

Abstract
DNA nanostructures have been shown viable for the creation of complex logic-enabled sensing motifs. To date, most of these types of devices have been limited to the interaction with strictly DNA-type inputs. Restriction endonuclease represents a class of enzyme with endogenous specificity to DNA, and we hypothesize that these can be integrated with a DNA structure for use as inputs to trigger structural transformation and structural rearrangement. In this work, we reconfigured a three-arm DNA switch, which utilizes a cyclic Förster resonance energy transfer interaction between three dyes to produce complex output for the detection of three separate input regions to respond to restriction endonucleases, and investigated the efficacy of the enzyme targets. We demonstrate the ability to use three enzymes in one switch with no nonspecific interaction between cleavage sites. Further, we show that the enzymatic digestion can be harnessed to expose an active toehold into the DNA structure, allowing for single-pot addition of a small oligo in solution.
Introduction
Sensors in their most basic form are devices that recognize a target and transduce that detection event into a signal that can be read and interpreted by an end user. A variety of sensors exist to accommodate a range of target types with chemical and biological targets being of increased interest. One challenge in particular for biosensing is interacting with the target using a probe that is relatively of the same size. Nanoscale structures have this advantage given many biomolecules are on the order of a few nanometers.1−9 DNA nanostructures, pioneered by Seeman,10 use the binding of Watson–Crick base pairs to program specific, non-native formations that can then perform various tasks at the nanoscale, and this technology is particularly suited for biosensing because it is a native biological material.11−14 DNA nanostructures have been shown to be highly modular for the creation of a range of two-dimensional and three-dimensional motifs, including both static and dynamic structures.15−18 The work within dynamic systems has often focused on induced motion through competitive DNA hybridization, such as multistep logic,19 hybridization chain reaction,20 DNA tweezers,21 and hinged lid boxes.22 This is a highly adaptable design regime that allows for rapid switching of states and a nearly limitless range of possible sequences to use. The strategy however is less adapted for non-nucleic acid sensor targets. The ability to adapt the modularity of DNA structural design to non-DNA molecules, such as proteins and enzymes, represents an area of interest and recent development.
Motifs, such as aptamers, can be easily added to other DNA structures, but their shape response is limited to a specific reaction phenotype.23−25 In a similar manner, DNAzymes use DNA/RNA chimeric sequences and act as a catalytic reaction center, allowing for the dynamic adjustment of the structure for both structural rearrangement.26−30 There has been minimal work, however, interacting DNA structures directly with enzymatic inputs for either sensing or directed structural rearrangement. Zuo et al. demonstrated a molecular beacon approach that uses exonuclease to generate an amplified fluorescent output, but this is neither the sensing target nor a means to structural change.31 It is our intent to explore the use of restriction enzymes as a directing molecule for the development of DNA-based sensors and enzyme-directed machines. We hypothesize that integration with these enzymes will enable multistep sensing.
Restriction enzymes, also called restriction endonucleases, are enzymes that cut DNA at specific sequences. Naturally found in bacteria to defend against viral pathogens, restriction enzymes have been harnessed by researchers and have proven a powerful asset for use in biotechnology applications, such as DNA cloning. These enzymes typically recognize sequences of DNA between 4 and 8 base pairs and can cut double-stranded DNA in a staggered manner, leaving a single-stranded overhang (sticky end) or they can cut at the same place on each strand producing a blunt end.32−34
Our previous efforts with DNA sensors produced a multiarm switch that enabled a logic-capable photonic output for three simultaneous single-stranded DNA targets.35 Linkers joining the arms can be added or removed by the addition of specific DNA sequences. This switch uses a base set of arms, each of which contains the molecular dye. From this base, the switch can be assembled with a range of different dyes and different linker lengths to produce a vast range of optical output.36 This modularity makes it an ideal candidate to explore restriction enzyme-enabled sensing and rearrangement. Although we have explored many dye triads as well as structural modification, to date, only DNA inputs have been used to modify the optical response. In this work, we investigate the role of restriction endonuclease to modulate our three-dye optical network. We test six different restriction enzymes with both sticky and blunt-end cleavage types and combine three of these into a single device capable of detecting rearrangement via single, double, and triple enzymatic digestion. The cleavage also results in the release of a seven base toehold, which we demonstrate can be used for the one-pot rearrangement and inclusion of a Cy5-containing DNA.
Results
Structural Design
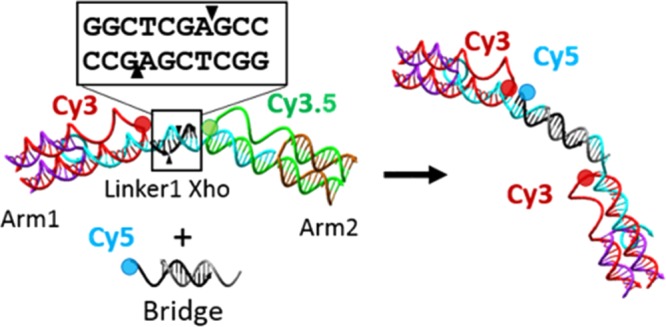
The three-arm switch, detailed in previous publications,35,36 was used as a basis for the underlying design of the enzyme-responsive DNA structure. This DNA-based structure positions three covalently linked fluorescent dyes and utilizes their overlapping excitation and emission profiles to form a Förster resonance energy transfer (FRET) triad that is responsive to changes in distance between each of the three fluorophores. This structure provides the demonstrated ability to detect three separate targets within one device, thus enabling complex detection.35 The underlying DNA structure consists of three double-crossover junctions formed by an arm strand annealed with a capping strand with a molecular dye covalently attached at the central end position of the arm strand. Each of these crossovers represents one of three arms, Arm1, Arm2, or Arm3, and are linked together via DNA linkers, which position the attached dyes into the FRET triad.
The controlling element in the device is the presence of an intact linker strand, which brings the dyes into proximity and turns on the FRET interaction. In Buckhout-White et al.,35 the linkers used were single-stranded and could be removed through toehold-mediated strand displacement, thus rendering this device a DNA sensor. With our goal to move into more complex, non-DNA targets, we have modified the original design of the linkers to include a double-stranded recognition sequence for restriction enzymes. This modification uses the basic structure detailed above, including the three-arm crossover structures, but replaces the single-stranded linker with a 10 base pair double-stranded linker, a long linker strand with a short 10 base compliment strand, which serves as the cleavage site. The overall structure is depicted in Figure 1A.
Figure 1.

(A) Schematic of the DNA switch structure and mechanism for rearrangement via enzymatic cleavage: (i) partial structure schematic indicating the arms and cleavage site. Each of these structures contains two molecular dyes, and the cleavage is transduced by simple FRET behavior; (ii) full three-arm structure, which contains three dyes and is transduced through examining each of the three acceptor-to-donor ratios. The linker between each arm of the structure is a 10 base double-stranded DNA and is coded to be a cleavage site for a specified restriction enzyme. The cleavage of one or more of these linker regions allows for the separation of the fluorescent dyes, which reduced the output of the donor-to-acceptor ratio. (B) Sequence of one of three linker sequences with the restriction site in bold and the cutting location indicated by the triangles. (C) Emission and excitation profiles of the Cy3, Cy3.5, and Cy5 molecular dyes. (D) Spectral overlap of these dyes form the basis of the multi-FRET-based optical output.
The modularity of the switch design allows for the structure to be assembled partially such that only two dye-containing arms are present, connected by one linker containing the recognition area, thus greatly simplifying the structure to allow for iterative analysis of the individual components. The partial structure used for testing each enzyme independently is depicted in Figure 1A,i, which shows one of the three dye pairs that can be formed using the partial structure: Cy3–Cy5, Cy3–Cy3.5, and Cy3.5–Cy5. The rearrangement induced by the cleavage from the restriction enzyme is shown on the full three-arm structure in Figure 1A,ii. The same process would proceed on the partial structure except the two arms would fully dissociate as no remaining DNA is left to bind the structure.
Each linker region is coded with a recognition site for a unique restriction enzyme. Six enzymes in total were tested: XbaI, EcoRI, BamHI, NcoI, SmaI, and XhoI. Figure 1B shows the sequences of all six restriction enzymes that were tested. Of these enzymes, XhoI, EcoRI, and SmaI were found to work in concert within a fully assembled switch. The bold portion of the sequence represents the restriction site, and the arrows indicate the cutting location. As can be seen in Figure 1B, we investigated both blunt-end cleavage and sticky-end cleavage. The sequences and melting temperatures for all oligos are listed in Table S1 in the Supporting Information (SI).
The transduction functionality of this switch is based on the cyclic FRET behavior of three spectrally overlapping and closely spaced molecular dyes. This is discussed in detail in Buckhout-White et al.35 The dye triad used for these studies were Cy3, Cy3.5, and Cy5. Figure 1C shows the excitation and emission curve for each dye, and the spectral overlap is shown in Figure 1D. The Förster distance, the distance which corresponds to a theoretical 50% transfer efficiency, is 5.3 nm for Cy3–Cy3.5, 5.4 nm for Cy3–Cy5, and 6.0 nm for Cy3.5–Cy5. For the original work, a spacing of nine bases, or ∼3 nm, would have theoretically yielded ∼95% efficiency. In practice, we measured 30% efficiency, which was sufficient to demonstrate change in the full three-arm configuration. This 10 base linker will have a 3.5 nm spacing, but will reduce some of the torsional contribution given the spacing is on par with the length of one full helical turn of DNA. As such, we expect similar performance from this configuration.
Although the activity of restriction enzymes is well documented and their use considered routine for many genetic engineering protocols, our application explores the limits of double-stranded DNA size and temperature conditions, for which these enzymes are optimized. To retain the close proximity of the dye molecules in the uncut “off” state, it is necessary to minimize the total length of the double-stranded linker. The recognition sites of many restriction enzymes, including those we worked with, are six bases, and linkers were designed to include two additional bases on either side of the recognition site. According to the enzyme manufacturers, it is recommended to include a minimum of six37 additional bases flanking the restriction site for optimal enzyme performance. The 10 base double-stranded linker is formed using a 10 base compliment to the single-stranded linker strand and thus naturally has a melting temperature ranging from 41.5 to 57.7 °C. The melt temperature defines the point at which half of the DNA is single-stranded. With 41.5 °C being the low range, we prefer that the enzyme be able to work at room temperature. Although several restriction enzymes, including SmaI, cut their recognition site at room temperature (25 °C), most restriction enzymes perform optimally at 37 °C. Due to the requirement that the enzymes function at room temperature and cut a 10 base pair DNA linker, it was critical to be able to test each restriction enzyme independently to determine which would function under these nonideal conditions.
Single-Enzyme Cleavage
Partial structures containing two arms and a single recognition sequence were used to assess the ability of each enzyme to cleave the 10 base pair linker at room temperature. The activity of each of the six enzymes, XhoI, NcoI, SmaI, XbaI, BamHI, and EcoRI, was compared against a positive and a negative control. The negative control comprised the partial structure without any enzyme present. This represents the condition with no enzymatic cleavage of the linker. The positive control is an approximation of a full cleavage by the enzyme and is represented experimentally by forming structures that lack the linker connecting the two dye-containing arms. The restriction enzymes XbaI, XhoI, NcoI, and SmaI were tested using the Arm1–Cy3, Arm2–Cy3.5 partial structure. EcoRI was tested using the Arm2–Cy3.5, Arm3–Cy5 partial structure, and SmaI and BamHI were tested using the Arm1–Cy3, Arm3–Cy5 partial structure. For all enzymes that were considered, five enzymes in four unique combinations emerged, showing ideal cutting behavior and no nonspecific cutting behavior in the presence of a nontargeted enzyme. Of the four combinations, NcoI-BamHI-XhoI, NcoI-EcoRI-XhoI, XhoI-SmaI-BamHI, and XhoI, EcoRI-SmaI, the latter was chosen to perform the full structured variations.
Figure 2A shows the analyzed fluorescent data, whereas Figure 2B shows the gel electropherogram. For each of the characterization methods, the samples were prepared in the same way in parallel runs with the positive and negative controls. The positive control solution was annealed according to the protocol specified in the Methods section in a batch large enough for 24, 20 μL sample to be produced. This sample contained equal molar ratios of each of two dye-containing arm strands and their corresponding capping strands as well as the linker and the compliment to the linker, which provides for the double-stranded cleavage site all in the provided CutSmart buffer. With CutSmart being a nonstandard buffer for DNA formation, a formation analysis was performed prior to begin the enzyme cutting and can be seen in the Supporting Information (SI) (Figure S1). Each of these samples was aliquoted into a 384-well plate in triplicate, where 2 μL of the specified enzyme was added and then mixed by gentle pipetting. For the negative control, the solution was annealed separately without the linker strand or its compliment. Both the positive control and negative control added 2 μL of buffer to account for the volume of the absent enzyme. The samples were allowed to digest for 1 h minimum before measuring the fluorescent output. All fluorescent measurements were excited at a wavelength of 515 nm, consistent with the shoulder of the Cy3 excitation peak and recorded between 530 and 800 nm. These parameters were set as such to allow for consistent readout once all three dyes were used in the full system. The samples for the gel electrophoresis were taken from the plate after the fluorescent data were obtained and mixed with the gel-loading buffer before pipetting the samples into the 3% agarose gel.
Figure 2.

Data from single-enzyme partial structures. (A) Ratios of the acceptor dye over the donor dye photoluminescence (PL) peak values. The three plots shown each represent a different programmed sequence designed for the enzyme specified. Each sequence was tested against six different enzymes with a negative, no enzyme, and positive, no linker, control. (B) Gel electropherogram on the partial structure before and after introduction of the specific enzyme.
To assess the efficacy of the cleavage via fluorescence, the maximum value of the deconstructed area of the acceptor peak in each of the dye pair is divided by the deconstructed donor peak maximum. XhoI and SmaI were initially analyzed using the Arm1–Cy3, Arm2–Cy3.5 partial structure and thus the ratio shown for these constructs is Cy3.5/Cy3. For EcoRI, the Arm2–Cy3.5, Arm3–Cy5 partial structure was used and the ratio is thereby Cy5/Cy3.5. These fluorescent PL ratios are plotted in Figure 2A for the three enzymes chosen for the triad assembly. The data for the enzymes not selected and the table showing all possible dye triads can be seen in the SI (Figure S2 and Table S2). The gray bars, first and last, in Figure 2A, represent the controls for each system, with the negative control indicating the maximum value for no enzyme activity and the positive control approximating a complete cleavage of the linker. All enzymes are shown, and based on fluorescent data alone, it is clear that these three enzymes show cutting behavior when presented with the specified enzyme and show no cutting behavior when presented with the nonspecified enzyme. It is of note that the value of the specified enzyme does not quite reach the value of the negative control. This may be due to a very small amount of incomplete digestion or may just reflect the fact that the negative control is an approximation because no portion of the linker or compliment is present.
Figure 2B shows the gel migration of the control sample as well as the specified enzyme digestion for the partial switch structure. The primary band at about 240 bp according to the ladder represents the fully formed structure. Once the enzyme is added, this band increases its migration consistent with 150 bp, roughly half. The intensity of the band also increases due to the doubling of the mass of DNA at that specific size. There is a small remnant of the full-structure band that is consistent with the thought that we are getting close to but not 100% digestion of these structures. This may also be a result of potential rehybridization of the sticky ends, or reassociation of the blunt ends that occur after the cleavage. In all, both characterization methodologies support the same conclusion that these three enzymes exhibit good cleavage with little to no nonspecific activity.
Kinetics of Enzyme Activity
With the activity confirmed, we assessed the kinetic rate of the enzymatic cleavage. To reiterate, these enzymes are demonstrating cleavage under nonideal conditions. The cleavage for all experiments occurs at room temperature, with most of these enzymes optimized for 37 °C. The cleavage site is also contained on a 10 base oligo with three-way junctions at both ends of that oligo. For these enzymes, it is recommended that there be a minimum of six37 base pairs on either side of the cleave site. Figure 3 shows the kinetic curves produced by measuring the same acceptor-to-donor ratio for each of the partial structures. This measurement was recorded every 20 min, with the exception of the first time point, which occurred 10 min after the initial plating. The black bars flanking the curves are averaged values of controls, positive, and negative, taken over the entire time. The variations in position of the positive and negative controls are directly due to the photophysical properties of the dyes in each system. The sharp line in the EcoRI indicated that full cleavage occurs within the 10 min time frame between initial plating and the addition and mixing of the enzyme.
Figure 3.
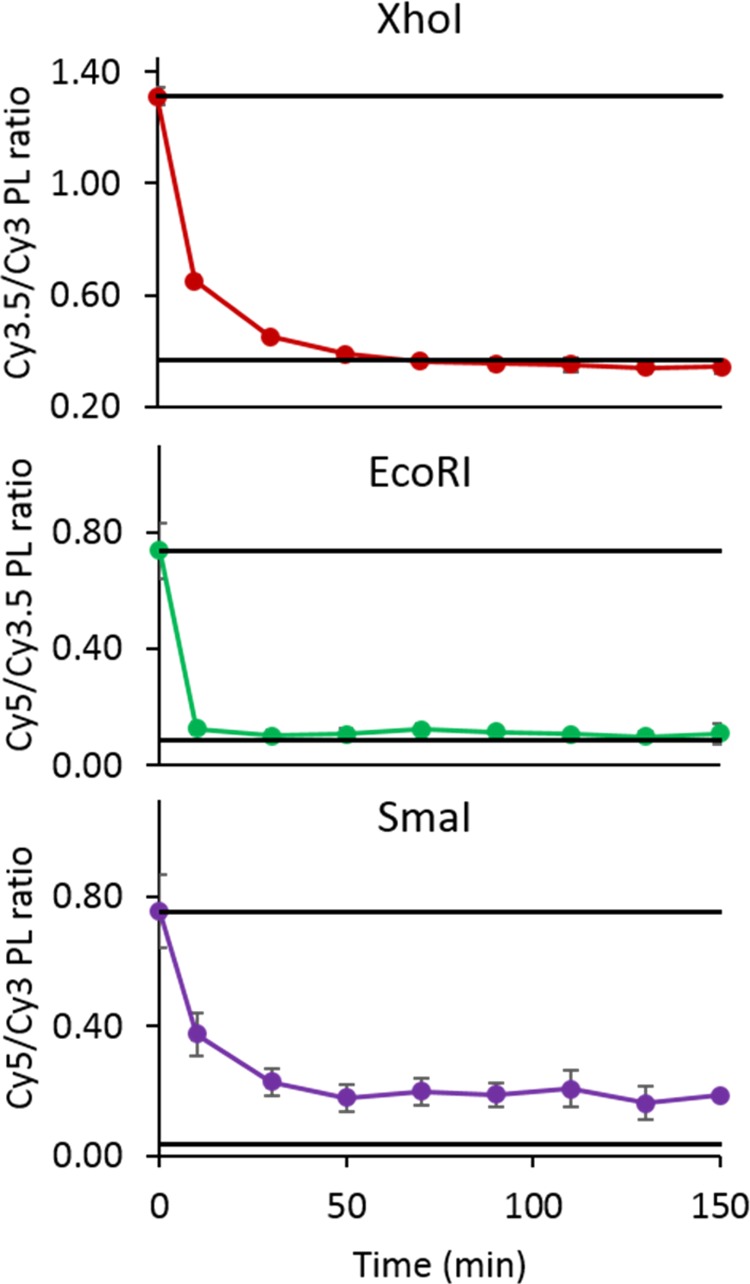
Assay of kinetic rate of cleavage for the partial structure cleavage. The top line in each plot represents the average of the positive control, and the bottom line is the average of the negative control.
Three-Arm Switch Performance
With the three enzymes determined to operate efficiently in the partial structure, we placed the three chosen enzymes’ recognition sequences into each of the three linker regions; XhoI in linker 1 between Cy3 and Cy3.5, EcoRI in linker 2 between Cy3.5 and Cy5, and SmaI in linker 3 between Cy3 and Cy5. Using the cutting of one linker as an input, we find eight potential permutations for the full three-arm switch. This is equivalent to the same eight potential permutations in Buckhout-White et al.35 The difference with the enzyme switch is the fragments of the cleaved linker is still present whereas the linker removal via strand displacement from the previous work, removes the entire sequence. The corollary between these two systems allows us to provide a set of positive controls such that we are mimicking the cleavage of one, two, or three of the linkers by removing the linker altogether.
To test the three-arm switch, we created a series of single, double, and triple digestions and compared them with the corresponding linker-removed structures. For example, the corollary to XhoI presence would be the removal of linker 1. For this series, we used XhoI single digestion, EcoRI single digestion, XhoI and EcoRI double digestion, and then the final XhoI EcoRI and SmaI triple digestion. For the three-arm structure, the FRET pathways are all interrelated and it is necessary to examine all three acceptor-to-donor ratios to get a full understanding of the spectral output. Figure 4 displays the acceptor-to-donor ratios for the enzyme digestion (A) and the linker-removed corollaries (B). Comparison shows general agreement between the two sets. As expected from the partial structure digestions, we do not fully reach the level of the positive controls. This again may be due to partial cutting, reassembly of the cut end, or inherent anomalies between a fully cut structure and the linker-removed version. It may also be due to the amount of glycerol in the system from the double and triple digestions. We chose to keep all variables constant, which meant the amount of glycerol inserted into the system was 10% for the double digestion and 15% for the triple digestion. Although this does not eliminate the desired reaction, it may limit completion of the reaction or full rearrangement of the structure. If we look at the trends of each ratio between samples, we see that in the case of the Cy3.5/Cy3 ratio the value decreases slightly, increases, and then subsequently decreases in the next two samples. All ratios follow the same trends with the exception of the Cy5/Cy3.5 ratio between the negative control and the XhoI enzyme addition. Here, the value roughly stays level in the enzyme sample and increases in the control. These similar trends can also be seen if we plot the dye contribution as a percentage of the total area. The ternary plot shown in Figure S3 displays this presentation.
Figure 4.

Plots of the three acceptor-to-donor ratios for each of the full three-arm structures. (A) Single-, double-, and triple-enzyme digest for the three-arm structures. (B) Comparable control structures in which a linker is removed where the corresponding enzyme structure would be cleaved.
Structural Rearrangement
One added benefit of the 10 base length of the recognition site compliment is that upon the sticky-end cleavage realized by both the XhoI and EcoRI enzymes we are left with a 7 base, 3 base split of the 10 base sequence. The three bases that remain of the linker strands do not have a sufficient melt temperature to allow this duplex to remain stable and thus the seven bases of the compliment dissociate into solution. The same is true of the other side where the three bases of the compliment do not have the energy to remain associated with the seven base linker strand. This end thereby leaves a seven base strand that is now suitable as a toehold for additional structural modification. This process is depicted schematically in Figure 5A. It is important to note that this toehold only exists after the restriction enzyme has fully cleaved the target sequence.
Figure 5.

(A) Schematic illustration of the partial structure enzyme cleavage and rearrangement via addition of an additional Cy5-containing DNA oligo. (B) PL Intensity of each of the structures depicted in (A), with XhoI + oligo and XhoI + bridge both being one-pot reactions. (C) PL peak height plots for each of the three dyes in the system. The addition was performed both as a one-pot reaction and a sequential reaction. The controls of the switch plus the oligo or bridge are done in the absence of enzyme and show little to no reaction without the presence of the enzyme.
To utilize this toehold display, we have formed the three-arm structure in full and partial form, with only the Cy3 and Cy3.5 dyes present on Arm1 and Arm2, respectively. For the full structure, Arm3 is present but in an unlabeled form to preserve the structural integrity but allow for the use of the Cy5 dye for our new addition. For this reconfiguration, we have designed two versions of a Cy5-labeled DNA that will interact with the exposed toehold. The oligo version is a simple single-stranded DNA of 15 bases with a Cy5 that will have the closest interaction with the Cy3. The bridge version is a partially duplex DNA with two identical binding regions that will bridge two of the three arm switches together. This is depicted in the bottom panel of Figure 5A.
Figure 5B shows the spectra of the original partial two-arm structure, the structure with the XhoI enzyme added, XhoI plus the bridge DNA, and XhoI plus the oligo DNA. In comparing the XhoI spectra to that where either the bridge or the oligo is added, there is a clear decrease in the Cy3 peak at 550 nm and a clear increase in the Cy5 peak at 650 nm. This change alone shows the inclusion of the new oligo, thus demonstrating the ability to modify the structure after the enzymatic cleavage has taken place.
Figure 5C shows further analysis in which each of the three acceptor-to-donor ratios is plotted. The control is shown in section I and is derived from the data from “original” in Figure 5B. Section II shows two controls where the oligo and the bridge are added to the control without enzyme. For the bridge structure, we see no increase in Cy5, but for the oligo structure, we see a slight increase in the Cy5 excitation, indicating some leakage. Section III is where XhoI is added, and we see the clear increase in the Cy3 excitation. For sections IV and V, we distinguished between a sequential addition method, where the enzyme is added to the full structure and allowed to cleave and then either the bridge or the oligo is added, and the one-pot addition, where the oligo or bridge and the enzyme are added simultaneously. In each case, the oligo or bridge is added in a 4-fold molar excess to the switch. For the sequential addition, we see minor increase from the oligo Cy5/Cy3 excitation signal but similar values with the other two ratios. For the one-pot addition, the bridge addition shows slight decrease in both the Cy5/Cy3 signal and the Cy3.5/Cy3 signal. Although the lower Cy5/Cy3 signal indicates a potentially lower rate of binding, the lower Cy3.5/Cy3 ratio may mean more disruption of the Cy3–Cy3.5 transfer. In all, this may be a direct result of the bridge molecule having asymmetric dye position of the Cy5.
Discussion and Conclusions
In this work, we utilized the inherent fit with restriction endonuclease to expand the range of detectable materials outside of the realm of simple DNA. We have clearly demonstrated the ability of a three-arm switch to be applied to enzymatic inputs. Furthermore, we have demonstrated that these enzymes can be used to rearrange the structure of the DNA, allowing multistep processes to occur, a necessary step toward the evolution of complex sensing. In investigating the use of restriction enzymes in this confined parametric space, we have also demonstrated the ability of these materials to engage effectively outside their optimal environment. We see no difference in cleavage rate between that of blunt-end- and sticky-end-type cleavage sites. In the steady-state measurement, we see no distinction in the cleavage efficiency between these cleavage types either. However, in the kinetic assay, the SmaI enzyme, a blunt-end restriction enzyme, completes the reaction with a higher ratio, indicating less complete cleavage than the control compared to either of the other two enzymes, XhoI or EcoRI, both of which are sticky-end cleavage types. This difference may relate to the affinity for the blunt-end nonspecific adhesion, which is documented in ref (38) or may be an artifact of the measurement parameter. Further work on a broader array of both cleavage type enzymes will help alleviate this question and expand the library of enzymes applicable to this type of activity environment.
We have presented the transfer from the partial structure analysis to the full-structure analysis and the multiplexing ability that this brings, as demonstrated by the successful single, double, and triple digestion. Although these do not reach their full-potential on–off range as demonstrated by the linker-removed controls, we still see clear distinction enough to determine which enzymes are acting. As demonstrated by the unique change particularly seen in the addition of the EcoRI enzyme, it is possible to determine, blindly, which enzyme is acting. Improvement on this may be available through dye change or condition optimization. Further, as a sensing modality, this multiplexing ability is quite powerful and may not be limited to simply three enzymes. The modular structure could easily be made more complex by the addition of arms to the unit. We have also shown that it can operate distinctly using different dye triads and that there are several unique combinations of enzymes.36 This means that we may potentially be able to have two separate switches in solution, each with unique recognition sites and optical output.
The structural rearrangement offers perhaps the most unexpected outcome of this work. As a byproduct of restriction enzyme cleavage of the target DNA, we reveal an active DNA toehold. Multistep reactions are not new within the field of DNA nanotechnology. Concentric FRET systems based on protease cleavage and quantum dot assembly show similar optical performance with regard to multienzyme detection, but they are limited to simple detection mechanisms.39 Other work demonstrates the ability to interact with an existing structure and expose a toehold that will continue the reaction in a prescribed manner.40−42 Our present demonstration, however, shows the clear ability of enzyme-directed chain reactions within a confined structural environment. This is evidenced by the appearance of the Cy5 excitation peak, which is not seen in either the control or the original structure. The effect seen in both the sequential and one-pot demonstrations further demonstrates that the secondary rearrangement with the toehold is only activated once the enzymatic cleavage has occurred.
With such little leakage seen, this simple demonstration portends to much greater potential application. The harnessing of multienzyme, multistep assemblies could have an impact on the fields of DNA nanorobotics and enzyme-directed nanofabrication. Within the context of complex sensing and theranostics, it is also conceivable that these systems may be adapted to a larger cagelike structure that upon interaction with the target enzymes can release payload or rearrangement to deliver targeted DNA codons.
In all, we have expanded the utility of the logic FRET-enabled three-arm DNA switch to the detection of restriction enzymes. We have shown six different enzymes in all and three enzymes in detail. These three enzymes have clear cleavage efficiency and no nonspecific cleavage, a necessary requirement for this three-target system to function. We have demonstrated single, double, and triple digestions with clear photonic distinction in the output, a necessary criterion to determine which enzymes are present. Finally, we show the ability of these enzymes to be used as triggers for site-specific rearrangement of these DNA structures, which may have broad-reaching potential for complex sensing and smart nanoscale systems.
Methods
DNA
The DNA sequences are based on Buckhout-White et al.35 with the exception of the linker sequences, which are designed to include cleavage sites for the specified restriction enzymes. All unlabeled and Cy3- and Cy5-labeled DNA are synthetic oligos ordered from Integrated DNA Technologies (Coralville, IA). The Cy3.5-containing oligos are sourced from Eurofins genomics (Louisville, KY). The oligos used were diluted to 20 μM working concentrations in water and analyzed for concentration using Thermo Fisher NanoDrop 2000.
Structural Assembly
All of the partial and full structures were assembled in a total volume of 80 μL with a 0.5 μM concentration unless specified otherwise. A small volume of 2 μL of each 20 μM DNA oligo was added to form either the partial or full structures. The CutSmart (New England Biolabs) buffer (8 μL) was used in each 80 μL sample unless specified otherwise. Each 80 μL sample was annealed on a ProFlex Thermal cycler using a program that heats the sample up to 95 °C for 5 min and is ramped down 1 °C every minute until 4 °C, at which temperature it is held.
Enzyme Digestion
All enzymes used (BamHI, EcoRI, NcoI, SmaI, XbaI, and XhoI) were from New England Biolabs and had a concentration of 20 000 U/mL. The high-fidelity versions of the enzymes BamHI, EcoRI, and NcoI were employed in the experiments. Single-enzyme digestions were conducted by inserting 1 μL of enzyme for every 10 μL of sample and gently mixing via repeated pipetting. Multienzyme digestions were conducted using the same DNA-to-enzyme ratio for each individual enzyme, with 1 μL of each enzyme per 10 μL volume of sample, i.e., three enzyme digest will contain 3 μL of enzyme solution per 10 μL of total solution volume. The solutions then sat out in a room-temperature environment for a minimum of 1 h prior to analysis.
Structural Rearrangement
For the structural rearrangement studies, Cy5 oligo and Cy5 bridge were prepared in a 20 μM dilution in the CutSmart buffer and annealed using the same thermal ramp protocol used to assemble the switch structure. A small volume of 1 μL of this preannealed solution was used for each addition, which represents a 4-fold molar excess to the assembled structure. The oligo is allowed to react with the switch structure at room temperature for a minimum of 10 min.
FRET Data Collection and Analysis
Fluorescence data were collected using the Tecan Infinite M100 dual monochromator multifunction plate reader that has a xenon flash lamp (Tecan, Research Triangle Park, NC). A small volume of 20 μL of each 0.5 μM concentration sample was inserted into a single well in a 384-well plate. All of the samples were run in triplicate unless otherwise specified. The DNA dye-labeled samples were excited at 515 nm. The data were recorded over an emission ranging from 530 to 800 nm. The raw data were deconstructed using model Cy3, Cy3.5, and Cy5 emission spectra. The maximum peak value of each spectra corresponding to 556, 606, and 664 nm was used to produce the donor-to-acceptor ratio values.
Gel Electrophoresis Data Collection and Analysis
Three percent agarose gels made with 1× tris acetate EDTA buffer were used to collect the gel electrophoresis data unless otherwise indicated. For every 100 mL of gel solution, 20 μL of GelRed stain (Biotium; Fremont, CA) was used. A DNA ladder (BioMarker EXT Plus, Bioventures Inc; Murfreesboro, TN) was used to determine the length of the double-stranded DNA bands. A volume of 10 μL of a 12 μL sample that consisted of 10 μL of digested sample (or control) and 2 μL of a loading dye was loaded into each well; 6 μL of ladder was inserted into one well that consisted of 5 μL of ladder and 1 μL of loading dye unless otherwise indicated. Each gel experiment was run at 80 V for 1 h. The Bio-Rad ChemiDoc XRS+ System was used to analyze the gel electrophoresis data.
Acknowledgments
Financial support from NRL and NRL-NSI is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01333.
DNA sequences, comparative buffer analysis, full enzyme analysis, enzyme triad table, and ternary plot for three dye assemblies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Teles F. R. R.; Fonseca L. P. Talanta 2008, 77, 606. 10.1016/j.talanta.2008.07.024. [DOI] [Google Scholar]
- Haes A. J.; Van Duyne R. P. J. Am. Chem. Soc. 2002, 124, 10596. 10.1021/ja020393x. [DOI] [PubMed] [Google Scholar]
- Liu S. P.; Su W. Q.; Li Z. L.; Ding X. T. Biosens. Bioelectron. 2015, 71, 57. 10.1016/j.bios.2015.04.006. [DOI] [PubMed] [Google Scholar]
- Medintz I. L.; Clapp A. R.; Mattoussi H.; Goldman E. R.; Fisher B.; Mauro J. M. Nat. Mater. 2003, 2, 630. 10.1038/nmat961. [DOI] [PubMed] [Google Scholar]
- Pumera M.; Sanchez S.; Ichinose I.; Tang J. Sens. Actuators, B 2007, 123, 1195. 10.1016/j.snb.2006.11.016. [DOI] [Google Scholar]
- Sheehan P. E.; Whitman L. J. Nano Lett. 2005, 5, 803. 10.1021/nl050298x. [DOI] [PubMed] [Google Scholar]
- Siwy Z.; Trofin L.; Kohli P.; Baker L. A.; Trautmann C.; Martin C. R. J. Am. Chem. Soc. 2005, 127, 5000. 10.1021/ja043910f. [DOI] [PubMed] [Google Scholar]
- Wang J. Anal. Chim. Acta 2003, 500, 247. 10.1016/S0003-2670(03)00725-6. [DOI] [Google Scholar]
- Wang J. Small 2005, 1, 1036. 10.1002/smll.200500214. [DOI] [PubMed] [Google Scholar]
- Wang J. Analyst 2005, 130, 421. 10.1039/b414248a. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. J. Theor. Biol. 1982, 99, 237. 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- Kerman K.; Kobayashi M.; Tamiya E. Meas. Sci. Technol. 2004, 15, R1. 10.1088/0957-0233/15/2/R01. [DOI] [Google Scholar]
- Brown C. W. 3rd; Buckhout-White S.; Diaz S. A.; Melinger J. S.; Ancona M. G.; Goldman E. R.; Medintz I. L. ACS Sens. 2017, 2, 401. 10.1021/acssensors.6b00778. [DOI] [PubMed] [Google Scholar]
- Li D.; Song S. P.; Fan C. H. Acc. Chem. Res. 2010, 43, 631. 10.1021/ar900245u. [DOI] [PubMed] [Google Scholar]
- Zhao M. Z.; Wang X.; Ren S. K.; Xing Y. K.; Wang J.; Teng N.; Zhao D. X.; Liu W.; Zhu D.; Su S.; Sho J. Y.; Song S.; Wang L. H.; Chao J.; Wang L. H. ACS Appl. Mater. Interfaces 2017, 9, 21942. 10.1021/acsami.7b05959. [DOI] [PubMed] [Google Scholar]
- Dietz H.; Douglas S. M.; Shih W. M. Science 2009, 325, 725. 10.1126/science.1174251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Dietz H.; Liedl T.; Hogberg B.; Graf F.; Shih W. M. Nature 2009, 459, 414. 10.1038/nature08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y.; Ye T.; Su M.; Zhang C.; Ribbe A. E.; Jiang W.; Mao C. D. Nature 2008, 452, 198. 10.1038/nature06597. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Nature 2006, 440, 297. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Seelig G.; Soloveichik D.; Zhang D. Y.; Winfree E. Science 2006, 314, 1585. 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Dirks R. M.; Pierce N. A. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275. 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurke B.; Turberfield A. J.; Mills A. P.; Simmel F. C.; Neumann J. L. Nature 2000, 406, 605. 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- Andersen E. S.; Dong M.; Nielsen M. M.; Jahn K.; Subramani R.; Mamdouh W.; Golas M. M.; Sander B.; Stark H.; Oliveira C. L. P.; Pedersen J. S.; Birkedal V.; Besenbacher F.; Gothelf K. V.; Kjems J. Nature 2009, 459, 73. 10.1038/nature07971. [DOI] [PubMed] [Google Scholar]
- Nutiu R.; Li Y. F. J. Am. Chem. Soc. 2003, 125, 4771. 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- Liu J. W.; Lu Y. Angew. Chem., Int. Ed. 2006, 45, 90. 10.1002/anie.200502589. [DOI] [Google Scholar]
- Wang F.; Lu C.-H.; Willner I. Chem. Rev. 2014, 114, 2881. 10.1021/cr400354z. [DOI] [PubMed] [Google Scholar]
- Brown C. W. 3rd; Lakin M. R.; Horwitz E. K.; Fanning M. L.; West H. E.; Stefanovic D.; Graves S. W. Angew. Chem., Int. Ed. 2014, 53, 7183. 10.1002/anie.201402691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Lu Y. J. Am. Chem. Soc. 2007, 129, 9838. 10.1021/ja0717358. [DOI] [PubMed] [Google Scholar]
- Liu J.; Lu Y. Angew. Chem., Int. Ed. 2007, 46, 7587. 10.1002/anie.200702006. [DOI] [PubMed] [Google Scholar]
- Liu J. W.; Lu Y. J. Am. Chem. Soc. 2003, 125, 6642. 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]
- Willner I.; Shlyahovsky B.; Zayats M.; Willner B. Chem. Soc. Rev. 2008, 37, 1153. 10.1039/b718428j. [DOI] [PubMed] [Google Scholar]
- Zuo X. L.; Xia F.; Xiao Y.; Plaxco K. W. J. Am. Chem. Soc. 2010, 132, 1816. 10.1021/ja909551b. [DOI] [PubMed] [Google Scholar]
- Loenen W. A. M.; Dryden D. T. F.; Raleigh E. A.; Wilson G. G.; Murray N. E. Nucleic Acids Res. 2014, 42, 3. 10.1093/nar/gkt990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A.; Wilson G. G.; Wende W. Nucleic Acids Res. 2014, 42, 7489. 10.1093/nar/gku447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5905. 10.1073/pnas.0500923102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckhout-White S.; Claussen J. C.; Melinger J. S.; Dunningham Z.; Ancona M. G.; Goldman E. R.; Medintz I. L. RSC Adv. 2014, 4, 48860. 10.1039/C4RA10580J. [DOI] [Google Scholar]
- Buckhout-White S.; Brown C. W.; Hastman D. A.; Ancona M. G.; Melinger J. S.; Goldman E. R.; Medintz I. L. RSC Adv. 2016, 6, 97587. 10.1039/C6RA23079B. [DOI] [Google Scholar]
- www.neb.com, 2017.
- Maffeo C.; Luan B. Q.; Aksimentiev A. Nucleic Acids Res. 2012, 40, 3812. 10.1093/nar/gkr1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey M.; Kim H.; Conroy E. M.; Algar W. R. J. Phys. Chem. C 2017, 121, 13345. 10.1021/acs.jpcc.7b02739. [DOI] [Google Scholar]
- Guo Y. H.; Yang K. L.; Sun J. C.; Wu J.; Ju H. X. Biosens. Bioelectron. 2017, 94, 651. 10.1016/j.bios.2017.03.066. [DOI] [PubMed] [Google Scholar]
- Kotani S.; Hughes W. L. J. Am. Chem. Soc. 2017, 139, 6363. 10.1021/jacs.7b00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Wang X. J.; Wei B.; Huang F. J.; Yao D. B.; Liang H. J. Nanoscale 2017, 9, 2981. 10.1039/C7NR00386B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.