

Abstract
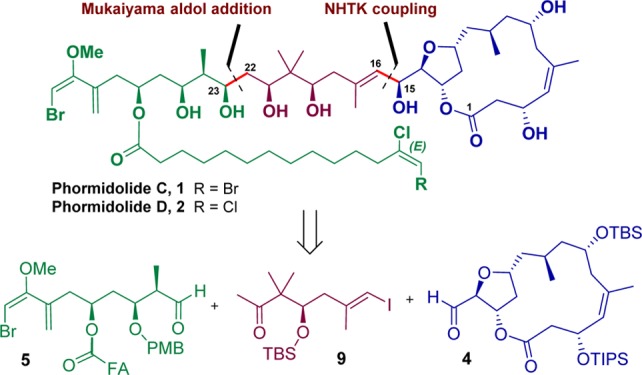
A convergent and stereoselective approach for the synthesis of marine natural product (MNP) phormidolide D (PM D) is proposed. Two main disconnections divided PM D in three molecular fragments: the macrocyclic core 4, the stapling iodoalkene 9 corresponding to the central part of PMs, and the east fragment 5 that includes the unusual bromo-methoxy-diene moiety and a tetradecanoic acid ended with a (E)-dichloro-ene functionality. Procedures for the preparation of compounds 5, 9, and the never-reported fatty acids 7 and 8, present in PMs C and D, respectively, have been afforded with good yields and high degree of stereoselectivity. The absolute configuration of all of the generated stereocenters has been established. The reaction to link iodoalkene 9 and formylmacrolactone 4, using the Nozaki–Hiyama–Takai–Kishi coupling, gave an advanced synthetic intermediate with total stereocontrol. Finally, a deeper study of protecting groups and reaction conditions for the last step of the synthesis is needed. All the information gathered in this publication will be of great value to continue performing synthetic studies for the preparation of these NPs. The versatility and the presence of a common polyol chain in oscillariolide and PMs A–C would allow applying the same retrosynthesis for the synthesis of the mentioned MNP.
Introduction
Polyketides, and more specifically macrolides, represent a family of marine natural products (MNPs) of great interest from a biological-therapeutical point of view,1 but at the same time, they show high complex molecular architectures that make their syntheses a real challenge. Phormidolides C and D (Scheme 1, PMs C and D, 1–2) are marine polyketides isolated by the biotech company PharmaMar that showed cytotoxic activity in three tumor cell lines with an unknown mechanism of action.2 From a structural point of view, PMs C and D present 13 stereocenters and 5 di- and trisubstituted double bonds distributed in the following moieties: a tetrahydrofuran (THF)-containing macrolide (C1–C14), a polyhydroxylated chain (C15–C31) terminated with a unusual bromo-metoxy-diene moiety, and two different tetradecanoic fatty acids (C39–C52) containing a terminal (E)-dichloro-ene or (E)-bomo-chloro-ene functionalities linked to the C27 hydroxylic position.a So far, and to the best of our knowledge, no total synthesis of PMs C and D or the related PM A3 and oscillariolide4 has been described.
Scheme 1. (a) First Retrosynthetic Analysis for the Preparation of PMs C and D and (b) Second-Generation Retrosynthesis Described in This Publication.

The structural and stereochemical elucidation of PMs C and D were carried out through mono- and bidimensional advanced NMR techniques (1H, 13C, 1D-TOCSY, gCOSY, gHSQC, and gHMBC) and high-resolution mass spectroscopy (HRMS).b The relative configuration of stereocenters present in the macrocyclic core was determined using rotational nuclear Overhauser effect spectroscopy combined with the J-based configuration analysis and nuclear Overhauser effect experiments. The two only stereocenters, with unclear configuration after the NMR analysis, were C3 and C14 positions. To solve this issue, our group reported the chemical syntheses of three different diastereomeric protected macrocyclic cores (C1–C15 fragment). NMR comparison of the synthetic and natural products suggested to us that the 3R,14R configuration was the most plausible one.2 A second-generation synthesis of the C1–C15 moiety with the mentioned stereochemistry, after the removal of the protecting groups, enabled a second NMR comparison with the NP which showed high similarity, hence confirming our 3R and 14R stereochemical hypothesis.5
Stereochemistry of the C15–C31 fragment, common to oscillariolide4 and PM A,3 was determined by the chemical shift and the coupling constant in comparison with PM A, whose absolute configuration was previously determined by Gerwick and co-workers. Recently, our group described the synthesis of the C19–C31 polyhydroxylated chain,6 including the tricky bromo-methoxy-diene motif.7 This allowed a new NMR chemical shift comparison between the synthetic and NPs showing high degree of similarity, thereby confirming the absolute configuration of the polyol chain by chemical synthesis.
Our initial proposed retrosynthesis (Scheme 1, a) explained during the enantioselective synthesis of the polyhydroxylated chain was based on a single disconnection through the C15–C16 bond.6 Both fragments 3 and 4 would be linked in the last step of the synthesis through a Nozaki–Hiyama–Takai–Kishi8 (NHTK) coupling reaction. However, this approach posed some clear disadvantages. First, a high number of linear synthetic steps for the preparation of 3 would be required, and the introduction of the C16 iodoalkene functionality would not be trivial. Second, a possible lack of chemoselectivity in the formation of C15–C16 bond using NHTK conditions could have occurred because of the presence of other reactive positions in 3 such as C31 or C52. For these reasons, a new retrosynthetic analysis that overcame all the above-mentioned problems was necessary.
The new synthetic approach (Scheme 1, b) envisions a disconnection through the C22–C23 bond to create it at the end of the route through a previously optimized6 Mukaiyama aldol addition from aldehyde 5 (C23–C31) and methyl ketone 6 (C1–C22). Fatty acids 7 and 8 present in different PMs would be synthesized separately and esterified at the C27 position during the synthetic route toward 5. Then, fragment 6 would be planned to be prepared by means of the NHTK8 coupling of enantiopure iodoalkene 9 (C16–C22) and the previously reported C1–C15 fragment aldehyde 4.5 A reliable preparation of central fragment 9 is crucial because of its useful bivalent nature (methyl ketone and iodoalkene) that allows its use as a “stapling” compound between macrocycle 4 and aldehyde 5.
Herein, we report the preparation of fatty acids 7 and 8, present on both PMs C and D, and the synthesis of aldehyde 5 and iodoalkene 9. Furthermore, with fragments 5, 9, and 4 in hand, the first synthetic approach for the synthesis of MNP PM D (PM D) is discussed.
Results and Discussion
Fatty Acids Preparation
Tetradecanoic acids containing (E)-dihalogenated terminal double bonds were prepared by direct dihalogenation of the corresponding terminal alkyne using the methodology developed by Negoro and Ikeda.9 The use of these halogenation conditions had important advantages such as good yields, easy experimental setup, mild conditions, stereospecificity toward the (E) double bond, and regioselectivity for the desired isomer in the case of bromochlorination.
The synthetic route was initially started using commercially available 11-bromo-1-dodecanol 10 through the protection of the hydroxyl prior to the alkyne introduction and through triple bond halogenation. The tetrahydropyranyl (THP) protecting group was chosen as the most convenient protecting group for the whole synthetic route.c After the THP protection of 10 and acetylide introduction, alkyne 11 was obtained in high yield. This compound is common to both synthetic routes for the preparation of fatty acids 7 and 8 (Scheme 2). The key reaction of 11 with tetrabutylammonium dichlorobromide (TBADCB) in CH2Cl2 at room temperature (rt) produced terminal alkene 12 as a single (E) double-bond isomer. (E) Stereochemistry was obtained due to the reaction mechanism.9 However, compound 12 was obtained as an 89:11 mixture of inseparable regioisomers.d The THP removal followed by a double Dess–Martin and Pinnick oxidation rendered fatty acid 7 found in PM C. Finally, 7 was protected as a methyl ester and purified to obtain 14 in good yield with a single final purification from 11. It is important to mention that this final protection was performed to facilitate the purification process and to enhance the stability of the compound for long-term storage.
Scheme 2. Preparation of the Fatty Acids Present in PMs C and D.

Reagents and conditions: (a) PPTS, CH2Cl2, 3,4-dihydro-2H-pyran, rt, 95%; (b) lithium acetylide–diethylamine complex, DMSO, rt, 99%; (c) TBADCB, CH2Cl2, rt; (d) tetrabutylammonium trichloride, CH2Cl2, rt; (e) p-TsOH, MeOH, rt; (f) DMP, CH2Cl2, rt; (g) NaClO2, NaH2PO4, 2-methyl-2-butene, tBuOH/H2O, rt; and (h) TMSCHN2, CH2Cl2, MeOH. Yields: 14 = 20% from 11; 15 = 12% from 11.
A similar synthetic route was followed for the preparation of 8, using tetrabutylammonium trichloride for the halogenation of 11. This salt was prepared by the reaction of tetrabutylammonium chloride with in situ generated chlorine.10 Protected alkene 13 was obtained as a single stereoisomer and then, a deprotection–oxidation sequence similar to that used for 7 delivered fatty acid 8. Finally, methyl ester protection and purification produced ester 15. These two simple synthetic routes allowed an easy, efficient, and scalable preparation of the fatty acids present in PMs C and D.
Preparation of Fragment C23–C31 (5)
With both fatty acids in hand, the challenging preparation of aldehyde 5, the east end of the molecule, was investigated. The route was started from the previously reported alcohol 16 (Scheme 3).6 t-Butyldimethylsilyl (TBS)-hydroxyl protection, DIBAL-H regioselective ketal reduction,11 and Swern oxidation afforded aldehyde 17 with good synthetic yields at the gram scale. Then, the Mukaiyama aldol addition of (E)-((4-bromo-3-methoxybuta-1,3-dien-2-yl)oxy)trimethylsilane(6) produced ketol 18 as an inseparable mixture of diastereomers (82:18) enriched in the undesired C27-(S) isomer.e Then, as it happened in our previous work,6 a three-step procedure was necessary to introduce the methylidene moiety including triethylsilane (TES) protection of 18, olefination using the Tebbe reagent and TES pyridinium p-toluenesulfonate (PPTS)-mediated removal to afford homoallylic alcohol 19 in good yield. At this point, C27-diastereomers were separated by column chromatography and the absolute configuration of the major isomer was confirmed using Mosher’s derivatization.12f Mitsunobu hydroxyl inversion13 of 19, followed by basic hydrolysis of the generated nitrobenzoate, cleanly delivered the C27-(S) isomer compound 20. Then, alcohol 20 was esterified with (E)-13,14-dichlorotetradec-13-enoic acid (8) to obtain the protected C23–C31 fragment of PM D, ester 21. tert-Butyldimethylsilyl (TBS) removal at position C23 and Dess–Martin oxidation gave us access to aldehyde 5 in 12 steps with useful yields from alcohol 16.
Scheme 3. Preparation of Compound 5, Fragment C23–C31.

Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, 86%; (b) DIBAL, CH2Cl2, −20 to 0 °C, 90%; (c) DMSO, (COCl)2, Et3N, DCM, −78 °C, 80%; (d) (E)-((4-bromo-3-methoxybuta-1,3-dien-2-yl)oxy)trimethylsilane, BF3·Et2O, CH2Cl2, −78 °C, 47%; (e) TESCl, imidazole, CH2Cl2, 86%; (f) Tebbe reagent, pyridine, THF, 50 °C, 81%; (g) PPTS, MeOH, 88%; (h) diastereomer column chromatography separation; (i) PPh3, p-nitrobenzoic acid, DIAD, THF; (j) K2CO3, MeOH, 59% for two steps; (k) 8, EDC·HCl, Et3N, DMAP, 61%; (l) TBAF, THF, 77%; and (m) DMP, NaHCO3, CH2Cl2, 93%.
Preparation of Fragment C16–C22 (9)
The synthesis of the central bifunctionalized C16–C22 fragment 9 (Scheme 4) began with the aldol addition of silylenolether 22(14) to aldehyde 23(15) to obtain ketol 24 as a C19 epimeric mixture. Several reported enantioselective strategies using N-Ts-l-valine or a tryptophan-derived oxazaborolidine as chiral inductors for this addition were tested unsuccessfully.16,17 The racemic mixture 24 was reacted with the chiral (R)-methoxyphenylacetic acid (MPA) to obtain the corresponding diastereomeric esters that were easily separated via column chromatography to isolate the desired C19-R epimer 25-R,R and its diastereomer 25-S,R. Taking advantage of the fact that 25-S,R is the enantiomer of 25-R,S, and therefore spectroscopically identical; it was possible to identify 25-R,R without additional derivatizations using Mosher’s model.18f
Scheme 4. Preparation of Compound 9, Fragment C16–C22.

Reagents and conditions: (a) BF3·Et2O, CH2Cl2, 63%; (b) (R)-MPA, EDC·HCl, DMAP, THF; (c) column chromatography to isolate C19 diastereomers 25-R,R, 41% and 25-S,R, 42%; (d) TMSOTf, 1,2-bis(trimethylsiloxy)ethane, −78 °C to rt, 97%, (e) LiOH·H2O, MeOH, 65 °C, 92%; (f) TBSOTf, Et3N, CH2Cl2, 74%; and (g) PPTS, MeOH, quant.
Ester basic hydrolysis at this point was nonviable probably because of methyl ketone known basic media instability; therefore, C21 position had to be protected. Mild protecting conditions20 to install the ketone 1,3-dioxolane protecting group were used and subsequent LiOH-mediated ester saponification rendered alcohol 26 in high yield for this two-step transformation. Finally, alcohol protection using TBSOTf and ketal removal to recover the methyl ketone delivered the desired iodoalkene 9 as a single (R) enantiomer, as needed.
Endgame of the Synthesis
Having synthesized fragments 5, 9, and 4,5 the pieces of the synthetic puzzle should be put together. First of all, the union of aldehyde 4 and iodoalkene 9 using the CrCl2–NiCl2-mediated NHTK coupling (Scheme 5, a) was attempted.8,21 This methodology allowed the selective formation of the C15–C16 bond in the presence of methyl ketone because of the well-known chemoselectivity of NHTK coupling toward aldehydes, thereby demonstrating the useful orthogonal reactivity of compound 9. Unfortunately, the reaction between 9 and 4 gave stereoselectively compound 6 possessing the configuration R-C15 with only 18% yield.8
Scheme 5. (a) NHTK Coupling To Obtain Alcohol 6, the C1–C21 Fragment and (b) Determination of C15 Absolute configuration Using VT Mosher Methodology19.

Reagents and conditions: (a) CrCl2, NiCl2, DMF, 48 h, 18% and (b) (R)-MPA, EDC·HCl, DMAP, THF, 48 h.
Because of the low amount of compound 6 available, it was decided to use the variable temperature (VT) Mosher determination that allows assigning the absolute configuration of a secondary alcohol performing a single derivatization.19 First, alcohol 6 was converted into its corresponding (R)-methoxyphenylacetic ester 27. Then, 1H NMR of compound 27 in CD2Cl2 was recorded at 25 and −60 °C, and the C15-(R) configuration was confirmed by calculating the increment in chemical shift (Scheme 5, below).g
When the synthetic route was planned, the formation of the C22–C23 bond was envisioned as its last step based on our previous outstanding results on the formation of this bond while working in the synthesis of the polyhydroxylated chain. As depicted in Scheme 6a, the Mukaiyama addition of silylenolether 29(22) to aldehyde 28 gave not only almost quantitative yield but also total stereoselectivity toward the desired C23-(R) diastereomer 30.6 Bearing this precedent in mind, methyl ketone and the hydroxyl present in compound 6 were simultaneously transformed into the corresponding silylenolether and protected hydroxyl in quantitative yield to render the nucleophile for the final Mukaiyama addition, compound 31 (Scheme 6, b). However, when the Mukaiyama addition of silylenolether 31 to aldehyde 5 (using the same conditions applied for the preparation of 30) was performed,6 no conversion toward the product took place, recovering both starting materials after column purification. Modification of the reaction conditions by increasing the equivalents of BF3·Et2O and the temperature to 0 °C resulted in no reaction either. After these negative results, it was concluded that the formation of the C22–C23 bond using the Mukaiyama methodology with the coupling partners 5 and 31 was not possible in this case.
Scheme 6. (a) Precedent Work in the Formation of the C22–C23 Bond and (b) Endgame of the Total Synthesis.

Regents and conditions: (a) BF3·Et2O, −78 °C, CH2Cl2 (5% Et2O), 2 h, 98%, dr = 98:2; (b) TMSOTf, Et3N, DMAP, CH2Cl2, rt, 4 h, quant.; and (c) BF3·Et2O, −78 °C, CH2Cl2 (5% Et2O), 2 h, no reaction.
The reason for this lack of reactivity is unknown, but by comparison with our reported precedents, it can be hypothesized that steric congestion around carbons C22 and C23 hinders reagents approximation. The presence of a tetradecanoic ester (C27) and p-methoxybenzyl (PMB) (C25) in aldehyde 5 reduces somehow the necessary carbonyl activation by BF3·Et2O. In addition, a secondary TBSO-protected hydroxyl (C19) in the large functionalized compound 31 could suppress its reactivity as a nucleophile.
Conclusions
Highly convergent strategies are the process of choice for the total synthesis of large NPs such as PM D. The synthetic approach reported herein is composed of a total of 73 reactions, but the longest synthetic sequence to achieve PM D is only 20 steps long (the preparation of 5 from the commercially available starting material plus Mukaiyama addition and C21 reduction). The use of highly convergent strategies normally relies on final complex reactions because large molecular entities have to be efficient and stereoselectively linked.
In summary, this publication reports a highly convergent approach toward the NP PM D. A robust and scalable synthesis of fatty acids 7 and 8, present in PMs C and D, using a stereoselective and regioselective halogenation of acetylene to obtain (E)-dihalogenated terminal double bonds with high yield under mild conditions is described. Stereoselective efficient strategies for the preparation of 5 (C23–C31 fragment) and 9 (C16–C22 fragment) are fully described setting up the conditions to achieve total synthesis. A notable NHTK coupling of macrocyclic fragment 4 and iodoalkene 9 was performed in useful yield and with total substrate-controlled diastereoselectivity.
Finally, the last C22–C23 bond formation using the Mukaiyama methodology to link aldehyde 5 and silylenolether 31 did not work despite our previous encouraging results with smaller molecules. These adverse results do not invalidate the proposed retrosynthetic strategy that probably could give good results using less hindered protecting groups. Therefore, a revision of the protecting groups in positions C19 and C25 should be carried out before embarking on the synthesis of these complex NPs. Consequently, all the information reported in this publication will be of great value in the development of the total synthesis of MNPs PMs B–D.
Experimental Section
General Procedures
THF and N,N-dimethylformamide (DMF) were dried using a PureSolv solvent purification system. All other solvents and reagents were used as purchased without further purification, unless otherwise indicated. Flash column chromatography was performed on silica gel (60A 35–70 μm) as a stationary phase. Analytical thin-layer chromatography (TLC) was performed on precoated silica gel 60 F254 plates (0.2 mm thick, 20 × 20 cm) and visualized under UV light (254 and 360 nm), with anisaldehyde in concd H2SO4 or with phosphomolybdic acid in ethanol. Chemical shifts are reported in parts per million (ppm) referenced to the appropriate residual solvent peaks (CDCl3), and coupling constants are reported in hertz. Multiplicity of the carbons was assigned with gHSQC experiments. Standard abbreviations for off-resonance decoupling were employed: s = singlet, d = doublet, t = triplet, q = quadruplet, bs = broad singlet, bd = broad doublet, and m = multiplet. The same abbreviations were also used for the multiplicity of signals in 1H NMR. HRMS was performed in an Agilent LC/MSD-TOF 2006 system using the electrospray ionization mass spectrometry (ESI-MS) technique.
2-((12-Bromododecyl)oxy)tetrahydro-2H-pyran
PPTS (282 mg, 1.3 mmol) was added to a stirred solution of commercial 12-bromododecan-1-ol (10) (2.00 g, 7.5 mmol) in CH2Cl2 (50 mL), the reaction mixture was stirred for 5 min, and 3,4-dihydro-2H-pyran (0.95 g, 11.3 mmol) was added. The reaction mixture was allowed to stir at rt overnight under N2. Then, it was quenched with a saturated solution of NaHCO3, and the organic layer was extracted three times with Et2O. The organic extract was washed with brine, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude obtained (2.75 g) was purified by silica gel column chromatography with hexane/EtOAc (95:5 to 90:10) to obtain the title compound (2.51 g, 85%). 1H NMR (400 MHz, CDCl3): δ 1.24–1.34 (m, 14H), 1.39–1.44 (m, 2H), 1.51–1.61 (m, 6H), 1.66–1.76 (m, 1H), 1.78–1.90 (m, 3H), 3.32–3.42 (m, 3H), 3.46–3.53 (m, 1H), 3.73 (dt, J = 9.6, 6.9 Hz, 1H), 3.82–3.90 (m, 1H), 4.55–4.60 (m, 1H). 13C NMR (100.6 MHz, CDCl3): δ 19.7 (t), 25.5 (t), 26.2 (t), 28.2 (t), 28.7 (t), 29.4 (t), 29.4 (t), 29.5 (t), 29.5 (t), 29.5 (t), 29.7 (t), 30.8 (t), 32.8 (t), 34.0 (t), 62.3 (t), 67.7 (t), 98.8 (d).
2-(Tetradec-13-yn-1-yloxy)tetrahydro-2H-pyran (11)
The lithium acetylide–ethylenediamine complex (1.37 g, 13.35 mmol) was stirred in dimethyl sulfoxide (DMSO, 10 mL) at 15–20 °C for 1 h. Then, a solution of the above-mentioned bromide (2.51 g, 6.36 mmol) in DMSO (10 mL) was added at rt for 2 h and under N2. The resulting mixture was stirred for 12 h. Then, it was quenched with water (10 mL), and the organic layer was extracted three times with hexane. The organic extract was washed several times with brine, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude obtained (2.10 g) was purified by silica gel column chromatography with hexane/EtOAc (99:1 to 97:3) to obtain 11 (1.47 g, 79%). 1H NMR (400 MHz, CDCl3): δ 1.22–1.39 (m, 15H), 1.47–1.61 (m, 9H), 1.64–1.74 (m, 1H), 1.76–1.86 (m, 1H), 1.92 (t, J = 2.7 Hz, 1H), 2.17 (td, J = 7.1, 2.7 Hz, 2H), 3.37 (dt, J = 9.5, 6.9 Hz, 1H), 3.45–3.52 (m, 1H), 3.72 (dt, J = 9.5, 6.9 Hz, 1H), 3.86 (ddd, J = 11.1, 6.9, 3.4 Hz, 1H), 4.53–4.58 (m, 1H). 13C NMR (100.6 MHz, CDCl3): δ 18.4 (t), 19.7 (t), 25.5 (t), 26.2 (t), 28.5 (t), 28.7 (t), 29.1 (t), 29.5 (t), 29.5 (t), 29.7 (t), 30.8 (t), 62.3 (t), 67.7 (t), 68.0 (d), 84.8 (s), 98.8 (d).
(E)-2-((14-Bromo-13-chlorotridec-13-en-1-yl)oxy)tetrahydro-2H-pyran (12)
TBADCB (4.16 g, 10.6 mmol) was added to a solution of alkyne 11 (880 mg, 2.98 mmol) in anhydrous CH2Cl2 (20 mL) at rt and under N2. The reaction mixture was stirred for 2.5 h. Then, it was quenched with water (10 mL), and the organic layer was extracted three times with CH2Cl2. The organic extract was washed with brine, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure to obtain 3.62 g of crude 12 as an 89:11 mixture of regioisomers which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.23–1.30 (m, 12H), 1.44–1.55 (m, 8H), 1.61–1.71 (m, 6H), 2.14 (td, J = 7.1, 2.7 Hz, 1H), 2.44–2.52 (m, 1H), 3.43–3.49 (m, 1H), 3.61 (t, J = 6.7 Hz, 1H), 3.66–3.71 (m, 1H), 3.79–3.87 (m, 1H), 4.52–4.56 (m, 1H), 6.18 (s, 1H).
(E)-14-Bromo-13-chlorotetradec-13-en-1-ol (S1)
p-TsOH (103 mg, 0.60 mmol) was added to a solution of alkene 12 (1.21 g, 2.97 mmol) in MeOH (20 mL), and the mixture was stirred for 3 h at rt. Then, it was quenched with a saturated solution of NaHCO3 (10 mL), and the organic layer was extracted three times with Et2O. The organic layer was washed with a saturated solution of NaHCO3 several times, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude of S1 (1.22 g) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.30 (m, 14H), 1.56 (d, J = 7.4 Hz, 6H), 2.36–2.44 (m, 1H), 2.48–2.55 (m, 1H), 3.64 (t, J = 6.6 Hz, 2H), 6.22 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 25.7 (t), 26.3 (t), 28.5 (t), 29.3 (t), 29.4 (t), 29.5 (t), 29.5 (t), 29.6 (t), 29.6 (t), 32.8 (t), 35.1 (t), 63.1 (t), 101.0 (d), 136.9 (s).
(E)-14-Bromo-13-chlorotetradec-13-enal (S2)
DMP (1.18 g, 2.70 mmol) was added to a solution of S1 (703 mg, 2.15 mmol) in dry CH2Cl2 (20 mL) at rt and under N2. The reaction mixture was stirred for 3 h. Then, it was quenched with a saturated solution of NaHCO3, and the saturated solution of NaS2O3 and the mixture were left stirring for 20 min. The aqueous layer was extracted three times with EtOAc, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude of S2 (751 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.30 (m, 14H), 1.57–1.66 (m, 4H), 2.40–2.45 (m, 2H), 2.53 (t, J = 7.4 Hz, 2H), 6.22 (s, 1H), 9.77 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 22.1 (t), 26.3 (t), 28.5 (t), 29.2 (t), 29.3 (t), 29.3 (t), 29.4 (t), 29.4 (t), 29.5 (t), 35.1 (t), 43.9 (t), 101.1 (d), 136.9 (s), 202.9 (s).
(E)-14-Bromo-13-chlorotetradec-13-enoic Acid (7)
NaClO2 (832 mg, 9.2 mmol) was added to a solution of aldehyde S2 (750 mg, 2.3 mmol) in tBuOH/H2O (10 mL/2 mL), in the presence of NaH2PO4 (1.4 g, 11.5 mmol) and 2-methyl-butene (806 mg, 11.5 mmol), at rt. The reaction mixture was stirred for 2 h and 30 min. Then, it was quenched with water and Na2S2O3. The organic phase was extracted three times with EtOAc and washed with brine. The organic extract was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The crude 7 (794 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.29–1.36 (m, 14H), 1.53–1.68 (m, 4H), 2.35 (t, J = 7.5 Hz, 2H), 2.50–2.55 (t, J = 7.4 Hz, 2H), 6.22 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 24.7 (t), 26.4 (t), 28.5 (t), 29.0 (t), 29.2 (t), 29.3 (t), 29.4 (t), 29.4 (t), 29.5 (t), 33.9 (t), 35.1 (t), 101.1 (d), 136.9 (s), 179.0 (s).
Methyl (E)-14-Bromo-13-chlorotetradec-13-enoate (14)
Trimethylsilyldiazomethane (TMSCHN2; 778 mg, 6.81 mmol) was added to a solution of 7 (774 mg, 2.27 mmol) in CH2Cl2/MeOH (9:1) at 0 °C and under N2. The reaction mixture was stirred for 3 h at 0–5 °C. The solvent was evaporated under reduced pressure, and the residue was purified in preparative RP-HPLC to obtain 14 (209 mg). Global yield from 11 to 14 20%. 1H NMR (400 MHz, CDCl3): δ 1.22–1.33 (m, 14H), 1.51–1.65 (m, 4H), 2.29 (t, J = 7.5 Hz, 2H), 2.46–2.55 (m, 2H), 3.66 (s, 3H), 6.21 (s, 1H). 13C NMR (100.6 MHz, CDCl3): δ 24.9 (t), 26.3 (t), 28.5 (t), 29.1 (t), 29.2 (t), 29.3 (t), 29.4 (t), 29.4 (t), 29.5 (t), 34.1 (t), 35.1 (t), 51.4 (q), 101.0 (d), 136.9 (s), 174.3 (s). HRMS (ESI+) m/z: calcd for C15H27BrClO2 (M + H), 353.0883; found, 353.0857.
(E)-2-((13,14-Dichlorotetradec-13-en-1-yl)oxy)tetrahydro-2H-pyran (13)
TBATC (3.96 g, 10.6 mmol) was added to a solution of alkyne 11 (880 mg, 2.98 mmol) in dry CH2Cl2 (20 mL) at rt and under N2. The reaction mixture was stirred for 2.5 h and quenched with water (10 mL). The organic layer was extracted three times with CH2Cl2. The organic extract was washed with brine, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. Crude 13 obtained (957 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.24–1.35 (m, 14H), 1.47–1.63 (m, 12H), 2.50 (t, J = 7.4 Hz, 2H), 3.38 (dt, J = 9.6, 6.7 Hz, 2H), 3.49 (m, 1H), 3.72 (dt, J = 9.6, 6.7 Hz, 1H), 3.87 (m, 1H), 4.57 (m, 1H), 6.13 (s, 1H).
(E)-13,14-Dichlorotetradec-13-en-1-ol (S3)
p-TsOH (262 mg, 1.52 mmol) was added to a solution of alkene 13 (926 mg, 2.54 mmol) in MeOH (20 mL) at rt. The reaction mixture was stirred for 1.5 h, then quenched with a saturated solution of NaHCO3 (10 mL), and extracted three times with Et2O. The organic layer was washed with a saturated solution of NaHCO3 several times, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude of S3 (819 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.24–1.34 (m, 14H), 1.41–1.51 (m, 4H), 1.52–1.62 (m, 2H), 2.50 (t, J = 7.4 Hz, 2H), 3.64 (t, J = 6.6 Hz, 2H), 6.13 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 25.7 (t), 26.3 (t), 28.5 (t), 29.3 (t), 29.4 (t), 29.5 (t), 29.5 (t), 29.6 (t), 29.6 (t), 32.8 (t), 33.1 (t), 63.1 (t), 113.5 (d).
(E)-13,14-Dichlorotetradec-13-enal (S4)
DMP (1.55 g, 3.5 mmol) was added to a solution of S3 (779 mg, 2.7 mmol) in dry CH2Cl2 (20 mL) at rt and under N2. The reaction was quenched with a saturated solution of NaHCO3, and the saturated solution of NaS2O3 and the mixture were left stirring for 20 min. The aqueous layer was extracted three times with EtOAc, dried over MgSO4, and filtered. The filtrate was evaporated under reduced pressure. The crude of S4 (605 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.29 (m, 14H), 1.44–1.66 (m, 4H), 2.35 (t, J = 7.5 Hz, 2H), 2.50 (t, J = 7.4 Hz, 2H), 6.13 (s, 1H), 9.76 (t, J = 2.0 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 22.1 (t), 26.3 (t), 28.5 (t), 29.1 (t), 29.3 (t), 29.3 (t), 29.4 (t), 29.4 (t), 29.5 (t), 33.1 (t), 43.9 (t), 113.5 (d), 136.6 (s), 203.0 (s).
(E)-13,14-Dichlorotetradec-13-enoic Acid (8)
NaClO2 (734 mg, 8.12 mmol) was added to a solution of aldehyde S4 (565 mg, 2.03 mmol) in tBuOH/H2O (14 mL/6 mL), containing NaH2PO4 (1.21 g, 10.15 mmol) and 2-methyl-butene (0.711 g, 10.15 mmol), at rt. The reaction mixture was stirred for 2 h and 30 min. The reaction was quenched with water and Na2S2O3. The organic phase was extracted three times with EtOAc and washed with brine. The organic extract was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. Crude 8 (602 mg) was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 1.31 (m, 14H), 1.45–1.62 (m, 4H), 2.35 (t, J = 7.5 Hz, 2H), 2.49 (d, J = 7.4 Hz, 2H), 6.13 (s, 1H). 13C NMR (101 MHz, chloroform-d): δ 24.7 (t), 26.3 (t), 28.5 (t), 29.0 (t), 29.2 (t), 29.4 (2C, t), 29.4 (t), 29.5 (t), 33.1 (t), 34.0 (t), 113.5 (d), 136.6 (s), 179.7 (s).
Methyl (E)-13,14-Dichlorotetradec-13-enoate (15)
TMSCHN2 (0.651 g, 5.7 mmol) was added to a solution of acid 8 (0.562 g, 1.9 mmol) in dichloromethane (DCM)/MeOH (9:1) at 0 °C and under N2. The reaction mixture was stirred for 3 h at 0–5 °C. The solvent was evaporated under reduced pressure, and the residue was purified in preparative RP-HPLC to obtain 15 (0.110 g, 0.35 mmol). Global yield from 11 to 15: 12%. 1H NMR (400 MHz, CDCl3): δ 1.30 (m, 14H), 1.59–1.65 (m, 4H), 2.30 (t, J = 7.5 Hz, 4H), 2.50 (t, J = 7.4 Hz, 3H), 3.66 (s, 3H), 6.13 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 25.0 (t), 26.3 (t), 28.5 (t), 29.1 (t), 29.2 (t), 29.3 (t), 29.4 (t), 29.4 (t), 29.5 (t), 33.1 (t), 34.1 (t), 51.4 (q), 113.5 (d), 136.4 (s), 174.3 (s). HRMS (ESI+) m/z: calcd for C15H27Cl2O2 (M + H), 309.1388; found, 309.1383.
tert-Butyl((2S)-2-((4S)-2-(4-methoxyphenyl)-1,3-dioxan-4-yl)propoxy)dimethylsilane (S5)
tert-Butyldimethylsilyl chloride (TBSCl; 1.25 g, 8.3 mmol) was added to a solution of alcohol 16(6) (1.70 g, 6.7 mmol) and imidazole (684 mg, 10.1 mmol) in CH2Cl2 (60 mL). The reaction mixture was stirred for 90 min. After this time, the mixture was washed with water, dried over MgSO4, and filtered and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (95:5) yielded S5 (2.0 g, 86%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.05 (s, 3H), 0.05 (s, 3H), 0.91 (s, 9H), 1.00 (d, J = 6.9 Hz, 3H), 1.45 (dtd, J = 13.2, 2.4, 1.4 Hz, 1H), 1.78 (pt, J = 6.9, 5.1 Hz, 1H), 1.95 (tdd, J = 13.2, 12.3, 11.3, 5.1 Hz, 1H), 3.55 (dd, J = 9.9, 5.1 Hz, 1H), 3.64 (dd, J = 9.9, 6.9 Hz, 1H), 3.80 (s, 3H), 3.83–4.01 (m, 2H), 4.27 (ddd, J = 11.3, 5.1, 1.4 Hz, 1H), 5.46 (s, 1H), 6.84–6.95 (m, 2H), 7.37–7.46 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.4 (q), 11.9 (q), 18.3 (s), 25.9 (q), 28.7 (t), 40.6 (d), 55.2 (q), 64.5 (t), 67.2 (d), 77.4 (d), 101.0, 113.4 (d), 127.2 (d), 131.7 (s), 159.7 (s). HRMS (ESI+) m/z: calcd for C20H34NaO4Si [M + Na]+, 389.2119; found, 389.2111.
(3S,4S)-5-((tert-Butyldimethylsilyl)oxy)-3-((4-methoxybenzyl)oxy)-4-methylpentan-1-ol (S6)
A solution of diisobutylaluminium hydride 1 M in heptane (1.3 mL, 1.3 mmol) was added to a solution of S5 (355 mg, 0.97 mmol) in CH2Cl2 (15 mL) at −20 °C. The reaction mixture was stirred at 0 °C for 2 h. Then, a saturated solution of Rochelle’s salt (15 mL) was added, and the mixture was stirred for 1 h until the formation of two layers. The organic solution was dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (8:2) yielded S6 (285 mg, 90% br s m) as a colorless oil and 43 mg of recovered starting material. 1H NMR (400 MHz, CDCl3): δ 0.05 (s, 6H), 0.90 (s, 9H), 0.95 (d, J = 6.9 Hz, 3H), 1.67–1.83 (m, 2H), 1.85–2.00 (m, 1H), 2.17 (m, 1H), 3.50 (dd, J = 9.9, 6.9 Hz, 1H), 3.62–3.78 (m, 4H), 3.80 (s, 3H), 4.46 (d, J = 11.0 Hz, 1H), 4.55 (d, J = 11.0 Hz, 1H), 6.83–6.92 (m, 2H), 7.22–7.31 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.4 (q), 12.6 (q), 18.3 (s), 25.9 (q), 33.6 (t), 39.0 (d), 55.3 (q), 61.2 (t), 64.7 (t), 71.9 (t), 79.2 (d), 113.8 (d), 129.5 (d), 130.7 (s), 159.2 (s). HRMS (ESI+) m/z: calcd for C20H37O4Si [M + H]+, 369.2456; found, 369.2446.
(3S,4S)-5-((tert-Butyldimethylsilyl)oxy)-3-((4-methoxybenzyl)oxy)-4-methylpentanal (17)
DMSO (0.031 mL, 0.44 mmol) was added to a solution of oxalyl chloride (0.019 mL, 0.22 mmol) in CH2Cl2 (2 mL) at −78 °C, and the reaction mixture was stirred for 30 min. A solution of alcohol S6 (40 mg, 0.11 mmol) in CH2Cl2 (1 mL) was added, and the mixture was stirred for 45 min at the same temperature. Et3N (0.123 mL, 0.88 mmol) was added, and the reaction mixture was stirred for 30 min at 0 °C. Then, it was quenched with NH4Cl saturated solution and extracted with CH2Cl2. The organic layer was washed with brine, dried over MgSO4, and evaporated. Purification by silica gel chromatography with hexane/EtOAc (9:1) yielded aldehyde 17 (32 mg, 80%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.05 (2s, 6H), 0.90 (s, 9H), 0.93 (d, J = 7.0 Hz, 3H), 1.81–1.92 (m, 1H), 2.56 (ddd, J = 16.4, 4.6, 2.0 Hz, 1H), 2.71 (ddd, J = 16.4, 7.8, 2.0 Hz, 1H), 3.53 (dd, J = 10.0, 5.7 Hz, 1H), 3.61 (dd, J = 10.0, 6.5 Hz, 1H), 3.80 (s, 3H), 4.03–4.10 (m, 1H), 4.49 (s, 2H), 6.83–6.88 (m, 2H), 7.20–7.25 (m, 2H), 9.76 (t, J = 2.0 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ −5.5 (q), −5.4 (q), 12.1 (q), 18.2 (s), 25.9 (q), 39.7 (d), 46.6 (t), 55.3 (q), 64.5 (t), 72.0 (t), 74.8 (d), 113.8 (d), 129.3 (d), 130.5 (s), 159.2 (s), 201.9 (s). HRMS (ESI+) m/z: calcd for C20H34NaO4Si [M + Na]+, 389.2119; found, 389.2130.
(5S,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-5-hydroxy-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methylnon-1-en-3-one (18)
A solution of silylenolether (E)-((4-bromo-3-methoxybuta-1,3-dien-2-yl)oxy)trimethylsilane(6) (527 mg, 2.1 mmol) in CH2Cl2 (10 mL) was added to a cooled (−78 °C) solution of aldehyde 17 (503 mg, 1.37 mmol) in CH2Cl2 (30 mL), and then BF3·OEt2 (155 μL, 1.26 mmol) was added dropwise. The reaction was stirred at −78 °C for 60 min, quenched with the saturated solution of NaHCO3 (30 mL), and extracted three times with CH2Cl2. The organic solution was dried over Na2SO4, filtrated, and concentrated under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (8:2) afforded 18 (312 mg, 47%) as an 82:17 mixture of diastereomers. Mayor diastereomer data: 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H), 0.89 (s, 9H), 0.94 (d, J = 6.9 Hz, 3H), 1.60–1.64 (m, 2H), 1.87–1.93 (m, 1H), 2.76–2.85 (m, 2H), 3.44–3.52 (m, 1H), 3.63 (s, 3H), 3.66 (dd, J = 9.9, 5.8 Hz, 1H), 3.80 (s, 3H), 3.80–3.81 (m, 1H), 4.23–4.35 (m, 1H), 4.46–4.57 (m, 2H), 5.66 (s, 1H), 6.82–6.89 (m, 2H), 7.24–7.28 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.3 (q), 12.5 (q), 18.3 (s), 25.9 (q), 38.3 (t), 39.4 (d), 47.1 (t), 55.3 (q), 56.0 (q), 64.7 (d), 64.8 (t), 72.4 (t), 76.8 (d), 84.4 (d), 113.8 (d), 129.5 (d), 130.9 (s), 152.9 (s), 159.2 (s), 197.7 (s). HRMS (ESI+) m/z: calcd for C25H41BrNaO6Si [M + Na]+, 567.1748; found, 567.1744.
(5S,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-5-((triethylsilyl)oxy)non-1-en-3-one (S7)
Imidazole (140 mg, 2 mmol), TESCl (0.14 mL, 0.85 mmol), and DMAP (6 mg, 0.05 mmol) were added sequentially to a solution of 18 (290 mg, 0.53 mmol) in CH2Cl2 (10 mL). The cloudy solution was stirred 45 min until the TLC analysis showed complete conversion of the starting material. The reaction was quenched with the saturated solution of NH4Cl (10 mL) and extracted three times with CH2Cl2. The organic layers were dried over MgSO4 and filtered, and the solvent was evaporated under reduced pressure. Purification with deactivated (1% Et3N) silica gel column chromatography with hexane/EtOAc (95:5) afforded S7 (300 mg, 86%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.03 (s, 6H), 0.56–0.63 (m, 6H), 0.89 (s, 9H), 0.89–0.96 (m, 12H), 1.68 (dt, J = 6.6, 5.4 Hz, 2H), 1.89 (qd, J = 6.9, 3.2 Hz, 1H), 2.78 (dd, J = 16.3, 5.4 Hz, 1H), 2.95 (dd, J = 16.3, 6.6 Hz, 1H), 3.41 (dd, J = 9.8, 7.0 Hz, 1H), 3.59 (s, 3H), 3.61–3.67 (m, 2H), 3.80 (s, 3H), 4.31–4.38 (m, 1H), 4.39–4.51 (m, 2H), 5.58 (s, 1H), 6.83–6.88 (m, 2H), 7.22–7.26 (m, 2H). HRMS (ESI+) m/z: calcd for C31H56BrO6Si2 [M + H]+, 659.2793; found, 659.2762.
(5R,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylene-5-((triethylsilyl)oxy)non-1-ene (S8)
A solution of the Tebbe reagent in toluene (0.5 M, 3.62 mL, 1.81 mmol) was added to a solution of S7 (290 mg, 0.45 mmol) and pyridine (0.150 mL, 1.81 mmol) in THF (12 mL) at 0 °C. The reaction mixture was warmed to rt and then heated up at 50 °C for 90 min. Then, it was quenched at rt with the saturated solution of Rochelle’s salt (3 mL) and extracted three times with Et2O. The organic phase was dried over MgSO4 and filtered, and the solvent was evaporated under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (95:5) yielded alkene S8 (235 mg, 81%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.03 (s, 6H), 0.54–0.64 (m, 6H), 0.87–0.90 (m, 12H), 0.92–0.97 (m, 9H), 1.43–1.50 (m, 1H), 1.76–1.85 (m, 1H), 1.92 (qd, J = 6.8, 3.2 Hz, 1H), 2.29 (dd, J = 13.8, 9.0 Hz, 1H), 2.61 (dd, J = 13.8, 4.2 Hz, 1H), 3.41 (dd, J = 9.8, 7.2 Hz, 1H), 3.49 (s, 3H), 3.62–3.72 (m, 2H), 3.80 (s, 3H), 3.88–3.96 (m, 1H), 4.42 (d, J = 11.1 Hz, 1H), 4.50 (d, J = 11.1 Hz, 1H), 5.26 (s, 1H), 5.33 (s, 1H), 5.38 (s, 1H), 6.79–6.88 (m, 2H), 7.23–7.26 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 5.3 (t), 7.2 (q), 12.1 (q), 26.1 (q), 38.9 (t), 39.4 (d), 43.4 (t), 55.3 (q), 55.4 (q), 65.1 (t), 68.5 (d), 71.0 (t), 77.0 (d), 78.4 (d), 113.5 (d), 121.3 (t), 128.6 (d). HRMS (ESI+) m/z: calcd for C32H58BrO5Si2 [M + H]+, 657.3001; found, 657.2991.
(5R,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-ol (19)
PPTS (17 mg, 0.07 mmol) was added to a solution of S8 (235 mg, 0.357 mmol) in MeOH (13 mL) at 0 °C, and the reaction mixture was stirred for 45 min at rt. After this time, the reaction mixture was quenched with the saturated solution of NaHCO3 (5 mL) and extracted three times with CH2Cl2. The organic phase was dried over MgSO4 and filtered, and the solvent was evaporated under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (85:15) yielded alcohol 19 (170 mg, 88%) as a colorless oil (mixture of isomers). Separation of diastereomers was performed at this point via silica column chromatography. 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H), 0.90 (s, 9H), 0.94 (d, J = 6.9 Hz, 3H), 1.52–1.65 (m, 2H), 1.84–1.93 (m, 1H), 2.28 (dd, J = 13.8, 9.0 Hz, 1H), 2.44 (dd, J = 13.8, 4.0 Hz, 1H), 3.46 (dd, J = 9.9, 6.9 Hz, 1H), 3.56 (s, 3H), 3.67 (dd, J = 9.9, 5.8 Hz, 1H), 3.80 (s, 3H), 3.83 (s, 1H), 3.86 (s, 1H), 4.48 (d, J = 10.9 Hz, 1H), 4.54 (d, J = 10.9 Hz, 1H), 5.32 (s, 1H), 5.38 (s, 2H), 6.84–6.89 (m, 2H), 7.25–7.28 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.3 (q), 12.5 (q), 18.3 (s), 25.9 (q), 38.4 (t), 39.5 (d), 43.1 (t), 55.3 (q), 55.6 (q), 64.9 (t), 66.5 (d), 72.5 (t), 77.1 (d), 78.6 (d), 113.8 (d), 122.1 (t), 129.5 (d), 131.1 (s), 139.7 (s), 159.1 (s), 159.1 (s). HRMS (ESI+) m/z: calcd for C26H43BrNaO5Si [M + Na]+, 565.1955; found, 565.1943.
General Procedure for Derivatization with MPA
α-MPA (5 equiv) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)·HCl (5 equiv) were added to a solution of alcohol 10(5R) (1 equiv) in THF, then DMAP (0.1 equiv) was added, and the solution was stirred for 2 h at 40 °C. The solution was filtered through Celite 545, poured into Et2O, and washed with NH4Cl saturated solution. The organic residue was dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (90:10) yielded the corresponding esters as colorless oils.
(5R,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-yl-(R)-2-methoxy-2-phenylacetate (S9)
Alcohol 19 (4 mg, 0.007 mmol) and (R)-MPA (5 equiv) afforded (R)-MPA ester S9 (4 mg, 83%). 1H NMR (400 MHz, CDCl3): δ 0.01 (s, 6H), 0.67 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 1.55–1.58 (m, 2H), 1.64–1.69 (m, 1H), 2.49 (dd, J = 14.5, 6.4 Hz, 1H), 2.58 (dd, J = 14.5, 5.8 Hz, 1H), 2.79 (dt, J = 8.5, 3.9 Hz, 1H), 3.21–3.32 (m, 1H), 3.41 (s, 3H), 3.49–3.52 (m, 1H), 3.53 (s, 3H), 3.79 (s, 3H), 3.85 (d, J = 10.3 Hz, 1H), 4.07 (d, J = 10.3 Hz, 1H), 4.72 (s, 1H), 5.18–5.26 (m, 1H), 5.30 (s, 1H), 5.33 (d, J = 1.4 Hz, 1H), 5.42 (d, J = 1.4 Hz, 1H), 6.81–6.85 (m, 2H), 7.15–7.20 (m, 2H), 7.30–7.37 (m, 3H), 7.43–7.47 (m, 2H). HRMS (ESI+) m/z: calcd for C35H55BrNO7Si [M + NH4]+, 708.2926; found, 708.2876.
(5R,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-yl-(S)-2-methoxy-2-phenylacetate (S10)
Alcohol 19 (4 mg, 0.007 mmol) and (S)-MPA (5 equiv) afforded (S)-MPA ester S10 (5 mg, quant.). 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H), 0.86 (d, J = 6.9 Hz, 3H), 0.90 (s, 9H), 1.68–1.74 (m, 2H), 1.76–1.83 (m, 1H), 2.35 (dd, J = 14.5, 5.7 Hz, 1H), 2.47 (dd, J = 14.5, 6.7 Hz, 1H), 3.32–3.36 (m, 1H), 3.38–3.41 (m, 1H), 3.41 (s, 3H), 3.45 (s, 3H), 3.61 (dd, J = 9.8, 6.0 Hz, 1H), 3.79 (s, 3H), 4.20 (d, J = 10.5 Hz, 1H), 4.32 (d, J = 10.5 Hz, 1H), 4.72 (s, 1H), 5.03 (d, J = 1.4 Hz, 1H), 5.21 (s, 3H), 6.82–6.86 (m, 2H), 7.22–7.26 (m, 2H), 7.30–7.35 (m, 3H), 7.42–7.47 (m, 2H). HRMS (ESI+) m/z: calcd for C35H55BrNO7Si [M + NH4]+, 708.2926; found, 708.2921.
(5S,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-ol (20)
Diisopropyl azodicarboxylate (DIAD; 0.22 mL, 1.06 mmol) was slowly added to a solution of 19 (105 mg, 0.19 mmol), p-nitrobenzoic acid (170 mg, 1 mmol), and PPh3 (294 mg, 1 mmol) in benzene (8 mL), and the yellowish solution was stirred for 4 h at 40 °C. The reaction mixture was quenched with the saturated solution of NaHCO3 (10 mL) and extracted three times with Et2O. The organic layer was dried over MgSO4, filtered, and evaporated under reduced pressure. The crude was filtered through a plug of silica using hexane/EtOAc 9:1 to eliminate the p-nitrobenzoic acid and PPh3O (Rf = 0.1). After evaporation, the resulting crude was dissolved in MeOH (10 mL), K2CO3 (180 mg, 1.30 mmol) was added, and the mixture was stirred for 2 h until TLC indicated total hydrolysis. The reaction mixture was quenched with the saturated solution of NH4Cl (10 mL) and extracted three times with AcOEt. The organic layer was dried over MgSO4, filtered, evaporated under reduced pressure, and purified by silica gel column chromatography with hexane/EtOAc 9:1 to obtain 20 as a colorless oil (60 mg, 59% for two steps). 1H NMR (400 MHz, CDCl3): δ 0.05 (s, 6H), 0.90 (s, 9H), 0.92 (d, J = 7.0 Hz, 3H), 1.59–1.72 (m, 2H), 1.91–2.00 (m, 1H), 2.35 (dd, J = 13.8, 6.4 Hz, 1H), 2.45 (dd, J = 13.8, 6.8 Hz, 1H), 3.46–3.51 (m, 1H), 3.57 (s, 3H), 3.67 (dd, J = 9.9, 5.9 Hz, 1H), 3.71–3.77 (m, 2H), 3.79 (s, 3H), 4.43 (d, J = 10.9 Hz, 1H), 4.55 (d, J = 10.9 Hz, 1H), 5.31 (s, 1H), 5.36 (s, 2H), 6.84–6.89 (m, 2H), 7.22–7.27 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.3 (q), 12.3 (q), 18.3 (s), 25.9 (q), 37.3 (t), 38.7 (d), 42.6 (t), 55.3 (q), 55.6 (q), 64.5 (t), 69.3 (d), 71.5 (t), 78.6 (d), 80.2 (d), 113.8 (d), 121.8 (t), 129.4 (d), 130.5 (s), 139.5 (s), 159.0 (s), 159.2 (s). HRMS (ESI+) m/z: calcd for C26H43BrNaO5Si [M + Na]+, 565.1955; found, 565.1959.
(5S,7S,8S,E)-1-Bromo-9-((tert-butyldimethylsilyl)oxy)-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-yl-(E)-13,14-dichlorotetradec-13-enoate (21)
(E)-13,14-Dichlorotetradec-13-enoic acid (8) (82 mg, 0.28 mmol) was added to a solution of EDC·HCl (268 mg, 1.4 mmol), Et3N (0.195 mL, 1.4 mmol), and DMAP (171 mg, 1.4 mmol) in CH2Cl2 (3 mL), and the solution was stirred for 20 min. Alcohol 20 (50 mg, 0.092 mmol) dissolved in CH2Cl2 (2.5 mL) was added, and the solution was stirred for 16 h. The reaction mixture was quenched with the saturated solution of NH4Cl (8 mL) and extracted three times with AcOEt. The organic layer was dried over MgSO4, filtered, evaporated under reduced pressure, and purified by silica gel column chromatography with hexane/EtOAc 95:5 to obtain ester 21 as a colorless oil (35 mg, 56% br s m) and recovered 20 (8 mg). 1H NMR (400 MHz, CDCl3): δ 0.03 (s, 6H), 0.86 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 1.25–1.31 (m, 14H), 1.55 (s, 4H), 1.76–1.91 (m, 3H), 2.23 (td, J = 7.6, 3.8 Hz, 2H), 2.47–2.59 (m, 4H), 3.44 (dd, J = 9.8, 6.5 Hz, 1H), 3.54 (s, 3H), 3.56–3.62 (m, 2H), 3.80 (s, 3H), 4.41 (d, J = 11.1 Hz, 1H), 4.48 (d, J = 11.1 Hz, 1H), 5.00 (p, J = 6.5 Hz, 1H), 5.31 (s, 1H), 5.34 (d, J = 1.3 Hz, 1H), 5.41 (d, J = 1.3 Hz, 1H), 6.13 (s, 1H), 6.84–6.88 (m, 2H), 7.25–7.27 (m, 2H). 13C NMR (101 MHz, CDCl3): δ −5.4 (q), −5.4 (q), 11.0 (q), 18.2 (s), 24.9 (t), 25.9 (q), 26.3 (t), 28.5 (t), 29.2 (t), 29.3 (t), 29.3 (t), 29.5 (t, 2C), 29.5 (t), 33.1 (t), 34.5 (t), 35.3 (t), 38.8 (t + d, 2C), 55.3 (q), 55.6 (q), 64.9 (t), 70.0 (d), 71.2 (t), 75.7 (d), 78.7 (d), 113.5 (d), 113.6 (d), 122.0 (t), 129.1 (d), 131.2 (s), 136.6 (s), 138.4 (s), 141.7 (s), 158.5 (s), 173.0 (s). HRMS (ESI+) m/z: calcd for C40H69BrCl2NO6Si [M + NH4]+, 836.3449; found, 836.3439.
(5S,7S,8S,E)-1-Bromo-9-hydroxy-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylenenon-1-en-5-yl-(E)-13,14-dichlorotetradec-13-enoate (S11)
A 1 M TBAF solution (0.071 mL, 0.071 mmol) was added to a solution of 21 (29 mg, 0.035 mmol) in THF (0.7 mL), and the solution was stirred at rt for 6 h. The reaction mixture was quenched with the saturated solution of NH4Cl (1 mL) and extracted three times with Et2O. The organic layer was dried over MgSO4, filtered, evaporated under reduced pressure, and purified by silica gel column chromatography with hexane/EtOAc 7:3 to obtain alcohol S11 as a colorless oil (19 mg, 77%). 1H NMR (400 MHz, CDCl3): δ 0.87 (d, J = 7.1 Hz, 3H), 1.25–1.31 (m, 14H), 1.55–1.59 (m, 4H), 1.78–1.86 (m, 1H), 1.87–1.95 (m, 1H), 2.04–2.08 (m, 1H), 2.19–2.28 (m, 2H), 2.46–2.56 (m, 4H), 3.56 (s, 3H), 3.50–3.64 (m, 3H), 3.80 (s, 3H), 4.40–4.52 (m, 2H), 5.02 (p, J = 6.5 Hz, 1H), 5.32 (s, 1H), 5.35 (d, J = 1.4 Hz, 1H), 5.43 (d, J = 1.4 Hz, 1H), 6.13 (s, 1H), 6.85–6.89 (m, 2H), 7.24–7.27 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 11.2 (q), 24.9 (t), 26.3 (t), 28.5 (d), 29.2 (t), 29.3 (t), 29.3 (t), 29.5 (t), 29.5 (t), 33.1 (t), 34.3 (t), 34.6 (t), 36.9 (d), 39.2 (t), 55.3 (q), 55.6 (q), 66.0 (t), 69.8 (d), 70.9 (t), 77.8 (d), 78.8 (d), 113.5 (d), 113.8 (d), 122.2 (t), 129.4 (d), 130.3 (s), 134.4 (s), 138.2 (s), 158.3 (s), 159.2 (s), 173.3 (s). HRMS (ESI+) m/z: calcd for C34H55BrCl2NO6 [M + NH4]+, 722.2584; found, 722.2588.
(5S,7S,8R,E)-1-Bromo-2-methoxy-7-((4-methoxybenzyl)oxy)-8-methyl-3-methylene-9-oxonon-1-en-5-yl-(E)-13,14-dichlorotetradec-13-enoate (5)
NaHCO3 (4 mg, 0.047 mmol) and DMP (5 mg, 0.118 mmol) were added to a solution of S11 (2.5 mg, 0.0035 mmol) in CH2Cl2 (0.8 mL), and the solution was stirred for 30 min. The reaction mixture was diluted with Et2O (10 mL) and quenched with saturated solutions of NaHCO3 and Na2S2O3 (2 + 2 mL). The crude was extracted twice with Et2O, and the organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (8:2) yielded aldehyde 5 (2.3 mg, 93%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 1.11 (d, J = 7.0 Hz, 3H), 1.27 (s, 14H), 1.56 (s, 4H), 1.85 (ddd, J = 14.5, 7.5, 4.2 Hz, 1H), 1.98 (ddd, J = 14.5, 8.5, 5.8 Hz, 1H), 2.19–2.27 (m, 2H), 2.46–2.61 (m, 5H), 3.56 (s, 3H), 3.79 (s, 3H), 3.91 (ddd, J = 7.5, 5.8, 3.0 Hz, 1H), 4.37 (d, J = 11.2 Hz, 1H), 4.44 (d, J = 11.2 Hz, 1H), 4.94–5.05 (m, 1H), 5.33 (s, 1H), 5.36 (s, 1H), 5.44 (s, 1H), 6.13 (s, 1H), 6.83–6.88 (m, 2H), 7.18–7.23 (m, 2H), 9.64 (s, 1H). 13C NMR (101 MHz, CDCl3): δ 7.5 (q), 24.9 (t), 26.3 (t), 28.5 (t), 29.2 (t), 29.3 (d), 29.5 (t), 29.5 (t), 33.1 (t), 34.5 (t), 35.3 (t), 39.2 (t), 49.2 (d), 55.3 (q), 55.6 (q), 69.3 (d), 70.8 (t), 74.3 (d), 78.9 (d), 113.5 (d), 113.8 (d), 122.4 (t), 129.3 (d), 130.1 (s), 136.6 (s), 138.0 (s), 158.2 (s), 159.2 (s), 173.6 (s), 204.1 (s). HRMS (ESI+) m/z: calcd for C34H53BrCl2NO6 [M + NH4]+, 720.2428; found, 720.2447.
(E)-4-Hydroxy-7-iodo-3,3,6-trimethylhept-6-en-2-one (24)
A solution of silylenolether 22(14) (1.15 g, 7.3 mmol) in CH2Cl2 (10 mL) was added to a cooled solution (−78 °C) of aldehyde 2323 (1.26 g, 6 mmol) in CH2Cl2 (65 mL). BF3·OEt2 (0.58 mL, 4.71 mmol) was added dropwise. The reaction mixture was stirred at −78 °C for 2 h and quenched with the saturated solution of NaHCO3 (75 mL). The residue was extracted three times with CH2Cl2, dried over Na2SO4, filtrated, and concentrated under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (8:2) afforded 1.13 g of 24 (63%) as a racemic mixture. 1H NMR (400 MHz, CDCl3): δ 1.14 (s, 3H), 1.17 (s, 3H), 1.89 (d, J = 0.9 Hz, 3H), 2.18 (s, 3H), 2.22–2.32 (m, 2H), 3.80–3.90 (m, 1H), 6.03 (q, J = 0.9 Hz, 1H). 13C NMR (101 MHz, CDCl3): δ 19.6 (q), 21.6 (q), 23.9 (q), 26.4 (q), 41.9 (t), 51.5 (s), 73.4 (d), 77.2 (d), 145.0 (s), 214.5 (s). HRMS (ESI+) m/z: calcd for C10H18IO2 [M + H]+, 297.0346; found, 297.0340.
(R,E)-1-Iodo-2,5,5-trimethyl-6-oxohept-1-en-4-yl-(R)-2-methoxy-2-phenylacetate (25)
(R)-(−)-α-MPA (565 mg, 3.4 mmol) and EDC·HCl (862 mg, 4.5 mmol) were added to a solution of alcohol 24 (596 mg, 2 mmol) in THF (15 mL), then DMAP (60 mg, 0.5 mmol) was added, and the solution was stirred at 45 °C for 16 h. The solution was poured into Et2O (15 mL) and washed with NH4Cl saturated solution (2 × 15 mL). The organic residue was dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (90:10) yielded the corresponding esters 25-R,R (366 mg, 41%) and 25-R,S (375 mg, 42%) as colorless oils.
25-R,R Characterization
1H NMR (400 MHz, CDCl3): δ 0.81 (s, 3H), 0.94 (s, 3H), 1.89 (d, J = 0.8 Hz, 3H), 1.90 (s, 3H), 2.20–2.35 (m, 2H), 3.43 (s, 3H), 4.65 (s, 1H), 5.36 (dd, J = 10.3, 2.5 Hz, 1H), 5.88 (br s, 1H), 7.31–7.42 (m, 5H). 13C NMR (101 MHz, CDCl3): δ 19.6 (q), 21.4 (q), 23.5 (q), 26.0 (q), 40.7 (t), 51.1 (s), 58.0 (q), 74.5 (d), 77.7 (d), 82.6 (d), 127.3 (d), 128.7 (d), 128.9 (d), 136.3 (s), 143.9 (s), 169.7 (s), 210.7 (s). HRMS (ESI+) m/z: calcd for C19H25INaO4 [M + Na]+, 467.0690; found, 467.0692.
25-R,S Characterization
1H NMR (400 MHz, CDCl3): δ 1.05 (s, 3H), 1.12 (s, 3H), 1.76 (d, J = 1.1 Hz, 3H), 2.12 (s, 3H), 2.13–2.15 (m, 2H), 3.40 (s, 3H), 4.69 (s, 1H), 5.33 (d, J = 1.1 Hz, 1H), 5.40 (t, J = 6.0 Hz, 1H), 7.32–7.40 (m, 5H).
(R,E)-6-Iodo-2,5-dimethyl-2-(2-methyl-1,3-dioxolan-2-yl)hex-5-en-3-yl-(R)-2-methoxy-2-phenylacetate (S12)
Trimethylsilyl trifluoromethanesulfonate (TMSOTf; 15 μL, 0.08 mmol) was added over a solution of ketone 25-R,R (360 mg, 0.81 mmol) and 1,2-bis(trimethylsiloxy)ethane (500 mg, 2.43 mmol) in CH2Cl2 at −78 °C, and the mixture was left to evolve to rt and stirred at rt for additional 48 h. The reaction mixture was quenched with 0.1 mL of pyridine, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (90:10) yielded ketal S12 (335 mg, 97%) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.62 (s, 3H), 0.73 (s, 3H), 1.12 (s, 3H), 1.87 (s, 3H), 2.43 (dd, J = 14.3, 11.3 Hz, 1H), 2.68 (dd, J = 14.3, 1.9 Hz, 1H), 3.43 (s, 3H), 3.69–3.88 (m, 4H), 4.63 (s, 1H), 5.19 (dd, J = 11.3, 1.9 Hz, 1H), 5.88 (s, 1H), 7.28–7.43 (m, 5H). 13C NMR (101 MHz, CDCl3): δ 17.7 (q), 20.0 (q), 20.5 (q), 23.6 (q), 41.4 (t), 45.8 (s), 58.2 (q), 64.4 (t), 64.7 (t), 75.0 (d), 76.9 (d), 82.7 (d), 112.4 (s), 127.4 (d), 128.4 (d), 128.6 (s), 136.5 (s), 145.1 (s), 169.7 (s). HRMS (ESI+) m/z: calcd for C21H29INaO5 [M + Na]+, 511.0952; found, 511.0962.
(R,E)-6-Iodo-2,5-dimethyl-2-(2-methyl-1,3-dioxolan-2-yl)hex-5-en-3-ol (26)
LiOH·H2O (315 mg, 8.3 mmol) was added to a solution of S12 (320 mg, 0.65 mmol) in MeOH (8 mL), and the solution was stirred at 65 °C for 16 h. The reaction mixture was added to a separatory funnel containing Et2O (10 mL) and H2O (10 mL), and it was extracted two times with Et2O (10 mL). The organic layer was washed with NaHCO3 saturated solution (15 mL) and dried over MgSO4, and the solvent was evaporated under reduced pressure yielding alcohol 26 (205 mg, 0.60 mmol) that was used in the next step without further purification.
(R,E)-tert-Butyl((6-iodo-2,5-dimethyl-2-(2-methyl-1,3-dioxolan-2-yl)hex-5-en-3-yl)oxy)dimethylsilane (S13)
Triethylamine (0.33 mL, 2.4 mmol) and TBSOTf (0.23 mL, 1 mmol) were sequentially added to a solution of crude alcohol 26 (205 mg, 0.60 mmol) in CH2Cl2 (8 mL) at −20 °C, and the mixture was stirred for 15 min at −20 °C and then 60 min at rt. The reaction mixture was quenched with NH4Cl saturated solution (10 mL), extracted with CH2Cl2 (2 × 10 mL), and dried over MgSO4, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (95:5) yielded S13 (200 mg, 68% for two steps) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ −0.05 (s, 3H), 0.03 (s, 3H), 0.88 (s, 9H), 0.94 (s, 3H), 0.97 (s, 3H), 1.29 (s, 3H), 1.82 (s, 3H), 2.32 (dd, J = 14.5, 9.0 Hz, 1H), 2.88 (dd, J = 14.5, 2.1 Hz, 1H), 3.70 (dd, J = 9.0, 2.1 Hz, 1H), 3.81–3.96 (m, 4H), 5.91 (s, 1H). 13C NMR (101 MHz, CDCl3): δ −3.5 (q), −3.2 (q), 18.4 (q), 20.8 (q), 23.0 (q), 23.9 (q), 26.2 (q), 30.3 (s), 45.0 (t), 47.4 (s), 64.3 (t), 64.7 (t), 75.2 (d), 77.9 (d), 113.2 (s), 145.8 (s). HRMS (ESI+) m/z: calcd for C18H35INaO3Si [M + Na]+, 477.1298; found, 477.1299.
(R,E)-4-((tert-Butyldimethylsilyl)oxy)-7-iodo-3,3,6-trimethylhept-6-en-2-one (9)
PTSA·H2O (3.8 mg, 0.02 mmol) was added to a solution of S13 (66 mg, 0.15 mmol) in acetone (5 mL), and the reaction mixture was stirred at rt for 30 min. Then, it was quenched with two drops of triethylamine, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (95:5) yielded enantiopure ketone 9 (59 mg, quant.) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ −0.01 (s, 3H), 0.02 (s, 3H), 0.87 (s, 9H), 1.10 (s, 6H), 1.82 (s, 3H), 2.14 (s, 3H), 2.21–2.24 (m, 2H), 4.05–4.09 (m, 1H), 5.94 (s, 1H). 13C NMR (101 MHz, CDCl3): δ −4.0 (q), −3.7 (q), 18.2 (s), 19.6 (q), 22.9 (q), 23.8 (q), 26.0 (q), 27.1 (q), 44.8 (t), 53.0 (s), 74.0 (d), 78.8 (d), 144.5 (s), 213.4 (s). HRMS (ESI+) m/z: calcd for C16H31INaO2Si [M + Na]+, 433.1036; found, 433.1039.
(1S,5R,9S,11S,13R,15R,Z)-9-((tert-Butyldimethylsilyl)oxy)-15-((1R,5R,E)-5-((tert-butyldimethylsilyl)oxy)-1-hydroxy-3,6,6-trimethyl-7-oxooct-2-en-1-yl)-7,11-dimethyl-5-((triisopropylsilyl)oxy)-2,14-dioxabicyclo[11.2.1]hexadec-6-en-3-one (6)
DMF was degassed using a freeze-thaw-pump technique. CrCl2 and NiCl2 were weighted in a glovebox. A mixture of iodoalkene 9 (52 mg, 0.13 mmol) and aldehyde 4(5) (50 mg, 0.083 mmol) in DMF (0.5 + 0.2 mL rinse) was cannulated to a solution of CrCl2 (202 mg, 1.64 mmol) and NiCl2 (2 mg, 1 m/m % CrCl2) in DMF (1 mL), and the solution was stirred at rt for 48 h. The reaction mixture was filtered through Celite and washed with Et2O. The organic ethereal phase was washed with H2O (2 × 10 mL) and brine (10 mL) and dried over MgSO4, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (8:2) yielded alcohol 6 (13.5 mg, 19%) as a single diastereomer as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 0.02 (s, 3H), 0.04 (s, 3H), 0.05 (s, 3H), 0.06 (s, 3H), 0.85 (s, 9H), 0.87 (s, 9H), 0.91 (d, J = 6.1 Hz, 3H), 1.03 (br s, 21H), 1.09 (s, 3H), 1.12 (s, 3H), 1.27 (m, 1H), 1.42 (m, 1H), 1.60 (m, 1H), 1.70 (m, 1H), 1.71 (s, 3H), 1.72 (s, 3H), 1.91 (m, 1H), 2.03–2.11 (m, 2H), 2.11 (m, 1H), 2.14 (s, 3H), 2.24 (m, 1H), 2.39 (m, 1H), 2.50 (m, 1H), 2.70 (m, 1H), 2.73 (m, 1H), 3.89 (dd, J = 4.6, 1.3 Hz, 1H), 4.06 (m, 1H), 4.19 (t, J = 6.1 Hz, 1H), 4.33 (m, 1H), 4.43 (m, 1H), 5.05 (m, 1H), 5.15 (d, J = 5.6 Hz, 1H), 5.42 (m, 1H), 5.44 (m, 1H). 13C NMR (101 MHz, CDCl3): δ −4.4 (q), −4.1 (q), −3.9 (q), 12.3 (d), 17.4 (q), 17.7 (s), 17.9 (q), 18.1 (s), 19.3 (q), 20.7 (q), 23.0 (q), 23.1 (q), 25.9 (q), 26.0 (q), 26.7 (d), 27.1 (q), 33.9 (t), 38.3 (t), 40.9 (t), 45.0 (t), 46.0 (t), 46.9 (t), 53.1 (s), 68.4 (d), 69.0 (d), 70.3 (d), 74.1 (d), 76.1 (d), 78.5 (d), 87.3 (d), 125.7 (d), 132.7 (d), 138.9 (s), 143.4 (s), 171.1 (s), 213.9 (s). HRMS (ESI+) m/z: calcd for C48H92NaO8Si3 [M + Na]+, 903.5992; found, 903.5993.
(1R,5R,E)-5-((tert-Butyldimethylsilyl)oxy)-1-((1S,5R,9S,11S,13R,15S,Z)-9-((tert-butyldimethylsilyl)oxy)-7,11-dimethyl-3-oxo-5-((triisopropylsilyl)oxy)-2,14-dioxabicyclo[11.2.1]hexadec-6-en-15-yl)-3,6,6-trimethyl-7-oxooct-2-en-1-yl-(R)-2-methoxy-2-phenylacetate (27)
(R)-(−)-α-MPA (7 mg, 0.04 mmol) and EDC·HCl (10 mg, 0.05 mmol) were added to a solution of alcohol 6 (4.3 mg, 0.005 mmol) in THF (0.25 mL), then DMAP (catalytic) was added, and the solution was stirred at 35 °C for 2 h. The solution was poured into Et2O (5 mL) and washed with NH4Cl saturated solution (2 × 3 mL). The organic residue was dried over Na2SO4 and filtered, and the solvent was removed under reduced pressure. Purification by silica gel column chromatography with hexane/EtOAc (90:10) yielded the corresponding ester 27 (2.2 mg, 48% br s m) and 6 (0.5 mg). 1H NMR (500 MHz, CDCl2-d2): δ 0.04 (s, 6H), 0.08 (s, 6H), 0.82 (d, J = 6.1 Hz, 3H), 0.86 (s, 9H), 0.90 (s, 9H), 1.07 (s, 21H), 1.09 (s, 3H), 1.11 (s, 3H), 1.13–1.16 (m, 1H), 1.23–1.27 (m, 1H), 1.36–1.42 (m, 1H), 1.51–1.54 (m, 1H), 1.55–1.57 (m, 1H), 1.65 (d, J = 13.9 Hz, 1H), 1.73 (d, J = 1.4 Hz, 3H), 1.79 (d, J = 1.3 Hz, 3H), 1.86–1.91 (m, 1H), 2.08–2.11 (m, 1H), 2.14 (s, 3H), 2.21–2.29 (m, 2H), 2.44 (dd, J = 13.8, 7.4 Hz, 1H), 2.62–2.69 (m, 2H), 3.40 (s, 3H), 3.75 (ddd, J = 12.0, 7.9, 4.5 Hz, 1H), 3.88 (dd, J = 3.0, 1.2 Hz, 1H), 4.05 (dd, J = 9.9, 3.8 Hz, 1H), 4.23 (t, J = 5.8 Hz, 1H), 4.70 (s, 1H), 4.97–5.03 (m, 1H), 5.06 (dd, J = 6.0, 1.2 Hz, 1H), 5.40–5.45 (m, 1H), 5.49–5.54 (m, 1H), 5.75 (dd, J = 9.3, 3.0 Hz, 1H), 7.32–7.49 (m, 5H). 13C NMR (126 MHz, CDCl2-d2): δ −4.3 (q), −3.9 (q), −3.5 (q), 12.8 (d), 17.9 (q), 18.1 (q), 18.2 (q), 18.4 (s), 18.5 (s), 20.1 (q), 20.8 (q), 22.1 (q), 23.3 (q), 26.1 (q), 26.2 (q), 27.0 (d), 27.0 (q), 33.9 (t), 38.7 (t), 41.6 (t), 45.0 (t), 46.2 (t), 47.1 (t), 57.7 (q), 68.8 (d), 70.8 (d), 71.9 (d), 74.5 (d), 75.7 (d), 78.9 (d), 83.1 (d), 86.0 (d), 121.5 (d), 128.2 (d), 129.0 (d), 129.2 (d), 132.9 (d), 133.5 (s), 137.1 (s), 142.1 (s), 169.9 (s), 170.8 (s), 213.0 (s). HRMS (ESI+) m/z: calcd for C57H104NO10Si3 [M + NH4]+, 1046.6963; found, 1046.6961.
(1S,5R,9S,11S,13R,15S,Z)-9-((tert-Butyldimethylsilyl)oxy)-15-((4R,8R,E)-8-((tert-butyldimethylsilyl)oxy)-2,2,6,9,9,12,12-heptamethyl-10-methylene-3,11-dioxa-2,12-disilatridec-5-en-4-yl)-7,11-dimethyl-5-((triisopropylsilyl)oxy)-2,14-dioxabicyclo[11.2.1]hexadec-6-en-3-one (31)
Et3N (14 μL, 0.1 mmol) and TMSOTf (11 μL, 0.06 mmol) were sequentially added to a solution of ketone 6 (8 mg, 0.01 mmol) in CH2Cl2 (0.7 mL) at 0 °C. The reaction mixture was stirred for 3 h at rt, diluted with CH2Cl2 (2 mL), and quenched with diluted NH4Cl (1 mL). The organic layer was washed three times with diluted NH4Cl, dried over MgSO4, and concentrated under reduced pressure to obtain silylenolether 31 as a brownish oil (10 mg, quant). The crude was pure enough to continue the synthesis. 1H NMR (400 MHz, CDCl3): δ 0.00 (s, 3H), 0.03 (s, 3H), 0.06 (s, 6H), 0.07 (s, 9H), 0.21 (s, 9H), 0.85 (s, 9H), 0.86 (s, 9H), 0.88 (d, J = 2.9 Hz, 3H), 0.93 (s, 3H), 1.02 (s, 3H), 1.05 (s, 21H), 1.28–1.29 (m, 1H), 1.66 (s, 3H), 1.72 (s, 3H), 1.73–1.76 (m, 1H), 1.93–2.04 (m, 3H), 2.07–2.17 (m, 3H), 2.20–2.30 (m, 2H), 2.47–2.58 (m, 2H), 2.68 (dd, J = 14.6, 3.7 Hz, 1H), 3.94–3.96 (m, 2H), 3.96–4.00 (m, 2H), 4.10 (d, J = 1.6 Hz, 1H), 4.29–4.38 (m, 1H), 4.44 (dd, J = 9.1, 2.7 Hz, 1H), 4.89–4.95 (m, 1H), 5.21 (d, J = 5.7 Hz, 1H), 5.24–5.27 (m, 1H), 5.40–5.43 (m, 1H).
Acknowledgments
This study was partially funded by the MINECO-FEDER (CTQ2015-67870P) and the Generalitat de Catalunya (2014 SGR 137). A.G. thanks the Spanish Ministry of Education for the FPI Grant. A.G. acknowledges Prof. Mark Bradley and Dr. Lorna Murray for the possibility of performing VT NMR experiments using University of Edinburgh’s facilities. M.G. acknowledges the Erasmus grant to do her internship at ChemBio Lab. ChemBio Lab acknowledges PharmaMar for all the support given to carry out this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00125.
Absolute configuration determinations and NMR spectra of new compounds (PDF)
The authors declare no competing financial interest.
Footnotes
From now onward in this publication, carbon numeration used in the structure determination of PMs B–D will be used for every molecular structure.
The structural and stereochemical characterization was performed in the R + D department of PharmaMar.
Other protecting groups such as TBS or PMB were tested with worst synthetic results.
Two singlets at 6.18 and 6.25 ppm with 89:11 relative area were observed in the 1H NMR.
The proportion of diastereomers was determined by the NMR analysis of the purified ketol.
The C15-(R) configuration is the opposite to the one present in the NPs PMs C and D. See Supporting Information.
Supplementary Material
References
- Lorente A.; Makowski K.; Albericio F. Bioactive Marine Polyketides as Potential and Promising Drugs. Ann. Med. Chem. Res. 2014, 1, 1003–1013. [Google Scholar]
- Lorente A.; Gil A.; Fernández R.; Cuevas C.; Albericio F.; Álvarez M. Phormidolides B and C, Cytotoxic Agents from the Sea: Enantioselective Synthesis of the Macrocyclic Core. Chem.—Eur. J. 2015, 21, 150–156. 10.1002/chem.201404341. [DOI] [PubMed] [Google Scholar]
- Williamson R. T.; Boulanger A.; Vulpanovici A.; Roberts M.; Gerwick W. H. Structure and Absolute Stereochemistry of Phormidolide, a New Toxic Metabolite from the Marine Cyanobacterium Phormidium Sp. J. Org. Chem. 2002, 67, 7927–7936. 10.1021/jo020240s. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Matsuda H.; Makabe K.; Yamaguchi K. Oscillariolide, a Novel Macrolide from a Blue-Green Alga Oscillatoria Sp. Tetrahedron Lett. 1991, 32, 2391–2394. 10.1016/s0040-4039(00)79931-x. [DOI] [Google Scholar]
- Gil A.; Lorente A.; Albericio F.; Álvarez M. Stereoselective Allylstannane Addition for a Convergent Synthesis of a Complex Molecule. Org. Lett. 2015, 17, 6246–6249. 10.1021/acs.orglett.5b03252. [DOI] [PubMed] [Google Scholar]
- Gil A.; Lamariano-Merketegi J.; Lorente A.; Albericio F.; Álvarez M. Enantioselective Synthesis of the Polyhydroxylated Chain of Oscillariolide and Phormidolides A–C. Org. Lett. 2016, 18, 4485–4487. 10.1021/acs.orglett.6b02014. [DOI] [PubMed] [Google Scholar]
- Gil A.; Lamariano-Merketegi J.; Lorente A.; Albericio F.; Álvarez M. Synthesis of (E)-4-Bromo-3-Methoxybut-3-En-2-One, the Key Fragment in the Polyhydroxylated Chain Common to Oscillariolide and Phormidolides A-C. Chem.—Eur. J. 2016, 22, 7033–7035. 10.1002/chem.201600770. [DOI] [PubMed] [Google Scholar]
- Gil A.; Albericio F.; Álvarez M. Role of the Nozaki–Hiyama–Takai–Kishi Reaction in the Synthesis of Natural Products. Chem. Rev. 2017, 117, 8420–8446. 10.1021/acs.chemrev.7b00144. [DOI] [PubMed] [Google Scholar]
- Negoro T.; Ikeda Y. Regio- and Stereochemistry of Bromochlorinations of Alkynes with Molecular Bromine Chloride and Dichlorobromate(1−) Ion. Bull. Chem. Soc. Jpn. 1986, 59, 3515–3518. 10.1246/bcsj.59.3515. [DOI] [Google Scholar]
- De Meyer M.; Levert J. M.; Vanclef A. Étude Vibrationnelle Des Trichlorures Organiques: (N-C4H9)4NCl3, ϕ4AsCl3, C5H6NCl3, ϕ4PCl3. Bull. Soc. Chim. Belg. 2010, 92, 699–710. 10.1002/bscb.19830920804. [DOI] [Google Scholar]
- Oikaw Y.; Yoshioka T.; Yonemitsu O. Protection of Hydroxy Groups by Intramolecular Oxidative Formation of Methoxybenzylidene Acetals with DDQ. Tetrahedron Lett. 1982, 23, 889–892. 10.1016/s0040-4039(00)86975-0. [DOI] [Google Scholar]
- Hoye T. R.; Jeffrey C. S.; Shao F. Mosher Ester Analysis for the Determination of Absolute Configuration of Stereogenic (Chiral) Carbinol Carbons. Nat. Protoc. 2007, 2, 2451–2458. 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- Swamy K. C. K.; Kumar N. N. B.; Balaraman E.; Kumar K. V. P. P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- Blanco L.; Amice P.; Conia J.-M. Reaction of Chloromethylcarbene with Trimethylsilyl Enol Ethers; Preparation of α-Methylene Ketones and α-Methyl-Α,β-Unsaturated Ketones and Aldehydes. Synthesis 1981, 291–293. 10.1055/s-1981-29421. [DOI] [Google Scholar]
- Amans D.; Bareille L.; Bellosta V.; Cossy J. Synthesis of the Monomeric Counterpart of Marinomycin A. J. Org. Chem. 2009, 74, 7665–7674. 10.1021/jo900945x. [DOI] [PubMed] [Google Scholar]
- Bülow L.; Naini A.; Fohrer J.; Kalesse M. A Kiyooka Aldol Approach for the Synthesis of the C(14)–C(23) Segment of the Diastereomeric Analog of Tedanolide C. Org. Lett. 2011, 13, 6038–6041. 10.1021/ol202515x. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Cywin C. L.; Roper T. D. Enantioselective Mukaiyama-Aldol and Aldol-Dihydropyrone Annulation Reactions Catalyzed by a Tryptophan-Derived Oxazaborolidine. Tetrahedron Lett. 1992, 33, 6907–6910. 10.1016/s0040-4039(00)60892-4. [DOI] [Google Scholar]
- Seco J. M.; Quiñoá E.; Riguera R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004, 104, 17–118. 10.1021/cr000665j. [DOI] [PubMed] [Google Scholar]
- Latypov S. K.; Seco J. M.; Quiñoá E.; Riguera R. Are Both the (R)- and the (S)-MPA Esters Really Needed for the Assignment of the Absolute Configuration of Secondary Alcohols by NMR? The Use of a Single Derivative. J. Am. Chem. Soc. 1998, 120, 877–882. 10.1021/ja9700055. [DOI] [Google Scholar]
- Yen C.-F.; Liao C.-C. Concise and Efficient Total Synthesis of Lycopodium Alkaloid Magellanine. Angew. Chem. 2002, 114, 4264–4267. . [DOI] [PubMed] [Google Scholar]
- Lamariano-Merketegi J.; Lorente A.; Gil A.; Albericio F.; Álvarez M. Addition of Vinylmetallic Reagents to Chiral 2-Formyltetrahydrofuran. Eur. J. Org. Chem. 2015, 235–241. 10.1002/ejoc.201402987. [DOI] [Google Scholar]
- Pulukuri K. K.; Chakraborty T. K. Formal Synthesis of Actin Binding Macrolide Rhizopodin. Org. Lett. 2014, 16, 2284–2287. 10.1021/ol5008179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.