

Abstract

The FDA-approved drug for the treatment of asthma, zafirlukast, is synthesized engaging multiple catalytic reactions including a new method for the construction of 3-aroylindoles via oxidative cyclization. The highlights include transition-metal and peroxide free C–H bond activation using the stoichiometric amount of sodium persulfate as an oxidizing agent in the construction of 3-aroylindole, avoiding transition metal, with over 28% overall yield. The complete process has a turnaround time of 28 h to get the target molecule starting from substituted aniline, with practically no protecting groups.
Introduction
Substituted indoles play a pivotal role in drug discovery, culminating in launch of several drugs, and many molecules are in clinical pipeline.1−5 The discovery and development of drugs to treat allergic conditions and asthma has been of paramount importance in the pharmaceutical industry.2 The success rate in this area of research has been significant because the human intralobar airways have a single receptor for peptido leukotrienes.6−8 There has been a great progress reported in identifying selective peptide leukotriene antagonists.9 Indole and indazole compounds have been extensively studied as selective antagonists of the leukotriene pathway.10−13 These efforts have culminated in launching zafirlukast (1) as the oral leukotriene receptor antagonist for the maintenance treatment of asthma, prescribed in combination with bronchodilators and steroids.14−23 Other marketed products which have indole skeleton are panobinostat (2),24 roxindole (3),25,26 tropisetron (4),27,28 etc (Figure 1).
Figure 1.
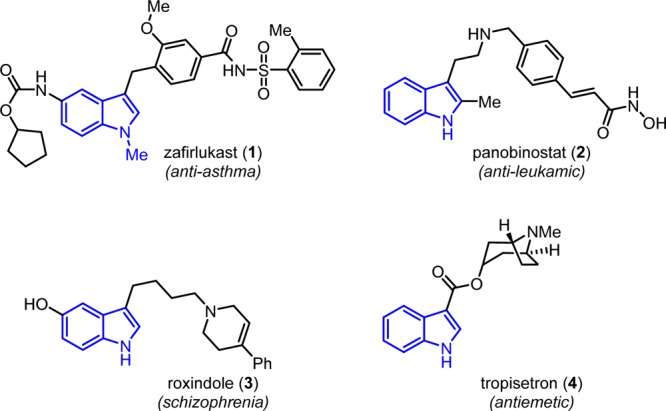
Representative drugs with 3-substituted indoles.
Zafirlukast attracted our attention owing to its biological profile along with our own interest to develop new strategies to build heterocyclic scaffolds for ongoing medicinal chemistry programmes.29−31 Traditionally, these classes of compounds are synthesized from prefabricated indole which offers lesser flexibility in substitutions in the benzene ring of indole.1
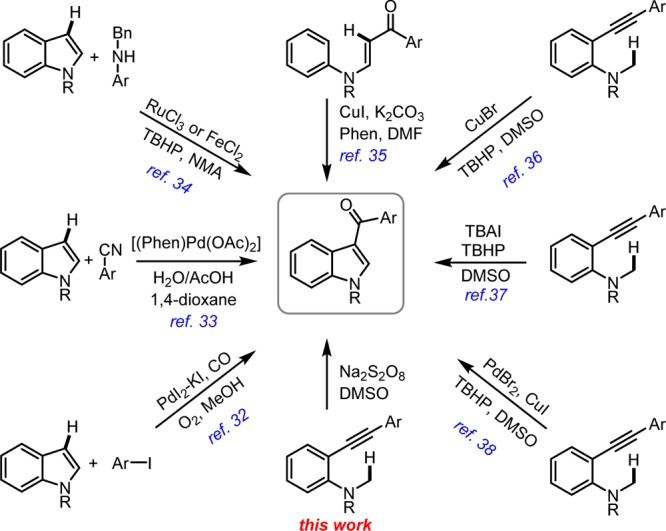
Thus, a nonconventional strategy engaging indole construction as the key step of synthesis, unlike using substituted indoles as starting materials, was envisaged, thereby avoiding Fischer indole synthesis. In this regard, we aimed at the synthesis of 3-aroylindoles owing to easy manipulation of its carbonyl functionality. Few literature representative methods for the synthesis of 3-aroylindoles have been shown in Scheme 1.32−38 Recently, Patel et al. have reported an intramolecular copper-catalyzed sp3 C–H activation followed by C–C and C–O bond formation reactions to construct indole scaffold from alkynyl aniline.36,37 Simultaneously, Liang and co-workers reported a palladium–copper-cocatalyzed 3-aroylindole synthesis.38 We desired a peroxide-free and preferably transition metal-free cyclization protocol which prompted us to explore various oxidants to trigger the cyclization process of alkynyl N-methyl anilines to substituted indoles, which could in turn be transformed to zafirlukast.
Scheme 1. Representative Methods for 3-Aroylindoles.
Initially, easily accessible N,N-dimethyl-2-(phenylethynyl)aniline 5a(39) was subjected to sodium persulfate catalyzed oxidation to produce the indole 6a in 75% yield. The optimum yields were obtained with three equivalents of sodium persulfate (see Table 1). After establishing the optimized conditions, we studied the scope and generality of the reaction with substituted N,N-dimethyl-2-(phenylethynyl)anilines 5 and obtained corresponding 3-aroylindoles (6a–e, Table 2) in good yields (68–91%). In general, aryl rings substituted with electron-withdrawing groups gave higher yields (6d, 91% yield) compared to electron-donating groups. The procedure is also applicable to electron-rich aniline 5e to give required product 6e in 60% yield. The scalability of this reaction is proved by the preparation of 3-aroylindole 6d in a large scale.
Table 1. Optimization of Conditions for sp3 C–H Bond Activationa.

| entry | Na2S2O8 (equiv) | time (h) | yieldb |
|---|---|---|---|
| 1 | 0.1 | 4 | 10 |
| 2 | 0.2 | 4 | 18 |
| 3 | 0.3 | 4 | 24 |
| 4 | 0.4 | 4 | 32 |
| 5 | 0.5 | 4 | 42 |
| 6 | 1.0 | 4 | 51 |
| 7 | 2.0 | 4 | 63 |
| 8 | 2.5 | 4 | 68 |
| 9 | 3.0 | 5 | 75 |
| 10 | 4.0 | 4 | 48 |
The reaction was carried out with 5a (0.1 mmol) in DMSO (2 mL) under an inert atmosphere at 80 °C.
Yield of the isolated product 6a.
Table 2. Synthesis of Various 3-Aroylindolesa,b.
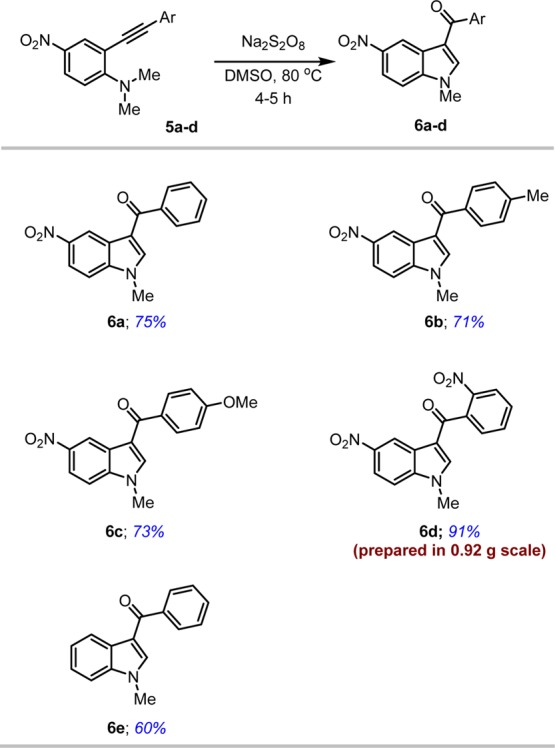
The reaction was carried out with 5 (0.1 mmol) and Na2S2O8 (0.3 mmol) in DMSO (2 mL) under an inert atmosphere at 80 °C.
Yield of the isolated product 6.
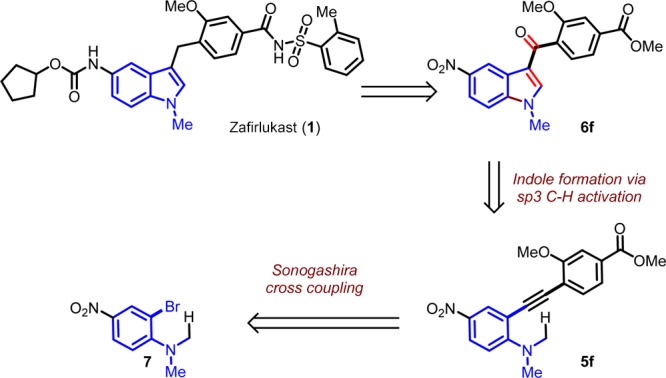
The synthesis of substituted indoles gave us the confidence to choose molecules containing indole skeleton and we narrowed down our search to zafirlukast. A retrosynthetic analysis of zafirlukast gave the key intermediate 6f, which can be synthesized from the alkyne 5f by extending our methodology. The alkyne can in turn be synthesized from 2-bromoaniline 7 (Scheme 2).
Scheme 2. Retrosynthetic Analysis of Zafirlukast.
To add value to this reaction, appropriately substituted alkynyl aniline derivative 5f was prepared in a one-pot three-step process. 2-bromoaniline 7 was subjected to Sonogashira coupling with trimethylsilyl-acetylene using palladium catalyst, followed by desilylation with the addition of K2CO340 and subsequent Sonogashira coupling in the same flask with methyl 4-iodo-3-methoxybenzoate 10 furnished 5f in 58% overall yield over three steps (Scheme 3). To our satisfaction, sodium persulfate in hot dimethyl sulfoxide (DMSO), in absence of any external catalyst, facilitated the entire sequence of sp3 C–H activation of N–Me group and formation of two new covalent C–C and C–O bonds in excellent yield (88%), to generate key intermediate 6f.
Scheme 3. Synthesis of Zafirlukast.

The task of converting 6f to zafirlukast was accomplished following four easy synthetic transformations (Scheme 3). Initially, 6f was hydrogenated with Pd/C and H2 which under acidic workup provided the indole acid 8 in 76% yield.41 The coupling of indole acid 8 with sulphonamide 11 in the presence of ZnCl2 catalyst afforded 9 in 92% yield.42 The reduction of the nitro group in 9 was accomplished with Ra–Ni under a H2 atmosphere in EtOAc to yield 5-amino indole derivative in quantitative yield. This was converted to zafirlukast (1) by conjugating with cyclopentyl chloroformate 12 following reported procedure, using N-methyl morpholine, in excellent yield (89%).
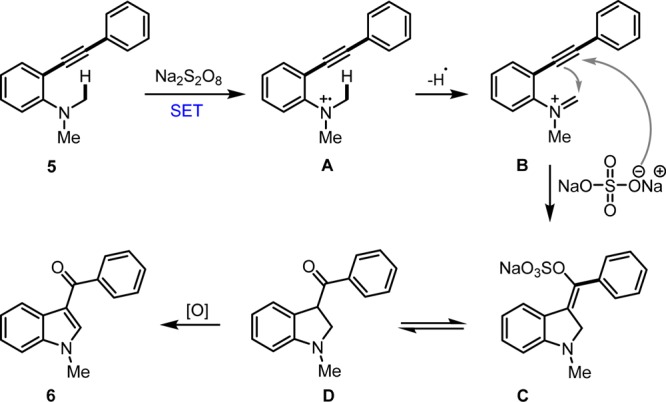
On the basis of literature reports, a plausible mechanism has been proposed for this transformation (Scheme 4). The aminyl radical cation A is formed by a single electron transfer process from the nitrogen atom of the N,N-dimethyl-2-(phenylethynyl)aniline (5). Abstraction of a hydrogen radical from N–Me gives an iminium intermediate B. The intramolecular cyclization of intermediate B via nucleophilic attack of the alkynyl group at the iminium carbon and subsequent addition of sodium persulfate gave enolate intermediate C. Then, ketonization of C afforded ketone D, which underwent further oxidation to provide 3-arolyindole 6.
Scheme 4. Plausible Mechanism.
In summary, an FDA-approved drug zafirlukast is synthesized in six steps starting from commercially available 2-bromo-N,N-dimethyl-4-nitroaniline engaging all catalytic transformations and almost with no protective groups. The key features are (i) one pot, three transformations to build biaryl alkynes, (ii) overall yield of 28%, and (iii) all the six steps requiring only 28 h overall for completing the process, which constitutes a very fast approach to the target.
General Details
General Information
All the reagents and solvents were of reagent grade and used without purification unless specified otherwise. All the dry reactions were performed under an atmosphere of nitrogen in flame-dried or oven-dried glassware with magnetic stirring. For reactions carried out using dry solvents, dimethylformamide (DMF), DMSO, CH2Cl2, were freshly distilled over calcium hydride and methanol was dried over calcium oxide. Technical grade ethyl acetate and hexane used for column chromatography were distilled prior to use. Column chromatography was carried out using silica gel (60–120 and 100–200 mesh) packed in glass columns. 1H and 13C NMR spectra were recorded at 400, 500 and 100, 125 MHz, respectively. Chemical shifts (δ) are reported in ppm, using the residual solvent peak in CDCl3 (H: δ = 7.26 and C: δ = 77.16 ppm) as the internal standard, and coupling constants (J) are given in Hz. High-resolution mass spectra (HRMS) were obtained by electron spray ionization (ESI) using an ESI-TOF mass spectrometer in positive ion mode (M + H or M + Na) as indicated.
Experimental Procedures and Analytical Data
General Procedure for Intramolecular Oxidative Coupling via sp3 C–H Bond Activation (6a–e)
To a stirred solution of 5a–e (0.1 mmol) in DMSO (3 mL), Na2S2O8 (0.3 mmol) was added into this mixture, and finally, the mixture was heated at 80 °C for 4 to 5 h. After completion of the reaction monitored by thin-layer chromatography (TLC), the reaction mixture was quenched with water (5 mL) and the product was extracted with ethyl acetate (2 × 5 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 3-aroylindole products (6a–e).
(1-Methyl-5-nitro-1H-indol-3-yl)(phenyl)methanone 6a
By following the general procedure, compound 6a was prepared (71 mg, 68%) as a yellow solid; Rf = 0.5 (silica, EtOAc/hexane = 1:9); mp = 134–136 °C; IR (neat): 3485, 2954, 2926, 2854, 1726, 1634, 1459, 1220, 894, 772, 688 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.29 (d, J = 2.6 Hz, 1H), 8.15 (dd, J = 9.2, 2.6 Hz, 1H), 7.57–7.51 (m, 2H), 7.42–7.36 (m, 3H), 6.58 (d, J = 9.2 Hz, 1H), 5.47 (s, 1H), 3.05 (d, J = 5.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 158.0, 139.0, 131.5, 131.3, 129.0, 127.0, 125.2, 123.1, 115.0, 111.0, 95.5, 88.0, 43.0; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C16H13N2O2, 281.1690; found, 281.1702.
(1-Methyl-5-nitro-1H-indol-3-yl)(p-tolyl)methanone 6b
By following the general procedure, compound 6b was prepared (75 mg, 71%) as a yellow solid; Rf = 0.6 (silica, EtOAc/hexane = 1:9); IR (neat): 2954, 2854, 2209, 1597, 1511, 1444, 1323, 1269, 1079, 906, 820, 772, 750 cm–1; mp = 113–118 °C; 1H NMR (500 MHz, CDCl3): δ 8.58 (d, J = 2.2 Hz, 1H), 8.15 (dd, J = 9.0, 2.2 Hz, 1H), 7.42–7.39 (m, 2H), 7.36 (d, J = 9.0 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 6.69 (d, J = 0.7 Hz, 1H), 3.79 (s, 3H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 145.1, 142.0, 141.1, 139.0, 130.0, 129.4, 129.0, 127.3, 118.0, 117.3, 109.5, 104.0, 32.0, 21.5; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C17H15N2O3, 295.1083; found, 295.1084.
(4-Methoxyphenyl)(1-methyl-5-nitro-1H-indol-3-yl)methanone 6c
By following the general procedure, compound 6c was prepared (77 mg, 73%) as a yellow solid; Rf = 0.5 (silica, EtOAc/hexane = 3:7); mp = 142–144 °C; IR (neat): 3415, 2928, 2838, 1738, 1627, 1534, 1416, 1318, 1219, 894, 772, 685 cm–1; 1H NMR (400 MHz, CDCl3): δ 9.25 (d, J = 1.9 Hz, 1H), 8.22 (dd, J = 9.0, 1.5 Hz, 1H), 7.85 (t, J = 5.5 Hz, 2H), 7.69 (s, 1H), 7.41 (d, J = 9.0 Hz, 1H), 7.02–6.98 (m, 2H), 3.93 (s, 3H), 3.90 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 189.0, 163.0, 144.0, 140.2, 139.1, 132.5, 131.1, 121.0, 120.0, 119.1, 117.5, 114.0, 110.0, 57.0, 34.1; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C17H15N2O4+, 311.1032; found, 311.1039.
(1-Methyl-5-nitro-1H-indol-3-yl)(2-nitrophenyl)methanone 6d
By following the general procedure, compound 6d was prepared (95 mg, 91%) as a yellow solid; Rf 0.6 (silica, EtOAc/hexane = 2:8); mp = 188–190 °C; IR (neat): 3423, 3098, 2946, 1740, 1614, 1577, 1524, 1326, 1142, 1068, 855, 756, 670 cm–1; 1H NMR (500 MHz, CDCl3): δ 8.58 (d, J = 2.2 Hz, 1H), 8.19–8.14 (m, 2H), 7.77 (td, J = 7.5, 1.3 Hz, 1H), 7.70 (td, J = 7.9, 1.5 Hz, 1H), 7.56 (dd, J = 7.5, 1.5 Hz, 1H), 7.38 (d, J = 9.1 Hz, 1H), 6.66 (d, J = 0.6 Hz, 1H), 3.60 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 149.5, 142.1, 140.4, 139.4, 134.0, 133.3, 131.0, 127.0, 125.0, 118.1, 118.0, 110.0, 105.0, 31.2; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C16H11N3O5, 326.1489; found, 598.1474.
(1-Methyl-1H-indol-3-yl)(phenyl)methanone 6e
By following the general procedure, compound 6e was prepared (64 mg, 60%) as a viscous liquid; Rf 0.7 (silica, EtOAc/hexane = 1:19); IR (neat): 3479, 3392, 2926, 2856, 1642, 1523, 1459, 1220, 1065, 772, 695 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.46–8.40 (m, 1H), 7.81–7.76 (m, 2H), 7.54–7.49 (m, 1H), 7.49–7.42 (m, 3H), 7.32 (dd, J = 5.7, 3.6 Hz, 3H), 3.78 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 190.9, 140.9, 138.1, 137.6, 131.1, 128.7, 128.3, 123.7, 122.7, 122.7, 115.5, 109.7, 33.6; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C16H14NO, 236.1075; found, 236.1063.
Experimental Procedures for the Total Synthesis of Zafirlukast
One-Pot Synthesis of Intermediate 5f
To a mixture of aryl bromide 7 (5.0 g, 20.41 mmol), PdCl2 (PPh3)2 (143 mg, 10 mol %), CuI (39 mg, 10 mol %) and Et3N (4.3 mL, 30.61 mmol) was added in dry DMF (80 mL); then, trimethylsilyl acetylene (5.5 mL, 20.41 mmol) was added slowly at room temperature, and the resultant mixture was stirred for 2 h at 80 °C. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature, and then, K2CO3 (3.9 g, 30.61 mmol) was added to this mixture then heating was continued until the silylated acetylene disappeared (monitored by TLC). Once again, the reaction was allowed to come to room temperature, then added with PdCl2 (PPh3)2 (143 mg, 10 mol %), Et3N (4.3 mL, 30.61 mmol), and CuI (39 mg, 10 mol %), and continued stirring for 30 min at room temperature. Finally, aryl iodide 10 (5.9 g, 20.41 mmol) in DMF (10 mL) was added dropwise in residual mixture, and then resultant mixture was heated for 3 h at 80 °C. The reaction mixture was re-cooled to room temperature and quenched with cold water (70 mL), and the solid was filtered using with EtOAc (150 mL). Then, the organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to provide the biaryl acetylene 5f (3.9 g, 58% in 3 steps) as a yellow solid; Rf = 0.7 (silica, EtOAc/hexane = 3:7); mp = 152–154 °C; IR (neat): 3476, 3226, 2951, 2925, 2850, 1723, 1622, 1582, 1435, 1293, 1108, 767 cm–1; 1H NMR (500 MHz, CDCl3): δ 8.38 (d, J = 2.8 Hz, 1H), 8.05 (dd, J = 9.3, 2.8 Hz, 1H), 7.64 (dd, J = 7.9, 1.3 Hz, 1H), 7.57 (d, J = 1.0 Hz, 1H), 7.51 (d, J = 7.9 Hz, 1H), 6.76 (d, J = 9.4 Hz, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.29 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 167.0, 160.1, 157.4, 138.5, 133.0, 132.0, 131.2, 125.5, 122.0, 117.2, 115.0, 111.4, 110.1, 94.5, 91.3, 56.1, 52.5, 43.0; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C19H19N2O5, 355.1294; found, 355.1299.
Methyl 3-Methoxy-4-(1-methyl-5-nitro-1H-indole-3-carbonyl) Benzoate 6f
To a stirred solution of methyl 4-((2-(dimethyl amino)-5-nitrophenyl)ethynyl)-3-methoxybenzoate 5f (3.0 g, 8.47 mmol) in dry DMSO (50 mL), was added Na2S2O8 (6.05 g, 25.42 mmol). The resultant mixture was heated to 80 °C for 5 h. The resultant reaction mixture was quenched with water (60 mL) and extracted with ethyl acetate (2 × 50 mL). The organic phase was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give methyl 3-methoxy-4-(1-methyl-5-nitro-1H-indole-3-carbonyl) benzoate 6f (2.75 g, 88%) as a yellow solid; Rf = 0.5 (silica, EtOAc/hexane = 4:6); mp = 198–200 °C; IR (neat): 2924, 2854, 1724, 1634, 1527, 1464, 1371, 1293, 1232, 1142, 1026, 952, 770 cm–1; 1H NMR (400 MHz, CDCl3): δ 9.29 (d, J = 2.2 Hz, 1H), 8.23 (dd, J = 9.0, 2.3 Hz, 1H), 7.73 (dd, J = 7.7, 1.4 Hz, 1H), 7.69 (d, J = 1.2 Hz, 1H), 7.45 (d, J = 5.6 Hz, 1H), 7.42 (d, J = 1.0 Hz, 1H), 7.38 (d, J = 17.6 Hz, 1H), 3.97 (s, 3H), 3.87 (s, 3H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 189.0, 166.5, 157.0, 144.3, 141.0, 140.4, 134.3, 133.0, 129.0, 126.0, 122.0, 120.0, 119.4, 118.5, 113.0, 110.1, 56.2, 53.0, 34.2; HRMS (ESI-TOF) (m/z): calcd for [M + H]+ C19H17N2O6, 369.1087; found, 369.1085.
3-Methoxy-4-(1-methyl-5-nitro-1H-indole-3-carbonyl) Benzoic Acid 8
To a stirred solution of compound 6f (2.0 g, 5.43 mmol) in MeOH (20 mL), were added 5% Pd–C (0.2 g, 5 mol %) and concd HCl (0.2 equiv) sequentially. The flask was evacuated and pressurized with H2 (balloon) atmosphere, and the reaction mixture was stirred for 6 h. After completion of the reaction, the solvent was removed under reduced pressure, and the pH value of the reaction mixture was adjusted to 2 with aq HCl (2.0 M) and stirred at room temperature for 3 h. The reaction mixture was filtered using diethyl ether and dried over anhydrous sodium sulfate. The organic layer was concentrated under reduced pressure and purified by column chromatography to give compound 8 (1.4 g, 76%) as a yellow solid; Rf = 0.5 (silica, EtOAc/hexane = 7:3); mp = 183–185 °C; IR (neat): 3417, 2951, 2852, 1725, 1622, 1661, 1583, 1435, 1293, 1047, 995, 825, 769 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.51 (d, J = 2.2 Hz, 1H), 8.02 (dd, J = 9.1, 2.3 Hz, 1H), 7.59 (d, J = 9.1 Hz, 1H), 7.50–7.45 (m, 2H), 7.36 (s, 1H), 7.25 (d, J = 7.8 Hz, 1H), 4.11 (s, 2H), 3.91 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 167.2, 157.0, 140.3, 139.4, 134.1, 132.0, 130.1, 130.0, 126.5, 122.0, 116.4, 116.0, 115.3, 111.0, 110.3, 55.5, 33.0, 24.4; HRMS (ESI-TOF) (m/z): calcd for [M + H]− calcd for C18H15N2O5, 339.0981; found, 339.0967.
3-Methoxy-4-(1-methyl-5-nitro-1H-indole-3-carbonyl)-N-(o-tolylsulfonyl) Benzamide 9
To a stirred solution of anhydrous ZnCl2 (2 mg, 30 mol %) in anhydrous dichloromethane (DCM; 2 mL), acid 8 (300 mg, 0.88 mmol) was added followed by the addition of benzoic anhydride (0.12 mL, 1.05 mmol) under a nitrogen atmosphere at room temperature. After 10 min, a solution of sulphonamide 11 (16 mg, 0.88 mmol) in DCM (1 mL) was added dropwise, and the resulting reaction mixture was stirred at room temperature for 2 h. After completion of the reaction, the reaction mixture was quenched with water (5 mL), and the mixture was extracted with DCM (2 × 10 mL); then, the organic layer was washed with brine, and the combined organic layer were dried over sodium sulfate and evaporated under reduced pressure. The crude compound was purified by column chromatography to afford title product 9 (406 mg, 92%) as a yellow solid; Rf = 0.5 (silica, EtOAc/hexane = 8:2); mp = 152–154 °C; IR (neat): 3479, 2925, 2850, 1723, 1622, 1582, 1435, 1293, 1108, 767, 671 cm–1; 1H NMR (400 MHz, CDCl3): δ 9.43 (s, 1H), 8.51 (d, J = 2.2 Hz, 1H), 8.25 (dd, J = 8.0, 1.1 Hz, 1H), 8.10 (dd, J = 9.1, 2.2 Hz, 1H), 7.52 (td, J = 7.5, 1.2 Hz, 1H), 7.40 (dd, J = 13.9, 5.9 Hz, 2H), 7.31–7.26 (m, 3H), 7.14 (d, J = 7.8 Hz, 1H), 6.92 (s, 1H), 4.10 (s, 2H), 3.88 (s, 3H), 3.78 (s, 3H), 2.68 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 164.2, 157.7, 141.4, 140.0, 138.0, 137.0, 136.0, 134.2, 133.0, 132.0, 130.5, 130.4, 130.1, 127.2, 127.0, 120.0, 117.5, 117.0, 116.0, 110.1, 109.3, 56; HRMS (ESI-TOF) (m/z): calcd for [M + Na]+ C25H23N3O6SNa, 516.1988; found, 516.1975.
Cyclopentyl(3-(2-methoxy-4-((o-tolylsulfonyl)carbamoyl)benzyl)-1-methyl-1H-indol-5-yl)carbamate (1)
A solution of N-{3-methyl-4-(N-methyl-5-nitro-1H-3-indolyl)methyl benzoyl}-2-methyl-1-benzenesulfonamide 9 (100 mg, 0.20 mmol) in MeOH (2 mL) and Ra–Ni (2 mg, 5 mol %) was placed under hydrogen balloon pressure at room temperature for 2 h. The reaction mass was filtered through a celite bed, and the catalyst was washed with diethyl ether (2 × 10 mL); the combined filtrates were concentrated under reduced pressure and dried using high vacuum for 1 h. The obtained residue and N-methyl morphine (26 μL, 0.24 mmol) were dissolved in DCM (2 mL), and then, cyclopentyl chloroformate was added slowly (25 μL, 0.20 mmol) in 0 °C. The resulting reaction mixture was stirred at room temperature for 3 h. After completion of the reaction, the solvent was evaporated under reduced pressure, and the solid precipitate was washed with water (5 mL) and then extracted with DCM (10 mL) and dried over sodium sulfate and evaporated in under reduced pressure. The crude compound was purified by column chromatography to afford title product 1 (102 mg, 89%) as a white solid; Rf = 0.7 (silica, EtOAc/hexane = 4:6); mp = 137–141 °C; IR (neat): 3476, 2951, 2925, 2850, 1723, 1689, 1554, 1499, 1431, 1270, 1163, 1062, 1036, 872, 758 cm–1; 1H NMR (500 MHz, CDCl3): δ 9.18 (s, 1H), 8.26 (dd, J = 8.0, 1.3 Hz, 1H), 7.51 (td, J = 7.5, 1.3 Hz, 2H), 7.40 (t, J = 7.5 Hz, 1H), 7.31 (d, J = 1.5 Hz, 1H), 7.29 (d, J = 7.5 Hz, 1H), 7.19 (d, J = 8.6 Hz, 1H), 7.16 (dd, J = 7.9, 1.6 Hz, 1H), 7.09 (d, J = 7.9 Hz, 2H), 6.77 (s, 1H), 6.51 (s, 1H), 5.18 (ddd, J = 8.9, 5.9, 2.6 Hz, 1H), 4.03 (s, 2H), 3.85 (s, 3H), 3.70 (s, 3H), 2.68 (s, 3H), 1.86 (s, 2H), 1.74 (d, J = 12.6 Hz, 4H), 1.60 (s, 2H); 13C NMR (125 MHz, CDCl3): δ 164.4, 158.0, 138.0, 137.0, 137.0, 134.4, 134.1, 133.0, 138.0, 130.1, 130.0, 128.4, 128.1, 127.0, 119.4, 115.2, 112.0, 110.0, 110.0, 56.0, 33.0, 33.0, 25.4, 24.0, 21.0; HRMS (ESI-TOF) (m/z): calcd for [M + Na]+ C31H33N3O6SNa, 598.1988; found, 598.1975.
Acknowledgments
The authors thank the Council of Scientific and Industrial Research (CSIR), New Delhi, and S.P. thanks University Grants Commission (UGC), New Delhi, for a research fellowship. S.C. thanks DST for J. C. Bose fellowship (SB/S2/JCB-002/2015) and SERB research grant (EMR/2014/000338) for financial support. This work is dedicated to Prof. S. P. Singh, Emeritus Professor, Kurukshetra University, India, on his 80th birthday acknowledging his contributions to organic chemistry.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00476.
General details; experimental procedures and analytical data for biaryl alkynes; and 1H and 13C NMR spectra of the obtained compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kaushik N.; Kaushik N.; Attri P.; Kumar N.; Kim C.; Verma A.; Choi E. Biomedical Importance of Indoles. Molecules 2013, 18, 6620–6662. 10.3390/molecules18066620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta R. G.; Liu J.; Constantinou A.; Thomas C. F.; Hawthorne M.; You M.; Gerhäuser C.; Pezzuto J. M.; Moon R. C.; Moriarty R. M. Cancer chemopreventive activity of brassinin, a phytoalexin from cabbage. Carcinogenesis 1995, 16, 399–404. 10.1093/carcin/16.2.399. [DOI] [PubMed] [Google Scholar]
- Ferreira S. H.; Moncada S.; Vane J. R. Indomethacin and aspirin abolish prostaglandin release from the spleen. Nature (London), New Biol. 1971, 231, 237–239. 10.1038/newbio231237a0. [DOI] [PubMed] [Google Scholar]
- Johnson I. S.; Armstrong J. G.; Gorman M.; Burnett J. P. The vinca alkaloids: A new class of oncolytic agents. Cancer Res. 1963, 23, 1390–1427. [PubMed] [Google Scholar]
- Duflos A.; Kruczynski A.; Barret J.-M. Novel aspects of natural and modified vinca alkaloids. Curr. Med. Chem. Anti Canc. Agents 2002, 2, 55–70. 10.2174/1568011023354452. [DOI] [PubMed] [Google Scholar]
- Hogaboom G. K.; Mong S.; Wu H.-L.; Crooke S. T. Peptidoleukotrienes: Distinct receptors for leukotriene C4 and D4 in the guinea-pig lung. Biochem. Biophys. Res. Commun. 1983, 116, 1136–1143. 10.1016/s0006-291x(83)80261-7. [DOI] [PubMed] [Google Scholar]
- Krell R. D.; Aharony D.; Buckner C. K.; Keith R. A.; Kusner E. J.; Snyder D. W.; Bernstein P. R.; Matassa V. G.; Yee Y. K.; Brown F. J.; Hesp B.; Giles R. E. The preclinical pharmacology of ICI204, 219. A peptide leukotriene antagonist. Am. Rev. Respir. Dis. 1990, 141, 978–987. 10.1164/ajrccm/141.4_pt_1.978. [DOI] [PubMed] [Google Scholar]
- Smith L. J.; Geller S.; Ebright L.; Glass M.; Thyrum P. T. Inhibition of leukotriene D4-induced bronchoconstriction in normal subjects by the oral LTD4 receptor antagonist ICI 204, 219. Am. Rev. Respir. Dis. 1990, 141, 988–992. 10.1164/ajrccm/141.4_pt_1.988. [DOI] [PubMed] [Google Scholar]
- Brown F. J.; Bernstein P. R.; Cronk L. A.; Dosset D. L.; Hebbel K. C.; Maduskuie T. P. Jr.; Shapiro H. S.; Vacek E. P.; Yee Y. K.; Willard A. K.; Krell R. D.; Snyder D. W. Hydroxyacetophenone derived antagonists of the peptidoleukotrienes. J. Med. Chem. 1989, 32, 807–826. 10.1021/jm00124a014. [DOI] [PubMed] [Google Scholar]
- Brown F. J.; Yee Y. K.; Cronk L. A.; Hebbel K. C.; Krell R. D.; Snyder D. W. Evolution of a series of peptidoleukotriene antagonists: Synthesis and structure-activity relationships of 1,6-disubstituted indoles and indazoles. J. Med. Chem. 1990, 33, 1771–1781. 10.1021/jm00168a036. [DOI] [PubMed] [Google Scholar]
- Matassa V. G.; Maduskuie T. P. Jr.; Shapiro H. S.; Hesp B.; Snyder D. W.; Aharony D.; Krell R. D.; Keith R. A. Evolution of a series of peptidoleukotriene antagonists: Synthesis and structure/activity relationships of 1,3,5-substituted indoles and indazoles. J. Med. Chem. 1990, 33, 1781–1790. 10.1021/jm00168a037. [DOI] [PubMed] [Google Scholar]
- Yee Y. K.; Bernstein P. R.; Adams E. J.; Brown F. J.; Cronk L. A.; Hebbel K. C.; Vacek E. P.; Krell R. D.; Snyder D. W. A novel series of selective leukotriene antagonists: exploration and optimization of the acidic region in 1,6-disubstituted indoles and indazoles. J. Med. Chem. 1990, 33, 2437–2451. 10.1021/jm00171a018. [DOI] [PubMed] [Google Scholar]
- Yee Y. K.; Brown F. J.; Hebbel K. C.; Cronk L. A.; Snyder D. W.; Krell R. D. Structure-activity relationships based on the peptide leukotriene receptor antagonist ICI 198, 615 enhancement of potency. Ann. N.Y. Acad. Sci. 1988, 524, 458–461. 10.1111/j.1749-6632.1988.tb38588.x. [DOI] [Google Scholar]
- Phipatanakul W.; Eggleston P. A.; Conover-Walker M. K.; Kesavanathan J.; Sweitzer D.; Wood R. A. A randomized, double blind, placebo controlled trial of the effect of zafirlukast on upper and lower respiratory responses to cat challenge. J. Allergy Clin. Immunol. 2000, 105, 704–710. 10.1067/mai.2000.105123. [DOI] [PubMed] [Google Scholar]
- Bernstein P. R.; Brown F. J.; Matassa V. G.; Yee Y. K.. Heterocyclic amide derivatives and pharmaceutical use. U.S. Patent 5,338,734 A, 1989.
- Gutman A.; Nisnevich G.; Zaltzman I.; Ponomarev V.; Sotrihin M.. Process for the preparation of zafirlukast. WO Patent 2002046153A2, 2002.
- Ancell C. L.; Derrick I.; Moseley J. D.; Stott J. A. Investigation into the acidification process of zafirluskast nitroacid leads to a surprising improvement in product quality. Org. Process Res. Dev. 2004, 8, 808–813. 10.1021/op049911+. [DOI] [Google Scholar]
- Srinivas K.; Srinivasan N.; Krishna M. R.; Reddy C. R.; Arunagiri M.; Lalitha R.; Reddy K. S. R.; Reddy B. S.; Reddy G. M.; Reddy P. P.; Kumar M. K.; Reddy M. S. A Novel and Efficient Route to Zafirlukast. Org. Process Res. Dev. 2004, 8, 952–954. 10.1021/op049869i. [DOI] [Google Scholar]
- Jiang X.; Tiwari A.; Thompson M.; Chen Z.; Cleary T. P.; Lee T. B. K. A Practical Method for N-Methylation of Indoles Using Dimethyl Carbonate. Org. Process Res. Dev. 2001, 5, 604–608. 10.1021/op0102215. [DOI] [Google Scholar]
- Goverdhan G.; Reddy A. R.; Sampath A.; Srinivas K.; Himabindu V.; Reddy G. M. An Improved and Scalable Process for Zafirlukast: An Asthma Drug. Org. Process Res. Dev. 2009, 13, 67–72. 10.1021/op800137b. [DOI] [Google Scholar]
- Anumula R. R.; Gilla G.; Alla S.; Kurella S.; Kopparapu J. S. R.; Medisetti R. K. V.; Maddula S. R.. Processes for preparing zafirlukast. U.S. Patent 20,090,149,662 A1, 2009.
- Brown F. J.; Bernstein P. R.; Yee Y. K.; Matassa V. G.. Heterocyclic amide derivatives. EP Patent 0199543 A2, 1986.
- Goverdhan G.; Reddy A. R.; Himabindu V.; Reddy G. M. Concise and Alternative synthesis of Zafirlukast, an anti-asthma drug. Synth. Commun. 2013, 43, 498–504. 10.1080/00397911.2011.603875. [DOI] [Google Scholar]
- Lee W.-Y.; Chen P.-C.; Wu W.-S. Panobinostat sensitizes KRAS-mutant non-small-cell lung cancer to gefitinib by targeting TAZ. Int. J. Cancer 2017, 141, 1921–1931. 10.1002/ijc.30888. [DOI] [PubMed] [Google Scholar]
- Klimke A.; Klieser E. Antipsychotic efficacy of the dopaminergic autoreceptor agonist EMD 49980 (Roxindol). Results of an open clinical study. Pharmacopsychiatry 1991, 24, 107–112. 10.1055/s-2007-1014451. [DOI] [PubMed] [Google Scholar]
- Müller W.; Stratz T. Local treatment of tendinopathies and myofascial pain syndromes with the 5-HT3 receptor antagonist tropisetron. Scand. J. Rheumatol. 2004, 33, 44–48. 10.1080/03009740410007032. [DOI] [PubMed] [Google Scholar]
- Macor J. E.; Gurley D.; Lanthorn T.; Loch J.; Mack R. A.; Mullen G.; Tran O.; Wright N. The 5-HT3 antagonist tropisetron (ICS 205-930) is a potent and selective α7 nicotinic receptor partial agonist. Bioorg. Med. Chem. Lett. 2001, 11, 319–321. 10.1016/s0960-894x(00)00670-3. [DOI] [PubMed] [Google Scholar]
- Cui R.; Suemaru K.; Li B.; Kohnomi S.; Araki H. Tropisetron attenuates naloxone-induced place aversion in single-dose morphine-treated rats: role of α7 nicotinic receptors. Eur. J. Pharmacol. 2009, 609, 74–77. 10.1016/j.ejphar.2008.12.051. [DOI] [PubMed] [Google Scholar]
- Lade D. M.; Pawar A. B.; Mainkar P. S.; Chandrasekhar S. Total Synthesis of Lamellarin D Trimethyl Ether, Lamellarin D and Lamellarin H. J. Org. Chem. 2017, 82, 4998–5004. 10.1021/acs.joc.7b00636. [DOI] [PubMed] [Google Scholar]
- Nagaraju K.; Chegondi R.; Chandrasekhar S. Expanding Diversity without Protecting Groups. (+)-Sclareolide to Indolosesquiterpene Alkaloid Mycoleptodiscin A and Analogues. Org. Lett. 2016, 18, 2684–2687. 10.1021/acs.orglett.6b01145. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S.; Patro V.; Chavan L. N.; Chegondi R.; Grée R. Multicomponent reactions in PEG-400. Ruthenium-catalyzed synthesis of substituted pyrroles. Tetrahedron Lett. 2014, 55, 5932–5935. 10.1016/j.tetlet.2014.08.105. [DOI] [Google Scholar]
- Gabriele B.; Veltri L.; Mancuso R.; Salerno G.; Costa M. A General synthesis of indole-3-carboxylic esters by palladium-catalyzed direct oxidative carbonylation of 2-alkynylaniline derivatives. Eur. J. Org. Chem. 2012, 2549–2559. 10.1002/ejoc.201200120. [DOI] [Google Scholar]
- Ma Y.; You J.; Song F. Facile Access to 3-Acylindoles through Palladium-Catalyzed Addition of Indoles to Nitriles. The One-Pot Synthesis of Indenoindolones. Chem.—Eur. J. 2013, 19, 1189–1193. 10.1002/chem.201203354. [DOI] [PubMed] [Google Scholar]
- Wu W.; Su W. Mild and Selective Ru-Catalyzed Formylation and Fe-Catalyzed Acylation of Free(N-H)Indoles Using Anilines as the Carbonyl Source. J. Am. Chem. Soc. 2011, 133, 11924–11927. 10.1021/ja2048495. [DOI] [PubMed] [Google Scholar]
- Bernini R.; Fabrizi G.; Sferrazza A.; Cacchi S. Copper-Catalyzed CC Bond Formation through C-H Functionalization. Synthesis of Multisubstituted Indoles from N-Aryl Enaminones. Angew. Chem., Int. Ed. 2009, 48, 8078–8081. 10.1002/anie.200902440. [DOI] [PubMed] [Google Scholar]
- Gogoi A.; Guin S.; Rout S. K.; Patel B. K. A Copper-Catalyzed Synthesis of 3-Aroylindoles via a sp3 C-H Bond Activation Followed by C-C and C-O Bond Formation. Org. Lett. 2013, 15, 1802–1805. 10.1021/ol400692b. [DOI] [PubMed] [Google Scholar]
- Gogoi A.; Modi A.; Guin S.; Rout S. K.; Das D.; Patel B. K. A metal free domino synthesis of 3-aroylindoles via two sp3 C–H activation. Chem. Commun. 2014, 50, 10445–40447. 10.1039/c4cc04407j. [DOI] [PubMed] [Google Scholar]
- Xia X.-F.; Zhang L.-L.; Song X.-R.; Niu Y.-N.; Liu X.-Y.; Liang Y.-M. Palladium-copper-cocatalyzed intramolecular oxidative coupling. An efficient and atom-economical strategy for the synthesis of 3-acylindoles. Chem. Commun. 2013, 49, 1410–1412. 10.1039/c2cc37805a. [DOI] [PubMed] [Google Scholar]
- For synthesis of biaryl alkynes 5: see Supporting Information.
- Ikeda A.; Omote M.; Kusumoto K.; Komori M.; Tarui A.; Sato K.; Ando A. A dramatic enhancing effect of InBr3 towards the oxidative Sonogashira cross-coupling reaction of 2-ethynylanilines. Org. Biomol. Chem. 2016, 14, 2127–2133. 10.1039/c5ob02558c. [DOI] [PubMed] [Google Scholar]
- Reddy A. S.; Chegondi R.; Chandrasekhar S. Total Synthesis of a Natural Product in Poly(ethylene glycol): (±)-Centrolobine. Proc. Indian Natl. Sci. Acad. 2015, 81, 1171–1175. 10.16943/ptinsa/2015/v81i5/48337. [DOI] [Google Scholar]
- Reddy C. R.; Mahipal B.; Yaragorla S. R. A new and efficient method for the facile synthesis of N-acyl sulfonamides under Lewis acid catalysis. Tetrahedron Lett. 2007, 48, 7528–7532. 10.1016/j.tetlet.2007.08.048. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.