interactions between a pyridinium-based ionic liquid [C4Py][SCN] and dimethyl sulfoxide. Chem. Eng. Sci. 2015, 121, 169–179. 10.1016/j.ces.2014.07.024. [DOI] [Google Scholar]
- Hansen W. H. Expanded formulas for attenuated total reflection and the derivation of absorption rules for single and multiple ATR spectrometer cells. Spectrochim. Acta 1965, 21, 815–833. 10.1016/0371-1951(65)80039-x. [DOI] [Google Scholar]
- Zhou Y.; Xu J.; Wang N.-N.; Yu Z.-W. Excess spectroscopy: Concept and applications. Acta Phys.-Chim. Sin. 2016, 32, 239–248. 10.3866/PKU.WHXB201511241. [DOI] [Google Scholar]
- Li Q.; Wu G.; Yu Z. The role of methyl groups in the formation of hydrogen bond in DMSO-methanol mixtures. J. Am. Chem. Soc. 2006, 128, 1438–1439. 10.1021/ja0569149. [DOI] [PubMed] [Google Scholar]
- Zhang Q.-G.; Wang N.-N.; Yu Z.-W. The hydrogen bonding interactions between the ionic liquid 1-ethyl-3-methylimidazolium ethyl sulfate and water. J. Phys. Chem. B 2010, 114, 4747–4754. 10.1021/jp1009498. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Wu P. Investigation of the hydrogen-bond structure of cellulose diacetate by two-dimensional infrared correlation spectroscopy. Carbohydr. Polym. 2008, 74, 509–513. 10.1016/j.carbpol.2008.04.005. [DOI] [Google Scholar]
- Morita S.; Shinzawa H.; Noda I.; Ozaki Y. Perturbation-correlation moving-window two-dimensional correlation spectroscopy. Appl. Spectrosc. 2006, 60, 398–406. 10.1366/000370206776593690. [DOI] [PubMed] [Google Scholar]
- Noda I. Generalized two-dimensional correlation method applicable to infrared, raman, and other types of spectroscopy. Appl. Spectrosc. 1993, 47, 1329–1336. 10.1366/0003702934067694. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D01; Gaussian, Inc.: Wallingford, CT, 2009.
- Jürgens B.; Irran E.; Schnick W. Syntheses, vibrational spectroscopy, and crystal structure determination from X-ray powder diffraction data of alkaline earth dicyanamides M[N(CN)2]2 with M=Mg, Ca, Sr, and Ba. J. Solid State Chem. 2001, 157, 241–249. 10.1006/jssc.2000.9036. [DOI] [Google Scholar]
- Tait S.; Osteryoung R. A. Infrared study of ambient-temperature chloroaluminates as a function of melt acidity. Inorg. Chem. 1984, 23, 4352–4360. 10.1021/ic00193a049. [DOI] [Google Scholar]
- Fumino K.; Wulf A.; Ludwig R. Strong, localized, and directional hydrogen bonds fluidize ionic liquids. Angew. Chem., Int. Ed. 2008, 47, 8731–8734. 10.1002/anie.200803446. [DOI] [PubMed] [Google Scholar]
- Ward M. D. Design of crystalline molecular networks with charge-assisted hydrogen bonds. Chem. Commun. 2005, 5838–5842. 10.1039/b513077h. [DOI] [PubMed] [Google Scholar]
- Kiefer J.; Molina M. M.; Noack K. The peculiar nature of molecular interactions between an imidazolium ionic liquid and acetone. ChemPhysChem 2012, 13, 1213–1220. 10.1002/cphc.201100845. [DOI] [PubMed] [Google Scholar]
- Glendening E. D.; Landis C. R.; Weinhold F. Natural bond orbital methods. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 1–42. 10.1002/wcms.51. [DOI] [Google Scholar]
- Jürgens B.; Höppe H. A.; Schnick W. Synthesis, crystal structure, vibrational spectroscopy, and thermal behaviour of lead dicyanamide Pb[N(CN)2]2. Solid State Sci. 2002, 4, 821–825. 10.1016/s1293-2558(02)01325-0. [DOI] [Google Scholar]
- Zheng Y.-Z.; Wang N.-N.; Luo J.-J.; Zhou Y.; Yu Z.-W. Hydrogen-bonding interactions between [BMIM][BF4] and acetonitrile. Phys. Chem. Chem. Phys. 2013, 15, 18055–18064. 10.1039/c3cp53356e. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Zheng Y.-Z.; Sun H.-Y.; Deng G.; Yu Z.-W. Hydrogen bonding interactions in ethanol and acetonitrile binary system: A near and mid-infrared spectroscopic study. J. Mol. Struct. 2014, 1069, 251–257. 10.1016/j.molstruc.2014.02.027. [DOI] [Google Scholar]
- Noda I.; Ozaki Y.. Two-Dimensional Correlation Spectroscopy: Applications in Vibrational and Optical Spectroscopy; John Wiley & Sons, 2005. [Google Scholar]
- Su B.-M.; Zhang S.; Zhang Z. C. Structural elucidation of thiophene interaction with ionic liquids by multinuclear NMR spectroscopy. J. Phys. Chem. B 2004, 108, 19510–19517. 10.1021/jp049027l. [DOI] [Google Scholar]
- Wasserscheid P.; Welton T.. Ionic Liquids in Synthesis; Wiley Online Library, 2008;, Vol. 1. [Google Scholar]
- Wulf A.; Fumino K.; Michalik D.; Ludwig R. IR and NMR properties of ionic liquids: Do they tell us the same thing?. ChemPhysChem 2007, 8, 2265–2269. 10.1002/cphc.200700508. [DOI] [PubMed] [Google Scholar]
- Bondi A. Van der waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Katayanagi H.; Nishikawa K.; Shimozaki H.; Miki K.; Westh P.; Koga Y. Mixing schemes in ionic liquid-H2O systems: A thermodynamic study. J. Phys. Chem. B 2004, 108, 19451–19457. 10.1021/jp0477607. [DOI] [Google Scholar]
- Li Y.; Liu X.; Zhang S.; Yao Y.; Yao X.; Xu J.; Lu X. Dissolving process of a cellulose bunch in ionic liquids: A molecular dynamics study. Phys. Chem. Chem. Phys. 2015, 17, 17894–17905. 10.1039/c5cp02009c. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu X.; Zhang Y.; Jiang K.; Wang J.; Zhang S. Why only ionic liquids with unsaturated heterocyclic cations can dissolve cellulose: A simulation study. ACS Sustainable Chem. Eng. 2017, 5, 3417–3428. 10.1021/acssuschemeng.7b00073. [DOI] [Google Scholar]
- Zhang Y.-P.; Wang H.-t.; Zhang L.; Yang L.; Guo X.-d.; Bai Y.; Sun H. Measurements of IR absorption across section and spectrum simulation of lewisite. Spectrosc. Spectral Anal. 2015, 35, 466–469. 10.3964/j.issn.1000-0593(2015)02-0466-04. [DOI] [PubMed] [Google Scholar]
- Fumino K.; Wulf A.; Ludwig R. Hydrogen bonding in protic ionic liquids: Reminiscent of water. Angew. Chem., Int. Ed. 2009, 48, 3184–3186. 10.1002/anie.200806224. [DOI] [PubMed] [Google Scholar]
- Kashinski D. O.; Chase G. M.; Nelson R. G.; Di Nallo O. E.; Scales A. N.; VanderLey D. L.; Byrd E. F. C. Harmonic vibrational frequencies: Approximate global scaling factors for TPSS, M06, and M11 functional families using several common basis sets. J. Phys. Chem. A 2017, 121, 2265–2273. 10.1021/acs.jpca.6b12147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.