

Abstract
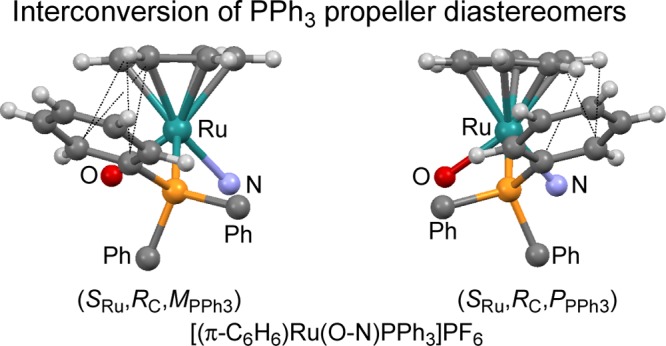
Chiral-at-metal compounds (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)PPh3]PF6 and (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)PPh3]PF6 were prepared using anions 1O-2N– and 2O-1N– of the Schiff bases, derived from the hydroxynaphthaldehydes and (S)-1-phenylethylamine. The pure (RRu,SC)-diastereomers were obtained by crystallization. In the unit cell of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, there are three independent molecules, which differ in the propeller sense of the PPh3 ligand. Molecules [1] and [2] have (MPPh3)-configuration and molecule [3] has (PPPh3)-PPh3 configuration. PPh3 diastereoisomerism is discussed including other pairs of compounds, differing only in the PPh3 configuration. A conformational analysis reveals an internal stabilization inside the PPh3 ligand by a system of attractive CH/π interactions and a new bonding motif PhPPh3 face-on π-Ar, both characteristic features of [(π-Ar)LL′MPPh3] compounds. The propeller diastereomers interconvert via a low-energy pathway and a high-energy pathway, corroborated by density functional theory calculations.
Introduction
In half-sandwich compounds of type [(π-Ar)LL′MPPh3], π-Ar = η6-C6H6, η5-C5H5, the triphenylphosphine ligand accounts for about half of the molecule. Figure 1 shows a hypothetical staggered conformation A in a Newman projection looking along P-M, which differentiates the phenyl rings into gauche and trans with respect to π-Ar. In such a conformation, the inner ortho-hydrogen atoms of the phenyl rings would approximate each other to unacceptably short distances. The phenyl rings avoid this steric hindrance by rotation around their P–Cipso bonds, adopting a propeller structure B in Figure 1. When steric hindrance disappears, weak attractive forces such as CH/π interactions in the internal PPh3 stabilization (see below) come into play.
Figure 1.

Newman projection of [(π-Ar)LL′MPPh3] looking along P-M. Hypothetical staggered conformation (A). Propeller conformation (B). PhPPh3 face-on π-Ar bonding conformation (C).
In a 1983 paper, we showed that in half-sandwich compounds [(π-Ar)LL′MPPh3] there is an additional rotation about the P-M bond, differentiating the gauche phenyl rings into close and distant to π-Ar (C in Figure 1).1 The phenyls close to π-Ar have rotation angles |0 < ρ < 60°|, and the phenyls distant to π-Ar have |60 < ρ < 120°|. Subsequently, we will show that this rotation is part of bonding motif PhPPh3 face-on π-Ar.
In the present paper, we describe the synthesis and characterization of compounds (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)PPh3]PF6 (Cy = cymene, 1-isopropyl-4-methylbenzene, 1O-2N– = (S)-2-[[(1-phenylethyl)imino]methyl]-1-naphthalenolate) and (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)PPh3]PF6 (2O-1N– = (S)-1-[[(1-phenylethyl)imino]methyl]-2-naphthalenolate). In the crystal, chiral-at-metal compound (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 forms diastereomers, which differ only in the configuration of the triphenylphosphine propeller. In this context, we discuss eight pairs of such propeller diastereomers, develop the PhPPh3 face-on π-Ar bonding concept, and reveal low- and high-energy pathways of the interconversion of the PPh3 propeller.
Results and Discussion
Synthesis and X-ray Characterization
Diastereomerically pure compounds (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 and (RRu,SC)-[CyRu(2O-1N)PPh3]PF6 (Scheme 1) were obtained in the following sequence of reactions. Deprotonated ligands 1OH-2N and 2OH-1N2 were reacted with [CyRuCl]2Cl2 to give (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)Cl] and (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)Cl], respectively. Treatment with PPh3 and NH4PF6 afforded products (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)PPh3]PF6 and (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)PPh3]PF6. In both cases, the pure (RRu,SC) diastereomers were obtained by crystallization from CH2Cl2 as red crystals suitable for X-ray analysis (Table S1 in the Supporting Information). In the unit cell of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, there are two different molecules [1] and [2] with the (M,M,M)-configuration of the PPh3 ligand and one molecule [3] with the (P,P,P)-PPh3 configuration.
Scheme 1. (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, 1O-2N– = (S)-2-[[(1-phenylethyl)imino]methyl]-1-naphthalenolate, and (RRu,SC)-[CyRu(2O-1N)PPh3]PF6, 2O-1N– = (S)-1-[[(1-phenylethyl)imino]methyl]-2-naphthalenolate.

The molecular structures will be discussed first for (RRu,SC)-[CyRu(2O-1N)PPh3]PF6 and then for the diastereomers of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6. The π-stack between the substituted phenyl ring of the naphthyl system and one of the phenyl rings of the PPh3 ligand is a striking feature in the structure of (RRu,SC)-[CyRu(2O-1N)PPh3]PF6 (Figure 2, left side). The distance between the carbon atoms [(Np)C2-Ci(Ph) = 3.21 Å] is considerably shorter than the distance between the layers in graphite (3.35 Å). The distances (Np)C2-Co(Ph) and (Np)C1-Co(Ph) are 3.27 and 3.16 Å.
Figure 2.

Molecular structures of (RRu,SC)-[CyRu(2O-1N)PPh3]PF6 (left side) and molecule (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 [3] with (P,P,P)-configuration of the PPh3 propeller (right side). Hydrogen atoms omitted for clarity.
Two independent molecules [1] and [2] of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 with the same propeller sense show similar π-stacks with corresponding distances, e.g., (Np)C1-Ci(Ph) = 3.20 and 3.21 Å. On the other hand, there is no such π-stack in the third independent molecule [3] of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, having the opposite PPh3 propeller configuration (Figure 2, right side).
Internal CH/π Stabilization within the PPh3 Propeller
The architecture of the PPh3 propeller is determined by CH/π interactions of the type found in the archetypal T-shaped benzene dimer.5−9 Contrary to those in the T-shaped benzene dimer, the CH/π interactions in PPh3 are intramolecular and thus entropically almost neutral. In the PPh3 ligand, there are six Co-H bonds, three inside the propeller (inCoH) and three outside (outCoH). It is the interaction between the inCo-H bonds and Ci, inCo, and outCo atoms of neighboring phenyl rings (i/o/p = ipso/ortho/para) that adds up to an appreciable stabilization, as discussed in refs (10) and (11) (Figure 3).
Figure 3.

CH/π interactions Ph3 → Ph1, Ph2 → Ph3, and Ph1 → Ph2 looking along the P-M axis.
In the following discussion, the torsion angles |Co-Ci-P-M|< 90° of phenyls Ph1, Ph2, and Ph3 will be called propeller angles τ. Table 1 contains these τ angles and distances CoH-Ci and CoH-Co of the CH/π interactions inside the PPh3 propeller. As in our former analyses,10,11 we ordered the propeller angles according to the smallest angle in the phenyl ring called Ph1. In addition to the four new molecules of the present paper, we added the pair of diastereomers of (RRu,SC)-[(π-C6H6)Ru(O-N)PPh3]PF6, O-N = anion of the Schiff base derived from salicylaldehyde and (S)-1-phenylethylamine, for which both diastereomers HEDYIY and HEDYOE differ only in the PPh3 propeller sense.12 (RRu,SC)-[(π-C6H6)Ru(O-N)PPh3]PF6 is the parent benzene/phenyl compound of the cymene/naphthyl compounds of the present paper.
Table 1. Internal Stabilization in Compounds (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, (RRu,SC)-[CyRu(2O-1N)PPh3]PF6, and (RRu,SC)-[(π-C6H6)Ru(O-N)PPh3]PF6a.
| entry varianta | CSD symbol or CCDC numberb | formula | 3 → 1 (Å) | 1 → 2 (Å) | 2 → 3 (Å) | |||
|---|---|---|---|---|---|---|---|---|
| M-P-Ci-Co | inCoH-Ci | M-P-Ci-Co | inCoH-Ci | M-P-Ci-Co | inCoH-Ci | |||
| Ph1 (deg) | inCoH-Coa | Ph2 (deg) | inCoH-Coa | Ph3 (deg) | inCoH-outCoa | |||
| 1 | 1519531 [1] | (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 | 6.4 | 2.75 | 89.5 | 2.78 | 44.7 | 2.77 |
| A/B | 2.67in | 2.93out | 2.56out | |||||
| 2 | 1519531 [2] | (RRu,SC)-[CyRu(1O- 2N)PPh3]PF6 | 14.2 | 2.71 | 82.6 | 2.77 | 44.0 | 2.72 |
| B | 2.77out | 2.97out | 2.58out | |||||
| 3 | HEDYOEc | (RRu,SC)-[(π- C6H6)Ru(O-N)PPh3]PF6 | –16.0 | 2.57 | –67.6 | 2.80 | –62.1 | 2.78 |
| A | 2.79in | 2.64out | ||||||
| 4 | 1519532 | (RRu,SC)-[CyRu(2O- 1N)PPh3]PF6 | 25.6 | 2.62 | 86.2 | 2.74 | 44.3 | 2.77 |
| A/B | 2.79in | 2.73out | 2.63out | |||||
| 5 | HEDYIYc | (RRu,SC)-[(π- C6H6)Ru(O-N)PPh3]PF6 | 23.9 | 2.58 | 77.9 | 2.63 | 53.7 | 2.76 |
| A/B | 2.86in | 2.78out | 2.54out | |||||
| 6 | 1519531 [3] | (RRu,SC)-[CyRu(1O- 2N)PPh3]PF6 | –25.2 | 2.59 | –61.9 | 2.58 | –69.5 | 2.73 |
| B | 3.02out | 2.54out |
Variants A and B, torsion angles M-P-Ci-Co < 90°, and distances inCoH-Ci, inCoH-inCo, and inCoH-outCo. Variant A refers to inCoH-inCo distances and variant B refers to inCoH-outCo distances for Ph1 and Ph2, respectively (see refs (10) and (11)).
Brackets [ ] indicate independent molecules.
See ref (12).
Each of the three phenyls plays a specific role in interactions Ph3 → Ph1 = inCoH(3) → Ci/o(1), Ph2 → Ph3 = inCoH(2) → Ci/o(3), and Ph1 → Ph2 = inCoH(1) → Ci/o(2), represented by the arrows in Figure 3. The differentiation into dashed and bold inCoH → Co interactions is relevant.10,11 All of the 18 inCoH-Ci distances are appreciably below 3.0 Å, the sum of the van der Waals radii of the hydrogen atom and the sp2-hybridized carbon atom,10,11,13 and thus within the bonding range of CH/π interactions. The same is true for 15 of the 18 inCoH-Co distances (Table 1). The approximation of the ortho-CH bonds to the ipso- and ortho-carbon atoms of neighboring phenyl rings to distances far below the sum of the van der Waals radii shows the internal stabilization in the PPh3 ligands. The Ph1 propeller angles in Table 1 span a broad range from |6.4°| to |25.2°|. For all of these τ(Ph1) angles, phenyls Ph2 and Ph3 find propeller angles to establish the necessary CH/π interactions for the internal stabilization.10,11
Propeller Chirality
Each of the three M-P-Ph systems in a PPh3 ligand is an independent element of chirality.10,11 The propeller angles Co-Ci-P-M < 90° are measures of the chirality of the M-P-Ph entities. They define (P)/(M) chirality of the M-P-Ph blades of the PPh3 propeller according to the helicity rule of the CIP system.14 Negative propeller angles correspond to (P) chirality, and positive propeller angles correspond to (M) chirality.
In (RRu,SC)-[CyRu(2O-1N)PPh3]PF6, the propeller angles Co-Ci-P-M < 90° in the PPh3 ligand are +25.6, +86.2, and +44.3° (Table 1). As all torsion angles are positive, the (M,M,M)-configuration has to be assigned to the PPh3 ligand. In (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, the unit cell contains three independent molecules. Two of them, [1] and [2], have (M,M,M)-configuration due to positive torsion angles +14.2, +82.6, and +44.0° and +6.4, +89.5 (−88.0), and +44.7°. In the phenyl ring, with the highest torsion angle of the (M,M,M)-diastereomers of 2O-1N and 1O-2N, the two ortho positions are almost equivalent (large thermal ellipsoids). Use of one or the other will interchange the symbols (M) and (P). The torsion angles of [1] and [2] are very similar to those of (RRu,SC)-[CyRu(2O-1N)PPh3]PF6. However, the third molecule [3] in the unit cell of (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 is very different. Its PPh3 ligand has (P,P,P)-configuration due to its negative torsion angles −25.2, −61.9, and −69.5° (Table 1).
Our recent analysis of 119 compounds of type [(π-Ar)LL′MPPh3] had shown that propeller configurations can be divided into two subgroups (P,P,P)/(M,M,M) (∼90% abundance) and (M,P,P)/(P,M,M) (∼10% abundance).10,11 As all of the new cymene/naphthyl compounds and their parent benzene/phenyl compounds HEDYOE and HEDYIY belong to the (P,P,P) or (M,M,M) type, we will subsequently use symbols (PPPh3) and (MPPh3) for the propeller configuration of the PPh3 ligand.
Rotation Angles ρ
In ref (1), we demonstrated that 11 [(π-Ar)LLMPPh3] compounds and 17 [(π-Ar)LL′MPPh3] compounds adopted structures of type C, in Figure 1, with rotation angles far below 60°. This rotation brings the gauche phenyl |0 < ρ < 60°| close and face-on toward π-Ar, whereas the gauche phenyl |60 < ρ < 120°| becomes distant and edge-on toward π-Ar. It was argued that the steric hindrance of the π-Ar ligand with the ortho-CH bond of the edge-exposed phenyl is responsible for the rotation,1 which is wrong (see below).
Figure 4 shows Newman projections of the six salicylaldiminato compounds. They clearly subdivide into two types, which have surprisingly similar conformations, irrespective of their π-Ar and O-N substituents. In all of the compounds, Ph3 is face-exposed to π-Ar with rotation angles ρ below |60°|, whereas Ph1 is edge-exposed with rotation angles ρ above |60°|. Taking into account the +/– signs of the rotation angles, the entire configurational symbols are (RRu,SC,MPPh3) for the compounds on the left and (RRu,SC,PPPh3) for those on the right of Figure 4. The rotation angles of Phface concentrate in the narrow range from |28.7°| to |47.0°|. With |89.2°| to |76.7°|, the rotation angles, ρ, of Phedge add up to 120° (Table S2 in the Supporting Information). The average of rotation angles ρ(Phface) is |40.0°|.
Figure 4.

Newman projections of (RRu,SC)-[CyRu(2O-1N)PPh3]PF6, (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 [1], [2], [3], HEDYIY, and HEDYOE looking along P-M.
In Table S2, we included another five pairs of diastereomers, which differ only in the propeller sense of the PPh3 ligand: VOWTUW,15 GIRYIP,16 ZINXOJ,17 FOMZEN,18 and RCMXFE.19 These compounds are of types [CpFe(CO)(R)PPh3] and [CpRe(NO)(R)PPh3]X. In the unit cell of these compounds, there are two independent molecules with the same metal configuration and opposite PPh3 configurations. This is similar to the four cymene/naphthyl compounds of the present paper, although they have an additional chiral center in the chelate ligand. For diastereomers HEDYIY and HEDYOE of (RRu,SC)-[(π-C6H6)Ru(O-N)PPh3]PF6, however, the situation is different. We could isolate the diastereomers of this compound as separate single crystals.12 Thus, HEDYOE and HEDYIY are two different modifications of (RRu,SC)-[(π-C6H6)Ru(O-N)PPh3]PF6. We also included SEPZUI in Table S2. Its two diastereomers differ in the metal configuration, having the same PPh3 propeller sense (PPPh3).20
The rotation angles of Phface and Phedge of the CpFe(CO) and CpRe(NO) compounds hook up with the salicyliminato compounds, except for (MPPh3) diastereomer GIRYIP[1], which is not used for average calculations (Table S2). The overall average of rotation angles ρ(Phface) is −36.8° for the (PPPh3) diastereomers and 40.0° for the (MPPh3) diastereomers. The face-on approximation of Phface to π-Ar is an indication of a bonding attraction, considered next.
Bonding Motif PhPPh3 Face-On π-Ar
The bonding system PhPPh3 face-on π-Ar includes elements of the T-shape as well as of the π-stack benzene dimer, and it contains the Ru and the P atom. In addition to rotation angles ρ, angle φ between the planes of π-Ar and Phface is a measure of the π-Ar/Phface interaction. Figure 5 shows the arrangement of π-Ar and PhPPh3 in HEDYIY (φ = 27.6°, left side) and HEDYOE (φ =28.6°, right side). In HEDYIY, distances CAr-Ci 3.32 Å and CAr-Co 3.30 Å are below the graphite distance of 3.35 Å, indicating a π-stack interaction. T-shape benzene dimer interactions show up in distances such as (π-Ar)CH-Co = 2.76 Å and (π-Ar)CH-Ci = 2.91 Å. The corresponding distances of propeller diastereomer HEDYOE are similar. In HEDYIY and HEDYOE, rotation angles ρ = 37.3 and −28.7° of the face-on phenyls enforce rotation angles of ρ = −82.4 and 89.2° for the corresponding edge-on phenyls.
Figure 5.

Bonding system PhPPh3 face-on π-Ar in HEDYIY, HEDYOE, and PIGJOG.
In Figure 6, rotation angles ρ of the face-on and edge-on phenyls of the 18 diastereomers, differing only in the propeller configuration of the PPh3 ligand, are shown as a function of π-Ar/Phface angles φ. They crowd around the averages of ρ and φ, which are ρav= −34.3° and φav = 23.6° for the (PPPh3) diastereomers and ρav = 40.0° and φav = 24.2° for the (MPPh3) diastereomers. The averages of ρ and φ of the edge-on phenyls are ρav = −81.0°/φav = 55.3° for the (PPPh3) diastereomers and ρav = 81.8°/φav = 46.1° for the (MPPh3) diastereomers.
Figure 6.
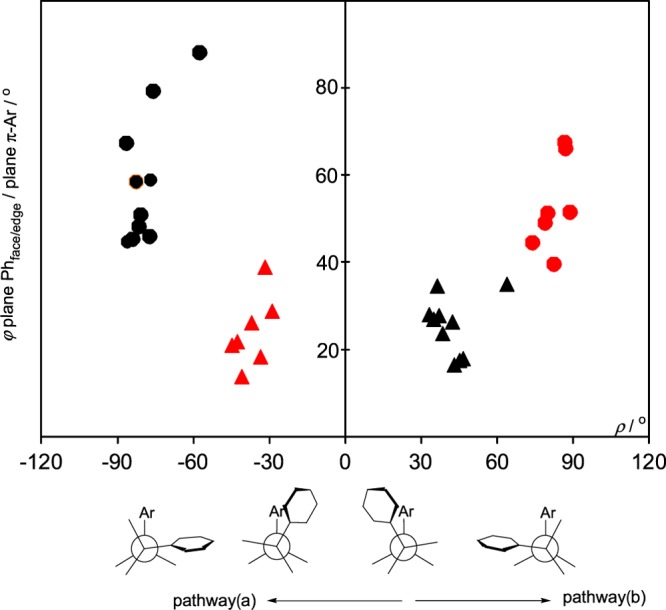
Plot of the rotation angle ρ versus angle φ plane Phface/edge/plane π-Ar for 18 diastereomers, differing only in the PPh3 propeller configuration: Rotation angles ρ of PPh(face) (red ▲), MPh(face) (▲), PPh(edge) (red ●), and MPh(edge) (●) versus angles φ plane Phface/edge/plane π-Ar. Bottom: Pathways a and b for diastereomer interconversion.
The turning of PhPPh3 face-on to π-Ar is a general phenomenon. In the histogram of Figure 7, this is shown for 140 cases of 119 compounds of type [(π-C6R6)RuLL′PPh3], obtained in a CSD search for [(π-C6R6)RuPPh3].21 The sample points concentrate around averages ρav = −39.0° and φav = 27.7° for the (PPPh3) diastereomers and ρav = 39.3° and φav = 25.7° for the (MPPh3) diastereomers. This is surprising because L and L′ and the substituents in the π-Ar ligand of the (π-Ar)LL′Ru fragments vary considerably. In all of the 140 cases of Figure 7, there is no exception with ρ > 60° such as GIRYIP[1] in Figure 6.
Figure 7.
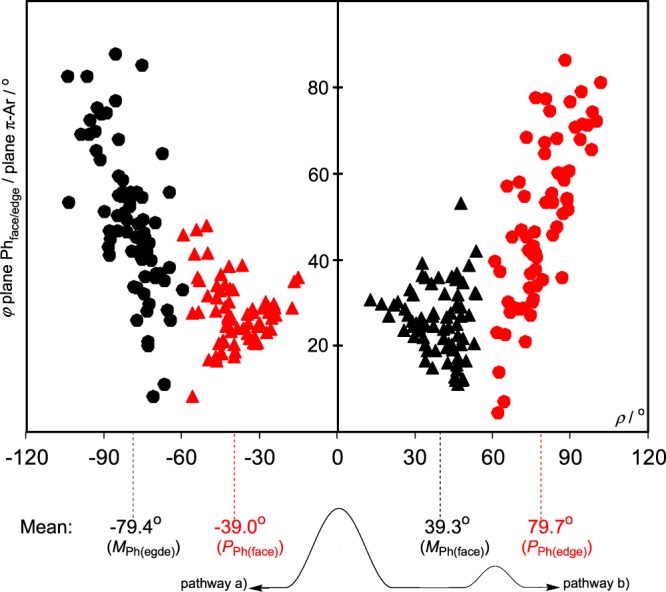
Plot of rotation angle ρ versus angle φ plane Phface/edge/plane π-Ar for 140 cases of 119 compounds of type [(π-C6R6)RuLL′PPh3] according to Table S3 (Supporting Information): Rotation angles ρ of PPh(face) (red ▲), MPh(face) (▲), PPh(edge) (red ●), and MPh(edge) (●) versus angles φ plane Phface/edge/plane π-Ar. Bottom: Pathways a and b and transition states for the interconversion of HEDYIY and HEDYOE.
With a rotation angle of ρ = −5.1°, PIGJOG22 is almost in the middle between HEDYIY and HEDYOE (Figure 5). PIGJOG is even more perfectly stabilized than HEDYIY and HEDYOE, as apparent from distances CAr-Ci = 3.28 and 3.31 Å as well as (π-Ar)CH-Co = 2.68 and 2.75 Å and (π-Ar)CH-Ci = 2.84 and 2.89 Å. When PIGJOG is very highly stabilized, the question arises, why do the rotation angles ρ of Phface concentrate around ±40° and not around 0°? The reason is the eclipsing interaction of the other two phenyls with substituents L and L′ in three-legged sandwich fragment (π-Ar)LL′M. PIGJOG’s substituent L = H2BHNMe3 is in a plane with Arcent, Ru, and P perpendicular to the plane of the paper, and the two phenyls stagger L perfectly (Figure 5). Thus, ±40° is a compromise of Phface to establish PhPPh3 face-on π-Ar stabilization and to avoid eclipsing of the other two phenyls with L and L′.
Interconversion of Propeller Diastereomers
The interconversion of diastereomers HEDYIY and HEDYOE, differing only in the propeller configuration, can occur by two different pathways: (a) Phface of HEDYIY is converted to Phface of HEDYOE via a transition state about ρ = 0° and vice versa and (b) Phface of HEDYIY is converted to Phedge of HEDYOE via a transition state about ρ = 60° and vice versa (Figures 6 and 7). Both pathways require only small intramolecular rotations of ρ and τ, far below full phenyl rotations.
Pathway a inverts the chirality of Ru-Phface from (MPh) in HEDYIY to (PPh) in HEDYOE and exchanges inCo/m of Phface to outCo/m. In addition, it brings Phtrans of HEDYIY up into the position of Phedge of HEDYOE and it moves Phedge of HEDYIY down to the position of Phtrans of HEDYOE. In pathway a, Phface passes through conformations with rotation angles ρ around 0° similar to the conformation of PIGJOG in Figure 5. Because these conformations are highly stabilized, pathway a would be energetically favorable for Phface. However, as discussed above, rotation angles of Phface around 0° imply the eclipsing of the other two phenyls with substituents L and L′, which makes the area of Phface around 0° a transition state.
Pathway b, although interchanging diastereomers HEDYIY and HEDYOE, does not change the (MPh) chirality, and it does not exchange inCo/m of Phface to outCo/m of the phenyl in question. In addition, this rotation brings Phedge of HEDYIY into the position of Phface of HEDYOE and it converts Phtrans of HEDYIY to Phtrans of HEDYOE. Thus, Phtrans stays Phtrans, but it inverts its chirality. Pathway b does not involve the eclipsing situation of pathway a.
It is well known that sample points of conformations, retrieved from the Cambridge Crystallographic Data file, concentrate in low-energy areas and thin out toward transition states.23 Therefore, the high population of the areas at about ρ = |40°| in Figures 6 and 7 by sample points means that these structures are favorable molecular conformations. On the other hand, the thinning out of sample points on the two sides of the energy minimum ρ = |40°| indicates the approximation to transition states. Furthermore, the distribution of sample points in the areas of the two transition states allows a differentiation between pathways a and b of the (PPh)/(MPh) interconversion of Phface. At about ρ = 0°, sample points not only thin out but disappear completely (Figures 6 and 7). That means, rotation angles about ρ = 0° correspond to a high-lying transition state. On the other hand, sample points of (MPh)-Phface and (PPh)-Phedge about ρ = 60° overlap, indicating a low-lying transition state.
The process of diastereomer interconversion along pathways a and b is shown at the bottom of Figure 7 on the right side. In pathway b, starting with HEDYIY at ρ = 37.3°, the transition state is reached at about ρ = 60° to finally arrive at HEDYOE with ρ = 80°. The process on the right side of Figure 6 would be similar. This low-energy pathway, far below full rotations around the Ci-P and P-Ru axes, is corroborated by the experimental sample points in Figure 7. The use of such experimental data to find reaction pathways has been pioneered by Dunitz et al.23
Whereas in the crystal, the PPh3 propeller configurations are fixed, in solution, they rapidly interconvert. For the 18 propeller diastereomers, pathway b seems to be the easiest mechanism of interconversion. This discussion concentrated on Phface and did not take into account a detailed consideration of Phedge and Phtrans. In addition, it must be kept in mind that each of the three phenyls has to carry out its duty in the internal stabilization of the PPh3 propeller.
Density Functional Theory (DFT) Calculations
We checked the results, obtained in the analysis of CSD sample points, by DFT calculations24 (RI25-B3LYP26/def2-TZVP25b,27). Using the cif files, we calculated the ground-state structures of HEDYIY and HEDYOE. HEDYOE turned out to be more stable than HEDYIY by 2.68 kJ/mol. The energy difference of 2.68 kJ/mol would account for a ratio HEDYIY/HEDYOE = 1:3 at 20 °C. Going from the conformation in the crystal to the conformation in the gas phase, the rotation angle changes for HEDYIY from ρ = 37.3 to 47.4° and for HEDYOE from ρ = −28.7 to −21.6°.
Our sample point analysis had predicted a low-lying transition state for the conversion of Phface of HEDYIY to Phedge of HEDYOE, resulting in the interconversion of the two diastereomers. This low transition state was reached after a counter-clockwise rotation of the PPh3 ligand in HEDYIY, which moved Phface from its position ρ = 37° in the crystal to 60° (pathway b). We calculated the relative energies of HEDYIY with the PPh3 ligand rotated from its gas phase ground state ρ = 47.7 to 50.3° and 59.4°. The relative energies rose from 0 via 0.75 to 4.66 kJ/mol (Figure 8), supporting a low-lying transition state at 60°.
Figure 8.

Relative energies of HEDYIY in its ground state at rotation angle ρ = 47.4° and on its way to the low transition state at ρ = 60° and the high transition state at ρ = 0°.
In the sample point analysis, we had assigned a high-lying transition state to a clockwise rotation of Phface from ρ = 37.3 to 0°, which converts Phface of HEDYIY to Phface of HEDYOE (pathway a). The calculation of the relative energies of HEDYIY with the PPh3 ligand rotated from ρ = 47.7 to 21.9° and 1.1° gave relative energies from 0 via 13.50 to 24.31 kJ/mol (Figure 8). Thus, the transition state of pathway a is much higher than that of pathway b and the results of sample point analysis and DFT calculations are fully in accord. As expected, the relative energies of the transition states are much higher than the ground state energies of HEDYIY and HEDYOE.
α- and β-Effects
In a recent paper, we reported CH/π interactions between cyclopentadienyl and phenyl rings in compounds of type CpM-L-E-Ph (Figure 9, left side), e.g., CpMo(CO)2-amidinato and -thioamidato complexes.28 These compounds were among the earliest examples, for which CH/π interactions have been observed. The Cp/Ph attraction had been termed the β-phenyl effect because of the β-position of Ph in the ligands.29 In comparison, the PhPPh3 face-on π-Ar system of the present paper is an α-phenyl effect.
Figure 9.
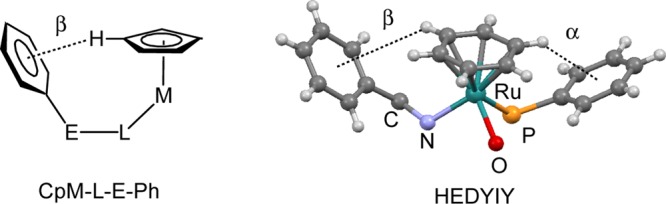
β-Phenyl effect in CpM-L-E-Ph compounds and α- and β-phenyl effects in HEDYIY.
In new compounds (RRu,SC)-[CyRu(2O-1N)PPh3]PF6, (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 [1], [2], and [3] and in HEDYIY and HEDYOE, CH/π interactions are established between Ar and the phenyl ring of the CHMePh substituent, resulting in short (π-Ar)CH-Ci and (π-Ar)CH-Co contacts far below the sum of the van der Waals radii. The dashed lines in Figure 9, right side, show the C6H6/Ph interactions in HEDYIY. An analysis according to ref (26) is given in Table S4 (Supporting Information).
The results in Table S4 reveal interesting differences between the compounds with and without π-stack stabilization. The two compounds (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 [3] and HEDYOE, lacking π-stacks, have appreciably shorter (π-Ar)CH-Ci and (π-Ar)CH-Co distances than those in the four compounds containing π-stacks. Obviously, the π-stacks prevent a perfect build-up of the β-CH/π interactions and the better β-CH/π stabilization seems to be a compensation for the absence of π-stack formation.
Conclusions
Chiral-at-metal half-sandwich compounds [(π-Ar)LL′MPPh3] form diastereomers, which differ in the propeller sense of the triphenylphosphine ligand. The inside of the PPh3 ligand is stabilized by a system of attractive CH/π interactions, in which each phenyl ring plays a specific role. One of the phenyl rings orients face-on toward the π-arene ligand, establishing a ubiquitous PhPPh3 face-on π-Ar bonding motif. Interconversion of the propeller diastereomers occurs by a low-energy pathway, which exchanges Phface and Phedge of the diastereomers.
Experimental Section
General Methods
For IR, JASCO FT/IR4100ST was used. For 1H/31P{1H} NMR, Bruker Avance 400 (400/162 MHz, T = 293 K) or Bruker Avance III 500 (500/202 MHz, T = 293 K) were used. Tetramethylsilane was used as the internal standard, and H3PO4 was used as the external standard. For MS, Finnigan MAT 95 (EI, 70 eV) or ThermoQuest Finnigan TSQ 7000 was used. All manipulations were carried out in purified nitrogen or argon. The Cambridge Structural Database ver. 5.38 (update May 31, 2017) for the 140 compounds of type [(π-C6R6)RuLL′PPh3] was used.21 The OLEX2,30 Mercury CSD ver. 3.9,31 and ConQuest ver. 1.1932 programs were used for structural analyses.
Preparation and Characterization
(RRu,SC)/(SRu,SC)-Chloro[η6-1-methyl-4-(1-methylethyl)benzene][1-[[(1-phenylethyl)imino-κN]methyl]-2-naphthalenolato-κO]ruthenium, (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)Cl]
To a solution of (S)-1-[[(1-phenylethyl)imino]methyl]-2-naphthalenol2 (200 mg, 0.73 mmol) in dichloromethane (20 mL) was added potassium t-butoxide (98 mg, 0.88 mmol). The solution was stirred for 1 h at room temperature and then cooled to −78 °C. [(η6-p-Cymene)RuCl]2Cl2 (250 mg, 0.36 mmol) was added to the cooled solution. The mixture was slowly warmed up to room temperature, stirred for 16 h, and then filtered on a short Celite column. After evaporation of the solvent, the residue was chromatographed on silica gel using EtOAc/hexane as an eluent. A reddish-brown band was collected and evaporated to give (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)Cl] 88:12 as a red powder in 70% yield (280 mg). Mp 125 °C (color changed from red to brown) > 200 °C. IR (KBr): ν 1614 cm–1 (N=C). 1H NMR (293 K, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets, if distinguishable): δ 8.75 (s, 1H, N=CH) [8.39 (s, 1H, N=CH)], 7.69 (d, 1H, 3JH-H = 8.4 Hz, nap-H) [7.83 (d, 1H, 3JH-H = 7.6 Hz, nap-H)], 7.62 (t, 2H, m-Ph-H), 7.56–7.07 (m, 8H, nap-H and Ph-H), 5.96 (q, 1H, 3JH-H = 7.1 Hz, N-CH) [5.73 (q, 1H, 3JH-H = 7.1 Hz, N-CH)], 5.25 (d, 1H, 3JH-H = 6.0 Hz, Cy-H) [5.52 (d, 1H, 3JH-H = 6.4 Hz, Cy-H)], 5.10 (d, 1H, 3JH-H = 6.0 Hz, Cy-H) [5.44 (d, 1H, 3JH-H = 6.4 Hz, Cy-H)], 5.02 (d, 1H, 3JH-H = 5.7 Hz, Cy-H) [5.41 (d, 1H, 3JH-H = 6.4 Hz, Cy-H)], 4.77 (d, 1H, 3JH-H = 5.7 Hz, Cy-H) [5.20 (d, 1H, 3JH-H = 6.4 Hz, Cy-H)], 2.63 (septet, 1H, 3JH-H = 6.8 Hz, iPr-CH) [2.83 (septet, 1H, 3JH-H = 7.0 Hz, iPr-CH)], 2.05 (s, 3H, Cy-CH3) [2.13 (s, 3H, Cy-CH3)], 1.81 (d, 3H, 3JH-H = 7.1 Hz, CH3), 1.14 (d, 3H, 3JH-H = 6.9 Hz, iPr-CH3) [1.14 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3)], 0.98 (d, 3H, 3JH-H = 6.9 Hz, iPr-CH3). MS (ESI, CH2Cl2/MeOH/NH4OAc): m/z 510 ([CyRu(2O-1N)]+; 100). Anal. Calcd for C29H30ClNORu (545.1): C, 63.90; H, 5.55; N, 2.57. Found: C, 63.90; H, 5.58; N, 2.45.
(RRu,SC)/(SRu,SC)-Chloro[η6-1-methyl-4-(1-methylethyl)benzene]-[2-[[(1-phenylethyl)imino-κN]methyl]-1-naphthalenolato-κO]ruthenium, (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)Cl]
In a procedure as above, the reaction of (S)-2-[[(1-phenylethyl)imino]methyl]-1-naphthalenol2 and [(η6-p-cymene)RuCl]2Cl2 gave (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)Cl] 86:14 as a red powder in 86% yield. Crystallization from dichloromethane/diethyl ether afforded red crystals of (RRu,SC)-[CyRu(1O-2N)Cl]. Mp 146 °C (color changed from red to brown) > 200 °C. IR (KBr): ν 1596 cm–1 (N=C). 1H NMR (400 MHz, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets, if distinguishable): δ 8.64 (d, 1H, 3JH-H = 7.6 Hz, nap-H) [8.61 (1H, 3JH-H = 8.1 Hz, nap-H)], 8.01 (s, 1H, N=CH) [7.65 (s, 1H, N=CH)], 7.58 (d, 1H, 3JH-H = 8.0 Hz, nap-H) [7.73 (d, 1H, 3JH-H = 8.0 Hz, nap-H)], 7.55–7.35 (m, 6H, Nap-H and Ph-H), 6.99 (d, 1H, 3JH-H = 8.6 Hz, nap-H) [7.75 (d, 1H, 3JH-H = 9.0 Hz, nap-H)], 6.84 (d, 1H, 3JH-H = 8.6 Hz, nap-H) [6.73 (d, 1H, 3JH-H = 9.0 Hz, nap-H)], 5.93 (q, 1H, 3JH-H = 7.0 Hz, N-CH) [5.66 (q, 1H, 3JH-H = 7.0 Hz, N-CH)], 5.32 (d, 1H, 3JH-H = 6.0 Hz, Cy-H) [5.59 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.23 (d, 1H, 3JH-H = 6.0 Hz, Cy-H) [5.49 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.04 (d, 1H, 3JH-H = 5.8 Hz, Cy-H) [5.18 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 4.87 (d, 1H, 3JH-H = 5.8 Hz, Cy-H), 2.68 (septet, 1H, 3JH-H = 7.0 Hz, iPr-CH) [2.84 (septet, 1H, 3JH-H = 7.0 Hz, iPr-CH)], 2.12 (s, 3H, Cy-CH3) [2.18 (s, 3H, Cy-CH3)], 1.81 (d, 3H, 3JH-H =7.1 Hz, CH3) [2.03 (d, 3H, 3JH-H = 6.9 Hz, CH3)], 1.12 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3) [1.23 (d, 3H, 3JH-H = 6.9 Hz, iPr-CH3)], 0.97 (d, 3JH-H = 6.9 Hz, iPr-CH3) [1.08 (d, 3H, 3JH-H = 6.9 Hz, iPr-CH3)]. MS (EI): m/z 545 ([CyRu(1O-2N)Cl]+, 4), 510 ([CyRu(1O-2N)]+, 6). Anal. Calcd for C29H30ClNORu (545.1): C, 63.90; H, 5.55; N, 2.57. Found: C, 63.93; H, 5.46; N, 2.61.
(RRu,SC)/(SRu,SC)-[η6-1-Methyl-4-(1-methylethyl)benzene][1-[[(1-phenylethyl)imino-κN]methyl]-2-naphthalenolato-κO](triphenylphosphanyl)ruthenium hexafluorophosphate, (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)PPh3]PF6
To a solution of (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)Cl] (87 mg, 0.16 mmol) in chloroform (20 mL) was added PPh3 (42 mg, 0.16 mmol). The mixture was stirred for 3 h at room temperature. Then, [NH4]PF6 (26 mg, 0.16 mmol) was added while stirring for 12 h. The reaction mixture was filtered on a short Celite column. After evaporation of the solvent, the residue was washed with diethyl ether to give (RRu,SC)/(SRu,SC)-[CyRu(2O-1N)PPh3]PF6 97:3 in 70% yield (102 mg). Crystallization from dichloromethane afforded orange crystals of pure diastereomer (RRu,SC)-[(CyRu(2O-1N)Ph)PPh3]PF6 suitable for X-ray structure analysis. Mp 143 °C (color changed from orange to brown) > 200 °C. IR (KBr): ν 1616 (N=C), 1435 (PPh3), 838 cm–1 (P-F). 1H NMR (400 MHz, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets, if distinguishable): δ 8.68 (s, 1H, N=CH) [8.90 (s, 1H, N=CH)], 7.59–7.19 (m, 25H, nap-H and Ph-H), 6.93 (d, 1H, 3JH-H = 9.1 Hz, nap-H), 5.56 (q, 1H, J = 7.0 Hz, N-CH), 5.49 (dd, 1H, 3JH-H = 6.5 Hz, 3JP-H = 1.3 Hz, Cy-H) [6.30 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.29 (d, 1H, 3JH-H = 6.5 Hz, Cy-H) [6.11 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.26 (d, 1H, 3JH-H = 6.0 Hz, Cy-H), 4.71 (br d, 1H, 3JH-H = 6.0 Hz, Cy-H), 2.35 (septet, 1H, 3JH-H = 7.0 Hz, iPr-CH) [2.70 (septet, 1H, 3JH-H = 7.0 Hz, iPr-CH)], 1.58 (s, 3H, Cy-CH3) [1.76 (s, 3H, Cy-CH3)], 1.39 (d, 3H, 3JH-H = 7.0 Hz, CH3) [2.09 (d, 3H, 3JH-H = 6.7 Hz, CH3)], 1.07 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3) [1.15(d, 3H, 3JH-H = 7.2 Hz, iPr-CH3)], 0.83 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3) [1.14 (d, 3H, 3JH-H = 7.2 Hz, iPr-CH3)]. 31P{1H} NMR (162 MHz, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets): δ 32.80 (s, 1P, PPh3) [29.60 (s, 1P, PPh3)], −142.81 (septet, 1P, 1JP-F = 713.5 Hz, PF6). MS (ESI, MeOH): m/z 772 ([(CyRu(2O-1N)Ph)PPh3]+, 100), 510 ([CyRu(2O-1N)Ph]+, 10). Anal. Calcd for C47H45F6NOP2Ru (916.87): C, 61.57; H, 4.75; N, 1.53. Found: C, 61.37; H, 4.88; N, 1.40.
(RRu,SC)/(SRu,SC)-[η6-1-Methyl-4-(1-methylethyl)benzene][2-[[(1-phenylethyl)imino-κN]methyl]-1-naphthalenolato-κO](triphenylphosphanyl)ruthenium hexafluorophosphate, (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)PPh3]PF6
The reaction of (RRu,SC)-[CyRu(1O-2N)Cl] (105 mg, 0.19 mmol) and PPh3 (50 mg, 0.19 mmol) was carried out as described above. After filtration on a short Celite column and evaporation, the residue was chromatographed on silica gel using EtOAc/hexane as an eluent. The orange fraction gave (RRu,SC)/(SRu,SC)-[CyRu(1O-2N)PPh3]PF6 96:4 in 68% yield (118 mg). Crystallization from dichloromethane afforded red crystals of pure diastereomer (RRu,SC)-[CyRu(1O-2N)PPh3]PF6 suitable for X-ray structure analysis. Mp 165 °C (color changed from red to brown) > 200 °C. IR (KBr): ν 1595 (N=C), 1431 (PPh3), 839 cm–1 (P-F). 1H NMR (400 MHz, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets, if distinguishable): δ 8.30 (d, 1H, 3JH-H = 8.4 Hz, nap-H), 7.83 (d, 1H, 4JP-H = 2.1 Hz, N=CH) [8.05 (d, 1H, 4JP-H = 2.0 Hz, N=CH)], 7.70–7.28 (m, 25H), 5.64 (d, 1H, 3JH-H = 6.4Hz, Cy-H) [6.37 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.54 (q, 1H, 3JH-H = 7.0 Hz, CH), 5.31 (d, 1H, 3JH-H = 6.1 Hz, Cy-H) [6.07 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 5.25 (d, 1H, 3JH-H = 6.1 Hz, Cy-H) [5.70 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 4.71 (d, 1H, 3JH-H = 6.4 Hz, Cy-H) [4.95 (d, 1H, 3JH-H = 6.0 Hz, Cy-H)], 2.31 (septet, 1H, 3JH-H = 6.8 Hz, iPr-CH), 1.68 (s, 3H, Cy-CH3) [1.77 (s, 3H, Cy-CH3)], 1.35 (d, 3H, 3JH-H = 7.0 Hz, CH3) [2.05 (d, 3H, 3JH-H = 7.0 Hz, CH3)], 0.97 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3) [1.07 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3)], 0.74 (d, 3H, 3JH-H = 6.9 Hz, iPr-CH3) [1.06 (d, 3H, 3JH-H = 7.0 Hz, iPr-CH3)]. 31P{1H} NMR (162 MHz, CDCl3, major (RRu,SC)-diastereomer, minor (SRu,SC)-diastereomer in brackets): δ 34.22 (s, 1P, PPh3) [31.06 (s, 1P, PPh3)], −142.79 (septet, 1P, 1JP-F = 713.5 Hz, PF6). MS (ESI, MeOH): m/z 772 ([CyRu(1O-2N)PPh3]+, 100), 510 ([CyRu(1O-2N)]+, 10). Anal. Calcd for C47H45F6NOP2Ru (916.87): C, 61.57; H, 4.75; N, 1.53. Found: C, 61.53; H, 4.76; N, 1.62.
X-ray Analyses
Crystal and refinement data are given in Table S1 (the Supporting Information). X-ray data were collected on a Rigaku RAXIS-RAPID imaging plate diffractometer using Mo Kα (graphite monochromated, λ = 0.71073 Å, fine focus tube, ω-scan) radiation at 173 K or an Oxford Diffraction Gemini Ultra diffractometer (Cu Kα radiation, λ = 1.54184 Å, ω-scan) at 123 K. The structures were solved by SIR200433 or SIR9734 and refined by full-matrix least squares on F2 by SHELX 2016/6.35 All H atoms were included at calculated positions. CCDC 1519530 {for (RRu,SC)-[CyRu(1O-2N)Cl]}, 1519531 {for (RRu,SC)-[(CyRu(1O-2N)PPh3)PF6]}, and 1519532 {for (RRu,SC)-[(CyRu(2O-1N)PPh3)PF6]} contain the supplementary crystallographic data for this paper.
DFT Calculations
All calculations have been performed with the TURBOMOLE program package at the RI24-B3LYP25/def26-TZVP25b,27 level of theory. To speed up the geometry optimization, the Multipole Accelerated Resolution-of-the-Identity24,36 approximation has been used. The relative energies have been calculated using the SCF energies without corrections.
Acknowledgments
Dedicated to Dr. Ralf Oeschey, who separated HEDYIY and HEDYOE.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b01460.
Table S1, crystallographic data of three complexes (RRu,SC)-[CyRu(1O-2N)Cl], (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, and (RRu,SC)-[CyRu(2O-1N)PPh3]PF6; 1H NMR spectra and 31P{1H} NMR spectra of the complexes; Table S2, rotation angles ρ and angles φ plane Ph/plane π-Ar for the compounds in Table 1; Table S3, rotation angles ρ and angles φ plane Ph/plane π-Ar for the 140 cases of the 119 compounds [(π-C6R6)RuLL′PPh3]; Table S4, CH/π interactions between π-Ar and Ph of the CHMePh substituent (β-phenyl effect) in compounds [(π-Ar)Ru(O-N)PPh3]PF6; Tables S5–S8, computational details (PDF)
Crystallographic data of three complexes (RRu,SC)-[CyRu(1O-2N)Cl], (RRu,SC)-[CyRu(1O-2N)PPh3]PF6, and (RRu,SC)-[CyRu(2O-1N)PPh3]PF6 (CIF)
The authors declare no competing financial interest.
Notes
CCDC 1519530–1519532 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/structures, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Supplementary Material
References
- Brunner H.; Hammer B.; Krüger C.; Angermund K.; Bernal I. Solid-state conformations of compounds (arene)L2MP(C6H5)3 and (arene)LL’MP(C6H5)3. Organometallics 1985, 4, 1063–1068. 10.1021/om00125a019. [DOI] [Google Scholar]
- Tsuno T.; Iwabe H.; Brunner H. Synthesis and structural characterization of isomeric palladium(II) complexes with chiral N,O-bidentate ligands. Inorg. Chim. Acta 2013, 400, 262–266. 10.1016/j.ica.2013.02.023. [DOI] [Google Scholar]
- Lecomte C.; Dusausoy Y.; Protas J.; Tirouflet J.; Dormond A. Structure cristalline et configuration relative d’un complexe du titanocene presentant une chiralite plane et une chiralite centree sur l’atome de titane. J. Organomet. Chem. 1974, 73, 67–76. 10.1016/S0022-328X(00)80382-4. [DOI] [Google Scholar]
- Brunner H. New “sequence rules”-higher priority of J. Am. Chem. Soc. versus other journals and of English versus other languages?. Enantiomer 1997, 2, 133–134. [Google Scholar]
- Dunitz J. D.; Gavezzotti A. Molecular Recognition in organic crystals: directedintermolecular bonds or nonlocalized bonding?. Angew. Chem., Int. Ed. 2005, 44, 1766–1787. 10.1002/anie.200460157. [DOI] [PubMed] [Google Scholar]
- Tummanapelli A. K.; Vasudevan S. Comunication: Benzene dimer—The free energy landscape. J. Chem. Phys. 2013, 139, 201102 10.1063/1.4834855. [DOI] [PubMed] [Google Scholar]
- van der Avoird A.; Podeszwa R.; Ensing B.; Szalewicz K. Comment on “Communication: Benzene dimer—The free energy landscape” [J. Chem. Phys. 139, 201102 (2013)]. J. Chem. Phys. 2014, 140, 227101 10.1063/1.4882015. [DOI] [PubMed] [Google Scholar]
- Schnell M.; Bunker P. R.; von Helden G.; Grabow J.-U.; Meijer G.; van der Avoird A. Stark effect in the benzene dimer. J. Phys. Chem. A 2013, 117, 13775–13778. 10.1021/jp408076q. [DOI] [PubMed] [Google Scholar]
- Podeszwa R.; Bukowski R.; Szalewicz K. Potential energy surface for the benzene dimer and perturbational analysis of π-π Interactions. J. Phys. Chem. A 2006, 110, 10345–10354. 10.1021/jp064095o. [DOI] [PubMed] [Google Scholar]
- Brunner H.; Tsuno T. CH/π-stabilization controls the architecture of the PPh3 propeller in transition-metal complexes. CH/π- and Cl/π-interactions determine its orientation within the molecule. Inorg. Chim. Acta 2016, 446, 132–142. 10.1016/j.ica.2016.02.039. [DOI] [Google Scholar]
- Brunner H.; Tsuno T. Comment on “Conformational analysis of triphenylphosphine ligands in stereogenic monometallic complexes: tools for predicting the preferred configuration of the triphenylphosphine rotor” by J. F. Costello, S. G. Davies, E. T. F. Gould and J. E. Thomson, Dalton Trans., 2015, 44, 5451. Dalton Trans. 2017, 46, 5103–5109. 10.1039/C7DT00474E. [DOI] [PubMed] [Google Scholar]
- Brunner H.; Oeschey R.; Nuber B. Optically active transition metal complexes. 105. Propeller isomerism of triphenylphosphine ligand in half-sandwich RuII complexes. Angew. Chem., Int. Ed. Engl. 1994, 33, 866–868. 10.1002/anie.199408661. [DOI] [Google Scholar]
- Nishio M.; Hirota M.; Umezawa Y.. The CH/π Interaction; Wiley-VCH: New York, 1998. [Google Scholar]
- Cahn R. S.; Ingold C.; Prelog V. Specification of molecular chirality. Angew. Chem., Int. Ed. Engl. 1966, 5, 385–415. 10.1002/anie.196603851. [DOI] [Google Scholar]
- Stolz F.; Strazewski P.; Tamm C.; Neuberger M.; Zehnder M. New chiral α-benzyloxyacryliron(II) complex for asymmetric synthesis of α,α-dialkyl-α-hydroxycarbonyl compounds. Angew. Chem., Int. Ed. 1992, 31, 193–196. 10.1002/anie.199201931. [DOI] [Google Scholar]
- Bruce M. I.; Liddell M. J.; Snow M. R.; Tiekink E. R. T. Stability of the cyclobutenyl group in Fe(C=CFCF2CF2)(CO)2)(η-C5H5) towards isomerisation by ring-opening. X-ray crystals structures of Fe(C=CFCF2CF2)(CO)(L)(η-C5H5) (L=CO and PPh)3. J. Organomet. Chem. 1988, 354, 103–115. 10.1016/0022-328X(88)80644-2. [DOI] [Google Scholar]
- Cagle P. C.; Meyer O.; Weickhardt K.; Arif A. M.; Gladysz J. A. Enantioselective synthesis of organosulfur compounds via [2,3] rearrangements of yliedes derived from di(allyl) and di(propargyl) sulfide complexes. Control of carbon configuration by an easily resolved and recycled chiral transition metal auxiliary. J. Am. Chem. Soc. 1995, 117, 11730–11744. 10.1021/ja00152a014. [DOI] [Google Scholar]
- Nakazawa H.; Itazaki M.; Owaribe M. Carbonyl(η5-cyclopentadienyl)(isocyanotriphenylborato-κC)(triphenyphosphine-κP)iron(II). Acta Crystallogr., Sect. E: Crystallogr. Commun. 2005, 61, m1166–m1168. 10.1107/S1600536805015369. [DOI] [Google Scholar]
- Chou C.-K.; Miles D. L.; Bau R.; Flood T. C. Crystallographic determination of the absolute configuration at iron of a series of chiral iron alkyls. Empirical circular dichroism spectroscopic correlates. J. Am. Chem. Soc. 1978, 100, 7271–7278. 10.1021/ja00491a025. [DOI] [Google Scholar]
- Kulawiec R. J.; Faller J. W.; Crabtree R. H. Binding and activation of halocarbons by iron(II) and ruthenium(II). Organometallics 1990, 9, 745–755. 10.1021/om00117a033. [DOI] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addy D. A.; Bates J. I.; Kelly M. J.; Riddlestone I. M.; Aldridge S. Aminoborane σ complexes: significance of hydride co-ligands in dynamic processes and dehydrogenative borylene formation. Organometallics 2013, 32, 1583–1586. 10.1021/om400040q. [DOI] [Google Scholar]
- Bye E.; Schweizer B.; Dunitz J. D. Chemical reaction paths. 8. Stereoisomerization path for triphenylphosphine oxide and related molecules: indirect observation of the structure of the transition state. J. Am. Chem. Soc. 1982, 104, 5893–5898. 10.1021/ja00386a008. [DOI] [Google Scholar]
- a Furche F.; Ahlrichs R.; Hättig C.; Klopper W.; Sierka M.; Weigend F. Turbomole. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 91–100. 10.1002/wcms.1162. [DOI] [Google Scholar]; b Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]; c Treutler O.; Ahlrichs R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. 10.1063/1.469408. [DOI] [Google Scholar]; d TURBOMOLE, v6.4; a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH. http://www.turbomole.com.
- a Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Auxiliary basis sets to approximate Coulomb potentials (Chem. Phys. Letters 240 (1995) 283. Chem. Phys. Lett. 1995, 242, 652–660. 10.1016/0009-2614(95)00838-U. [DOI] [Google Scholar]; b Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. 10.1007/s002140050244. [DOI] [Google Scholar]
- a Dirac P. A. M. Quantum mechanics of many-electron systems. Proc. R. Soc. London, Ser. A 1929, 123, 714–733. 10.1098/rspa.1929.0094. [DOI] [Google Scholar]; b Slater J. C. A simplification of the Hartree-Fock method. Phys. Rev. 1951, 81, 385–390. 10.1103/PhysRev.81.385. [DOI] [Google Scholar]; c Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]; d Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; e Becke A. D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; f Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Brunner H.; Tsuno T. Cyclopentadienyl/phenyl attraction in CpM-L-E-Ph compounds by CH/π interactions. Organometallics 2015, 34, 1287–1293. 10.1021/acs.organomet.5b00022. [DOI] [Google Scholar]
- Brunner H. Rhodium catalysts for enantioselective hydrosilylation - a new concept for development of asymmetric catalysts. Angew. Chem., Int. Ed. Engl. 1983, 22, 897–907. 10.1002/anie.198308973. [DOI] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0 – new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Bruno I. J.; Cole J. C.; Edgington P. R.; Kessler M.; Marcrae C. F.; McCabe P.; Pearson J.; Taylor R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr., Sect. B: Struct. Sci. 2002, 58, 389–397. 10.1107/S0108768102003324. [DOI] [PubMed] [Google Scholar]
- Burla M. C.; Caliandro R.; Camalli M.; Carrozzini B.; Cascarano G. L.; De Caro L.; Giacovazzo C.; Polidoria G.; Spagnac R. SIR2004: an improved tool for crystal structure determination and refinement. J. Appl. Crystallogr. 2005, 38, 381–388. 10.1107/S002188980403225X. [DOI] [Google Scholar]
- Altomare A.; Burla M. C.; Camalli M.; Cascarano G. L.; Giacovazzo C.; Guagliardi A.; Moliterni A. G. G.; Polidori G.; Spagna R. SIR. 97: a new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119. 10.1107/S0021889898007717. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierka M.; Hogekamp A.; Ahlrichs R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. 10.1063/1.1567253. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.