

Abstract
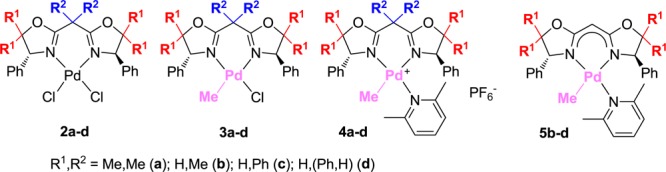
A series of neutral and cationic palladium(II) complexes containing C2-symmetric bis(oxazoline) (BOX) ligands, (BOX)PdCl2 (2a–d), (BOX)Pd(Me)Cl (3a–d), and [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d) [BOX: 2,2′-(2-propylidene)bis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline}, 2,2′-methylenebis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline}, 2,2′-methylenebis{(4R)-4,5,5-triphenyl-2-oxazoline}, and 2,2′-methylenebis{(4R,5S)-4,5-diphenyl-2-oxazoline}], were prepared, and their structures were determined by X-ray crystallography. It was found that substituents at the 5-position (Ph, Me) in addition to substituents on the bridgehead carbon directly affect the structure around palladium, especially the BOX bite angle and the dihedral angles between the phenyl rings at the 4-position and the N2Pd plane. Treatment of the bridged methylene proton in the BOX ligand (1b–d) with KH afforded the anionic BOX ligand; also, the neutral Pd complexes, (BOX)PdMe(2,6-Me2C5H3N) (5b–d), could thus be prepared by reaction with Pd(Me)Cl(cod) (cod = 1,5-cyclooctadiene); 5b–d showed strong coordination to Pd, as demonstrated by X-ray crystallographic analysis.
Introduction
Carbon–carbon bond formation is one of the most important reactions in synthetic chemistry; transition metal–alkyl complexes play roles as reagents or intermediates in stoichiometric/catalytic organic reactions and olefin polymerization.1−6 The synthesis and reaction chemistry of the metal alkyls are thus important in the design of efficient catalysts as well as to better understand the reaction mechanisms. Palladium complexes are widely used as catalysts in efficient carbon–carbon bond formation reactions, such as coupling reactions and olefin oligomerization/polymerization.7−16 In particular, palladium complexes containing α-diimine ligands17−24 and phosphine–sulfonate ligands25−30 are known to be effective catalysts for ethylene copolymerization, not only with α-olefin, but also with olefins containing polar functional groups.31−33
The synthesis and reaction chemistry of transition metal complexes containing optically active C2-symmetric ligands are an important subject for the design of asymmetric catalytic reactions. Zirconocene complexes containing C2-symmetric ligands, exemplified as [Me2Si(indenyl)2]ZrCl2,34,35 and nickel complexes containing C2-symmetric α-diimine ligands36−38 have been used as the catalysts for the isospecific polymerization of α-olefins. Chiral bis(oxazoline) (BOX, 1) has also been employed as a highly attractive neutral C2-symmetric bidentate ligand in various transition metal catalysts (such as Cu, Pd, Ni, Zn, Fe, Rh, Mg, Li, Mn, and Co);39−41 various palladium-catalyzed organic transformations are also known.42−64 However, in contrast to their wide applications in organic transformations, the synthesis and structural analyses of a series of metal complexes with different ligand substituents have not yet been explored in detail. Such a study would provide important information for the design of efficient catalysts and also increase understanding of catalysis. Some studies have been published concerning the effects of modifications (such as Ph, t-Bu, benzyl, or naphthyl) at the 4-position of BOX ligands, adjacent to the metal center, on the selectivity/activity of catalytic reactions.39−41 In contrast, we herein focus on modification at the 5-position because we assumed at an early stage that modification at this position should also play a role in asymmetric induction (the structure around the metal center) and thus in the reactivity, probably on the basis of the stabilities of the catalytic intermediates. In fact, the introduction of substituents (methyl and phenyl groups) at the 5-position in BOX affects its reactivity in nickel-catalyzed ethylene polymerization and ethylene/1-hexene copolymerization.65,66 However, reports concerning the effects of the substituents on the basis of structural analysis (isolated complexes) remain limited.
Therefore, in this article, we have explored the synthesis and structural analysis of a series of neutral and cationic palladium(II) complexes containing BOX ligands (shown in Chart 1), (BOX)PdCl2 (2a–d), (BOX)Pd(Me)Cl (3a–d), and [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d) [BOX: 2,2′-(2-propylidene)bis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline}, 2,2′-methylenebis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline}, 2,2′-methylenebis{(4R)-4,5,5-triphenyl-2-oxazoline}, and 2,2′-methylenebis{(4R,5S)-4,5-diphenyl-2-oxazoline}]. Moreover, we have prepared a series of novel neutral (BOX)PdMe(2,6-Me2C5H3N) (5b–d) (Chart 1) by deprotonation of the bridgehead methylene protons using KH. Through this research, we sought to obtain basic information concerning the effects of ligands on their structures (coordination modes and electronic states) for better understanding the performance of catalysts.
Chart 1. Neutral or Cationic Pd Complexes Containing C2-Symmetric BOX Ligands 2–5.

Results and Discussion
Synthesis and Molecular Structure of C2-Symmetric BOX Ligands (1a–d)
We focused on C2-symmmetric BOX ligands containing methyl or phenyl substituents at the 5-position in the oxazoline ring and at the bridgehead carbon between two oxazolines to explore their influences on the structures of a series of BOX–palladium(II) complexes. Because optically active amino alcohol derivatives (6) bearing substituents (R1) can be synthesized by the reaction of amino ester hydrochloride salt with Grignard reagents (Scheme 1), various substituents (R1) can be thus introduced in BOX ligands using these amino alcohols as a starting material. First, optically active amino alcohols, (R)-1-amino-2-methyl-1-phenylpropan-2-ol (6a) and (R)-2-amino-1,1,2-triphenylethanol (6c), were prepared using modifications of reported procedures (Scheme 1).67,68 The reaction of chiral amino alcohol 6a with oxalyl chloride in the presence of triethylamine afforded N1,N3-bis((R)-2-hydroxy-2-methyl-1-phenylpropyl)-2,2-dimethylmalonamide (7a); then, intramolecular dehydration condensation of 7a using Ti(OiPr)4 as a catalyst afforded 2,2′-(2-propylidene)bis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline} (1a). One-pot syntheses were conducted for 2,2′-methylenebis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline} (1b) and 2,2′-methylenebis{(4R)-4,5,5-triphenyl-2-oxazoline} (1c). Condensation of amino alcohol 6a or 6c with dimethylmalonate afforded the corresponding bisamide alcohol (7b or 7c) in situ; then, the addition of Ti(OiPr)4 afforded BOX (1b or 1c) in good overall yields. Furthermore, the detailed structures of BOX (1b, 1c) and commercially available BOX (1d) were confirmed by X-ray crystallography (Figure 1). There were no significant differences in the bond lengths or angles, as shown in Table 1.
Scheme 1. Synthesis of C2-Symmetric BOX Ligands (1a–c).

Figure 1.

Oak ridge thermal ellipsoid plot (ORTEP) drawings for BOX 1b–d. Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.
Table 1. Selected Bond Distances (Å) and Angles (deg) for BOX 1b–d.
| BOXs |
|||
|---|---|---|---|
| 1b | 1c | 1d | |
| Selected Bond Distances (Å) | |||
| C(1)–C(2) | 1.501(3) | 1.492(3) | 1.500(5) |
| C(1)–C(5) | 1.493(3) | 1.504(3) | 1.506(4) |
| N(1)–C(2) | 1.268(3) | 1.262(3) | 1.277(4) |
| N(2)–C(5) | 1.272(3) | 1.267(3) | 1.277(4) |
| Selected Bond Angles (deg) | |||
| C(2)–C(1)–C(5) | 111.24(14) | 110.69(16) | 109.4(3) |
| N(1)–C(2)–C(1) | 125.21(16) | 126.80(18) | 126.5(3) |
| N(2)–C(5)–C(1) | 126.03(16) | 126.30(18) | 126.2(3) |
Synthesis and Characterization of Neutral (BOX)PdCl2 (2a–d)
Reactions of BOX (1a–d) with equimolar amounts of PdCl2(CH3CN)2 in CH2Cl2 at room temperature (25 °C) afforded the corresponding dichloro complexes, (BOX)PdCl2 (2a–d), in high yields (92–94%, Scheme 2); these were identified by NMR spectra and elemental analyses (shown in the Experimental Section). The structures of 2a–d were also determined by X-ray crystallography (shown below).69
Scheme 2. Synthesis of (BOX)PdCl2 (2a–d).

Figure 2 shows ORTEP drawings of complexes 2a–d; their selected bond lengths and angles are summarized in Table 2. These complexes fold around palladium in a square planar geometry consisting of two chlorine ligands and two nitrogen atoms in the BOX ligand. The total bond angles around palladium are close to 360° [sums of the bond angles of N(1)–Pd(1)–N(2), Cl(1)–Pd(1)–Cl(2), N(1)–Pd(1)–Cl(2), and N(2)–Pd(1)–Cl(1): 2a: 360.03°, 2b: 360.07°, 2c: 360.46°, 2d: 359.95°], suggesting that palladium, two chlorine ligands, and two nitrogen atoms exist almost on the same plane [mean deviations from the plane in 2a: 0.0193 Å, 2b: 0.0292 Å, 2c: 0.0000 Å, 2d: 0.0228 Å]. It was found that the bridge angles of C(2)–C(1)–C(5) in 2b–d [2b: 114.3(3)°, 2c: 115.9(4)°, 2d: 115.3(3)°] are larger than those in 1b–d [1b: 111.24(14)°, 1c: 110.69(16)°, 1d: 109.4(3)°]. These angles are influenced by the coordination of the BOX ligand to palladium, whereas no significant differences in the bond lengths between (BOX)PdCl2 (2b–d) and BOX (1b–d) were observed (as shown in Tables 1 and 2). It is noteworthy that complex 2a [87.17(16)°] has a smaller coordination angle in the BOX ligand than that of complexes 2b–d [88.88(11)–88.98(13)°], whereas no significant differences in the angles were observed in 2b–d. The bridge angle of C(2)–C(1)–C(5) in 2a [110.1(4)°] is also smaller than that in 2b–d [114.3(3)–115.9(4)°], probably because of the geminal dimethyl groups at the bridgehead carbon C(1) in the BOX ligand. These data suggest that the substituents at the bridgehead carbon (not at the 5-position) control the BOX coordination angle, which should influence both the catalytic reactivity and selectivity.70−72
Figure 2.

Oak ridge thermal ellipsoid plot (ORTEP) drawings for (BOX)PdCl2 (2a–d). Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.
Table 2. Selected Bond Distances (Å) and Angles (deg) for (BOX)PdCl2 (2a–d)a.
| (BOX)PdCl2 |
||||
|---|---|---|---|---|
| 2a | 2b | 2c | 2d | |
| Selected Bond Distances (Å) | ||||
| C(1)–C(2) | 1.519(7) | 1.494(6) | 1.488(5) | 1.481(5) |
| C(1)–C(5) | 1.522(6) | 1.493(5) | 1.488(5), C(1)–C(2i) | 1.492(5) |
| N(1)–C(2) | 1.285(7) | 1.285(4) | 1.277(4) | 1.276(5) |
| N(2)–C(5) | 1.265(7) | 1.276(5) | 1.277(4), N(1i)–C(2i) | 1.280(4) |
| Pd(1)–N(1) | 2.035(5) | 2.031(3) | 2.030(3) | 2.032(3) |
| Pd(1)–N(2) | 2.030(4) | 2.041(3) | 2.030(3), Pd(1)–N(1i) | 2.037(3) |
| Pd(1)–Cl(1) | 2.2975(19) | 2.2936(10) | 2.2877(12) | 2.2951(12) |
| Pd(1)–Cl(2) | 2.2796(16) | 2.2827(11) | 2.2877(12), Pd(1)–Cl(1i) | 2.2792(11) |
| Selected Bond Angles (deg) | ||||
| C(2)–C(1)–C(5) | 110.1(4) | 114.3(3) | 115.9(4) | 115.3(3) |
| C(2)–C(1)–C(2i) | ||||
| N(1)–C(2)–C(1) | 127.8(5) | 127.5(3) | 129.3(4) | 129.7(3) |
| N(2)–C(5)–C(1) | 127.6(4) | 127.5(4) | 129.3(4) | 128.7(3) |
| N(1i)–C(2i)–C(1) | ||||
| N(1)–Pd(1)–N(2) | 87.17(16) | 88.98(13) | 88.94(12) | 88.88(11) |
| N(1)–Pd(1)–N(1i) | ||||
| Cl(1)–Pd(1)–Cl(2) | 89.10(7) | 89.25(4) | 88.38(5) | 87.69(4) |
| Cl(1)–Pd(1)–Cl(1i) | ||||
| N(1)–Pd(1)–Cl(2) | 92.28(12) | 91.00(10) | 91.57(10) | 91.38(9) |
| N(1)–Pd(1)–Cl(1) | ||||
| N(2)–Pd(1)–Cl(1) | 91.48(13) | 90.84(10) | 91.57(10) | 92.00(9) |
| N(1i)–Pd(1)–Cl(1i) | ||||
| Dihedral Angles (deg) | ||||
| Ph1–N2PdCl2 | 95.905 | 87.827a | 89.910 | 81.246 |
| Ph2–N2PdCl2 | 100.819 | 99.487a | 89.910 | 100.088 |
| Distances between Pd and p-C (Ph) (Å) | ||||
| Pd–p-C(Ph1) | 5.994 | 5.813a | 5.773 | 5.705 |
| Pd–p-C(Ph2) | 6.119 | 6.222a | 5.773 | 6.066 |
Mean values of four structures.
The geometry of the substituent at the 4-position in the oxazoline ring should affect the selectivity and reactivity of BOX-metal catalysts because the substituents influence the size of the coordination site on the metal (Figure 3). In fact, the dihedral angle (the mean value of the Ph1–N2PdCl2 and Ph2–N2PdCl2 dihedral angles, Table 2) in 2a–d decreases in the following order: 2a (98.362°) > 2b (93.707°) > 2d (90.667°) > 2c (89.910°). The distance between Pd and the para-carbon (p-C) of the phenyl groups at the 4-position (mean value of Pd–p-C in the Ph1 and Ph2 distances, Table 2) also decreases in the following order: 2a (6.057 Å) > 2b (6.017 Å) > 2d (5.886 Å) > 2c (5.773 Å). These results highly suggest that steric bulk in the substituents at the 5-position decreases the dihedral angles as well as the Pd–p-C distances, whereas the geminal dimethyl groups at the bridgehead carbon increase these angles and distances (Figure 3). Consequently, the reactivities and selectivities of catalytic reactions should be attributed to the introduction of substituents, especially at the 5-position, in BOX. In fact, the enhanced reactivities and/or selectivities have been reported for several reactions using (BOX)metal catalysts (palladium: allylic alkylation48 and hydroarylation;57 nickel: polymerization65,66 and conjugate addition;73 copper: cyclopropanation74−76 and dearomatization;77 and lanthanide: hydroamination/cyclization).78
Figure 3.

Left: X-ray structure of (BOX)PdCl2 (2c). Right: Steric repulsion between the Ph groups at the 4-position and the substituents (R1) at the 5-position in the oxazoline ring.
Synthesis and Characterization of Neutral (BOX)Pd(Me)Cl (3a–d)
Reaction of 1a–d with Pd(Me)Cl(cod) (cod = 1,5-cyclooctadiene) in CH2Cl2 at room temperature afforded (BOX)Pd(Me)Cl (3a–d) in high yields (86–95%, Scheme 3); these were identified by NMR spectra and elemental analysis (shown in the Experimental Section). The 1H NMR spectrum of each complex shows resonance of the corresponding protons of the methyl group bound to palladium in a high magnetic field (3a: 0.07 ppm, 3b: 0.10 ppm, 3c: 0.25 ppm, 3d: 0.22 ppm in CD2Cl2 at 25 °C).
Scheme 3. Synthesis of (BOX)Pd(Me)Cl (3a–d).

Single crystals of (BOX)Pd(Me)Cl (3a, 3c) were grown by slow diffusion of n-hexane into dichloromethane solution, and the structures were confirmed by X-ray crystallography (Figure 4, Table 3). In crystallographic analyses of 3a,c, the R1 and wR2 values are a little higher than those of complexes 2a–d due to the disordered Me and Cl groups bonding to Pd (3a: R1 = 0.0550, wR2 = 0.1442; 3c: R1 = 0.0493, wR2 = 0.1229; 2a–d: R1 = 0.0238–0.0279, wR2 = 0.0571–0.0731). Similar to complexes 2a–d, complexes 3a and 3c fold around palladium in a square planar geometry consisting of two nitrogen atoms in the BOX ligand and chlorine and methyl ligands. The total bond angles around palladium are close to 360° (sums of the bond angles of N(1)–Pd(1)–N(2), Cl(1)–Pd(1)–C(1), N(1)–Pd(1)–Cl(1), and N(2)–Pd(1)–C(1): 3a: 360.1° and 3c: 360.0°), suggesting that the methyl group attached to palladium exists on the plane consisting of palladium, chlorine, and two nitrogen atoms (mean deviations from the plane in 3a, 0.0211 Å and in 3c, 0.0289 Å). The bond lengths of Pd(1)–C(1) [3a, 3c: 2.13(2) and 2.018(9) Å, respectively] are shorter than those of Pd(1)–Cl(1) [3a, 3c: 2.236(5) and 2.301(2) Å, respectively]. Moreover, the C(3)–C(2)–C(6) and N(1)–Pd(1)–N(2) bond angles in 3a [108.6(9) and 86.9(4)°, respectively] are smaller than those in 3c [108.6(9) and 86.9(4)°, respectively], as observed in 2a–d (Table 2). The dihedral angle in 3c is smaller than that in 3a, probably influenced by the steric bulk of the substituents at the 5-position (Ph1–N2PdCCl and Ph2–N2PdCCl: 3a, 89.308 and 91.958°, respectively; 3c, 82.621 and 81.874°, respectively).
Figure 4.

Oak ridge thermal ellipsoid plot (ORTEP) drawings for (BOX)Pd(Me)Cl (3a, 3c). Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.
Table 3. Selected Bond Distances (Å) and Angles (deg) for (BOX)Pd(Me)Cl (3a, 3c), Cationic [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a), and Neutral (BOX)PdMe(2,6-Me2C5H3N) (5b).
| Pd complex |
||||
|---|---|---|---|---|
| 3a | 3c | 4a | 5b | |
| Selected Bond Distances (Å) | ||||
| C(2)–C(3) | 1.517(15) | 1.485(7) | 1.500(4), C(1)–C(2) | 1.402(5), C(1)–C(2) |
| C(2)–C(6) | 1.508(17) | 1.502(6) | 1.522(4), C(1)–C(5) | 1.392(5), C(1)–C(5) |
| N(1)–C(3) | 1.292(14) | 1.270(7) | 1.271(4), N(1)–C(2) | 1.316(5), N(1)–C(2) |
| N(2)–C(6) | 1.297(14) | 1.274(7) | 1.283(4), N(2)–C(5) | 1.316(5), N(2)–C(5) |
| Pd(1)–N(1) | 2.110(9) | 2.114(4) | 2.134(3) | 2.108(3) |
| Pd(1)–N(2) | 2.090(10) | 2.070(4) | 2.060(3) | 2.036(3) |
| Pd(1)–X | 2.236(5) | 2.301(2) | 2.043(3) | 2.063(3) |
| X = Cl(1) or N(3) | Pd(1)–Cl(1) | Pd(1)–Cl(1) | Pd(1)–N(3) | Pd(1)–N(3) |
| Pd(1)–C(1) | 2.13(2) | 2.018(9) | 2.032(3), Pd(1)–C(26) | 2.037(4), Pd(1)–C(24) |
| Selected Bond Angles (deg) | ||||
| C(3)–C(2)–C(6) | 108.6(9) | 115.7(5) | 114.7(3) | 122.5(3) |
| C(2)–C(1)–C(5) | C(2)–C(1)–C(5) | |||
| N(1)–C(3)–C(2) | 126.0(10) | 128.2(5) | 129.6(3) | 128.1(3) |
| N(1)–C(2)–C(1) | N(1)–C(2)–C(1) | |||
| N(2)–C(6)–C(2) | 126.2(10) | 128.0(4) | 132.1(3) | 130.3(3) |
| N(2)−C(5)−C(1) | N(2)−C(5)−C(1) | |||
| N(1)–Pd(1)–N(2) | 86.9(4) | 87.48(16) | 87.81(9) | 90.06(12) |
| X–Pd(1)–C(1) | 92.2(6) | 86.5(3) | 86.28(11) | 88.18(14) |
| X = Cl(1) or N(3) | Cl(1)–Pd(1)–C(1) | Cl(1)–Pd(1)–C(1) | N(3)–Pd(1)–C(26) | N(3)–Pd(1)–C(24) |
| N(1)–Pd(1)–X | 91.9(3) | 91.66(13) | 91.95(9) | 90.89(12) |
| X = Cl(1) or N(3) | N(1)–Pd(1)–Cl(1) | N(1)–Pd(1)–Cl(1) | N(1)–Pd(1)–N(3) | N(1)–Pd(1)– N(3) |
| N(2)–Pd(1)–C(1) | 89.1(6) | 94.4(3) | 93.88(12) | 90.89(14) |
| N(2)–Pd(1)–C(26) | N(2)–Pd(1)–C(24) | |||
| Dihedral Angles (deg) | ||||
| Ph1–N2PdCX | 89.308a | 82.621 | 77.612 | 82.004 |
| (X = Cl or N) | Ph1–N2PdCCl | Ph1–N2PdCCl | Ph1–N2PdCN | Ph1–N2PdCN |
| Ph2–N2PdCX | 91.958a | 81.874 | 90.714 | 96.131 |
| (X = Cl or N) | Ph2–N2PdCCl | Ph2–N2PdCCl | Ph2–N2PdCN | Ph2–N2PdCN |
Mean values of two structures.
Synthesis and Characterization of Cationic [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d)
The corresponding cationic [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d) could be prepared by treating 3a–d, prepared in situ according to the procedure described above, with AgPF6 and 2,6-lutidine (Scheme 4). The resulting complexes (4a–d) were identified by NMR spectroscopy and elemental analysis; the structure of complex 4a was determined by X-ray crystallography (Figure 5, Table 3). In the 13C NMR spectra, resonances ascribed to the methyl groups bound to palladium appear at lower magnetic fields than those of the corresponding neutral (BOX)Pd(Me)Cl (3) (4a: −4.16 ppm, 4b: −3.65 ppm, 4c: −3.72 ppm, 4d: −3.07 ppm, 3a: −5.85 ppm, 3b: −5.30 ppm, 3c: −5.12 ppm, 3d: −4.26 ppm in CD2Cl2 at 25 °C). These spectral data suggest low electron density at the palladium center in complex 4.
Scheme 4. Synthesis of [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d).

Figure 5.

Oak ridge thermal ellipsoid plot (ORTEP) drawings for [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a). Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity.
In complex 4a, a square planar geometry is folded around palladium, consisting of three nitrogen atoms in the BOX ligand and 2,6-lutidine and a methyl ligand (sum of the bond angles of N(1)–Pd(1)–N(2), N(3)–Pd(1)–C(26), N(1)–Pd(1)–N(3), and N(2)–Pd(1)–C(26) in 4a: 359.92°; mean deviation from the plane in 4a: 0.0339 Å). Pd(1)–N(3) [2.043(3) Å] is relatively short, which implies that the 2,6-lutidine ligand strongly coordinates to Pd. The dihedral angle of Ph1–N2PdCN close to the less bulky methyl ligand is much smaller than that of Ph2–N2PdCN close to the bulky 2,6-lutidine (Ph1–N2PdCN: 77.612°, Ph2–N2PdCN: 90.714°).
Synthesis and Characterization of Neutral (BOX)PdMe(2,6-Me2C5H3N) (5b–d)
Interestingly, it was found that a treatment of the BOX ligands (1b–d) with KH in tetrahydrofuran (THF) afforded the potassium salts of the anionic BOX ligands (1b–d-K) by abstraction of the hydrogen attached to the bridgehead carbon (Scheme 5). Pd(Me)Cl(cod) was then treated with the obtained K-salt (1b–d-K) in the presence of 2,6-lutidine to afford novel neutral palladium complexes with anionic BOX ligands (5b–d); these complexes were identified by NMR spectroscopy and elemental analysis. In the 13C NMR spectra, resonances ascribed to the methyl groups bound to palladium in 5a–d appear at higher magnetic fields than those in the corresponding cationic [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4) and neutral (BOX)Pd(Me)Cl (3) (5b: −5.74 ppm, 5c: −5.93 ppm, 5d: −5.67 ppm in C6D6 at 25 °C, 4b: −3.65 ppm, 4c: −3.72 ppm, 4d: −3.07 ppm in CD2Cl2 at 25 °C, 3b: −5.30 ppm, 3c: −5.12 ppm, 3d: −4.26 ppm in CD2Cl2 at 25 °C). Moreover, the methylene proton signal of 5c (−0.39 ppm) in the 1H NMR spectrum (in CDCl3 at 25 °C) is observed in a higher magnetic field than that of complexes 4c and 3c (0.17 and 0.25 ppm, respectively). These spectral properties of complex 5 compared to those of the other complexes (3, 4) suggest that the palladium in 5 possesses higher electron density because of electron donation from the anionic BOX ligand.
Scheme 5. Synthesis of (BOX)PdMe(2,6-Me2C5H3N) (5b–d).

Single crystals of 5b suitable for X-ray crystallography were grown by slow diffusion of Et2O into dichloromethane solution. The ORTEP drawing is shown in Figure 6, and the selected bond distances and angles are summarized in Table 3 (together with 3a, 3c, and 4a for comparison). Complex 5b also folds in a distorted square planar geometry around palladium with BOX, 2,6-lutidine, and methyl ligands (sum of the bond angles of N(1)–Pd(1)–N(2), N(3)–Pd(1)–C(24), N(1)–Pd(1)–N(3), and N(2)–Pd(1)–C(24): 360.02°; mean deviation from the plane: 0.0250 Å). The bond lengths of C(1)–C(2), C(1)–C(5), N(1)–C(2), and N(2)–C(5) are between those of single and double bonds [C(1)–C(2): 1.402(5) Å, C(1)–C(5): 1.392(5) Å, N(1)–C(2): 1.316(5) Å, N(2)–C(5): 1.316(5) Å] in contrast to those in BOX (1) and complexes 2–4 [C(1)–C(2) or C(2)–C(3): 1.481(5)–1.519(7) Å, C(1)–C(5) or C(2)–C(6): 1.488(5)–1.522(6) Å, N(1)–C(2) or N(1)–C(3): 1.270(7)–1.292(14) Å, N(2)–C(5) or N(2)–C(6): 1.265(7)–1.297(14) Å] (Tables 1–3). This structural feature suggests a resonance hybrid of the two local structures, A and B, in complex 5b (Figure 6). The high planarity of the plane consisting of Pd(1), C(1), C(2), C(5), N(1), and N(2) (mean deviation from the plane: 0.0208 Å) also indicates a resonance hybrid structure. The Pd–N bond lengths are shorter than those in the corresponding methyl palladium complexes 3 and 4, indicating strong coordination of the BOX ligand to the metal in 5 [Pd(1)–N(1), Pd(1)–N(2) in 5b: 2.108(3) and 2.036(3) Å, 3a: 2.110(9) and 2.090(10) Å, 3c: 2.114(4) and 2.070(4) Å, 4a: 2.134(3) and 2.060(3) Å, respectively]. The large C(2)−C(1)−C(5) bond angle in 5b [122.5(3)°] is derived from the sp2 hybridization of the bridgehead carbon. In turn, the large bite angle of N(1)–Pd(1)–N(2) [90.06(12)°] is derived from the large bridge angle of C(2)−C(1)−C(5) [122.5(3)°]. The dihedral angle of Ph1–N2PdCN is much smaller than that of Ph2–N2PdCN, similar to that in complex 4a (Ph1–N2PdCN, Ph2–N2PdCN in 5b: 82.004 and 96.131°, 4a: 77.612 and 90.714°, respectively). However, the dihedral angles in 5b are larger than those in complex 4a because of strong coordination of the BOX ligand to Pd in 5b [Pd(1)–N(1), Pd(1)–N(2) in 5b: 2.108(3), 2.036(3) Å, 4a: 2.134(3), 2.060(3) Å, respectively]. Azabis(oxazoline) ligands with electron-donating amino groups in the central bridge are also known to coordinate strongly to metals, resulting in greatly improved stability of the metal complexes;79,80 this results in higher reactivity, selectivity, and reusability not only for homogeneous catalytic reactions but also for heterogeneous reactions.41,79−83 Both the spectral and structural features of complex 5 suggest higher stability as well as higher electron density at the palladium center derived from the anionic BOX ligand, similar to that in azabis(oxazoline) metal complexes. Thus, a series of complexes 5 are expected to be applied as new catalytic intermediates, in contrast to complexes 2–4.
Figure 6.

Left: Oak ridge thermal ellipsoid plot (ORTEP) drawings for (BOX)PdMe(2,6-Me2C5H3N) (5b). Thermal ellipsoids are drawn at the 50% probability level, and H atoms are omitted for clarity. Right: Local structures of A and B.
Conclusions
As summarized in Chart 2, a series of neutral palladium complexes containing C2-symmetric BOX ligands, (BOX)PdCl2 (2a–d) and (BOX)Pd(Me)Cl (3a–d), have been synthesized by reacting PdCl2(CH3CN)2 or Pd(Me)Cl(cod) with BOX ligands (1a–d) [2,2′-(2-propylidene)bis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline} (1a), 2,2′-methylenebis{(4R)-4-phenyl-5,5-dimethyl-2-oxazoline} (1b), 2,2′-methylenebis{(4R)-4,5,5-triphenyl-2-oxazoline} (1c), and 2,2′-methylenebis{(4R,5S)-4,5-diphenyl-2-oxazoline} (1d)]; the cationic methyl complexes, [(BOX)PdMe(2,6-Me2C5H3N)]+PF6– (4a–d), have been synthesized by in situ preparation of 3a–d, followed by addition of AgPF6. Moreover, novel neutral palladium–methyl complexes containing anionic ligands (5b–d) have also been prepared by treatment of Pd(Me)Cl(cod) with the ligand potassium salt prepared by hydrogen abstraction of the methylene bridgehead. The X-ray crystal structures reveal that all complexes fold in a distorted square planar geometry around Pd; steric bulk in the substituents at the 5-position in the BOX ligand decreases the dihedral angles between the Ph ring at the 4-position and the N2Pd plane. The bite angle in 2a (with geminal dimethyl groups at the bridgehead carbon in the BOX ligand) is smaller than that in 2b–d (with no substituents at the bridgehead carbon). Cationic [(BOX)PdMe(NC6H3Me2)]+PF6– (4) reveals lower electron density at the palladium center compared to that in the corresponding neutral (BOX)Pd(Me)Cl (3); this was supported by the 13C NMR spectra. Neutral (BOX)PdMe(2,6-Me2C5H3N) (5) also demonstrates higher electron density because of electron donation from the anionic BOX ligand. The high planarity of the plane constructed with Pd(1), C(1), C(2), C(5), N(1), and N(2), the large bridge and bite angles of C(2)−C(1)−C(5) and N(1)–Pd(1)–N(2), and the shorter Pd–N bond lengths indicate a resonance hybrid structure in the BOX ligand. These electronic and steric features are different from those of complexes 2–4; however, they are similar to those of azabis(oxazoline) metal complexes. Because these palladium complexes are highly promising as precatalysts for efficient organic transformations (C–C bond formation, etc.), the information introduced here may be important for the design of more efficient catalysts and may also increase the general understanding of catalysis.
Chart 2. New Palladium(II) Complexes Containing Neutral or Anionic BOX Ligands.

Experimental Section
General Procedures
All experimental procedures were carried out under an atmosphere of dry nitrogen using standard Schlenk techniques or using a Vacuum Atmospheres drybox unless otherwise specified. All chemicals used were of reagent grades and were purified by standard purification procedures. Diethyl ether (anhydrous grade, Kanto Chemical Co., Inc.), dichloromethane (anhydrous grade, Kanto Chemical Co., Inc.), xylene (anhydrous grade, Kanto Chemical Co., Inc.), and THF (anhydrous grade, Kanto Chemical Co., Inc.) were used as received. (R)-Methyl 2-amino-2-phenylacetate hydrochloride (Sigma-Aldrich Co.), 2,2-dimethylmalonyl chloride (Sigma-Aldrich Co.), dimethylmalonate (Wako Pure Chemical Industries Ltd.), Ti(OiPr)4 (Wako Pure Chemical Industries Ltd.), 2,2′-methylenebis[(4R,5S)-4,5-diphenyl-2-oxazoline] (1d) (Sigma-Aldrich Co.), PdCl2(CH3CN)2 (Sigma-Aldrich Co.), 2,6-lutidine (Tokyo Chemical Industry Co.), and AgPF6 (Sigma-Aldrich Co.) were used as received without further purification. KH (30 wt % dispersion in mineral oil, Sigma-Aldrich Co.) was washed three times with dry hexanes and dried before the preparation of potassium salt of anionic BOX ligands (1-K). (Cycloocta-1,5-diene)chloromethylpalladium(II), Pd(Me)Cl(cod), was prepared according to the literature procedure.84
Elemental analyses were performed by using an EAI CE-440 CHN/O/S Elemental Analyzer (Exeter Analytical, Inc.). All 1H and 13C NMR spectra were recorded on a Bruker AV500 spectrometer (500.13 MHz for 1H NMR, 125.77 MHz for 13C NMR) or a JEOL ECS-400 spectrometer (399.8 MHz for 1H NMR, 100.5 MHz for 13C NMR). All spectra were obtained in the solvent indicated at 25 °C unless otherwise noted. Chemical shifts are given in parts per million (ppm) and are referenced to SiMe4 (δ 0.00 ppm, 1H NMR, 13C NMR). Coupling constants are given in hertz (Hz).
Synthesis of (R)-1-Amino-2-methyl-1-phenylpropan-2-ol (6a)
Methylmagnesium bromide (3.0 M in diethyl ether, 100 mL, 0.300 mol) was added slowly to a suspension of (R)-methyl 2-amino-2-phenylacetate hydrochloride (12.2 g, 60.5 mmol) in diethyl ether (200 mL) at −40 °C. After addition, the reaction mixture was stirred at −40 °C for 1 h and then allowed to reach at room temperature with vigorous stirring overnight. Saturated aqueous NH4Cl was added dropwise at 0 °C until the precipitate was dissolved. The aqueous phase was separated from the organic phase and extracted with ethyl acetate (3 × 150 mL). The combined organic extract was washed with water (2 × 100 mL) and dried over Na2SO4. The organic solution was filtered and concentrated under reduced pressure to give yellow oil. Recrystallization of the oil from AcOEt–n-hexane gave a pale yellow solid (9.2 g, 92%). 1H NMR (500.1 MHz, CDCl3): δ 7.34–7.28 (m, 5H, Ph), 3.81 (s, 1H, CH), 2.02 (brs, 2H, NH, OH), 1.22 (s, 3H, CH3), 1.00 (s, 3H, CH3). The spectral data were identical to the same compound.85
Synthesis of (R)-2-Amino-1,1,2-triphenylethanol (6c)
(R)-Methyl 2-amino-2-phenylacetate hydrochloride (6.0 g, 30.0 mmol) was added in small portions to methylmagnesium bromide solution in diethyl ether (3.0 M, 100 mL, 0.300 mol) at 0 °C for 30 min. After addition, the reaction mixture was stirred at 0 °C for 30 min and then allowed to reach room temperature with vigorous stirring overnight. The reaction mixture was poured slowly into the iced water (120 mL), and concentrated HCl (45 mL) was added dropwise. After stirring for 1 h, the generated precipitate was collected by filtration and the remaining yellow solid was washed with water three times. The solid was dissolved in aqueous NaOH (2.0 M, 110 mL); then, the solution was stirred for 30 min. The aqueous phase was extracted with diethyl ether (3 × 150 mL), and the combined organic extract was washed with brine (100 mL) and dried over Na2SO4. The organic solution was filtered and concentrated under reduced pressure to give a yellow solid. Recrystallization of the oil from CH2Cl2–n-hexane gave a pale yellow solid (7.7 g, 89%). 1H NMR (500.1 MHz, CDCl3): δ 7.75 (d, J = 7.55 Hz, 2H, o-Ph), 7.41 (dd, J = 7.55, 7.25 Hz, 2H, m-Ph), 7.28 (t, J = 7.25 Hz, 1H, p-Ph), 7.15–7.09 (m, 7H, Ph), 7.06–6.98 (m, 3H, Ph), 5.01 (s, 1H, CH), 4.66 (brs, 1H, OH), 1.59 (brs, 2H, NH2). The spectral data were identical to the same compound.86
Synthesis of N1,N3-bis((R)-2-hydroxy-2-methyl-1-phenylpropyl)-2,2-dimethylmalonamide (7a)
A solution of amino alcohol 6a (1.50 g, 7.53 mmol) and Et3N (1.25 mL, 8.96 mmol) in CH2Cl2 (anhydrous, 13 mL) was cooled to −10 °C. 2,2-Dimethylmalonyl chloride (0.65 g, 3.77 mmol) was then added dropwise over 3 min. The reaction mixture was allowed to warm to 20 °C and stirred for 6 h. Subsequently, aqueous HCl (1 N, 15 mL) was added in one portion. The organic phase was separated, washed with aqueous NaHCO3 (5%, 20 mL), washed with H2O (5 mL), dried over Na2SO4, and concentrated under reduced pressure to give a white solid 7a, which was used in the next step without further purification (1.64 g, quant). 1H NMR (270.1 MHz, DMSO-d6): δ 7.73 (d, J = 8.56 Hz, 2H, NH), 7.21 (brs, 10H, Ph), 4.76 (brs, 2H, OH), 4.64 (d, J = 8.75 Hz, 2H, CH), 1.34 (s, 6H, CH3), 1.14 (s, 6H, CH3), 0.89 (s, 6H, CH3). 13C NMR (100.5 MHz, DMSO-d6): δ 171.97 (CO), 140.48 (i-Ph), 128.38 (Ph), 127.35 (Ph), 126.56 (p-Ph), 70.69 (qC), 61.12 (CH), 48.99 (qC), 27.89 (CH3), 27.50 (CH3), 23.72 (CH3).
Synthesis of 2,2′-(2-Propylidene)bis[(4R)-4-phenyl-5,5-dimethyl-2-oxazoline] (1a)
7a (0.859 g, 2.01 mmol) and xylene (anhydrous, 50 mL) were charged into a 2-necked flask equipped with a Dean–Stark apparatus; then, the reaction mixture was heated to reflux to dissolve the bisamide alcohol completely. Ti(OiPr)4 (57.1 mg, 0.201 mmol) was then added to the solution in one portion. The reaction mixture was refluxed for 48 h with removal of the water byproduct. After the reaction mixture was cooled to room temperature, the solution was concentrated under reduced pressure. The resulting pale yellow oil was purified by column chromatography (neutral almina, hexane/AcOEt = 10:1 to 2:1) to give a white solid 1a (0.579 g, 74% yield 2 step based on 6a). 1H NMR (500.1 MHz, CDCl3): δ 7.31–7.20 (m, 10H, Ph), 4.85 (s, 2H, CH), 1.68 (s, 6H, CH3), 1.56 (s, 6H, CH3), 0.86 (s, 6H, CH3). 13C NMR (125.8 MHz, CDCl3): δ 168.88 (N=C), 138.63 (i-Ph), 127.75 (Ph), 127.07 (Ph), 126.96 (Ph), 87.14 (qC), 77.60 (CH), 38.58 (qC), 28.71 (CH3), 23.65 (CH3), 23.35 (CH3). Anal. Calcd for C25H30N2O2: C, 76.89; H, 7.74; N, 7.17. Found: C, 76.69; H, 7.86; N, 7.06.
Synthesis of 2,2′-Methylenebis[(4R)-4-phenyl-5,5-dimethyl-2-oxazoline] (1b)
Dimethylmalonate (0.245 g, 1.86 mmol), amino alcohol 6a (0.652 g, 3.95 mmol), and xylene (anhydrous, 60 mL) were charged into a 2-necked flask equipped with a Dean–Stark apparatus; then, the reaction mixture was heated to reflux for 13 h. After cooling the reaction mixture at room temperature, Ti(OiPr)4 (113 mg, 0.398 mmol) was then added to the solution in one portion. The reaction mixture was refluxed for 43 h with removal of the water byproduct. After the reaction, the mixture was concentrated under reduced pressure. The resulting red-yellow oil was purified by column chromatography (neutral alumina, hexane/AcOEt = 9:1 to 1:1) to give pale yellow oil. Recrystallization from dichloromethane and hexane gave a white solid 1b (0.414 g, 61% yield 2 step based on dimethylmalonate). Single crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into dichloromethane solution. 1H NMR (500.1 MHz, CDCl3): δ 7.33–7.24 (m, 10H, Ph), 4.90 (s, 2H, CH), 3.53 (s, 2H, CH2), 1.60 (s, 6H, CH3), 0.88 (s, 6H, CH3). 13C NMR (125.8 MHz, CDCl3): δ 162.21 (N=C), 138.69 (i-Ph), 128.12 (Ph), 127.43 (Ph), 127.20 (Ph), 88.06 (qC), 78.14 (CH), 29.64 (CH2), 29.07 (CH3), 23.79 (CH3). The spectral data were identical to the same compound.77
Synthesis of 2,2′-Methylenebis[(4R)-4,5,5-triphenyl-2-oxazoline] (1c)
The synthetic procedure for 1c is similar to that for 1b, except that dimethylmalonate (0.315 g, 2.38 mmol), amino alcohol 6c (1.38 g, 4.77 mmol) in place of 6a, xylene (anhydrous, 50 mL), and Ti(OiPr)4 (68.0 mg, 0.239 mmol) were used. Recrystallization from dichloromethane and hexane gave a white solid 1c (0.945 g, 68% yield 2 step based on dimethylmalonate). Single crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into dichloromethane solution. 1H NMR (500.1 MHz, CDCl3): δ 7.68 (d, J = 7.25 Hz, 4H, o-Ph), 7.40 (dd, J = 7.25, 6.95 Hz, 4H, m-Ph), 7.34 (t, J = 6.95 Hz, 2H, p-Ph), 7.12–7.10 (m, 4H, Ph), 7.04–6.88 (m, 16H, Ph), 6.05 (s, 2H, CH), 3.91 (s, 2H, CH2). 13C NMR (125.8 MHz, CDCl3): δ 161.34 (N=C), 144.33 (i-Ph), 139.91 (i-Ph), 138.18 (i-Ph), 128.44 (Ph), 128.32 (Ph), 128.09 (Ph), 127.59 (Ph), 127.11 (Ph), 127.07 (Ph), 126.81 (Ph), 126.57 (Ph), 126.43 (Ph), 95.23 (qC), 79.38 (CH), 29.54 (CH2). Anal. Calcd for C43H34N2O2: C, 84.56; H, 5.61; N, 4.59. Found: C, 84.26; H, 5.66; N, 4.56.
Synthesis of (BOX)PdCl2 (2a)
PdCl2(CH3CN)2 (40.1 mg, 0.155 mmol) and BOX (1a) (64.8 mg, 0.166 mmol) were dissolved with 2 mL of anhydrous CH2Cl2 at room temperature in a glove box; then, the reaction mixture was stirred at room temperature for 19 h. The mixture was filtered through a pad of celite, and the filtrate was concentrated to 0.5 mL. n-Hexane (ca. 10 mL) was added dropwise to the concentrated solution with stirring. The resultant orange solid was collected by filtration and washed with n-hexane three times. The solid was dried in vacuo to afford an orange powder 2a (81.8 mg, 93%). Single crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into dichloromethane solution. 1H NMR (500.1 MHz, CDCl3): δ 7.35 (dd, J = 7.25, 6.85, 6H, m-Ph), 7.31 (d, J = 7.25 Hz, 2H, p-Ph), 6.99 (d, J = 6.85 Hz, 4H, o-Ph), 5.47 (s, 2H, CH), 1.90 (s, 6H, CH3), 1.56 (s, 6H, CH3), 1.00 (s, 6H, CH3). 13C NMR (125.8 MHz, CDCl3): δ 170.94 (N=C), 136.01 (i-Ph), 128.63 (Ph), 128.27 (Ph), 127.08 (Ph), 91.45 (qC), 76.60 (CH), 40.54 (qC), 27.93 (CH3), 25.21 (CH3), 23.03 (CH3). Anal. Calcd for C25H30Cl2N2O2Pd·(H2O)0.3: C, 52.37; H, 5.39; N, 4.89%. Found: C, 52.35; H, 5.30; N, 4.78%.
Synthesis of (BOX)PdCl2 (2b)
Synthesis of 2b was conducted by a similar procedure as that for 2a, except that BOX 1b (60.2 mg, 0.166 mmol) was used in place of BOX 1a. A pale yellow powder 2b was obtained (76.7 mg, 92%). 1H NMR (500.1 MHz, CDCl3): δ 7.40 (dd, J = 7.25, 7.25 Hz, 4H, m-Ph), 7.31 (t, J = 7.25 Hz, 2H, p-Ph), 7.05 (d, J = 7.25 Hz, 4H, o-Ph), 5.43 (s, 2H, CH), 3.93 (s, 2H, CH2), 1.57 (s, 6H, CH3), 0.98 (s, 6H, CH3). 13C NMR (125.8 MHz, CDCl3): δ 164.41 (N=C), 136.12 (i-Ph), 128.61 (Ph), 128.22 (Ph), 127.29 (Ph), 92.37 (qC), 76.60 (CH), 28.55 (CH2), 28.41 (CH3), 23.33 (CH3). Anal. Calcd for C23H26Cl2N2O2Pd: C, 51.18; H, 4.85; N, 5.19%. Found: C, 51.47; H, 5.06; N, 4.99%.
Synthesis of (BOX)PdCl2 (2c)
Synthesis of 2c was conducted by a similar procedure as that for 2a, except that BOX 1c (101.2 mg, 0.166 mmol) was used in place of BOX 1a. A pale yellow powder 2c was obtained (114.6 mg, 94%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.63–7.60 (m, 4H, Ph), 7.52–7.50 (m, 6H, Ph), 7.11 (t, J = 7.25 Hz, 2H, p-Ph), 7.06 (dd, J = 7.25, 7.25 Hz, 4H, m-Ph), 7.03–7.00 (m, 6H, Ph), 6.89–6.86 (m, 4H, Ph), 6.76 (d, J = 7.25 Hz, 4H, o-Ph), 6.56 (s, 2H, CH), 4.16 (s, 2H, CH2). 13C NMR (125.8 MHz, CD2Cl2): δ 164.25 (N=C), 141.13 (Ph), 137.64 (Ph), 135.36 (Ph), 130.03 (Ph), 129.39 (Ph), 128.42 (Ph), 128.32 (Ph), 128.27 (Ph), 128.01 (Ph), 127.96 (Ph), 127.59 (Ph), 126.78 (Ph), 99.60 (qC), 75.01 (CH), 28.98 (CH2). Anal. Calcd for C43H34Cl2N2O2Pd·(H2O): C, 64.07; H, 4.50; N, 3.48. Found: C, 63.91; H, 4.35; N, 3.43.
Synthesis of (BOX)PdCl2 (2d)
Synthesis of 2d was conducted by a similar procedure as that for 2a, except that BOX 1d (155.3 mg, 0.339 mmol) was used in place of BOX 1a. A pale yellow powder 2d was obtained (199.9 mg, 92%). 1H NMR (500.1 MHz, CDCl3): δ 7.16–7.10 (m, 12H, Ph), 6.90–6.86 (m, 8H, Ph), 6.15 (d, J = 8.55 Hz, 2H, CH), 6.08 (d, J = 8.55 Hz 2H, CH), 4.31 (s, 2H, CH2). 13C NMR (125.8 MHz, CDCl3): δ 165.75 (N=C), 134.35 (Ph), 132.09 (Ph), 128.70 (Ph), 128.24 (Ph), 128.14 (Ph), 128.03 (Ph), 127.75 (Ph), 126.54 (Ph), 88.82 (CH), 72.81 (CH), 28.63 (CH2). Anal. Calcd for C31H26Cl2N2O2Pd: C, 58.55; H, 4.12; N, 4.41. Found: C, 58.46; H, 4.00; N, 4.53.
Synthesis of (BOX)Pd(Me)Cl (3a)
Pd(Me)Cl(cod) (46.0 mg, 0.174 mmol) and BOX (1a) (70.6 mg, 0.181 mmol) were dissolved with 2 mL of anhydrous dichloromethane under a nitrogen atmosphere; then, the reaction mixture was stirred for 2 h. The mixture was filtered through a pad of celite, and the filtrate was concentrated to 0.5 mL. The concentrated solution was added dropwise to n-hexane (ca. 10 mL) with stirring. The resultant pale yellow solid was collected by filtration and washed with n-hexane three times. The solid was dried in vacuo to afford a pale yellow powder 3a (82.0 mg, 86%). Single crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into dichloromethane solution. 1H NMR (500.1 MHz, CD2Cl2): δ 7.42–7.30 (m, 6H, Ph), 7.06–7.02 (m, 4H, o-Ph), 5.32 (s, 1H, CH), 4.89 (s, 1H, CH), 1.98 (s, 3H, CH3), 1.75 (s, 3H, CH3), 1.57 (s, 3H, CH3), 1.55 (s, 3H, CH3), 0.96 (s, 3H, CH3), 0.95 (s, 3H, CH3), 0.07 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 172.87 (N=C), 169.42 (N=C), 138.12 (Ph), 137.16 (Ph), 129.01 (Ph), 128.61 (Ph), 128.55 (Ph), 128.06 (Ph), 127.81 (Ph), 90.29 (qC), 89.69 (qC), 78.41 (CH), 75.79 (CH), 40.52 (qC), 28.59 (CH3), 28.54 (CH3), 26.88 (CH3), 24.04 (CH3), 23.47 (CH3), 23.148 (CH3), −5.85 (CH3). Anal. Calcd for C26H33ClN2O2Pd·(H2O)0.5: C, 56.12; H, 6.16; N, 5.03. Found: C, 56.34; H, 6.10; N, 5.03.
Synthesis of (BOX)Pd(Me)Cl (3b)
Synthesis of 3b was conducted by a similar procedure as that for 3a, except that BOX 1b (63.8 mg, 0.176 mmol) was used in place of BOX 1a. A pale yellow powder 3b was obtained (75.5 mg, 91%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.43–7.31 (m, 6H, Ph), 7.11–7.06 (m, 4H, Ph), 5.27 (s, 1H, CH), 4.89 (s, 1H, CH), 3.85 (d, J = 20.50, 1H, CH2), 3.73 (d, J = 20.50, 1H, CH2), 1.60 (s, 3H, CH3), 1.58 (s, 3H, CH3), 0.95 (s, 3H, CH3), 0.93 (s, 3H, CH3), 0.10 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 165.87 (N=C), 162.29 (N=C), 138.12 (Ph), 136.91 (Ph), 129.00 (Ph), 128.71 (Ph), 128.53 (Ph), 128.07 (Ph), 127.81 (Ph), 91.18 (qC), 90.38 (qC), 78.07 (CH), 76.01 (CH), 29.04 (CH2), 28.84 (CH3), 28.37 (CH3), 23.78 (CH3), 23.38 (CH3), −5.30 (CH3). Anal. Calcd for C24H29ClN2O2Pd: C, 55.50; H, 5.63; N, 5.39. Found: C, 55.41; H, 5.64; N, 5.34.
Synthesis of (BOX)Pd(Me)Cl (3c)
Synthesis of 3c was conducted by a similar procedure as that for 3a, except that BOX 1c (107.5 mg, 0.176 mmol) was used in place of BOX 1a. A pale yellow powder 3c was obtained (111.0 mg, 90%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.65–7.61 (m, 4H, Ph), 7.52–7.43 (m, 6H, Ph), 7.08–6.97 (m, 12H, Ph), 6.94–6.92 (m, 2H, Ph), 6.94–6.88 (m, 2H, Ph), 6.84–6.82 (m, 2H, Ph), 6.74 (d, J = 7.55 Hz, 2H, o-Ph), 6.47 (s, 1H, CH), 5.95 (s, 1H, CH), 4.10 (d, J = 20.50 Hz, 1H, CH2), 4.00 (d, J = 20.50 Hz, 1H, CH2), 0.25 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 165.44 (N=C), 161.59 (N=C), 142.74 (Ph), 141.99 (Ph), 139.00 (Ph), 137.78 (Ph), 136.86 (Ph), 135.69 (Ph), 129.76 (Ph), 129.38 (Ph), 129.31 (Ph), 129.13 (Ph), 128.79 (Ph), 128.39 (Ph), 128.27 (Ph), 128.17 (Ph), 128.03 (Ph), 127.98 (Ph), 127.88 (Ph), 127.73 (Ph), 127.70 (Ph), 127.41 (Ph), 127.36 (Ph), 127.17 (Ph), 126.94 (Ph), 126.84 (Ph), 98.27 (qC), 97.70 (qC), 77.19 (CH), 74.67 (CH), 28.49 (CH2), −5.12 (CH3). Anal. Calcd for C44H37ClN2O2Pd·(H2O): C, 67.26; H, 5.00; N, 3.57. Found: C, 67.17; H, 4.74; N, 3.47.
Synthesis of (BOX)Pd(Me)Cl (3d)
Synthesis of 3d was conducted by a similar procedure as that for 3a, except that BOX 1d (76.1 mg, 0.166 mmol) was used in place of BOX 1a. A pale yellow powder 3d was obtained (93.1 mg, 95%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.16–7.10 (m, 12H, Ph), 6.99–6.85 (m, 8H, Ph), 6.09 (brd, J = 9.45 Hz, 2H, CH), 6.01 (brd, J = 9.10 Hz, 1H, CH), 5.53 (brd, J = 9.15 Hz, 1H, CH), 4.21 (d, J = 21.61 Hz, 1H, CH2), 4.13 (d, J = 21.61 Hz, 1H, CH2), 0.22 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 167.17 (N=C), 163.49 (N=C), 136.46 (Ph), 135.07 (Ph), 134.27 (Ph), 133.04 (Ph), 128.81 (Ph), 128.50 (Ph), 128.40 (Ph), 128.35 (Ph), 128.32 (Ph), 128.30 (Ph), 128.23 (Ph), 128.05 (Ph), 127.77 (Ph), 127.59 (Ph), 126.86 (Ph), 126.71 (Ph), 88.34 (CH), 87.73 (CH), 73.91 (CH), 72.05 (CH), 28.20 (CH2), −4.26 (CH3). Anal. Calcd for C32H29ClN2O2Pd·(C6H14)0.1: C, 62.74; H, 4.91; N, 4.49. Found: C, 62.98; H, 4.71; N, 4.58.
Synthesis of [(BOX)PdMe(2,6-Me2C5H3N)]+PF6− (4a)
Pd(Me)Cl(cod) (53.7 mg, 0.203 mmol) and BOX (1a) (79.1 mg, 0.203 mmol) were dissolved with 2 mL of anhydrous CH2Cl2 at room temperature in a glove box; then, the reaction solution was stirred for 3 h. 2,6-Lutidine (109.0 mg, 1.02 mmol) and AgPF6 (52.0 mg, 0.206 mmol) were added to the solution. The reaction mixture was stirred for additional 3 h at room temperature and filtered through a pad of celite. The volatilities were removed under reduced pressure, and the resultant solid was dissolved with 2.0 mL of chloroform. The yellow suspension was filtered through a pad of celite, and the filtrate was concentrated to 0.5 mL. n-Hexane (10 mL) was added dropwise to the concentrated solution with stirring. The resultant pale yellow solid was collected by filtration with a Hirsch funnel and washed with n-hexane three times. The solid was dried in vacuo to afford a pale yellow powder 4a (113.7 mg, 76%). Single crystals suitable for X-ray crystallography were grown by slow diffusion of hexane into dichloromethane solution. 1H NMR (500.1 MHz, CD2Cl2): δ 7.50–7.42 (m, 3H, Ph), 7.38–7.29 (m, 1H, Ph), 7.20 (t, J = 7.55 Hz, 1H, Ph), 7.12 (d, J = 7.60 Hz, 2H, o-Ph), 7.06 (d, J = 7.55 Hz, 1H, o-Ph), 6.92 (brs, 1H, lutidine), 6.80 (brs, 1H, lutidine), 6.54 (d, J = 7.60 Hz, 1H, o-Ph), 6.18 (brs, 1H, lutidine), 4.96 (s, 1H, CH), 4.11 (s, 1H, CH), 2.67 (s, 3H, CH3), 2.37 (s, 3H, CH3), 2.19 (s, 3H, CH3), 1.75 (s, 3H, CH3), 1.65 (s, 3H, CH3), 1.58 (s, 3H, CH3), 1.08 (s, 3H, CH3), 0.89 (s, 3H, CH3), 0.02 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 173.34 (N=C), 172.546 (N=C), 159.81 (Ar), 158.72 (Ar), 138.82 (Ar), 136.49 (Ar), 135.11 (Ar), 129.44 (Ar), 129.25 (Ar), 129.22 (Ar), 129.10 (Ar), 128.73 (Ar), 127.64 (Ar), 125.78 (Ar), 123.25 (Ar), 123.11 (Ar), 91.35 (qC), 90.84 (qC), 78.03 (CH), 77.16 (CH), 40.61 (qC), 29.00 (CH3), 28.87 (CH3), 28.68 (CH3), 27.80 (CH3), 27.02 (CH3), 23.63 (CH3), 22.74 (CH3), 22.24 (CH3), −4.16 (CH3). Anal. Calcd for C33H42F6N3O2PPd: C, 51.87; H, 5.54; N, 5.50. Found: C, 51.67; H, 5.54; N, 5.43.
Synthesis of [(BOX)PdMe(2,6-Me2C5H3N)]+PF6− (4b)
Synthesis of 4b was conducted by a similar procedure as that for 4a, except that BOX 1b (43.0 mg, 0.119 mmol) was used in place of BOX 1a. A pale yellow powder 4b was obtained (82.5 mg, 96%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.49–7.41 (m, 3H, Ph), 7.38 (t, J = 7.73 Hz, 1H, Ph), 7.22 (t, J = 7.40 Hz, 1H, Ph), 7.17 (d, J = 6.95 Hz, 2H, o-Ph), 7.05 (d, J = 7.85 Hz, 1H, o-Ph), 6.99 (brs, 3H, Ar), 6.56 (d, J = 7.60 Hz, 1H, o-Ph), 6.17 (brs, 1H, Ar), 4.95 (s, 1H, CH), 4.10 (d, J = 19.85 Hz, 1H, CH2), 4.06 (s, 1H, CH), 3.83 (d, J = 19.85 Hz, 1H, CH2), 2.78 (s, 3H, CH3), 2.34 (s, 3H, CH3), 1.68 (s, 3H, CH3), 1.58 (s, 3H, CH3), 1.04 (s, 3H, CH3), 0.87 (s, 3H, CH3), −0.01 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 166.68 (N=C), 165.26 (N=C), 160.39 (Ar), 158.70 (Ar), 138.78 (Ar), 136.57 (Ar), 135.05 (Ar), 129.19 (Ar), 129.15 (Ar), 129.13 (Ar), 129.07 (Ar), 127.59 (Ar), 123.28 (Ar), 122.98 (Ar), 91.64 (qC), 91.25 (qC), 78.03 (CH), 76.98 (CH), 29.19 (2C, CH2, CH3), 28.61 (CH3), 27.79 (CH3), 27.18 (CH3), 24.00 (CH3), 23.00 (CH3), −3.65 (CH3). Anal. Calcd for C31H38F6N3O2PPd: C, 50.59; H, 5.20; N, 5.71%. Found: C, 50.35; H, 5.13; N, 5.96%.
Synthesis of [(BOX)PdMe(2,6-Me2C5H3N)]+PF6− (4c)
Synthesis of 4c was conducted by a similar procedure as that for 4a, except that BOX 1c (134.6 mg, 0.220 mmol) was used in place of BOX 4a. A pale yellow powder 4c was obtained (198.2 mg, 92%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.64–7.48 (m, 10H, Ph), 7.43 (t, J = 7.58 Hz, 1H, Ph), 7.16–7.11 (m, 2H, Ph), 7.04–6.87 (m, 16H, Ar), 6.73 (d, J = 7.55 Hz, 2H, Ar), 6.57 (d, J = 7.60 Hz, 1H, Ar), 6.37 (brs, 1H, lutidine), 6.04 (s, 1H, CH), 5.16 (s, 1H, CH), 4.41 (d, J = 19.22 Hz, 1H, CH2), 4.31 (d, J = 19.22 Hz, 1H, CH2), 2.57 (s, 3H, CH3), 2.26 (s, 3H, CH3), 0.17 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 166.11 (N=C), 165.14 (N=C), 160.31 (Ar), 158.71 (Ar), 143.05 (Ar), 141.97 (Ar), 139.01 (Ar), 137.91 (Ar), 137.42 (Ar), 135.73 (Ar), 134.28 (Ar), 129.88 (Ar), 129.81 (Ar), 129.60 (Ar), 129.49 (Ar), 128.80 (Ar), 128.70 (Ar), 128.67 (Ar), 128.65 (Ar), 128.60 (Ar), 128.09 (Ar), 128.03 (Ar), 127.79 (Ar), 126.61 (Ar), 126.58 (Ar), 126.38 (Ar), 125.72 (Ar), 123.44 (Ar), 123.17 (Ar), 98.44 (qC), 97.88 (qC), 78.30 (CH), 76.73 (CH), 28.75 (CH2), 27.57 (CH3), 26.96 (CH3), −3.72 (CH3). Anal. Calcd for C51H46F6N3O2PPd·CH2Cl2: C, 58.41; H, 4.52; N, 3.93. Found: C, 58.30; H, 4.46; N, 3.98.
Synthesis of [(BOX)PdMe(2,6-Me2C5H3N)]+PF6− (4d)
Synthesis of 4d was conducted by a similar procedure as that for 4a, except that BOX 1d (83.5 mg, 0.182 mmol) was used in place of BOX 1a. A pale yellow powder 4d was obtained (97.7 mg, 65%). 1H NMR (500.1 MHz, CD2Cl2): δ 7.37 (t, J = 7.55 Hz, 1H, Ph), 7.24–7.09 (m, 8H, Ph), 7.05–6.80 (m, 13H, Ar), 6.50 (d, J = 7.90 Hz, 1H, o-Ph), 6.21 (d, J = 9.75 Hz, 2H, CH), 5.64 (d, J = 9.75 Hz, 1H, CH), 4.79 (d, J = 9.75 Hz, 1H, CH), 4.43 (d, J = 21.16 Hz, 1H, CH2), 4.29 (d, J = 21.16 Hz, 1H, CH2), 2.97 (s, 3H, CH3), 2.24 (s, 3H, CH3), 0.06 (s, 3H, CH3). 13C NMR (125.8 MHz, CD2Cl2): δ 168.05 (N=C), 166.26 (N=C), 160.57 (Ar), 159.25 (Ar), 138.67 (Ar), 135.18 (Ar), 134.14 (Ar), 133.51 (Ar), 133.20 (Ar), 128.90 (Ar), 128.63 (Ar), 128.59 (Ar), 128.48 (Ar), 128.46 (Ar), 128.42 (Ar), 128.27 (Ar), 128.19 (Ar), 127.98 (Ar), 127.56 (Ar), 127.05 (Ar), 126.42 (Ar), 123.18 (Ar), 123.04 (Ar), 87.76 (CH), 87.72 (CH), 73.57 (CH), 72.73 (CH), 28.29 (CH2), 27.65 (CH3), 27.47 (CH3), −3.07 (CH3). Anal. Calcd for C39H38F6N3O2PPd·H2O: C, 55.10; H, 4.74; N, 4.94. Found: C, 54.96; H, 4.59; N, 4.82.
Synthesis of Potassium Salt of Anionic BOX (1b-K)
BOX (1b) (111.0 mg, 0.306 mmol) was dissolved with 2.1 mL of anhydrous THF at room temperature in a glove box, and KH (15.0 mg, 0.374 mmol) was added to the solution. The reaction mixture was heated at reflux temperature using a dryer and then stirred at room temperature for 4.5 h. During the reaction, the mixture was additionally heated at reflux temperature three times. The red solution was filtered through a celite pad, and the filtrate was concentrated to 0.5 mL. n-Hexane (ca. 10 mL) was added dropwise to the concentrated solution with stirring. The resultant yellow solid was collected by filtration with a glass filter and washed with n-hexane three times. The solid was dried in vacuo to afford a yellow powder 1b-K; this was used in the next step without further purification (107.0 mg, 87%). Other potassium salts of anionic BOX (1c-K and 1d-K) were prepared by a similar procedure as that for 1b-K.
Synthesis of Neutral (BOX)PdMe(2,6-Me2C5H3N) (5b)
A THF solution (4.3 mL) containing 2,6-lutidine (143.1 mg, 1.335 mmol) was added to a mixture of Pd(Me)Cl(cod) (71.0 mg, 0.268 mmol) and 1b-K (107.0 mg, 0.267 mmol) at −30 °C in a glove box. The reaction mixture was warmed slowly to room temperature; then, the mixture was stirred for 5 h. The orange suspension was filtered through a pad of celite, and the filtrate was concentrated to 0.5 mL. n-Hexane (ca. 10 mL) was added dropwise to the concentrated solution with stirring. The resultant pale yellow solid was collected by filtration and washed with n-hexane three times. The solid was dried in vacuo to afford a pale yellow powder 5b (133.3 mg, 84%). Single crystals suitable for X-ray crystallography were grown by slow diffusion of Et2O into dichloromethane solution. 1H NMR (500.1 MHz, C6D6): δ 7.85 (brs, 1H, Ar), 7.55 (d, J = 7.60 Hz, 1H, Ar), 7.23 (brs, 4H, Ar), 7.00–6.91 (m, 2H, Ar), 6.71–6.62 (m, 2H, Ar), 6.33 (d, J = 7.55, 1H, Ar), 6.05 (d, J = 7.55, 1H, Ar), 5.66 (d, J = 7.25 Hz, 1H, Ar), 4.90 (s, 1H, CH), 4.86 (s, 1H, CH), 3.41 (s, 1H, CH), 3.00 (s, 3H, CH3), 2.08 (s, 3H, CH3), 1.46 (s, 3H, CH3), 1.32 (s, 3H, CH3), 0.90 (s, 3H, CH3), 0.75 (s, 3H, CH3), 0.12 (s, 3H, CH3). 13C NMR (125.8 MHz, C6D6): δ 169.43 (N=C), 169.22 (N=C), 161.81 (Ar), 159.64 (Ar), 144.22 (Ar), 142.69 (Ar), 136.36 (Ar), 128.72 (Ar), 128.58 (Ar), 127.24 (Ar), 126.89 (Ar), 126.76 (Ar), 122.07 (Ar), 121.34 (Ar), 83.88 (qC), 83.52 (qC), 76.64 (CH), 76.15 (CH), 55.68 (CH), 28.90 (CH3), 28.66 (CH3), 27.10 (CH3), 26.87 (CH3), 24.63 (CH3), 23.82 (CH3), −5.75 (CH3). Anal. Calcd for C31H37N3O2Pd: C, 63.10; H, 6.32; N, 7.12%. Found: C, 62.93; H, 6.27; N, 7.02%.
Synthesis of Neutral (BOX)PdMe(2,6-Me2C5H3N) (5c)
Synthesis of 5c was conducted by a similar procedure as that for 5b, except that 1c-K (131.0 mg, 0.202 mmol) was used in place of 1b-K. A pale yellow powder 5c was obtained (135.6 mg, 83%). 1H NMR (500.1 MHz, C6D6): δ 7.94 (d, J = 7.85 Hz, 2H), 7.74 (d, J = 7.85 Hz, 2H, Ar), 7.74 (d, J = 7.85 Hz, 2H, Ar), 7.23–7.14 (m, 13H, Ar), 7.11 (d, J = 8.20 Hz, 2H, Ar), 7.04 (t, J = 7.25, 1H, Ar), 6.94–6.81 (m, 5H, Ar), 6.75–6.65 (m, 6H, Ar), 6.40 (d, J = 7.60 Hz, 1H, Ar), 6.05 (d, J = 7.55 Hz, 1H, Ar), 6.02 (s, 1H, CH), 5.25 (s, 1H, CH), 4.47 (s, 1H, CH), 2.73 (s, 3H, CH3), 1.94 (s, 3H, CH3), 0.22 (s, 3H, CH3). 13C NMR (125.8 MHz, C6D6): δ 168.96 (N=C), 168.76 (N=C), 161.91 (Ar), 160.10 (Ar), 145.91 (Ar), 145.12 (Ar), 142.67 (Ar), 141.67 (Ar), 141.44 (Ar), 140.85 (Ar), 136.55 (Ar), 128.91 (Ar), 128.54 (Ar), 128.42 (Ar), 128.35 (Ar), 128.16 (Ar), 127.97 (Ar), 127.66 (Ar), 127.58 (Ar), 127.40 (Ar), 127.33 (Ar), 127.25 (Ar), 126.85 (Ar), 126.64 (Ar), 126.48 (Ar), 126.449 (Ar), 122.11 (Ar), 121.33 (Ar), 92.34 (qC), 92.08 (qC), 77.06 (CH), 74.69 (CH), 56.54 (CH), 27.08 (CH3), 26.80 (CH3), −5.93 (CH3). Anal. Calcd for C51H45N3O2Pd·(H2O)0.5: C, 72.29; H, 5.47; N, 4.96. Found: C, 72.46; H, 5.44; N, 4.92.
Synthesis of Neutral (BOX)PdMe(2,6-Me2C5H3N) (5d)
Synthesis of 5d was conducted by a similar procedure as that for 5b, except that 1d-K (93.0 mg, 0.187 mmol) was used in place of 1b-K. A pale yellow powder 5d was obtained (86.7 mg, 68%). 1H NMR (500.1 MHz, C6D6): δ 7.27 (brs, 2H, Ar), 7.08 (d, J = 7.58 Hz, 2H, Ar), 6.97 (t, J = 7.25 Hz, 1H, Ar), 6.93 (d, J = 6.60, 2H, Ar), 6.89–6.72 (m, 13H, Ar), 6.62 (t, J = 7.73 Hz, 1H, Ar), 6.35 (d, J = 7.55, 1H, Ar or CH), 6.01 (d, J = 7.90, 1H, CH), 5.64 (d, J = 7.60, 1H, Ar), 5.54 (d, J = 7.55 Hz, 1H, Ar or CH), 5.36 (d, J = 7.55 Hz, 1H, Ar or CH), 5.28 (s, 1H, CH), 4.06 (d, J = 7.90 Hz, 1H, CH), 3.07 (s, 3H, CH3), 2.15 (s, 3H, CH3), 0.23 (s, 3H, CH3). 13C NMR (125.8 MHz, C6D6): δ 170.65 (N=C), 170.45 (N=C), 161.76 (Ar), 159.71 (Ar), 141.64 (Ar), 140.73 (Ar), 137.04 (Ar), 136.98 (Ar), 136.52 (Ar), 128.36 (Ar), 128.33 (Ar), 128.16 (Ar), 127.97 (Ar), 127.76 (Ar), 127.68 (Ar), 127.65 (Ar), 127.40 (Ar), 127.19 (Ar), 127.07 (Ar), 126.77 (Ar), 122.10 (Ar), 121.38 (Ar), 84.95 (CH), 84.79 (CH), 72.88 (CH), 71.86 (CH), 55.66 (CH), 27.54 (CH3), 26.79 (CH3), −5.67 (CH3). Anal. Calcd for C39H37N3O2Pd: C, 68.27; H, 5.44; N, 6.12. Found: C, 68.27; H, 5.72; N, 5.88.
Crystallographic Analysis
The measurement was made on a Rigaku XtaLAB mini diffractometer using graphite monochromated Mo Kα radiation or a Rigaku XtaLAB P200 diffractometer using multilayer mirror monochromated Mo Kα radiation (XtaLAB mini for BOX 1b–d and complex 2a, 2c, 2d and XtaLAB P200 for complex 2b, 3a, 3c, 4a, 5b). The structures except for 3c were solved by direct methods87,88 expanded using Fourier techniques. Heavy-atom Patterson methods89 expanded using Fourier techniques were conducted for 3c. The nonhydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. All calculations were performed using the CrystalStructure crystallographic software package except for refinement,90−92 which was performed using SHELXL97, SHELXL2013, or SHELXL version 2014/7.93 Selected crystal collection parameters are listed in Tables S1–S3.
Acknowledgments
The present research was partly supported by the advanced research program (Tokyo Metropolitan Government). The authors express their heartfelt thanks to Dr. G. Suzukamo (GS Tech Lab.) and Dr. M. Itagaki (Sumitomo Chemical Co., Ltd.) for discussions and comments, and K.N. and K.T. thank Profs. A. Inagaki, S. Komiya, and Dr. S. Sueki (Tokyo Metropolitan University) for fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00457.
Additional data (NMR spectra) for complex 2–5, BOX 1, amino alcohol 6a, 6c, and bisamide alcohol 7a and selected crystal collection parameters for BOX 1b–d, complex 2a–d, 3a, 3c, 4a, 5b; these data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif (PDF)
CCDC Nos. 1533692, 1533912–1533921 contain the supplementary crystallographic data for this paper (TXT) (XYZ)
The authors declare no competing financial interest.
Supplementary Material
References
- The Organometallic Chemistry of the Transition Metals, 5th ed.; Crabtree R. H.; John Wiley & Sons, Inc.: Hoboken, New Jersey, 2009. [Google Scholar]
- Comprehensive Organometallic Chemistry III; Crabtree R. H., Mingos D. M. P., Eds.; Elsevier Science, 2006. [Google Scholar]
- Synthesis of Organometallic Compounds: A Practical Guide; Komiya S., Ed.; John Wiley & Sons, Inc.: West Sussex, England, 1997. [Google Scholar]
- Organometallics in Synthesis A Manual, 3rd ed.; Schlosser M., Ed.; John Wiley & Sons Ltd.: Hoboken, New Jersey, 2013. [Google Scholar]
- Astruc D.Organometallic Chemistry and Catalysis; Springer-Verlag: Berlin Heidelberg, Germany, 2007. [Google Scholar]
- Steinborn D.Fundamentals of Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Cross-Coupling Reactions: A Practical Guide; Miyaura N., Ed.; Springer: Berlin, Germany, 2002. [Google Scholar]
- Palladium in Organic Synthesis; Tsuji J. Ed.; Springer-Verlag: Berlin Heidelberg, Germany, 2005. [Google Scholar]
- Metal-Catalyzed Cross-Coupling Reactions and More; Meijere A. d., Bräse S., Oestreich M., Eds.; Wiley-VCH, Weinheim, 2014. [Google Scholar]
- Palladium Reagents and Catalysts: New Perspectives for the 21st Century, Tsuji J., Ed.; Wiley, Chichester, 2004. [Google Scholar]
- Seechurn C. C. C. J.; Kitching M. O.; Colacot T. J.; Snieckus V. Palladium-catalyzed cross-coupling: A historical contextual perspective to the 2010 Nobel Prize. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- Corbet J.-P.; Mignani G. Selected patented cross-coupling reaction technologies. Chem. Rev. 2006, 106, 2651–2710. 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- Hassan J.; Sévignon M.; Gozzi C.; Schulz E.; Lemaire M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002, 102, 1359–1470. 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- Knappke C. E. I.; von Wangelin A. J. 35 years of palladium-catalyzed cross-coupling with Grignard reagents: how far have we come?. Chem. Soc. Rev. 2011, 40, 4948–4962. 10.1039/c1cs15137a. [DOI] [PubMed] [Google Scholar]
- Catellani M.; Motti E.; Ca’ N. D.; Ferraccioli R. Recent developments in catalytic aryl coupling reactions. Eur. J. Org. Chem. 2007, 4153–4165. 10.1002/ejoc.200700312. [DOI] [Google Scholar]
- Miura M.; Satoh T.; Hirano K. Development of direct aromatic coupling reactions by transition-metal catalysis. Bull. Chem. Soc. Jpn. 2014, 87, 751–764. 10.1246/bcsj.20140099. [DOI] [Google Scholar]
- Ittel S. D.; Johnson L. K.; Brookhart M. Late-metal catalysts for ethylene homo- and copolymerization. Chem. Rev. 2000, 100, 1169–1204. 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- Mecking S.; Johnson L. K.; Wang L.; Brookhart M. Mechanistic studies of the palladium-catalyzed copolymerization of ethylene and α-olefins with methyl acrylate. J. Am. Chem. Soc. 1998, 120, 888–899. 10.1021/ja964144i. [DOI] [Google Scholar]
- Johnson L. K.; Mecking S.; Brookhart M. Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(II) catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. 10.1021/ja953247i. [DOI] [Google Scholar]
- Guan Z.; Cotts P. M.; McCord E. F.; McLain S. J. Chain Walking: A new strategy to control polymer topology. Science 1999, 283, 2059–2062. 10.1126/science.283.5410.2059. [DOI] [PubMed] [Google Scholar]
- Kochi T.; Noda S.; Yoshimura K.; Nozaki K. Formation of linear copolymers of ethylene and acrylonitrile catalyzed by phosphine sulfonate palladium complexes. J. Am. Chem. Soc. 2007, 129, 8948–8949. 10.1021/ja0725504. [DOI] [PubMed] [Google Scholar]
- Wanga F.; Tanaka R.; Li Q.; Nakayama Y.; Yuana J.; Shiono T. Synthesis and application of α-diimine Ni(II) and Pd(II) complexes with bulky steric groups to polymerization of ethylene and methyl methacrylate. J. Mol. Catal. A: Chem. 2015, 398, 231–240. 10.1016/j.molcata.2014.11.004. [DOI] [Google Scholar]
- Guo L.; Dai S.; Sui X.; Chen C. Palladium and nickel catalyzed chain walking olefin polymerization and copolymerization. ACS Catal. 2016, 6, 428–44. 10.1021/acscatal.5b02426. [DOI] [Google Scholar]
- Na Y.; Wang X.; Lian K.; Zhu Y.; Li W.; Luo Y.; Chen C. Dinuclear α-diimine NiII and PdII complexes that catalyze ethylene polymerization and copolymerization. ChemCatChem 2017, 9, 1062–1066. 10.1002/cctc.201601500. [DOI] [Google Scholar]
- Guironnet D.; Roesle P.; Rünzi T.; Göttker-Schnetmann I.; Mecking S. Insertion polymerization of acrylate. J. Am. Chem. Soc. 2009, 131, 422–423. 10.1021/ja808017n. [DOI] [PubMed] [Google Scholar]
- Ito S.; Munakata K.; Nakamura A.; Nozaki K. Copolymerization of vinyl acetate with ethylene by palladium/alkylphosphine-sulfonate catalysts. J. Am. Chem. Soc. 2009, 131, 14606–14607. 10.1021/ja9050839. [DOI] [PubMed] [Google Scholar]
- Rünzi T.; Fröhlich D.; Mecking S. Direct synthesis of ethylene-acrylic acid copolymers by insertion polymerization. J. Am. Chem. Soc. 2010, 132, 17690–17691. 10.1021/ja109194r. [DOI] [PubMed] [Google Scholar]
- Bouilhac C.; Rünzi T.; Mecking S. Catalytic copolymerization of ethylene with vinyl sulfones. Macromolecules 2010, 43, 3589–3590. 10.1021/ma100237s. [DOI] [Google Scholar]
- Shen Z.; Jordan R. F. Copolymerization of ethylene and vinyl fluoride by (phosphine-bis(arenesulfonate))PdMe(pyridine) catalysts: Insights into inhibition mechanisms. Macromolecules 2010, 43, 8706–8708. 10.1021/ma1017296. [DOI] [Google Scholar]
- Nakano R.; Chung L. W.; Watanabe Y.; Okuno Y.; Okumura Y.; Ito S.; Morokuma K.; Nozaki K. Elucidating the key role of phosphine–sulfonate ligands in palladium-catalyzed ethylene polymerization: Effect of ligand structure on the molecular weight and linearity of polyethylene. ACS Catal. 2016, 6, 6101–6113. 10.1021/acscatal.6b00911. [DOI] [Google Scholar]
- Boffa L. S.; Novak B. M. Copolymerization of polar monomers with olefins using transition-metal complexes. Chem. Rev. 2000, 100, 1479–1494. 10.1021/cr990251u. [DOI] [PubMed] [Google Scholar]
- Chen E. Y. Coordination polymerization of polar vinyl monomers by single-site metal catalysts. Chem. Rev. 2009, 109, 5157–5214. 10.1021/cr9000258. [DOI] [PubMed] [Google Scholar]
- Nakamura A.; Ito S.; Nozaki K. Coordination-insertion copolymerization of fundamental polar monomers. Chem. Rev. 2009, 109, 5215–5244. 10.1021/cr900079r. [DOI] [PubMed] [Google Scholar]
- Brintzinger H. H.; Fischer D.; Mülhaupt R.; Rieger B.; Waymouth R. M. Stereospecific olefin polymerization with chiral metallocene catalysts. Angew. Chem., Int. Ed. Engl. 1995, 34, 1143–1179. 10.1002/anie.199511431. [DOI] [Google Scholar]
- Coates G. W. Precise control of polyolefin stereochemistry using single-site metal catalysts. Chem. Rev. 2000, 100, 1223–1252. 10.1021/cr990286u. [DOI] [PubMed] [Google Scholar]
- Cherian A. E.; Lobkovsky E. B.; Coates G. W. Chiral anilines: development of C2-symmetric, late-transition metal catalysts for isoselective 2-butene polymerization. Chem. Commun. 2003, 2566–2567. 10.1039/b307659h. [DOI] [PubMed] [Google Scholar]
- Cherian A. E.; Rose J. M.; Lobkovsky E. B.; Coates G. W. A C2-Symmetric, living α-diimine Ni(II) catalyst: Regioblock copolymers from propylene. J. Am. Chem. Soc. 2005, 127, 13770–13771. 10.1021/ja0540021. [DOI] [PubMed] [Google Scholar]
- Rose J. M.; Deplace F.; Lynd N. A.; Wang Z.; Hotta A.; Lobkovsky E. B.; Kramer E. J.; Coates G. W. C2-Symmetric Ni(II) α-diimines featuring cumyl-derived ligands: Synthesis of improved elastomeric regioblock polypropylenes. Macromolecules 2008, 41, 9548–9555. 10.1021/ma8019943. [DOI] [Google Scholar]
- Rasappan R.; Laventine D.; Reiser O. Metal-bis(oxazoline) complexes: From coordination chemistry to asymmetric catalysis. Coord. Chem. Rev. 2008, 252, 702–714. 10.1016/j.ccr.2007.11.007. [DOI] [Google Scholar]
- Desimoni G.; Faita G.; Jørgensen K. A. Update 1 of: C2-Symmetric chiral bis(oxazoline) ligands in asymmetric catalysis. Chem. Rev. 2011, 111, PR284–PR437. 10.1021/cr100339a. [DOI] [PubMed] [Google Scholar]
- Rechavi D.; Lemaire M. Enantioselective catalysis using heterogeneous bis(oxazoline) ligands: Which factors influence the enantioselectivity?. Chem. Rev. 2002, 102, 3467–3494. 10.1021/cr020008m. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Stavenger R. A.; Faucher A.-M.; Edwards J. P. Cyclopropanation with diazomethane and bis(oxazoline)palladium(II) complexes. J. Org. Chem. 1997, 62, 3375–3389. 10.1021/jo970044z. [DOI] [PubMed] [Google Scholar]
- Perch N. S.; Kisanga P.; Widenhoefer R. A. Reductive Cyclization of dimethyl diallylmalonate catalyzed by palladium-bisoxazoline complexes in the presence of silane and water. Organometallics 2000, 19, 2541–2545. 10.1021/om000251g. [DOI] [Google Scholar]
- Perch N. S.; Pei T.; Widenhoefer R. A. Enantioselective diene cyclization/hydrosilylation catalyzed by optically active palladium bisoxazoline and pyridine-oxazoline complexes. J. Org. Chem. 2000, 65, 3836–3845. 10.1021/jo0003192. [DOI] [PubMed] [Google Scholar]
- Iwata T.; Miyake Y.; Nishibayashi Y.; Uemura S. Palladium(II) complex-catalysed enantioselective benzoylation of alcohols using carbon monoxide and an organobismuth(V) compound. J. Chem. Soc., Perkin Trans. 1 2002, 1548–1554. 10.1039/b203465d. [DOI] [Google Scholar]
- Pericas M. A.; Puigjaner C.; Riera A.; Vidal-Ferran A.; Gomez M.; Jimenez F.; Muller G.; Rocamora M. Modular bis(oxazoline) ligands for palladium catalyzed allylic alkylation: Unprecedented conformational behaviour of a bis(oxazoline) palladium η3-1,3-diphenylallyl complex. Chem. Eur. J. 2002, 8, 4164–4178. . [DOI] [PubMed] [Google Scholar]
- Bayardon J.; Sinou D. Enantiopure fluorous bis(oxazolines): Synthesis and applications in catalytic asymmetric reactions. J. Org. Chem. 2004, 69, 3121–3128. 10.1021/jo049853q. [DOI] [PubMed] [Google Scholar]
- Aït-Haddou H.; Hoarau O.; Cramailyre D.; Pezet F.; Daran J.-C.; Balavoine G. G. A. new dihydroxy bis(oxazoline) ligands for the palladium-catalyzed asymmetric allylic alkylation: Experimental investigations of the origin of the reversal of the enantioselectivity. Chem. Eur. J. 2004, 10, 699–707. 10.1002/chem.200204649. [DOI] [PubMed] [Google Scholar]
- Kato K.; Matsuba C.; Kusakabe T.; Takayama H.; Yamamura S.; Mochida T.; Akita H.; Peganova T. A.; Vologdinc N. V.; Gusev O. V. 2,20-Isopropylidenebis[(4S,5R)-4,5-di(2-naphthyl)-2-oxazoline] ligand for asymmetric cyclization–carbonylation of meso-2-alkyl-2-propargylcyclohexane-1,3-diols. Tetrahedron 2006, 62, 9988–9999. 10.1016/j.tet.2006.08.004. [DOI] [Google Scholar]
- Yasuhara S.; Sasa M.; Kusakabe T.; Takayama H.; Kimura M.; Mochida T.; Kato K. Cyclization–carbonylation–cyclization coupling reactions of propargyl acetates and amides with palladium(II)–bisoxazoline catalysts. Angew. Chem., Int. Ed. 2011, 50, 3912–3915. 10.1002/anie.201008139. [DOI] [PubMed] [Google Scholar]
- Kusakabe T.; Kawaguchi K.; Kawamura M.; Niimura N.; Shen R.; Takayama H.; Kato K. Cyclization-carbonylation-cyclization coupling reaction of propargyl ureas with palladium(II)-bisoxazoline catalyst. Molecules 2012, 17, 9220–9230. 10.3390/molecules17089220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookhart M.; Wagner M. I.; Balavoine G. G. A.; Haddou H. A. Polymers with main-chain chirality. Synthesis of highly isotactic, optically active poly(4-tert-butylstyrene-alt-CO) using Pd(II) catalysts based on C2-symmetric bisoxazoline ligands. J. Am. Chem. Soc. 1994, 116, 3641–3642. 10.1021/ja00087a077. [DOI] [Google Scholar]
- Brookhart M.; Wagner M. I. Synthesis of a stereoblock polyketone through ancillary ligand exchange. Synthesis of a stereoblock polyketone through ancillary ligand exchange. J. Am. Chem. Soc. 1996, 118, 7219–7220. 10.1021/ja9610141. [DOI] [Google Scholar]
- Binotti B.; Carfagna C.; Gatti G.; Martini D.; Mosca L.; Pettinari C. Mechanistic aspects of isotactic CO/styrene copolymerization catalyzed by oxazoline palladium(II) complexes. Organometallics 2003, 22, 1115–1123. 10.1021/om0208669. [DOI] [Google Scholar]
- Schätz A.; Scarel A.; Zangrando E.; Mosca L.; Carfagna C.; Gissibl A.; Milani B.; Reiser O. High stereocontrol and efficiency in CO/styrene polyketone synthesis promoted by azabis(oxazoline)-palladium complexes. Organometallics 2006, 25, 4065–4068. 10.1021/om060424n. [DOI] [Google Scholar]
- Hara K.; Tayama S.; Kano H.; Masuda T.; Takakusagi S.; Kondo T.; Uosaki K.; Sawamura M. Functionalization of silicon surfaces with catalytically active Pd complexes and application to the aerobic oxidation of benzylic alcohols. Chem. Commun. 2007, 4280–4282. 10.1039/b710820f. [DOI] [PubMed] [Google Scholar]
- Podhajsky S. M.; Iwai Y.; Cook-Sneathen A.; Sigman M. S. Asymmetric palladium-catalyzed hydroarylation of styrenes and dienes. Tetrahedron 2011, 67, 4435–4441. 10.1016/j.tet.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottumukkala A. L.; Matcha K.; Lutz M.; De Vries J. G.; Minnaard A. J. Palladium-catalyzed asymmetric quaternary stereocenter formation. Chem. Eur. J. 2012, 18, 6907–6914. 10.1002/chem.201200694. [DOI] [PubMed] [Google Scholar]
- Buter J.; Moezelaar R.; Minnaard A. J. Enantioselective palladium catalyzed conjugate additions of ortho-substituted arylboronic acids to β,β-disubstituted cyclic enones: total synthesis of herbertenediol, enokipodin A and enokipodin B. Org. Biomol. Chem. 2014, 12, 5883–5890. 10.1039/C4OB01085J. [DOI] [PubMed] [Google Scholar]
- De Crisci A. G.; Chung K.; Oliver A. G.; Solis-Ibarra D.; Waymouth R. M. Chemoselective oxidation of polyols with chiral palladium catalysts. Organometallics 2013, 32, 2257–2266. 10.1021/om4001549. [DOI] [Google Scholar]
- Ibrahim M. B.; Suleiman R.; Fettouhi M.; Ali B. E. A palladium–bisoxazoline supported catalyst for selective synthesis of aryl esters and aryl amides via carbonylative coupling reactions. RSC Adv. 2016, 6, 78826–78837. 10.1039/C6RA15506E. [DOI] [Google Scholar]
- Oliveira C. C.; Pfaltz A.; Correia C. R. D. Quaternary stereogenic centers through enantioselective Heck arylation of acyclic olefins with aryldiazonium salts: Application in a concise synthesis of (R)-verapamil. Angew. Chem., Int. Ed. 2015, 54, 14036–14039. 10.1002/anie.201507927. [DOI] [PubMed] [Google Scholar]
- Best D.; Kujawa S.; Lam H. W. Diastereo- and enantioselective Pd(II)-catalyzed additions of 2-alkylazaarenes to N-Boc imines and nitroalkenes. J. Am. Chem. Soc. 2012, 134, 18193–18196. 10.1021/ja3083494. [DOI] [PubMed] [Google Scholar]
- Motodate S.; Kobayashi T.; Fujii M.; Mochida T.; Kusakabe T.; Katoh S.; Akita H.; Kato K. Synthesis of β-methoxyacrylate natural products based on box-PdII-catalyzed intermolecular methoxycarbonylation of alkynoles. Chem. Asian J. 2010, 5, 2221–2230. 10.1002/asia.201000292. [DOI] [PubMed] [Google Scholar]
- Hirahata W.; Itagaki M.; Nomura K.. Transition metal compounds, catalyst components and catalysts for olefin polymerization, and process for producing olefinic polymer. Eur. Patent EP1998942010, 1998.
- Hirahata W.; Itagaki M.; Nomura K.. Transition metal compound, catalyst component for olefin polymerization, catalyst for olefin polymerization, and process for producing olefinic polymer. U.S. Patent US20010027162A1, 2001.
- Déry M.; Lefebvre L.-P. D.; Aissa K.; Spino C. N-Heteropolycyclic compounds from the formal intramolecular (4+1)-cycloaddition of chromium aminocarbenes. Org. Lett. 2013, 15, 5456–5459. 10.1021/ol4025887. [DOI] [PubMed] [Google Scholar]
- Da C.-S.; Che L.-P.; Guo Q.-P.; Wu F.-C.; Ma X.; Jia Y.-N. 2,4-Dinitrophenol as an effective cocatalyst: Greatly improving the activities and enantioselectivities of primary amine organocatalysts for asymmetric aldol reactions. J. Org. Chem. 2009, 74, 2541–2546. 10.1021/jo802758b. [DOI] [PubMed] [Google Scholar]
- Structural analysis of 2d was also reportedXu H.-W.; Zhang L.-X.; Li J.-X. Chiral bis(oxazoline)palladium(II) complexes with 2,2′-methylenebis(4S,5R)-4,5-diphenyl-2-oxazoline as ligand. Synth. React. Inorg., Met.-Org., Nano-Met. Chem. 2014, 44, 208–211. 10.1080/15533174.2013.769589. [DOI] [Google Scholar]
- Davies I. W.; Gerena L.; Castonguay L.; Senanayake C. H.; Larsen R. D.; Verhoeven T. R.; Reider P. J. The influence of ligand bite angle on the enantioselectivity of copper(II)-catalysed Diels–Alder reactions. Chem. Commun. 1996, 1753–1754. 10.1039/CC9960001753. [DOI] [Google Scholar]
- Davies I. W.; Deeth R. J.; Larsen R. D.; Reider P. J. A CLFSE/MM study on the role of ligand bite-angle in Cu(ll)-catalyzed Diels-Alder reactions. Tetrahedron Lett. 1999, 40, 1233–1236. 10.1016/S0040-4039(98)02596-9. [DOI] [Google Scholar]
- Denmark S. E.; Stiff C. M. Effect of ligand structure in the bisoxazoline mediated asymmetric addition of methyllithium to imines. J. Org. Chem. 2000, 65, 5875–5878. 10.1021/jo0007175. [DOI] [PubMed] [Google Scholar]
- Kwak Y.-S.; Corey E. J. Catalytic enantioselective conjugate addition of trimethylsilylacetylene to 2-cyclohexen-1-one. Org. Lett. 2004, 6, 3385–3388. 10.1021/ol048623v. [DOI] [PubMed] [Google Scholar]
- Itagaki M.; Masumoto K.; Yamamoto Y. Asymmetric cyclopropanation of 2,5-dimethyl-2,4-hexadiene by copper catalysts bearing new bisoxazoline ligands. J. Org. Chem. 2005, 70, 3292–3295. 10.1021/jo050009p. [DOI] [PubMed] [Google Scholar]
- Itagaki M.; Masumoto K.; Suenobu K.; Yamamoto Y. Studies of copper-bisoxazoline-catalyzed asymmetric cyclopropanation of 2,5-dimethyl-2,4-hexadiene. Org. Process Res. Dev. 2006, 10, 245–250. DFT calculations of the bis(oxazoline)-copper intermediates suggested that the gem-dimethyl groups at the 5-position affected the space around metal center. 10.1021/op050203d. [DOI] [Google Scholar]
- Alexander K.; Cook S.; Gibson C. L. cis-Selective cyclopropanations using chiral 5,5-diaryl bis(oxazoline) catalysts. Tetrahedron Lett. 2000, 41, 7135–7138. 10.1016/S0040-4039(00)01232-6. [DOI] [Google Scholar]
- Liu C.; Yi J.-C.; Liang X.-W.; Xu R.-Q.; Dai L.-X.; You S.-L. Copper(I)-catalyzed asymmetric dearomatization of indole acetamides with 3-indolylphenyliodonium salts. Chem. Eur. J. 2016, 22, 10813–10816. 10.1002/chem.201602229. [DOI] [PubMed] [Google Scholar]
- Hong S.; Tian S.; Metz M. V.; Marks T. J. C2-Symmetric bis(oxazolinato)lanthanide catalysts for enantioselective intramolecular hydroamination/cyclization. J. Am. Chem. Soc. 2003, 125, 14768–14783. 10.1021/ja0364672. [DOI] [PubMed] [Google Scholar]
- Fraile J. M.; GarcÌa J. I.; HerrerÌas C. I.; Mayoral J. A.; Reiser O.; Socuÿllamos A.; Werner H. The role of binding constants in the efficiency of chiral catalysts immobilized by electrostatic interactions: The case of azabis(oxazoline)copper complexes. Chem. Eur. J. 2004, 10, 2997–3005. 10.1002/chem.200305739. [DOI] [PubMed] [Google Scholar]
- Geiger C.; Kreitmeier P.; Reiser O. Cobalt(II)-azabis(oxazoline)-catalyzed conjugate reduction of α,β-unsaturated carbonyl compounds. Adv. Synth. Catal. 2005, 347, 249–254. 10.1002/adsc.200404295. [DOI] [Google Scholar]
- Glos M.; Reiser O. Aza-bis(oxazolines): New chiral ligands for asymmetric catalysis. Org. Lett. 2000, 2, 2045–2048. 10.1021/ol005947k. [DOI] [PubMed] [Google Scholar]
- Werner H.; Herrerías C. I.; Glos M.; Gissibl A.; Fraile J. M.; Pérez I.; Mayoral J. A.; Reiser O. Synthesis of polymer bound azabis(oxazoline) ligands and their application in asymmetric cyclopropanations. Adv. Synth. Catal. 2006, 348, 125–132. 10.1002/adsc.200505197. [DOI] [Google Scholar]
- Lim J.; Riduan S. N.; Lee S. S.; Ying J. Y. Siliceous mesocellular foam-supported aza(bisoxazoline)copper catalysts. Adv. Synth. Catal. 2008, 350, 1295–1308. 10.1002/adsc.200700388. [DOI] [Google Scholar]
- Salo E. V.; Guan Z. Late-transition-metal complexes with bisazaferrocene ligands for ethylene oligomerization. Organometallics 2003, 22, 5033–5046. 10.1021/om034051r. [DOI] [Google Scholar]
- Tse M. K.; Bhor S.; Klawoon M.; Anilkumar G.; Jiao H.; Döbler C.; Spannenberg A.; Mägerlein W.; Hugl H.; Beller M. Ruthenium-catalyzed asymmetric epoxidation of olefins using H2O2, Part I: Synthesis of new chiral N,N,N-tridentate pybox and pyboxazine ligands and their ruthenium complexes. Chem. Eur. J. 2006, 12, 1855–1874. 10.1002/chem.200501261. [DOI] [PubMed] [Google Scholar]
- Fan G.; Liu Y. Titanium-mediated cross-coupling reactions of imines with ketones or aldehydes: an efficient route for the synthesis of 1,2-amino alcohols. Tetrahedron Lett. 2012, 53, 5084–5087. 10.1016/j.tetlet.2012.07.020. [DOI] [Google Scholar]
- SHELX97 (for 1b, 1d, 2a, 2b), SHELXS2013 (for 4a), SHELXS (for 3a, 5b):Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- SIR92 (for 1c, 2c, 2d):Altomare A.; Cascarano G.; Giacovazzo C.; Guagliardi A.; Burla M.; Polidori G.; Camalli M. SIR92 – a program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. 10.1107/S002188989400021X. [DOI] [Google Scholar]
- PATTY (for 3c):Beurskens P. T.; Admiraal G.; Behm H.; Beurskens G.; Smits J. M. M.; Smykalla C. The DIRDIF91 program package. Z. Kristallogr. Suppl. 1991, 4, 99. [Google Scholar]
- CrystalStructure 4.0 (for 1b–d, 2a, 2c, 2d): Crystal Structure Analysis Package; Rigaku Corporation: Tokyo, Japan, 2000–2010. [Google Scholar]
- CrystalStructure 4.1 (for 4a): Crystal Structure Analysis Package; Rigaku Corporation: Tokyo, Japan, 2000–2014. [Google Scholar]
- CrystalStructure 4.2 (for 2b, 3a, 3c, 5b): Crystal Structure Analysis Package; Rigaku Corporation: Tokyo, Japan, 2000–2015. [Google Scholar]
- SHELX97 (for 1b–d, 2a–d), SHELXL2013 (for 4a), SHELXL, version 2014/7 (for 3a, 3c, 5b), see ref 81.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.