

Abstract
Pectin-g-(sodium acrylate-co-3-(N-isopropylacrylamido) sodium propanoate-co-N-isopropylacrylamide) interpenetrating polymer networks (PANIPNs) were synthesized through systematic multistage optimization of equilibrium swelling ratio by response surface methodology for individual and/or synergistic removal(s) of cationic safranine (SF), anionic methyl orange, and M(II/III), such as Hg(II), Cd(II), and Cr(III). The relative effects of copolymer compositions on ligand-selective adsorption, strong/weak H-bonds, thermal stabilities, crystallinity, surface properties, swelling abilities, cross-link densities, network parameters, hydrophilic–hydrophobic characteristics, and adsorption capacities (ACs) were measured through extensive microstructural analyses of adsorbed and/or unadsorbed PANIPN41 and PANIPN21 bearing sodium acrylate and N-isopropylacrylamide (SA/NIPAm) in 4:1 and 2:1 ratios, respectively, using Fourier transform infrared, 1H and 13C NMR, X-ray photoelectron spectroscopy, thermogravimetric analysis, differential scanning calorimetry, X-ray diffraction, scanning electron microscopy, and energy dispersive X-ray spectroscopy, along with measuring lower critical solution temperature, % gel content (% GC), % −COOH, and pHPZC. Extensive UV–vis measurements were carried out at varying copolymer compositions, initial pH (pHi), and dyes, interpreted considering monomer–dimer and azonium–ammonium equilibrium of dye, dye–dye complexation, ligand-selective PANIPNs–dye adduct formation, π–π stacking interactions, and orientation effect of dyes. Thermodynamically feasible chemisorption processes showed the maximum ACs of 127.61, 96.78, 103.36, and 99.41 mg g–1 for SF, Hg(II), Cd(II), and Cr(III), respectively, under optimum conditions.
1. Introduction
Hydrogels possess the unique ability to imbibe a large amount of water without loss of structure owing to the H-bonding and electrostatic/covalent interactions. Depending on the use of different extent of ingredients, temperatures, and methods of synthesis, relative proportions of several hydrophilic functional groups demonstrate diversified swelling behaviors.1 Although hydrogels of synthetic polymers have adequate mechanical strengths, yet these suffer from poor biodegradability. Conversely, natural polymer-based hydrogels exhibit better biodegradability but lack mechanical strengths. However, the attainment of optimum balance between mechanical properties and biodegradability is possible via synthesizing interpenetrating polymer network (IPN)-based hydrogels comprising a homo-/co-/terpolymer, synthesized through polymerizing one or two synthetic monomer(s) with a natural polymer.2 Synthetic hydrogels find extensive applications as superadsorbents in tissues, ion exchangers, size exclusion chromatography, membrane-based applications, catheters, and contact lenses. Recently, hydrogels were used as biosensors for glucose, bile acids, and human metabolites, such as uric acid, cholesterol, and triglycerides. Currently, self-healing hydrogels, especially magnetic hydrogels, are under the utmost consideration for drug delivery and separation, image enhancement, and remote-controlled actuators and valves. In this context, Fe(II)- and Fe(III)-grafted poly(2-acrylamido-2-methyl-1-propansulfonic acid-co-vinylimidazole),3,4 carboxymethyl starch-g-polyvinyl imidazole/poly(vinyl alcohol) (PVA)/Fe3O4 mixture,5 Fe3O4-poly(l-cysteine/2-hydroxyethyl acrylate) (Fe3O4-p(Cys/HEA)),6 carboxymethyl-β-cyclodextrin polymer-modified Fe3O4 nanoparticles,7 Fe3O4 nanoparticle-coated polyacrylic acid,8 PVA-based nanocomposites,9 and PVA/chitosan/agar–agar tri-polymer-based magnetic nanohydrogels10 have been examined for the adsorptive exclusion of toxic dyes/metal ions. Moreover, biocompatible hydrogels are used as injectable cell therapy carriers and scaffolding materials for cartilage tissue engineering. Polyacrylic acid (PAA)-based hydrogels have been effectively used for drug delivery to specific regions of the gastrointestinal tract, in particular, for the colon-specific delivery of low-molecular-weight protein drugs. However, poor thermal and mechanical stabilities and high water solubility limit the usability of the PAA hydrogels. Moreover, the thermosensitive poly-NIPAm (PNIPAm) hydrogels have found diverse applications for making biosensors and contact lenses, drug delivery, and cell sheet engineering. The utilization of such hydrogels is limited because of the slow response rate and poor mechanical properties.
Pectin (PN), an anionic polysaccharide, is composed of poly(1,4-galacturonic acid) as a 3-fold helix, linked together by 1 → 4 glycosidic linkages. PN consists of two regions: (a) a smooth region, a linear chain of (1–4) linked α-d-galacturonic acid, and (b) a hairy region, containing galacturonic acid with or without rhamnose as the main chain and substituted by neutral sugars.11 The relative proportion of both the regions depends on the botanical source of PN and type of extraction. PN finds immense applications in food industries and biomedical fields.12
Response surface methodology (RSM) is widely used for analyzing synergistic effects of process variables on response(s) to physical/chemical studies through the minimum number of experimental runs. However, few studies have been devoted for RSM-based optimization of operational variables during adsorption for attaining the maximum adsorption capacity (AC).13,14 In the present study, RSM has been employed for systematic multistage optimization of variables for synthesis of a hydrogel possessing the maximum equilibrium swelling ratio (ESR).
The presence of toxic dyes/pigments and metal ions (M(II/III)) beyond the tolerance limit in the effluents of cosmetic, leather, pharmaceutical, food, textile, paper, plastic, plating, and mining industries are severely detrimental for the ecosystem and environment. In fact, such industries use more than 10 000 dissimilar nonbiodegradable aromatic synthetic dyes for conquering, intensifying, and maintaining the color of products, of which around 15 wt % of these are discharged as wastes.13 Among several popular synthetic dyes, safranine (SF) is one of the most extensively used dyes for dyeing natural fibers. SF is frequently used for dyeing wool, silk, and acrylic fibers and leather. SF poisoning may cause carcinogenicity, mitochondrial toxicity, mutagenicity, and nucleic acid damage.15 The possible oxidation of Cr(III) to Cr(VI) causes toxicity via carcinogenicity and/or mutagenicity, skin ulceration, liver damage, pulmonary congestion, and many other health problems. The toxicity of Cd(II) includes high blood pressure, renal dysfunction, lung cancer, destruction of red blood cell, and anemia. Hg(II), one of the most toxic heavy metal ions, can damage brain, nervous system, endocrine system, and other organs of human body. Elimination of such contaminants through an ecofriendly waste management system is gaining high insight.
Although the use of different hydrogels, such as homo-/co-/terpolymers16,17 and IPN1,2-based polymers is available in the literature for the adsorptive removal of dyes and M(II/III), unorthodox synthesis of natural polymer-grafted-terpolymer IPN hydrogels through in situ strategic attachment of a third monomer using optimized compositions of SA/NIPAm, temperature, and other ingredients, appearance of several physicochemical effects during synergistic removals of structurally distinct dyes and extensive characterization of loaded and/or unloaded hydrogel(s) to understand the changes in physicochemical properties of PANIPNs of varying copolymer compositions is comprehensively reported. In addition, the relative effects of varied copolymer compositions of PANIPNs on diversified interactions between M(II/III) and N- or O-donor ligands, such as −COO–, −COOH, and −CONH–, resulting in the formation of different coordinate and/or ionic bond(s) and, hence, varied ACs have thoroughly been investigated.
2. Experimental Section
2.1. Materials
Na2HPO4, Na2B4O7·10H2O, HCl, acrylic acid, NIPAm, PN (degree of esterification = 63–66%), N,N′-methylenebisacrylamide (MBA), potassium persulfate (PPS), and sodium bisulfite (SBS) of analytical grades were purchased from Merck and used without any further modification. SF, methyl orange (MO), and nitrate salts of Cd(II), Hg(II), and Cr(III), used in adsorption studies, were purchased from Sigma-Aldrich.
2.2. Synthesis of Hydrogels
PANIPNs were synthesized through free radical solution polymerization of SA and NIPAm (Scheme 1) using PPS and SBS as redox initiators and MBA as a cross-linker in a N2 atmosphere. The exact composition of ingredients and reaction temperature were optimized by RSM. In fact, two PANIPNs, one possessing the optimum composition of monomers (i.e., PANIPN41, SA/NIPAm = 4:1) and the other involving the 2-fold variation of SA/NIPAm (i.e., PANIPN21, SA/NIPAm = 2:1), were chosen for the comparative studies. Initially, a three-neck reactor was placed on a magnetic stirrer cum hot plate, followed by the gradual addition of 0.5 g of PN in 30 mL of water, 0.20/0.16 mol of SA in 27 mL of water at pHi = 5.5 and 0.05/0.08 mol of NIPAm in 25 mL of water with constant stirring at 300 rpm for synthesizing PANIPNs. Thereafter, 0.15 mmol of MBA solution in 8 mL of water was added at a constant temperature of 293 K and the resultant solution was kept stirring for 6 h for homogenization and then for another 2 h after purging N2 gas. After that, a solution mixture of PPS and SBS (i.e., 0.74 and 1.92 mmol in 10 mL of water) was added to initiate polymerization (Scheme 2). The as-prepared PANIPNs were washed several times with 1:3 methanol/water solutions for complete removal of unreacted components and water-soluble oligomers. Finally, PANIPNs were air dried for 3 days, followed by drying under vacuum for another 3 days at 323 K.
Scheme 1. Probable Steps of PANIPN Synthesis.

Scheme 2. Synthesis of PANIPN Using Pectin (1), SA (2), NIPAm (3), MBA (4), and Redox Pair of Initiators (K2S2O8 and NaHSO3) (5).

2.3. Characterization and Methodology
The characterization techniques for loaded and/or unloaded PANIPNs and the methodology used for adsorption isotherm studies are provided in the Supporting Information section of this article.
3. Results and Discussion
3.1. Experimental Design and Model Development for the Synthesis of PANIPN
The ESR of a hydrogel depends on several synthesis parameters, such as SA (wt %, A), initiator (wt %, B), PN (wt %, C), MBA (wt %, D), pHi (−, E), and temperature (K, F), individually or synergistically. The hydrogel having the maximum ESR was synthesized using optimum conditions of such variables via executing the minimum number of studies, obtained through two-factor interactions (2FI) between the variables. A fractional factorial design (Resolution-IV) was employed to filter the variable, imparting the most significant effect on ESR. Finally, such significant variables were optimized using central composite design (CCD) analysis.
3.1.1. Phase-1: Screening of Significant Variables of Synthesis
In phase-1, screening of variables was executed through nineteen experiments using Resolution-IV design (Table S1). The maximum/minimum levels were considered to be 66.67/95.24 wt %, 1.00/4.00 wt %, 0.10/0.50 wt %, and 0.10/0.40 wt %, 4.00/12.00, and 303/323 K for A, B, C, D, E, and F, respectively, for ESR within 129–225.75. However, A, B, E, AB, AD, and CF crossed the Bonferroni limit of 3.7283 in the Pareto chart (Figure S1). Thus, these six significant terms were considered for building the regression model. However, the adequacy of this model was evaluated through the following second-order polynomial equation
| 1 |
Indeed, the chosen model was considered to be highly significant because of close resemblance of Adj. and Pred. R2 (i.e., 0.9804/0.9713), very high Adj. R2 (i.e., 0.9804), and very low p-value (<0.0001) (Table S2). The individual effect of the significant variables was noted to be positive, whereas only AB imparted the antagonism effect among AB, AD, and CF. However, the optimum A, B, and E for attaining the maximum ESR of 225.75 were 95.24 wt %, 4.00 wt %, and 12, respectively.
3.1.2. Phase-2: CCD Optimization of the Three Most Significant Variables of Synthesis
The CCD was adopted for optimizing the three most significant process variables of synthesis, i.e., A, B, and E, considering the individual and interactive effects on the response, i.e., ESR. In fact, the ESRs of such analyses were scrutinized and interlinked with input variables for optimization through empirical second-order polynomial eq 2.
| 2 |
Here, Y, β0, βi, βii, and βij represent the predicted response, constant, linear, quadratic, and interaction coefficients, respectively. The adequacy of the predicted model was justified by ANOVA (Table S4), taking A, B, and E within 66.67–95.24 wt %, 2.00–4.00 wt %, and 5–10, respectively. The process variables and software-generated responses are listed in Table S3. In addition, sequential model sum of squares (type I) and model summary statistics tests were conducted for rational understanding of interactive effects of A, B, and E on the response via linear, 2FI, quadratic, and cubic models. However, the quadratic model was found to be the best because of higher Adj. R2 and finer correspondence between Adj. and Pred. R2. In this context, swelling data were fitted to eq 2 to obtain eq 3.
| 3 |
The response surface plots (Figure 1a–c) showed the interactive effects among BA, EA, and EB. The numerical optimization section, in which A, B, and E were kept in range and ESR became maximize, A, B, and E were found to be 84.43 wt %, 2.97 wt %, and 10, respectively.
Figure 1.

Three-dimensional response surface plots of ESR (−) vs (a) amounts of initiator (wt %, B) and SA (wt %, A), (b) pHi (−, E) and SA (wt %, A), and (c) pHi (−, E) and amount of initiator (wt %, B).
3.2. Swelling and pH/Temperature Reversibility Studies of PANIPNs
Hydrogels exhibit diversified swelling behaviors because of variation in hydrophilic functional groups, originated from different extents of added ingredients, temperatures, and methods of synthesis. At pHi > pHPZC, the repulsive force between −COO– groups caused an enhancement of chain relaxation. Moreover, the presence of relatively higher number of mobile counterions in the PANIPN network resulted in a higher osmotic pressure for better swelling. In fact, reverse effects were produced at pHi < pHPZC as a result of extensive protonation of −COO–. Thus, a relatively higher ESR of PANIPN41 and the respective terpolymer could be realized by the presence of a higher amount of hydrophilic SA (Figure S2a–f). Furthermore, the thermoresponsive nature of hydrogels was controlled via relative variations of hydrophilic to hydrophobic moieties, and they showed better swelling below the lower critical solution temperature (LCST) because of the better population of stronger H-bonds (Figure S2g). Again, the grafting of hydrophilic PN within the terpolymer network caused an enhanced swelling as well as chain flexibility, reflected via retention of structural integrity even after the completion of six swelling–deswelling cycles (Figure S2a,b).
3.3. Fourier Transform Infrared (FTIR) Analyses of Unloaded and SF/M(II/III)-Loaded PANIPNs
Grafting of PN with TerP41 and thereby formation of an ether linkage of type >CH–O–CH2– in PANIPN41 were confirmed from the appearance of new peaks at 1458 and 2855 cm–1 for −CH2– deformation and symmetric −CH2– str. of −O–CH2– in >CH–O–CH2–. Moreover, such C–O–C bond formation during grafting through free radical polymerization of PN and C=C of SA/NIPAm/MBA and formation of at least monobranched matrices was substantiated by the appearance of new skeletal vibration peaks at 473 (medium), 460 (weak), and 453 cm–1.18 Indeed, grafting-related structural alterations were also manifested from the conversion of PN-specific α-1,4- to β-1,4-glycosidic linkage in PANIPN41 (Table S6), along with the appearance of β-d-glucose peak at 874 cm–1 in PANIPN41. In fact, the grafting-related transformation of α-1,4- to β-1,4 glycosidic linkages in PANIPN41 was also corroborated from the absence of equatorial anomeric hydrogen-specific peaks within 825–860 cm–1 in PANIPN41. In addition, grafting and associated intimate interactions between PN and TerP41 were apprehended from the loss of several PN-specific peaks in PANIPN41 (Figure S3a and Table S6). Accordingly, significant changeover in stronger-/weaker-type H-bonding and associated changes in the H-bonding environment around NIPAm could be correlated with the alteration in NIPAm-specific doublet peaks for PANIPN41 and the increase in N–C=O in-plane bending vibration from 661 cm–1 of TerP41 to 669 cm–1 in PANIPN41.19 For M(II/III)-PANIPN41, the formation of M(II/III)–N and M(III)–O bonds, the relative variation of mutual O–H/N–H H-bonding, and several kinds of microstructural alterations are provided in the Supporting Information of the article (Page: S12–S15).
3.4. NMR Analyses
3.4.1. TerP21
1H NMR (300 MHz, CDCl3, δ, ppm): 0.83, 0.99, 1.25, 1.37, 1.42, 1.58, 1.96, 2.18, 2.35, 2.65, 3.49–3.53, 4.18, 4.31, 6.91, 6.98, 7.51, 7.60, 7.70 (Figure 2a); solid state 13C NMR (100 MHz, δ, ppm): 23.90, 42.80, 46.70, 176.50–179.40, 186.20, 192.10 (Figure 2d).
Figure 2.

1H NMR of (a) TerP21 and (b) PANIPN21 and 13C NMR of (c) PN, (d) TerP21, and (e) PANIPN21.
3.4.2. PANIPN21
1H NMR (300 MHz, CDCl3, δ, ppm): 0.84, 1.21, 1.54, 2.04–2.13, 2.32, 2.58, 2.92, 3.40–3.69, 4.04, 4.24, 5.08, 5.22, 6.87, 7.57 (Figure 2b); solid state 13C NMR (100 MHz, δ, ppm): 23.94, 42.83, 46.34, 65.21–81.54, 102.05, 104.61, 109.48, 165.71, 176.48–179.98, 185.71, 194.42 (Figure 2e).
3.4.3. PN
1H NMR (300 MHz, DMSO-d6, δ, ppm): 3.50–3.61, 3.72, 3.83, 4.37, 4.46, 4.71–4.79, 5.02, 5.14 (Figure S4a); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 61.62, 69.38, 71.16, 72.36, 73.82, 76.57, 82.08, 102.12, 104.62, 109.47, 165.72, 177.20 (Figure 2c).
3.4.4. NIPAm
1H NMR (300 MHz, CDCl3, δ, ppm):1.19 (6H, d), 4.16 (1H, m), 5.60 (1H, dd), 6.22 (2H, m), 6.61 (1H, s) (Figure S4b); 13C NMR (75 MHz, CDCl3, δ, ppm): 22.39, 41.16, 125.49, 131.30, 164.78 (Figure S4c).
3.4.5. AA
1H NMR (300 MHz, CDCl3, δ, ppm): 6.03 (1H, dd), 6.23 (1H, dd), 6.59 (1H, dd), 11.41 (1H, s) (Figure S4d); 13C NMR (75 MHz, CDCl3, δ, ppm): 127.87, 132.97, 171.62 (Figure S4e).
3.4.6. MBA
1H NMR (300 MHz, DMSO-d6, δ, ppm):4.49 (2H, t), 5.59 (2H, dd), 6.13 (4H, m), 8.73 (2H, t) (Figure S4f); 13C NMR (75 MHz, DMSO-d6, δ, ppm): 42.76, 125.53, 130.88, 164.41 (Figure S4g).
From 1H NMR, incorporation of NIPAm within TerP21 was ascertained from the appearance of characteristic peaks of −CH3 and −CH– at 1.25 and 4.18 ppm, respectively. However, such peaks were appeared at 1.21 and 4.04 ppm in PANIPN21 (Figure 2a,b). Moreover, the presence of peaks specific to −CH2– of MBA at 4.31 and 4.24 ppm in TerP21 and PANIPN21, respectively, confirmed the cross-linking by MBA (Figure 2a,b).20 In addition, the formation of −C(sp3)–C(sp3)– from C=C of SA, NIPAm, and MBA was inferred from the absence of olefinic proton-specific peaks at 6.03, 6.23, and 6.59 ppm of SA, 5.60 and 6.22 ppm of NIPAm, and 5.59 and 6.13 ppm of MBA. However, the symbolic peaks within 0.83–1.58 and 1.96–2.65 ppm confirmed the formation of −CH2– and >CH–, respectively, in TerP21, whereas such peaks appeared within 0.84–1.54 and 2.04–2.92 ppm in PANIPN.21 The presence of moieties of type −CH2–N(CHMe2)–CO–, generated via chain propagation between −CON(CHMe2)• of NIPAm and SA, was determined from the appearance of new broad peaks within 3.49–3.53 and 3.40–3.69 ppm in TerP21 and PANIPN21, respectively. The characteristic N–H peaks of NIPAm/MBA appeared at 6.91, 6.98, 7.51, 7.60, and 7.70 in TerP21 and at 6.87 and 7.57 ppm in PANIPN21. In addition, PANIPN21 also showed PN-specific peaks at 5.22 and 5.08 ppm. In this context, the characteristic peaks of PN at 5.14/3.83/5.02 and 3.72 ppm were attributed to the H1/H3/H5 ring protons of galacturonic acid (GalA) and −COOCH3 of methyl galacturonate, respectively (Figure S4a). Indeed, the δ of GalA ring protons were affected by the variegated arrangements of esterified (E) and nonesterified (G) GalA units in PN, realized from the H5-specific δ within 4.71–4.79 ppm for GGG-, EGG-, GGE-, or EGE-type arrangements. The characteristic H4 peaks appeared at 4.37 and 4.46 ppm for GG and EE or EG arrangements, respectively.22 However, residual peaks of PN moieties were appeared within 3.50–3.61 ppm.22
The peaks at 186.20 and 192.10 ppm of TerP21 and 185.71 and 194.42 ppm of PANIPN21 were assigned to −COOH/–COO– of PANIPNs (Figure 2d,e). The presence of different types of amides (−CONH−) was confirmed by the appearance of peaks within 176.50–179.40 and 176.48–179.98 ppm in TerP21 and PANIPN21, respectively. The free radical polymerization and cross-linking were inferred from the absence of olefinic carbon peaks at 127.87/132.97, 125.49/131.30, and 125.53/130.88 ppm of SA, NIPAm, and MBA, respectively, in TerP21/PANIPN21.
The prevalent sharp peaks at 23.90 and 23.94 ppm in TerP21 and PANIPN21, respectively, assigned to the −(CH3)2 fragment of NIPAm, inferred the incorporation of NIPAm in both networks. Moreover, the peaks of –CH– of NIPAm, −CH2– of MBA, −CH(COOH), −CH(COO–), −CHCONHCHMe2, and −CH2N(CHMe2)CO– moieties broadened the spectrum and appeared at 42.80 and 46.70 ppm (shoulder) in TerP21. However, such peaks were appeared at 42.83 and 46.34 ppm in PANIPN21. Additionally, several weak peaks at 165.71, 109.48, 104.61, 102.05, and from 65.21 to 81.54 ppm in PANIPN21 inferred the presence of −COOCH3 of GalA, C1 of GalA, C1 of α-l-arabinose, C1 of α-d-galactose, C2 to C5 of GalA, and rest of ring residues (esterified and nonesterified) of PN, respectively (Figure 2c).23
3.5. X-ray Photoelectron Spectroscopy (XPS) Analyses of Unloaded and SF/M(II/III)-Loaded PANIPN21
The in situ strategic introduction of the acrylamido moiety in PANIPNs and ligand-specific binding affinities of Hg(II), Cd(II), and Cr(III) with PANIPN21 were investigated via XPS analyses by measuring the shift of binding energies (BEs) of core-level electrons (i.e., O 1s, N 1s, and C 1s) and M(II/III) (i.e., Hg 4f7/2, Hg 4f5/2, Cd 3d5/2, and Cr 2p3/2) (Figure 3). The characteristic BEs before and after M(II/III) adsorption have been listed in Table 1.
Figure 3.

XPS analyses of C 1s (a, d, h, and l), N 1s (b, e, i, and m), and O 1s (c, f, j, and n) for PANIPN21 and Hg(II)-, Cd(II)-, and Cr(III)-PANIPN21; Hg 4f7/2 and 4f5/2 (g), Cd 3d5/2 (k) and Cr 2p3/2 (o) of Hg(II)-, Cd(II)-, and Cr(III)-PANIPN21, respectively.
Table 1. XPS Analyses of PANIPN21 and Hg(II)-/Cd(II)-/Cr(III)-PANIPN21.
| peaks
(eV) |
||||||
|---|---|---|---|---|---|---|
| center | PANIPN21 | Hg(II)-PANIPN21 | Cd(II)-PANIPN21 | Cr(III)-PANIPN21 | explanations | ref |
| O 1s | (i) 530.91 | (i) 531.19 | (i) 531.06 | (i) 531.57 | (i) C=O of −COOH | (24) |
| (ii) 532.14 | (ii) 533.48 | (ii) 532.38 | (ii) 533.90 | (ii) −COO–, ionic bonding with Cd(II), but coordinate bonding with Hg(II) and Cr(III) | (25) | |
| (iii) 533.36 | (iii) 535.63 | (iii) 533.46 | (iii) 534.51 | (iii) O–H of −COOH, ionic bonding with Cd(II), but coordinate bonding with Hg(II) and Cr(III) | (26) | |
| (iv) 540.16 | (iv) 540.54 | (iv) 540.74 | (iv) shake-up satellite band of the O atom in the –CO–N(CHMe2)–CH2– segment of PANIPN21 due to π–π* transition | (2) | ||
| C 1s | (i) 284.81 | (i) 285.77 | (i) 285.56 | (i) 284.94 | (i) C of hydrocarbon moieties (i.e., CxHy) | (27) |
| (ii) 286.82 | (ii) 287.01 | (ii) 286.75 | (ii) 286.23 | (ii) >CH–CO– or protonated C attached to N (i.e., −NH–CH< and −NH–CH2−) | ||
| (iii) 287.88 | (iii) 288.53 | (iii) 288.19 | (iii) 287.84 | (iii) C of C=O | ||
| N 1s | (i) 399.70 | (i) 398.70 | (i) 399.78 | (i) 399.91 | (i) N of secondary amides of both NIPAm side chains and MBA cross-links of PANIPN21; formation of the amido-Hg covalent bond was inferred from the decrease in BE for Hg(II)-PANIPN21; ionic bonding between Cr(III) and N-donors | (28) |
| (ii) 404.23 | (ii) coordinate bonding between Hg(II) and N-donor ligands | |||||
| Hg 4f7/2 | (i) 101.76 | significant shifting from (i) 102.58 and (ii) 106.68 eV, indicating covalent/coordinate interactions between Hg(II) and N-donor ligands | (29) | |||
| Hg 4f5/2 | (ii) 104.72 | |||||
| Cd 3d5/2 | (i) 404.75 | (i) formation of (−COO)2Cd species in Cd(II)-PANIPN21 | ||||
| (ii) 406.27 | (ii) ionic bonding | |||||
| Cr 2p3/2 | 585.53 | decreased with respect to the Cr(NO3)3 peak at 587.00 eV, indicating coordinate interaction with Cr(III) and −COO– | (30) | |||
From Table 1, the coexistence of both −COO– and −COOH in the unadsorbed PANIPN21 at pHi = 7.0 confirmed the presence of O 1s peaks at 530.91/533.36 and 532.14 eV for C=O/O–H of −COOH and −COO–, respectively (Figure 3c). In fact, the BE of O 1s for −COO– was intermediate between the BEs of O 1s for C=O and O–H of −COOH, which reflected the mutual equivalence of both O-atoms of −COO– due to resonance stabilization. Moreover, the peak at 540.16 eV was ascribed to the shake-up satellite band of the O atom in the −CO–N(CHMe2)–CH2– segment of PANIPN21 because of the π–π* transition. Among all of the M(II/III), preferential covalent interaction between Hg(II) and N-donor ligands over O-donors of PANIPN21 was realized from the significant lowering of the N 1s BE from 399.70 to 398.70 eV (Figure 3e). Indeed, such substantial lowering in BE of N 1s was attributed to the appearance of the amido-Hg covalent bond, in which Hg(II) shared electron clouds with the amidic N.28 Additionally, formation of ionic/coordinate bonds between Hg(II) and N-donor ligands was also rationalized from the significant increase in BE of N 1s from 399.782 to 404.23 eV and the decrease in BEs of Hg 4f7/2/Hg 4f5/2 from 102.58/106.68 to 101.76/104.72 eV (Figure 3g).29 In this regard, the O 1s peak for C=O of −COOH at 531.19 did not change significantly (Figure 3f), whereas the other two peaks increased significantly. However, none of these O 1s peaks were shifted significantly as compared to those of N 1s. Such findings again confirmed the preferential and intimate interaction of Hg(II) with N-donor over O-donor ligands. In this regard, considerable changes were also registered in all of the C 1s peaks (Figure 3d), reflecting the prevalence of coordinate/covalent bonds in Hg(II)-PANIPN21 rather than ionic interactions.
In contrast to other M(II/III), Cd(II), possessing full-filled d subshells, preferably interacted with PANIPN21 via ionic bonds, as evidenced from the characteristic XPS peak of Cd 3d5/2 at 406.27 eV (Figure 3k). However, chelating interaction via coordination was also apprehended from the appearance of the peak at 404.75 eV (Figure 3k), indicating the formation of (−COO)2Cd within Cd(II)-PANIPN21. Theoretically, it is believed that Cd(II), having full-filled d subshells, preferentially occupies the ionic environment rather than the coordination sphere. In this regard, predominantly, the ionic interaction between Cd(II) and PANIPN21 was also realized from the minimal shift of the characteristic peaks of N 1s and O 1s (Figure 3i,j). Moreover, lesser coordinating tendency of Cd(II) was further emphasized from the marginal change in BEs of the C 1s spectrum (Figure 3h).
In contrast to Hg(II), the privileged binding affinity of Cr(III) with N-donors was mainly of ionic type, as apprehended from the marginal increase in the N 1s peak from 399.70 to 399.91 eV (Figure 3m) (Table 1). However, the intimate coordinate bonding between the O-donor and Cr(III) was realized from the significant increase in all O 1s peaks (Figure 3n), especially peaks of −COO– and O–H of −COOH (Table 1), and the lowering in BE from 587.00 of Cr(NO3)3 to 585.53 eV of Cr(III)-PANIPN21 (Figure 3o). However, such significant interaction of Cr(III) with O-donor ligands resulted in the marginal shifts of all C 1s peaks (Figure 3l).
3.6. Thermogravimetric Analysis (TGA) of Unloaded and SF/M(II/III)-Loaded PANIPNs
From Figure 4a,b, it is clear that grafting of PN enhanced the thermal stability of TerP41, whereas relatively destabilized grafting was noted for PANIPN21 because of considerable lowering of heat-resistant cross-links. For Hg(II)-PANIPN41, the drastic two-step thermal degradation, within 160.13–229.80 and 229.80–315.32 °C, was associated with the mass losses of 45 and 36 wt %, respectively, followed by a gradual loss of up to 800 °C. However, Hg(II)-PANIPN21, comprising a relatively higher amount of NIPAm, showed substantially improved thermal resistance than Hg(II)-PANIPN41. The two-step thermal degradation of Hg(II)-PANIPN21, within 160.97–227.30 and 227.30–314.64 °C, was associated with mass losses of 39 and 37 wt %, respectively.
Figure 4.

TGA of (a) TerP21 and PANIPN21 and SF-, Hg(II)-, Cd(II)-, and Cr(III)-PANIPN21 and (b) TerP41 and PANIPN41 and SF-, Hg(II)-, Cd(II)-, and Cr(III)-PANIPN41.
Because the preferential interaction of Hg(II) with amides of PANIPNs led to the formation of Hg–N covalent bonds, inferred from FTIR and XPS analyses (Figures S3 and 3e), the two-stage drastic degradation of Hg(II)-PANIPNs could also be related to the possible breakdown of strong bonds, including amides from Hg(II)-PANIPNs, via reactive sublimation of the intermediates and subsequent evaporation of Hg, especially in PANIPN41.31−33 In fact, among all of the M(II/III)-adsorbed hydrogels, the highest residue of Cd(II)-PANIPNs could be attributed to the slowest evaporation rate of metallic Cd, especially above 800 °C.31,32 In fact, both Cd(II) and Cr(III) preferably interacted with −COO– than −CONH– and, hence, the thermal degradation of NIPAm moieties at 400 °C induced a moderate effect on the thermal degradation of Cd(II)-PANIPN41 (i.e., 23 wt % within 400–500 °C). However, rapid degradation of Cr(III)-PANIPN41 (i.e., 36 wt % within 400–500 °C) was noted to be more intense after the thermal decomposition of NIPAm moieties at 400 °C. The lowering in the available number of −COO– in PANIPN21 resulted in the formation of lower residues for both Cd(II)- and Cr(III)-PANIPN21, indicating the reduction of adsorption for Cd(II)-/Cr(III)-PANIPN21. As the extent of metallic residue (i.e., 33.72 wt %) of Cd(II)-PANIPNs was appreciably higher than that of Cr(III)-PANIPNs (i.e., 32.63 wt %), the AC of Cd(II)-PANIPNs was noted to be higher than that of Cr(III)-PANIPNs. In fact, the lower extent of prevalent −COO– in PANIPN21 envisaged the lowering of O-donor ligands in PANIPN21.
From Figure 4a, better thermal stability of SF-PANIPN21 than that of PANIPN21 within 150–230 °C could be attributed mostly to the formation of stronger H-bonds, as envisaged in the respective FTIR spectra (Figure S3b). Conversely, the initial thermal stability within 150–350 °C of PANIPN41 was found to be deteriorated extensively in SF-PANIPN41 via predominant ionic interactions (Figure 4b). However, SF-PANIPN41 produced lower residue as compared with the unloaded hydrogels. The residue was markedly lower for SF-PANIPN21 (i.e., 0.02 wt %) than for SF-PANIPN41 (i.e., 25.91 wt %), indicating the formation of better temperature-resistant ash during thermal degradation of SF-PANIPN41. In fact, extensive electrovalent bond formation between SF cations and polyanions of PANIPN41 resulted in improved thermal stability of SF-PANIPN41 over PANIPN41 within 380–570 °C. In the intermediate stage, especially within 340–430 °C, the inferior thermal stability of SF-PANIPN21 as compared to that of PANIPN21 could be ascribed to the thermal decomposition of thermally susceptible secondary amide containing NIPAm moieties. Finally, the rapid thermal decomposition beyond 450 °C was attributed to main chain splitting because of the lowering of the electrovalent interactions between adsorbed SF cations and polyanions of PANIPN21.
3.7. Differential Scanning Calorimetry of Unloaded and SF/M(II/III)-Loaded PANIPNs
From Figure S5a,b, manifold increase in the intensity of the exothermic peak of Hg-PANIPN21 than that of Hg-PANIPN41 was ascribed to the relative enhancement of the displacement reaction between higher amount of adsorbed Hg(II) in Hg-PANIPN21 and Al-pan. In addition, appearance of two small and broad endothermic peaks at 218 and 262 °C could be related to the formation of new phases via metal chelation.2 Notably, an intense peak at 371 °C for PANIPN41, related to the natural ordering of the PN-grafted terpolymer, disappeared in the thermogram of PANIPN21. Moreover, the pronounced alteration of peak from 371 to 330, 412, and 425 °C for Hg-PANIPN41, Cr-PANIPN41, and Cd-PANIPN41, respectively, could be linked with the structural changes, induced by variegated interactions. The peaks at 323 and 329 °C in Hg-PANIPN21 indicated the relatively higher extent of crystalline phases as Hg(II) interacted strongly with the increasing number of available NIPAm moieties within PANIPN21 (Figure S5a). The less intense peaks at 411 and 416 °C for Cr(III)-PANIPN21 and Cd(II)-PANIPN21, respectively, were lower than the peaks at 412 and 425 °C of Cr(III)-PANIPN41 and Cd(II)-PANIPN41, respectively, indicating the preferential binding of both Cr(III) and Cd(II) with −COO– rather than with the amides of NIPAm moieties.
3.8. X-ray Diffraction (XRD) of Unloaded and SF/M(II/III)-Loaded PANIPNs
From respective XRD spectra (Figure S5c), the increase in crystallinity of PANIPNs compared to that of TerPs could be apprehended from the extent of intermixing of phases via intercalation of the grafted PN through the TerP network. Several characteristic peaks at 20.40, 21.72, 29.91, and 45.24° of Hg(II)-PANIPNs indicated the presence of 001, 110, 111, and, 102 planes, related to the deposition of HgNH2Cl- and HgCl2-type crystals on the Hg(II)-PANIPNs surface. Conversely, a slightly closer packing in Cr(III)-PANIPN21 compared to that in Cr(III)-PANIPN41 could be apprehended by the shift of 2θ from 20.21 to 20.90. In contrast, adsorption of Cd(II) resulted in relative lowering of the crystalline domain at the hydrogel surface because of percolation of smaller hydrated Cd(II) in the bulk of PANIPN (Figure S5c,d). The XRD spectra of SF-PANIPN21 produced three distinct peaks at 32.06, 45.75, and 56.78° (Figure S5c), along with the disappearance of the characteristic PANIPN21 peak at 20.71°, reflecting the appearance of a new crystalline phase at the surface of SF-PANIPN21 as compared with PANIPN21. In contrast, the absence of any symbolic peak in both PANIPN41 and SF-PANIPN41 indicated retention of amorphous characteristics at the PANIPN41 surface even after the adsorption of SF. Moreover, sharp and intense peaks at 32.06° with spacing = 2.88 Å at n = 1 and at 45.75° with spacing = 1.99 Å at n = 1 could be linked with the distance between the carbon atoms, located at the opposite vertices of benzene hexagons of SF, and the spacing obtained from the XRD spectrum of liquid benzene (i.e., 2.00 Å), respectively.34 In this regard, diffusion of appreciable amount of stacks of rigid, flat, and relatively hydrophobic SF into the bulk of PANIPN41 through the micropores than the individual surface occupation could be realized by a higher number of −COO– in PANIPN41.35 In contrast, fewer −COO– in PANIPN21 could attach lesser amount of SF cations and, hence, a substantial amount of SF cations might be accumulated at the charge-compensated hydrogel in the form of organized stacks via hydrophobic interactions. In this context, the small peak at 56.78°, attributed to the spacing of 4.86 Å for n = 3, envisaged the presence of SF of dimension = 1.10 × 0.95 × 0.49 nm3 at the SF-PANIPN21 surface.36
3.9. Scanning Electron Microscopy (SEM) and Energy Dispersive X-ray Spectroscopy (EDX) Analyses
As observed from SEM photomicrographs, the bulk of PANIPNs was more porous than the surface (Figure S6a,b). Notably, the microporous surfaces of both Hg(II)-PANIPNs were populated with appreciably bigger crystals of mercuric compounds along with the pores of various dimensions (Figure S6c,d). Comparatively smooth surfaces of PANIPNs were roughened in Cd(II)-PANIPNs (Figure S6e,f), more so in Cd(II)-PANIPN41, with the generation of distinct folding and rough patches throughout the surface (Figure S6e,f). Moreover, almost all of the pores disappeared in Cd(II)-PANIPNs as a result of negligible deposition of Cd(II), which eventually covered up the entire micro-, meso-, and macropores of PANIPNs (Figure S6e,f). However, small particles of chromium compounds having indistinct edges and visible pores of variegated sizes (Figure S6g,h) were scattered in both Cr(III)-PANIPNs. The surface depositions of M(II/III) could also be evidenced from the appearance of intense peaks in EDX spectra (Figure S6i–k).
3.10. Adsorption Isotherm, Kinetics, and Thermodynamics Studies
The interaction between the adsorbate and adsorbent during isothermal adsorption could be understood via fitting of the experimental data with the adsorption isotherm models (eqs S11–S14). In the entire temperature and concentration ranges, the Langmuir model fitted the best with equilibrium adsorption data of SF and M(II/III) (Figures S7e–f and S8j–l). Ligand-selective adsorption by PANIPNs could be justified from higher ACs of Cd(II), Cr(III), and SF (Table 2) because of the formation of predominant ionic bonds between −COO– of PANIPN41 and M(II/III)/–NH3+ of SF. In contrast, higher AC of Hg(II) onto PANIPN21 supported better bonding with N-donors because NIPAm was in a higher extent in PANIPN21. Again, the separation factor (RL) within 0–1 indicated favorable adsorption.
Table 2. Adsorption Isotherm and Kinetics Parameters.
| temperature
(K) |
||||
|---|---|---|---|---|
| models/parameters | 293 | 303 | 313 | 323 |
| PANIPN41 | ||||
| Langmuir (Cd(II)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 91.45/7/5–55 | 96.53/7/5–55 | 99.72/7/5–55 | 103.36/7/5–55 |
| kL (L mg–1) | 0.58 | 0.44 | 0.37 | 0.33 |
| R2/F/χ2 | 0.9979/4453.42/2.16 | 0.9995/19611.03/0.49 | 0.9981/4806.60/1.98 | 0.9977/4064.90/2.36 |
| pseudosecond order (Cd(II)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 9.72/7/5 | 9.87/7/5 | 9.56/7/5 | 9.39/7/5 |
| qe,exp (mg g–1) | 9.32 ± 0.28 | 9.43 ± 0.29 | 9.26 ± 0.25 | 9.22 ± 0.21 |
| k2 (g mg–1 min–1) | 0.0051 | 0.0058 | 0.0096 | 0.0099 |
| R2/F/χ2 | 0.9949/12108.99/0.04 | 0.9960/15798.76/0.01 | 0.9973/23244.37/0.02 | 0.9981/34099.89/0.01 |
| Langmuir (Hg(II)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 67.98/7/5–55 | 74.74/7/5–55 | 76.28/7/5–55 | 78.44/7/5–55 |
| kL (L mg–1) | 1.92 | 1.29 | 1.25 | 1.13 |
| R2/F/χ2 | 0.9940/1729.04/4.53 | 0.9994/16307.55/0.52 | 0.9997/38545.76/0.23 | 0.9983/5921.10/1.50 |
| pseudosecond order (Hg(II)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 10.31/7/5 | 10.05/7/5 | 9.83/7/5 | 9.64/7/5 |
| qe,exp (mg g–1) | 9.82 ± 0.35 | 9.76 ± 0.34 | 9.72 ± 0.33 | 9.68 ± 0.31 |
| k2 (g mg–1 min–1) | 0.0038 | 0.0049 | 0.0076 | 0.0098 |
| R2/F/χ2 | 0.9963/13955.91/0.03 | 0.9909/5846.27/0.07 | 0.9906/5674.12/0.07 | 0.9841/3846.08/0.12 |
| Langmuir (Cr(III)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 88.10/7/5–55 | 94.75/7/5–55 | 98.09/7/5–55 | 99.41/7/5–55 |
| kL (L mg–1) | 0.64 | 0.47 | 0.38 | 0.37 |
| R2/F/χ2 | 0.9977/4111.10/2.29 | 0.9991/9978.40/0.96 | 0.9981/4947.31/1.92 | 0.9968/2900.53/3.28 |
| pseudosecond order (Cr(III)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 10.19/7/5 | 9.88/7/5 | 9.56/7/5 | 9.37/7/5 |
| qe,exp (mg g–1) | 9.74 ± 0.28 | 9.46 ± 0.24 | 9.28 ± 0.19 | 9.04 ± 0.17 |
| k2 (g mg–1 min–1) | 0.0046 | 0.0057 | 0.0096 | 0.0099 |
| R2/F/χ2 | 0.9971/20678.21/0.02 | 0.9979/30228.44/0.01 | 0.9974/24287.27/0.02 | 0.9955/14496.52/0.03 |
| Langmuir (SF) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 127.61/9/5–100 | 111.52/9/5–100 | 93.53/9/5–100 | 79.10/9/5–100 |
| kL (L mg–1) | 0.18 | 0.19 | 0.19 | 0.18 |
| R2/F/χ2 | 0.9767/742.96/33.98 | 0.9724/666.53/32.41 | 0.9673/283.34/28.33 | 0.9529/568.97/23.59 |
| pseudosecond order (SF) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 51.28/9/30 | 51.09/9/30 | 52.83/9/30 | 49.82/9/30 |
| qe,exp (mg g–1) | 51.97 ± 1.56 | 51.92 ± 1.54 | 52.96 ± 1.61 | 49.58 ± 1.51 |
| k2 (g mg–1 min–1) | 0.0036 | 0.0046 | 0.0059 | 0.0079 |
| R2/F/χ2 | 0.9892/27082.11/1.25 | 0.9798/15267.34/2.23 | 0.9963/89214.61/0.42 | 0.9917/43830.51/0.76 |
| PANIPN21 | ||||
| Langmuir (Cd(II)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 74.49/7/5–55 | 77.47/7/5–55 | 80.74/7/5–55 | 83.97/7/5–55 |
| kL (L mg–1) | 0.42 | 0.36 | 0.31 | 0.28 |
| R2/F/χ2 | 0.9939/1661.10/4.30 | 0.9999/67099.63/0.11 | 0.9982/5387.70/1.37 | 0.9972/3538.10/2.13 |
| pseudosecond order (Cd(II)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 9.97/7/5 | 9.61/7/5 | 9.23/7/5 | 9.17/7/5 |
| qe,exp (mg g–1) | 9.28 ± 0.24 | 9.27 ± 0.23 | 8.99 ± 0.19 | 9.04 ± 0.25 |
| k2 (g mg–1 min–1) | 0.0039 | 0.0054 | 0.0074 | 0.0092 |
| R2/F/χ2 | 0.9909/6174.39/0.07 | 0.9959/15849.90/0.03 | 0.9960/16375.92/0.03 | 0.9979/33754.26/0.01 |
| Langmuir (Hg(II)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 90.68/7/5–55 | 92.09/7/5–55 | 95.30/7/5–55 | 96.78/7/5–55 |
| kL (L mg–1) | 0.72 | 0.73 | 0.71 | 0.68 |
| R2/F/χ2 | 0.9926/1276.46/7.73 | 0.9975/3768.59/2.66 | 0.9981/4434.78/2.32 | 0.9985/6171.49/1.68 |
| pseudosecond order (Hg(II)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 10.09/7/5 | 10.09/7/5 | 9.84/7/5 | 9.82/7/5 |
| qe,exp (mg g–1) | 9.79 ± 0.27 | 9.71 ± 0.26 | 9.68 ± 0.22 | 9.61 ± 0.20 |
| k2 (g mg–1 min–1) | 0.0044 | 0.0055 | 0.0083 | 0.0091 |
| R2/F/χ2 | 0.9989/46795.04/0.09 | 0.9981/27447.82/0.02 | 0.9937/8132.33/0.05 | 0.9961/13550.82/0.03 |
| Langmuir (Cr(III)) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 71.44/7/5–55 | 76.12/7/5–55 | 79.48/7/5–55 | 83.11/7/5–55 |
| kL (L mg–1) | 0.47 | 0.39 | 0.33 | 0.29 |
| R2/F/χ2 | 0.9994/16002.60/0.43 | 0.9994/16246.01/0.45 | 0.9971/3349.13/2.19 | 0.9975/3939.60/1.90 |
| pseudosecond order (Cr(III)) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 10.19/7/5 | 9.79/7/5 | 9.53/7/5 | 9.35/7/5 |
| qe,exp (mg g–1) | 9.32 ± 0.29 | 9.24 ± 0.26 | 9.09 ± 0.21 | 9.04 ± 0.22 |
| k2 (g mg–1 min–1) | 0.0046 | 0.0050 | 0.0069 | 0.0086 |
| R2/F/χ2 | 0.9971/20678.21/0.02 | 0.9939/10176.44/0.04 | 0.9951/12750.37/0.03 | 0.9943/11369.41/0.04 |
| Langmuir (SF) | ||||
| qmax (mg g–1)/pHi/C0 (ppm) | 117.60/9/5–100 | 102.40/9/5–100 | 87.50/9/5–100 | 71.02/9/5–100 |
| kL (L mg–1) | 0.22 | 0.23 | 0.24 | 0.28 |
| R2/F/χ2 | 0.9827/1036.80/23.14 | 0.9810/1015.93/20.07 | 0.9718/739.82/22.33 | 0.9494/416.37/27.28 |
| pseudosecond order (SF) | ||||
| qe,cal (mg g–1)/pHi/C0 (ppm) | 52.13/9/30 | 51.99/9/30 | 46.21/9/30 | 44.66/9/30 |
| qe,exp (mg g–1) | 53.85 ± 1.76 | 51.99 ± 1.56 | 49.16 ± 1.43 | 47.51 ± 1.39 |
| k2 (g mg–1 min–1) | 0.0021 | 0.0018 | 0.0014 | 0.0013 |
| R2/F/χ2 | 0.9621/7776.66/4.79 | 0.9471/4009.04/7.14 | 0.9522/3866.67/6.40 | 0.9559/3846.83/5.88 |
The closeness of qe,exp and qe,cal and higher R2 and F (Table 2) indicated better applicability of pseudosecond order kinetics (eqs S15 and S16), supporting the prevalence of chemisorption for SF and M(II/III) (Figures S7a–d and S8a–i). In fact, the existence of chemisorption was also apprehended by the increasing trend of k2 with the rise in temperature. Again, higher k2 for Hg(II) onto PANIPN21 was related to the preferential formation of Hg–N bonds between Hg(II) and N-donor-populated PANIPN21. In contrast, k2 values of Cr(III), Cd(II), and SF were higher for PANIPN41 because of greater ionic interactions with a higher number of −COO– in PANIPN41.
Thermodynamic spontaneity of adsorption was justified from the −ΔG0 values (eqs S17–S19) in entire concentration and temperature ranges of the experiment (Table 3, Figures S8 and S9).13,37 The −ΔG0 values of SF, Cd(II), and Cr(III) were higher for PANIPN41 as compared to those for PANIPN21. However, higher −ΔG0 for Hg(II) onto PANIPN21 indicated the better chemical attachment between Hg(II) and the N-donor of the NIPAm moiety. The exothermic nature of adsorption was realized from negative ΔH0, whereas the positive ΔS0 (eq S19) indicated the fair affinity of SF/M(II/III) for PANIPNs and decrease in randomness at the solid–solution interface during adsorption. The activation energies (Ea) of chemisorption were 21.78/20.16, 20.44/25.65, and 23.97/22.13 kJ mol–1 for Cd(II)-, Hg(II)-, and Cr(III)-PANIPN41/21, respectively (Figure S10).2
Table 3. Adsorption Thermodynamics Parameters.
| concentration (ppm) of SF/M(II/III)/temperature (K) | –ΔG0 (kJ mol–1) of SF/Cd(II)/Hg(II)/Cr(III) | –ΔH0 (kJ mol–1) of SF/Cd(II)/Hg(II)/Cr(III) | ΔS0 (J mol–1 K–1) of SF/Cd(II)/Hg(II)/Cr(III) |
|---|---|---|---|
| PANIPN41 | |||
| 5/5/293 | 4.69/10.36/11.42/10.49 | 12.85/28.45/12.89/29.09 | –27.93/–63.08/–4.45/–64.84 |
| 5/5/303 | 4.21/8.81/10.83/8.96 | ||
| 5/5/313 | 3.87/8.49/11.33/8.45 | ||
| 5/5/323 | 3.48/8.37/11.01/8.48 | ||
| 10/15/293 | 6.37/8.79/10.81/8.86 | 7.71/8.93/2.13/7.35 | –4.59/–0.71/27.14/4.91 |
| 10/15/303 | 6.09/8.62/10.34/8.80 | ||
| 10/15/313 | 5.89/8.75/10.68/8.73 | ||
| 10/15/323 | 5.64/8.69/10.85/9.07 | ||
| 15/25/293 | 7.24/7.86/8.64/7.99 | 14.18/6.99/–1.99/7.51 | –23.53/2.80/36.23/1.43 |
| 15/25/303 | 6.89/7.81/8.94/7.87 | ||
| 15/25/313 | 6.36/7.79/9.34/7.88 | ||
| 15/25/323 | 5.95/7.96/9.72/8.05 | ||
| 20/35/293 | 7.34/6.76/6.06/6.66 | 14.61/3.65/–9.17/2.95 | –24.76/10.59/52.66/12.68 |
| 20/35/303 | 6.92/6.88/7.04/6.83 | ||
| 20/35/313 | 6.35/6.92/7.74/6.88 | ||
| 20/35/323 | 6.02/7.10/7.69/7.06 | ||
| 25/45/293 | 7.02/5.59/3.89/5.44 | –/2.54/–11.57/1.30 | –/10.26/53.20/14.10 |
| 25/45/303 | 6.59/5.62/4.76/5.57 | ||
| 25/45/313 | 6.09/5.76/5.07/5.71 | ||
| 25/45/323 | 4.94/5.89/5.54/5.86 | ||
| 30/55/293 | 6.64/4.28/3.01/4.11 | 21.56/–3.16/–4.97/–3.70 | –50.37/25.40/27.36/26.86 |
| 30/55/303 | 6.24/4.57/3.36/4.55 | ||
| 30/55/313 | 5.67/4.75/3.64/4.70 | ||
| 30/55/323 | 4.58/5.07/3.82/4.94 | ||
| PANIPN21 | |||
| 5/5/293 | 1.86/7.92/11.42/8.06 | 14.64/11.29/13.74/10.34 | –31.74/11.19/8.28/–7.78 |
| 5/5/303 | 1.84/8.18/11.08/8.03 | ||
| 5/5/313 | 1.52/7.52/11.09/7.80 | ||
| 5/5/323 | 1.29/7.76/11.17/7.89 | ||
| 10/15/293 | 2.57/7.13/9.42/7.47 | 11.99/0.87/–1.37/4.22 | –17.09/21.38/36.86/11.10 |
| 10/15/303 | 2.49/7.36/9.80/7.55 | ||
| 10/15/313 | 2.44/7.56/10.17/7.74 | ||
| 10/15/323 | 2.36/7.76/10.53/7.78 | ||
| 15/25/293 | 2.75/6.14/8.03/6.18 | 16.14/2.25/–3.58/2.41 | 29.16/13.31/39.82/12.90 |
| 15/25/303 | 2.72/6.29/8.57/6.33 | ||
| 15/25/313 | 2.57/6.41/8.92/6.43 | ||
| 15/25/323 | 2.46/6.54/9.23/6.58 | ||
| 20/35/293 | 2.73/5.47/7.45/5.03 | 15.37/5.49/–2.47/1.59 | –26.84/0.44/33.82/11.78 |
| 20/35/303 | 2.69/5.19/7.73/5.17 | ||
| 20/35/313 | 2.55/5.32/8.21/5.29 | ||
| 20/35/323 | 2.45/5.42/8.40/5.37 | ||
| 25/45/293 | 2.58/3.74/5.72/3.63 | 13.91/2.52/–6.17/–3.53 | –23.40/21.40/40.51/24.55 |
| 25/45/303 | 2.52/3.99/6.04/3.96 | ||
| 25/45/313 | 2.41/4.17/6.62/4.41 | ||
| 25/45/323 | 2.31/4.39/6.87/4.39 | ||
| 30/55/293 | 2.43/2.67/4.41/2.58 | 15.93/–6.10/–4.65/–6.62 | –31.56/29.99/30.90/31.53 |
| 30/55/303 | 2.37/3.02/4.71/2.99 | ||
| 30/55/313 | 2.22/3.26/5.03/3.24 | ||
| 30/55/323 | 1.99/3.59/5.33/3.54 | ||
3.11. Synergistic Adsorption of SF and MO on PANIPNs at Different pHi and SF/MO Values
3.11.1. PANIPN41
In simultaneous removal of SF–MO at pHi = 9, the extent of adsorption onto PANIPN41 was enhanced with the increase in [SF]/[MO] (Figure 5a–d). Indeed, the characteristic λmax values of SF and MO were merged together to produce intermediate λmax within SF and MO, attributed to the formation of SF–MO ionic adducts (Scheme 3a–d). The initial λmax was gradually red-shifted from 482 to 514 nm as [SF]/[MO] was increased from 1:2 to 4:1 (Figure 5a–d). In fact, such a red shift in initial λmax of the binary mixture was directly dependent on the relative proportion of SF in the mixture because the characteristic λmax of SF at 515 nm is higher than that of the MO anion at 465 nm. Moreover, the initial λmax of all individual SF–MO combinations was appreciably blue-shifted at the end of the adsorption process (Figure 5a–d). In fact, with the progress of adsorption, anionic MO was detached from the solid phase via continuous breakdown of SF–MO ionic adducts in the increasingly polar environment of the gradually diluted binary mixture.
Figure 5.

Synergistic removal of (a–d) SF–MO/pHi = 9/PANIPN41 and (e–h) SF–MO/pHi = 2/PANIPN41 (C0 = 30 ppm, T = 303 K, and adsorbent dose = 1 g L–1).
Scheme 3. Possible Structure of the MO–SF-PANIPN41 Adduct (a–h) at pHi 9 or 2 Involving Ionic Interaction between (I) SF Dimer and MO, (II) SF Dimer and PANIPN41, (III) SF and PANIPN41, (IV) SF and MO, (V) MO and the SA Moiety of PANIPN41, and (VI) MO and the NIPAm Moiety of PANIPN41.

Thus, SF cations, originated from SF–MO ionic adducts, should be adhered to polyanions of PANIPN41, leading to enhanced relative population of MO anions in solution, resulting in the observed blue shift of λmax in all of the combined visible spectra. Furthermore, for a binary mixture having [SF]/[MO] = 4:1, a shoulder peak at 460 nm emerged in the intermediate phase and disappeared completely prior to the end of the adsorption process (Figure 5d). Such phenomenon could be related to the preferential adsorption of SF cations by the polyanionic adsorbent, accentuated by 4-fold numerical advantage of SF over MO. Moreover, MO anions preferred to reside in the solution, envisaged from the emergence of the shoulder peak at 460 nm. In the later stages, once the nearly or completely neutralized polyanion-SF adduct was produced via adsorption of SF cations on the polyanions of PANIPN41, MO anions became capable to be attached with the polyanion-SF adducts, mostly via nonionic interactions, leading to the disappearance of the characteristic shoulder at 460 nm (Figure 5d).
At pHi = 2, the initial λmax of binary solution, pertaining to all SF–MO combinations, appeared within 510–515 nm (Figure 5e–h). The visible peaks exhibited a blue shift with respect to the initial λmax with the advancement of adsorption. Notably, the extent of such shifting was found to be directly proportional to the [SF]/[MO] ratio. Thus, the maximum blue shift from 510 to 475 nm was attained for [SF]/[MO] of 1:2. Such a radical hypsochromic shift of λmax might be attributed to the formation of either strong ion-pairs or weak metachromic compound (Scheme 3e–h), assisted by the larger population of MO zwitterions in the mixture (Figure 5e). In this context, ammonium tautomers of MO were detected from the appearance of a shoulder at 320 nm at a relatively higher concentration of MO (Figure 5e). However, the disappearance of such shoulder in the UV–vis spectra of SF-populated binary mixtures could be realized in two ways: first, through conversion of the ammonium tautomer (λmax = 320 nm) to the azonium tautomer (λmax = 510 nm), followed by the adsorption of ammonium tautomer by PANIPN41. In this context, the azonium tautomer appeared predominantly due to the conversion of the ammonium tautomer to the resonance-stabilized azonium tautomer in water or other polar solvent of low, acidic, pHi. Altogether, a satisfactory level of adsorption of both MO and SF was demonstrated in the acidic medium.
3.11.2. PANIPN21
At alkaline pHi, gradual adsorption of SF onto PANIPN21 could be envisaged from the time-dependent hypochromic effect at 516 nm. However, the adsorption of SF on PANIPN21 was noted to be incomplete as both of the characteristic SF peaks were visible in the UV–vis spectra even after a reasonable period of absorption. Interestingly, neither the symbolic peak of the SF cation nor the characteristic peak of the MO anion was observed in the combined UV–vis spectra of SF–MO. In fact, such combined peak was noted to fluctuate in between the characteristic peaks of SF and MO, ascribed to the ionic to weakly metachromic interactions between cationic and anionic dyes (Scheme S1a–d). The initial λmax of the combined absorption spectra was gradually red-shifted from 492 to 516 with the increase in [SF]/[MO] ratios from 1:1 to 4:1 (Figure S11b–d). The characteristic initial λmax of the binary mixture was found to depend directly on the relative proportion of SF. However, in contrast to those for PANIPN41, the initial λmax (i.e., 512 nm) and peak intensity were found to be unexpectedly higher for [SF]/[MO] = 1:2, attributed to the formation of clusters/aggregates, consisting of MO–SF ion pairs and excess MO anions (Figure S11a). However, the unusual red shift of λmax and the aggregate formation were not associated with adsorption onto PANIPN41. In fact, greater availability of −COO– in PANIPN41 played the pivotal role in controlling the faster and easier adsorption of SF cations that resulted in the lesser possibility of bigger aggregate formation. Nevertheless, similar to those for PANIPN41, the initial λmax values for all SF–MO combinations were appreciably blue-shifted toward the characteristic MO peak at the end of adsorption, suggesting the successive enhancement of MO anions in the solution via breakdown of MO–SF ion pairs in the continuously increasing polar environment via dilution. Altogether, in comparison to PANIPN41, PANIPN21 was noted to be reluctant in adsorbing the dyes, also apprehended from the poor overall time-dependent hypochromic effect in the visible region.
At pHi = 2, a hyperchromic effect followed by hypochromic effect could be noted up to 85 min in the individual absorption spectra of MO, showing an initial increase followed by the gradual decrease in peak intensities in both UV (i.e., λ = 275 and 317 nm) and visible (i.e., λ = 513 nm) regions (Figure S11e–h). It was realized that both azonium and ammonium tautomers of zwitterionic MO formed weak aggregates at the relatively higher concentrations, showing little tendency to be absorbed by the relatively negligible number of −COO– in PANIPN21 (Scheme S1e–h).
Interestingly, the combined absorption spectra of the 1:1 binary mixture of MO–SF envisaged a gradual hypochromic effect at 513 nm, whereas the initial hyperchromic shift of the peaks was noted at 275 and 248 nm. Similar effects were observed with greater extent of overall adsorption by the increase in the [SF]/[MO] ratio up to 1:2. In this regard, the peak at 250 nm should only be attributed to SF, whereas the other two peaks at 513 and 275 nm were characteristics of both MO and SF. The prevalence of two types of benzene rings was responsible for the appearance of two peaks at 248 and 275 nm in the UV–vis spectrum of SF. In comparison to that of MO, the relative lowering of SF onto the adsorbent was understood from the initial time-dependent hyperchromic effect at 248 nm. Because the λmax values of MO and SF were almost similar at acidic pHi, the hypochromic effect at 513 nm could be resulted by the combined adsorption of MO and SF. Thus, the attainment of poor adsorbing tendency of SF was realized by the preferential attachment of resonance hybrids of the azonium tautomer rather than those of the ammonium tautomer of MO to produce a time-dependent hypochromic effect at 513 nm. As compared to that of SF, better adsorbing behavior of MO in PANIPN21 was also realized from enhanced adsorption abilities by the increasing proportion of MO in MO–SF. Furthermore, the overall adsorption behavior of PANIPN21 became worse when SF became the major component in the mixture, reaffirming better adsorbing tendency of MO than that of SF onto PANIPN21. Thus, unlike the case for PANIPN41, SF cations at acidic pHi were not readily adsorbed by PANIPN21, owing to lesser availability of −COO– and −COOH and the enhanced cationic character of PANIPN21 because of a greater number of protonated secondary amides of NIPAm moieties.
3.12. Recyclability of PANIPNs
The cost-effectiveness of PANIPN superadsorbents was assessed via desorption of the SF- and M(II/III)-loaded PANIPNs at pHi = 2 and in 0.01 N solution of NaCl, respectively, and repetitive adsorption of SF and M(II/III) at the pHi = 9 and 7, respectively. Five consecutive adsorption–desorption cycles were carried out separately by taking 30 ppm solutions at pHi 9 and 7 for SF and M(II/III), respectively, using the regenerated PANIPNs (Figure 6). However, complete desorption beyond 90% was obtained in all of the cases.
Figure 6.
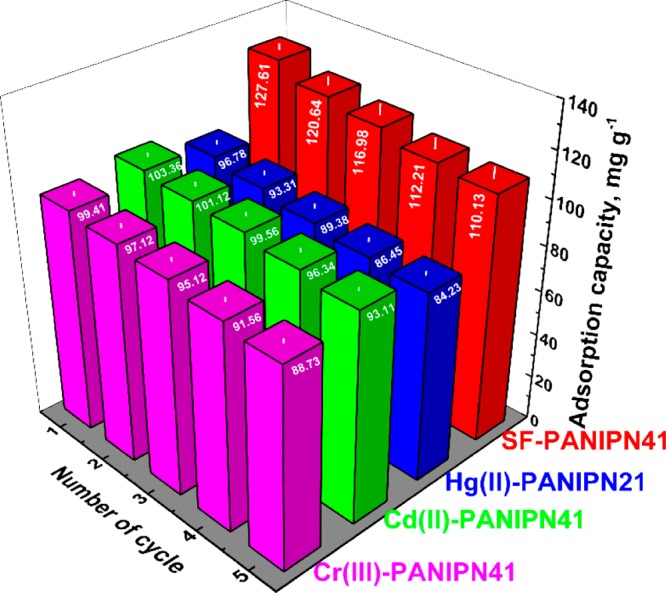
Recyclability of PANIPNs for SF, Hg(II), Cd(II), and Cr(III).
3.13. Comparison of the Results
Several micro-/nanomaterial-based adsorbents, homo-/co-/terpolymers, and IPN hydrogels have been applied for the adsorptive exclusion of Hg(II), Cd(II), Cr(III), and SF of varying initial concentrations (1–1000 ppm), temperatures (283–333 K), and pHis (Table S9). From Table S9, it could be observed that the ACs of PANIPNs were either closer to or much better than the already reported results within the definite working range.
4. Conclusions
The unorthodox in situ inclusion of acrylamido derivatives can also be attempted for the synthesis of terpolymer/IPN hydrogels without ex situ addition of third monomer. Hydrogels possessing various extents of N-/O-donor ligands have demonstrated relative variation of physicochemical properties and ACs for SF, Cd(II), Hg(II), and Cr(III). Indeed, the chemical structure of PANIPNs, grafting of PN, physicochemical changes, and possible modes of attachment between PANIPNs and dye/M(II/III) have rationally been understood by performing systematic characterization of both unadsorbed and adsorbed hydrogels using spectroscopic, thermal, diffractometric, and microscopic methods. The structure–property relationships of PANIPNs of varying comonomers have also been realized by the measurements of % GC, % −COOH, pHPZC, LCST, and network parameters, such as ρc and Mc, of IPNSs. Indeed, NMR and FTIR studies have indicated the prevalence of strong physicochemical interactions between terpolymer and grafted polysaccharide chains of PN, also harmonized from the appearance of phaseless morphology in the SEM photomicrographs. Moreover, the increasing trend of LCST from terpolymer to PANIPN has envisaged the relative increase in hydrophilicity of PANIPN by grafted PN within the hydrogel network. Furthermore, the formation of stronger Hg–N covalent bonds by the preferential adsorption of Hg(II) has been established by FTIR and XPS analyses, whereas variegated coordinative and ionic interactions for Cd(II) and Cr(III) have been inferred via analyses of high-resolution XPS spectra of all M(II/III)-PANIPN21. In this context, relatively stronger affinity and thus greater relative population of Hg–N bonds for PANIPN21 as compared to that for PANIPN41 have reflected the enhanced thermal stability of Hg(II)-PANIPN21 compared to that of Hg(II)-PANIPN41, supported by the appearance of higher AC, faster rate of adsorption, and higher −ΔG0 of PANIPN21 as compared to those of PANIPN41. In the synergistic removal of structurally dissimilar dyes, removal efficiencies have been found to depend strongly on the variation of concentration, time, dilution effect, and monomer–dimer/tautomeric equilibrium, comprehended through an extensive UV–vis analyses. This tailor-made hydrogel of excellent physicochemical properties can also be attempted for targeted drug delivery, tissue engineering, self-healing materials, membrane-based applications, and sensors. The present systematic design of PANIPNs exhibiting excellent recyclability, performance characteristics toward kinetically fast and feasible chemisorption of dye/M(II/III), very high adsorption efficiency (Table S9), and cost effectivity can be employed for developing a cost-effective/ecofriendly waste management system.
Acknowledgments
The authors gratefully acknowledge the Department of Science and Technology (DST), Government of India (YSS/2015/000886) and DST, Government of West Bengal (113(Sanc.)/ST/P/S&T/15G-2/2015), CSIR (Project No. 02(0250)/15/EMR-II) and UGC (Mid-Career Award to D.K.M.), Govt. of India, for providing financial assistance and the Department of Higher Education, Government of West Bengal, for giving the opportunity to participate in Inter-institutional collaboration with the University of Calcutta. M.M. and M.K. are grateful to the University Grants Commission (sr. no. 2061410291, ref no. 22/06/2014 (i) EU-V and roll no. 137632) and the DST (ref no. IF160386), Government of India, respectively, for providing fellowships.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00446.
Characterization; methodology; swelling and pH/temperature reversibility studies of PANIPNs; calculation of % −COOH, pHPZC, % GC, and network parameters of PANIPNs; adsorption isotherm studies of dye/M(II/III) onto PANIPN41 and PANIPN21; kinetics of adsorption; thermodynamics of adsorption; effect of temperature on adsorption kinetics; experimental design and model development for the synthesis of PANIPN; calculation of LCST, % −COOH, pHPZC, % GC, and network parameters of PANIPNs, and FTIR analyses (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Singha N. R.; Karmakar M.; Mahapatra M.; Dutta A.; Mondal H.; Chattopadhyay P. K. Synthesis of guar gum-g-(acrylic acid-co-acrylamide-co-3-acrylamido propanoic acid) IPN viain situ attachment of acrylamido propanoic acid for analyzing superadsorption mechanism of Pb(II)/Cd(II)/Cu(II)/MB/MV. Polym. Chem. 2017, 8, 6750–6777. 10.1039/C7PY01564J. [DOI] [Google Scholar]
- Singha N. R.; Dutta A.; Mahapatra M.; Karmakar M.; Mondal H.; Chattopadhyay P. K.; Maiti D. K. Guar gum-grafted terpolymer hydrogels for ligand-selective individual and synergistic adsorption: Effect of comonomer composition. ACS Omega 2018, 3, 472–494. 10.1021/acsomega.7b01682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozay O.; Ekici S.; Baran Y.; Kubilay S.; Aktas N.; Sahiner N. Utilization of magnetic hydrogels in the separation of toxic metal ions from aqueous environments. Desalination 2010, 260, 57–64. 10.1016/j.desal.2010.04.067. [DOI] [Google Scholar]
- Ozay O.; Ekici S.; Baran Y.; Aktas N.; Sahiner N. Removal of toxic metal ions with magnetic hydrogels. Water Res. 2009, 43, 4403–4411. 10.1016/j.watres.2009.06.058. [DOI] [PubMed] [Google Scholar]
- Pour Z. S.; Ghaemy M. Removal of dyes and heavy metal ions from water by magnetic hydrogel beads based on poly(vinyl alcohol)/carboxymethyl starch-g-poly(vinyl imidazole). RSC Adv. 2015, 5, 64106–64118. 10.1039/C5RA08025H. [DOI] [Google Scholar]
- Hua R.; Li Z. Sulfhydryl functionalized hydrogel with magnetism: Synthesis, characterization, and adsorption behavior study for heavy metal removal. Chem. Eng. J. 2014, 249, 189–200. 10.1016/j.cej.2014.03.097. [DOI] [Google Scholar]
- Badruddoza A. Z. M.; Shawon Z. B. Z.; Daniel T. W. J.; Hidajat K.; Uddin M. S. Fe3O4/cyclodextrin polymer nanocomposites for selective heavy metals removal from industrial wastewater. Carbohydr. Polym. 2013, 91, 322–332. 10.1016/j.carbpol.2012.08.030. [DOI] [PubMed] [Google Scholar]
- Huang S. H.; Chen D. H. Rapid removal of heavy metal cations and anions from aqueous solutions by an amino-functionalized magnetic nano-adsorbent. J. Hazard. Mater. 2009, 163, 174–179. 10.1016/j.jhazmat.2008.06.075. [DOI] [PubMed] [Google Scholar]
- Mahdavinia G. R.; Soleymani M.; Sabzi M.; Azimi H.; Atlasi Z. Novel magnetic polyvinyl alcohol/laponite RD nanocomposite hydrogels for efficient removal of methylene blue. J. Environ. Chem. Eng. 2017, 5, 2617–2630. 10.1016/j.jece.2017.05.017. [DOI] [Google Scholar]
- Sarkar N.; Sahoo G.; Das R.; Swain S. K. Three-dimensional rice straw structured magnetic nanoclay decorated tri-polymeric nanohydrogels as superabsorbent of dye pollutants. ACS Appl. Nano Mater. 2018, 1, 1188–1203. 10.1021/acsanm.7b00358. [DOI] [Google Scholar]
- Videcoq P.; Steenkeste K.; Bonnin E.; Garnier C. A multi-scale study of enzyme diffusion in macromolecular solutions and physical gels of pectin polysaccharides. Soft Matter 2013, 9, 5110–5118. 10.1039/c3sm00058c. [DOI] [Google Scholar]
- Neves S. C.; Gomes D. B.; Sousa A.; Bidarra S. J.; Petrini P.; Moroni L.; Barrias C. C.; Granja P. L. Biofunctionalized pectin hydrogels as 3D cellular microenvironments. J. Mater. Chem. B 2015, 3, 2096–2108. 10.1039/C4TB00885E. [DOI] [PubMed] [Google Scholar]
- Karmakar M.; Mahapatra M.; Dutta A.; Chattopadhyay P. K.; Singha N. R. Fabrication of semisynthetic collagenic materials for mere/synergistic adsorption: A model approach of determining dye allocation by systematic characterization and optimization. Int. J. Biol. Macromol. 2017, 102, 438–456. 10.1016/j.ijbiomac.2017.04.044. [DOI] [PubMed] [Google Scholar]
- Karmakar M.; Mahapatra M.; Singha N. R. Separation of tetrahydrofuran using RSM optimized accelerator-sulfur-filler of rubber membranes: Systematic optimization and comprehensive mechanistic study. Korean J. Chem. Eng. 2017, 34, 1416–1434. 10.1007/s11814-017-0021-5. [DOI] [Google Scholar]
- Teodoro F. S.; Elias M. M. C.; Ferreira G. M. D.; Adarme O. F. H.; Savedra R. M. L.; Siqueira M. F.; da Silva L. H. M.; Gil L. F.; Gurgel L. V. A. Synthesis and application of a new carboxylated cellulose derivative. Part III: Removal of auramine-O and safranin-T from mono- and bi-component spiked aqueous solutions. J. Colloid Interface Sci. 2018, 512, 575–590. 10.1016/j.jcis.2017.10.083. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Wang A. Superadsorbent with three-dimensional networks: From bulk hydrogel to granular hydrogel. Eur. Polym. J. 2015, 72, 661–686. 10.1016/j.eurpolymj.2015.02.031. [DOI] [Google Scholar]
- Walter J.; Sehrt J.; Vrabec J.; Hasse H. Molecular dynamics and experimental study of conformation change of poly(N-isopropylacrylamide) hydrogels in mixtures of water and methanol. J. Phys. Chem. B 2012, 116, 5251–5259. 10.1021/jp212357n. [DOI] [PubMed] [Google Scholar]
- Reis A. V.; Guilherme M. R.; Paulino A. T.; Muniz E. C.; Mattoso L. H. C.; Tambourgi E. B. Synthesis of hollow-structured nano- and microspheres from pectin in a nanodroplet emulsion. Langmuir 2009, 25, 2473–2478. 10.1021/la803313j. [DOI] [PubMed] [Google Scholar]
- Socrates G.Infrared and Raman characteristic group frequencies: Tables and charts. In The Carbonyl Group: C=O, 3rd ed.; John Wiley & Sons Ltd.: New York, 2001; pp 115–156. [Google Scholar]
- Karbarz M.; Romanski J.; Michniewicz K.; Jurczak J.; Stojek Z. Influence of polymer network-metal ion complexation on the swelling behaviour of new gels with incorporated α-amino acid groups. Soft Matter 2010, 6, 1336–1342. 10.1039/b920809g. [DOI] [Google Scholar]
- Prasannan A.; Tsai H. C.; Chen Y. S.; Hsiue G. H. A. Thermally triggered in situ hydrogel from poly(acrylic acid-co-N-isopropylacrylamide) for controlled release of anti-glaucoma drugs. J. Mater. Chem. B 2014, 2, 1988–1997. 10.1039/c3tb21360a. [DOI] [PubMed] [Google Scholar]
- Grasdalen H.; Bakdy O. E.; Larsen B. Determination of the degree of esterification and the distribution of methylated and free carboxyl groups in pectins by 1H-N.M.R. spectroscopy. Carbohydr. Res. 1988, 184, 183–191. 10.1016/0008-6215(88)80016-8. [DOI] [Google Scholar]
- Matharu A.; Houghton J. A.; Lucas-Torres C.; Moreno A. Acid-free microwave-assisted hydrothermal extraction of pectin and porous cellulose from mango peel waste-towards a zero waste mango biorefinery. Green Chem. 2016, 18, 5280–5287. 10.1039/C6GC01178K. [DOI] [Google Scholar]
- Dong Y.; Ma Y.; Zhai T.; Shen F.; Zeng Y.; Fu H.; Yao J. Silver nanoparticles stabilized by thermoresponsivemicrogel particles: Synthesis and evidence of an electron donor-acceptor effect. Macromol. Rapid Commun. 2007, 28, 2339–2345. 10.1002/marc.200700483. [DOI] [Google Scholar]
- Dai J.; Yan H.; Yang H.; Cheng R. Simple method for preparation of chitosan/poly(acrylic acid) blending hydrogel beads and adsorption of copper(II) from aqueous solutions. Chem. Eng. J. 2010, 165, 240–249. 10.1016/j.cej.2010.09.024. [DOI] [Google Scholar]
- Ling C.; Liu F. Q.; Long C.; Chen T. P.; Wu Q. Y.; Li A. M. Synergic removal and sequential recovery of acid black 1 and copper (II) with hyper-crosslinked resin and inside mechanisms. Chem. Eng. J. 2014, 236, 323–331. 10.1016/j.cej.2013.09.058. [DOI] [Google Scholar]
- Rittikulsittichai S.; Kolhatkar A.; Sarangi S.; Vorontsova M.; Vekilov P.; Brazdeikis A.; Lee T. R. Multi-responsive hybrid particles: thermo-, pH-, photo-, and magneto-responsive magnetic hydrogel cores with gold nanorod optical triggers. Nanoscale 2016, 8, 11851–11861. 10.1039/C5NR09235C. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Chen Y.; Li M.; Zhou S.; Xue A.; Xing W. Adsorption of Hg2+ from aqueous solution onto polyacrylamide/attapulgite. J. Hazard. Mater. 2009, 171, 640–646. 10.1016/j.jhazmat.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Liu J.; Du X. Fast removal of aqueous Hg(II) with quaternary ammonium-functionalized magnetic mesoporous silica and silica regeneration. J. Mater. Chem. 2011, 21, 6981–6987. 10.1039/c1jm10111k. [DOI] [Google Scholar]
- Desimoni E.; Malitesta C.; Zambonin P. G.; Riviere J. C. An X-ray photoelectron spectroscopic study of some chromium-oxygen systems. Surf. Interface Anal. 1988, 13, 173–179. 10.1002/sia.740130210. [DOI] [Google Scholar]
- Yilmaz V. T.; Topcu Y.; Yilmaz F.; Thoene C. Saccharin complexes of Co(II), Ni(II), Cu(II), Zn(II), Cd(II) and Hg(II) with ethanolamine and diethanolamine: Synthesis, spectroscopic and thermal characteristics. Crystal structures of [Zn(Ea)2(Sac)2] and [Cu2(μ-Dea)2(Sac)2]. Polyhedron 2001, 20, 3209–3217. 10.1016/S0277-5387(01)00930-5. [DOI] [Google Scholar]
- Golcuk K.; Altun A.; Kumru M. Thermal studies and vibrational analyses of m-methylaniline complexes of Zn(II), Cd(II) and Hg(II) bromides. Spectrochim. Acta, Part A 2003, 59, 1841–1847. 10.1016/S1386-1425(02)00415-8. [DOI] [PubMed] [Google Scholar]
- Onwudiwe D. C.; Ajibade P. A. Thermal studies of Zn(II), Cd(II) and Hg(II) complexes of some N-alkyl-N-phenyl-dithiocarbamates. Int. J. Mol. Sci. 2012, 13, 9502–9513. 10.3390/ijms13089502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmstadt H.; Roy C.; Kaliaguine S.; Xu G.; Auger M.; Tuel A.; Ramaswamy V. Solid state 13C-NMR spectroscopy and XRD studies of commercial and pyrolytic carbon blacks. Carbon 2000, 38, 1279–1287. 10.1016/S0008-6223(99)00259-6. [DOI] [Google Scholar]
- Satilmis B.; Budd P. M. Base-catalysed hydrolysis of PIM-1: Amide versus carboxylate formation. RSC Adv. 2014, 4, 52189–52198. 10.1039/C4RA09907A. [DOI] [Google Scholar]
- Sun S. P.; Hatton T. A.; Chan S. Y.; Chung T. S. Novel thin-film composite nanofiltration hollow fiber membranes with double repulsion for effective removal of emerging organic matters from water. J. Membr. Sci. 2012, 401–402, 152–162. 10.1016/j.memsci.2012.01.046. [DOI] [Google Scholar]
- Mahapatra M.; Karmakar M.; Dutta A.; Mondal H.; Roy J. S. D.; Chattopadhyay P. K.; Singha N. R. Microstructural analyses of loaded and/or unloaded semisynthetic porous material for understanding of superadsorption and optimization by response surface methodology. J. Environ. Chem. Eng. 2018, 6, 289–310. 10.1016/j.jece.2017.11.078. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.