

Abstract
Inflammation has recently gained tremendous attention as a key contributor in several chronic diseases. While physiological inflammation is essential to counter a wide variety of damaging stimuli and to improve wound healing, dysregulated inflammation such as in the myocardium and vasculature can promote cardiovascular diseases. Given the high severity, prevalence, and economic burden of these diseases, understanding the factors involved in the regulation of physiological inflammation is essential. Like other complex biological phenomena, RNA-based processes are emerging as major regulators of inflammatory responses. Among such processes are cis-regulatory elements in the mRNA of inflammatory genes, noncoding RNAs directing the production or localization of inflammatory cytokines/chemokines, or pathogenic RNA driving inflammatory responses. In this review, we describe several specific RNA-based molecular mechanisms by which physiological inflammation pertaining to cardiovascular diseases is regulated. These include the role of AU-rich element-containing mRNAs, long non-coding RNAs, microRNAs, and viral RNAs.
1. Introduction
Inflammation is a physiological response to injury aimed at regulated and sequential activation of pro- and anti-inflammatory pathways to facilitate wound healing [1]. While the stimuli that incite inflammation are abundant and diverse, the inflammatory responses to different insults follow similar general pathways. The initial injury, due to pathogens or sterile tissue damage, involves the recognition of pathogen associated molecular patterns (PAMPs) or damage associated molecular patterns (DAMPs), respectively [2]. These are sensed by the host immune system via pattern recognition receptors (PRRs). Activated PRRs alert the body that damage has occurred, and they promote the production of inflammatory cytokines and chemokines to activate body’s defenses and initiate repair processes [3]. Pro-inflammatory cytokines released from the injury site prompt nearby endothelial cells to express intercellular adhesion molecules (ICAMs) to bind circulating immune cells [3]. These cells then follow the chemokine gradients to transmigrate into the site of tissue injury through the endothelium. Immune cells can then phagocytose damaged/infected cells, release compounds (i.e. perforin or reactive oxygen species [ROS]) to fight invaders, or stimulate fibrosis in an attempt to repair the damage [2].
Although acute inflammatory responses play an important role in minimizing injury and initiating the recovery process, prolonged and excessive inflammation is often tissue-injurious and plays a causal role in the progression of cardiovascular diseases [3]. For example, excessive levels of monocytes, macrophages, dendritic cells and T-lymphocytes in the myocardium during chronic heart failure can enhance myocyte apoptosis, hypertrophy and interstitial fibrosis [4–8]. This ultimately leads to progressive cardiac dysfunction. Similarly, following aggregation and oxidation of low-density lipoproteins (LDLs) at the endothelium, inflammatory cells (primarily macrophages and T lymphocytes) are drawn to the site by ICAMs expressed on the endothelial cells during atherosclerosis [9–11]. Macrophages then engulf LDLs, become foam cells, and release inflammatory cytokines to initiate recruitment of other immune cells to the site [12]. This vicious cycle ultimately results in the buildup of atherosclerotic plaques, which alter arterial stiffness, vessel flexibility, blood transport efficiency, and regenerative capacity [9,10,12,13]. Notably, excessive immune activation and dysregulated inflammation during atherosclerosis is directly associated with increased severity of several other debilitating diseases, including aortic stenosis, dementia, diabetes, myocardial infarction, stable and unstable angina, ischemic and non-ischemic heart failure, and stroke [14–17]. Further supporting the concept that excessive inflammation can damage the cardiovascular system is the observation that anti-inflammatory treatments are capable of reducing the incidence of cardiovascular events [18].
It is clear that inflammation is essential to counter a wide variety of tissue-injurious stimuli, which, coupled with the fact that excessive inflammation is often harmful, suggests that the inflammatory response must be tightly regulated in terms of its initiation and duration. To this effect, studies in the last decade have shown that RNA-based mechanisms can represent a major cellular strategy to regulate complex immunological phenomena. An expanding array of noncoding RNA (ncRNA) species are being shown to regulate protein production and function, and mRNA molecules have been found to be densely packed with cis-regulatory elements to control transcript localization, stability, and translation. Given the extreme regulatory potential afforded by RNA-based processes and the need for sophisticated regulation during inflammation, it is not surprising that inflammatory responses are tightly controlled at the RNA level. In this review, we highlight several key RNA-based mechanisms by which cardiovascular inflammation is regulated. Specifically, we will discuss the role of AU-rich mRNA regulatory elements, long non-coding RNA, microRNA, and viral RNA in cardiovascular inflammation.
2. RNA-based mechanisms of cardiovascular inflammation regulation
2.1. AU-rich elements
The production and activity of pro-inflammatory genes is regulated at all levels, but the competing needs of responding to inflammatory stimuli quickly while avoiding overproduction of inflammatory proteins renders translation of mRNA a particularly important regulatory check-point. Many inflammatory mRNAs possess regulatory AU-rich elements (AREs), a group of sequence motifs rich in adenine and uracil found in the 3′UTR. AREs typically act as destabilizing elements, but they can also stabilize transcripts and directly regulate their translation (reviewed in ref. [19]). The effect of an ARE on the mRNA fate is primarily dictated by the presence of different ARE binding proteins (ARE-BPs). Additionally, different ARE-BPs can compete for the same ARE, a mechanism that allows a particular mRNA to be differentially regulated in different cellular contexts [20]. Lastly, even the same ARE-BP at the same ARE can direct different mRNA fates depending on the cell type or how the ARE-BP is post-translationally modified [21–23]. These characteristics reveal the complex nature of ARE-dependent mRNA regulation. It is important to keep this complexity in mind as we discuss several examples of ARE-based regulation, because the mechanistic details likely vary in different cell types and in response to different conditions.
Among the key inflammatory genes possessing AREs is tumor necrosis factor-alpha (TNF-α), a potent pro-inflammatory cytokine that regulates immune activation and has been shown to be associated with high morbidity and mortality in cardiovascular diseases [24–28]. TNF-α is a master regulator of inflammation that exerts its pro-inflammatory activity primarily by binding to TNFR1 (tumor necrosis factor receptor 1) on the membranes of target cells and initiating canonical MAPK (mitogen-activated protein kinase) signaling to activate the pro-inflammatory transcription factor nuclear factor-kappa B (NF-κB) [29]. The TNF-α ARE can be bound by many different ARE-BPs, including AU-rich element RNA-binding protein 1, KH-type splicing regulatory protein, T-cell intercellular antigen-1, T-cell intercellular antigen related protein, human antigen R, glyceraldehyde 3-phosphate dehydrogenase, and tristetraprolin [21,30–34]. Of these, tristetraprolin (TTP) is among the most well-studied. TTP destabilizes TNF-α mRNA by binding to the ARE and recruiting decay factors [35]. TTP knockout (KO) mice have increased circulating TNF-α and an inflammatory phenotype characterized by syndromes such as cachexia, arthritis, dermatitis, and conjunctivitis [36]. They also have cardiac inflammation characterized by fibrosis and inflammatory cell infiltration of the mitral and aortic valves [37]. The extreme inflammation associated with TTP KO is eliminated upon injecting the mice with TNF-α antibodies (to inactivate circulating TNF-α) or crossing them with mice that had the main TNF-α receptor TNFR1 knocked out [38]. This confirms that the observed inflammation is truly a consequence of TNF-α overproduction. This also confirms that improper regulation of AREs can lead to extreme inflammation.
Loss of TTP in multiple cell types could contribute to the overproduction of TNF-α and inflammatory phenotype of TTP KO mice. TTP KO mice have been shown to have higher TNF-α production in macrophages, neutrophils, and CD8+ T-cells, suggesting that TNF-α overproduction in immune cells could be the dominant cause of inflammation [39–41]. TTP has also been shown to play an anti-inflammatory role in endothelial cells during atherosclerosis, although this is thought to be at least partially independent from TNF-α misregulation [42,43]. It is clear that TTP plays a widespread anti-inflammatory role by binding to the TNF-α ARE and reducing TNF-α production.
While most TNF-α-binding ARE-BPs downregulate TNF-α production, several ARE-BPs can stabilize TNF-α mRNA and result in an increase in protein production. One such ARE-BP is human antigen R ([HuR]; also called ELAV-like protein 1). While it typically resides in the nucleus, different stimuli (including inflammatory stimuli) can cause HuR to translocate to the cytoplasm, where it influences the stability or translation of many different transcripts [44,45]. While HuR is expressed in many different cell types and likely has cell type-specific targets, its role in regulating TNF-α mRNA has been well-studied in macrophages. Following stimulation, HuR can compete with TTP for binding the TNF-α ARE, stabilize TNF-α mRNA, and increase its translation [46]. Additionally, inflammatory stimuli lead to phosphorylation of TTP which prevents it from recruiting deadenylases and further facilitates the shift to TNF-α mRNA stabilization [47]. It is important to note that this model in which inflammatory stimuli inactivate TTP and encourage HuR to bind and activate TNF-α mRNA is likely to be oversimplified, as observations inconsistent with the model have been made. For example, myeloid-specific HuR KO mice actually have a pro-inflammatory phenotype with increased TNF-α production [48]. This discrepancy could be a attributed to the fact that HuR also stabilizes TTP mRNA (and thus loss of HuR disrupts ARE-BP activities other than its own), or it could be a consequence of stress-specific differences in HuR function [46]. Nevertheless, it is clear that HuR plays a central role in TNF-α regulation.
HuR has been shown to promote cardiac inflammation and the development of atherosclerosis. More specifically, cardiac inflammation following myocardial infarction is attenuated when recombinant antiinflammatory IL-10 is injected post-infarction [49]. This anti-inflammatory response was found to result at least in part from a reduction in HuR protein abundance and corresponding reduction in TNF-α mRNA and protein. This effect of reduced HuR on cardiac inflammation is likely a consequence of more than just a reduction in TNF-α, as HuR regulates many other factors involved in myocarditis, including β-adrenergic receptors, angiotensin II receptors, endothelial nitric oxide synthase (eNOS), transforming growth factor-beta, interferon-gamma, and IL-6 [50,51]. HuR has also been shown to promote the expression of cell adhesion molecules in endothelial cells during shear stress and to increase monocyte binding [52]. Taken together, these studies show that HuR can drive cardiovascular inflammation by modulating immune cell trafficking and the expression of inflammatory genes. While HuR increases the activity of pro-inflammatory genes like TNF-α, it also decreases the activity of anti-inflammatory genes like eNOS, a classical example of reciprocal regulation of different genes by the same ARE-BP to achieve maximal activation of pro-inflammatory immunogenic pathways [52]. Importantly, misregulation of HuR has also been shown to contribute to cardiac arrhythmias, fibrosis, and pathogenic remodeling during HF [53,54]. This further supports the idea that ARE misregulation can lead to cardiovascular damage.
Our laboratory has recently shown that a protein called BEX1 (brain-expressed X-linked 1) also has the capacity to stabilize TNF-α mRNA [55]. Mice subjected to pressure-overload cardiac stress experience an increase in BEX1 expression in the heart, which correlates with cardiac inflammation. BEX1 gain and loss of function experiments reveal that BEX1 plays a causal role in the observed cardiac inflammation. The mechanism of BEX1-induced cardiac inflammation appears to involve the stabilization of ARE-containing transcripts, as observed for TNF-α in vivo in BEX1 overexpressing hearts and for several other mRNAs in vitro [55]. Interestingly, BEX1 appears to relocalize from the nucleus to the cytoplasm following hypertrophic stress in cardiomyocytes [55]. This could suggest that like HuR, BEX1 can be stimulated to translocate to the cytoplasm, where it stabilizes certain ARE-containing transcripts and promotes the inflammatory response. The role of BEX1 in regulating TNF-α mRNA stability was only investigated in cardiomyocytes, and an important future step will be to investigate its role in inflammatory cells.
Interleukin 6 (IL-6) is another important inflammatory cytokine whose production is regulated by AREs. IL-6 regulates the response to injury and infection, and exerts its pro-inflammatory role primarily by activating the JAK/STAT (Janus kinases/signal transducer and activator of transcription proteins) pathway [56]. Like TNF-α, IL-6 is negatively regulated by TTP. However, unlike with TNF-α, TTP KO mice do not have systemic increases in IL-6 expression [57]. Instead, TTP KO appears to increase IL-6 stability in a stimulus and cell type-specific manner, suggesting that IL-6 suppression is heavily influenced by other factors [58]. Regnase-1 (also called monocyte chemotactic protein-induced protein 1 [MCPIP-1]) appears to be one such factor. Regnase-1 is an RNase that binds to the 3′UTR of IL-6 and facilitates its degradation [59,60]. Mice deficient in Regnase-1 develop an inflammatory phenotype characterized by inflammatory cell infiltration of the spleen, lungs, and pancreas [59]. Regnase-1 KO macrophages overproduce pro-inflammatory cytokines (including IL-6) in response to the inflammatory stressor lipopolysaccharide (LPS) [59]. Importantly, Regnase-1 has also been implicated in numerous cardiovascular processes and diseases including myocarditis, ischemia, and myocardial regeneration [61–63].
Regnase-1 appears to be poised for rapid response to inflammatory stimuli, as LPS treatment leads to ubiquitination and degradation of Regnase-1 and a subsequent increase in IL-6 production [60]. However, rather than just passive stabilization as a consequence of Regnase-1 degradation, there are also factors that actively stabilize IL-6 mRNA in response to inflammatory stress. For example, a protein called Arid5a (AT-rich interactive domain containing 5a) has been shown to bind and stabilize IL-6 mRNA in macrophages [64]. After treatment with LPS, Arid5a translocates from the nucleus to the cytoplasm, where it binds to the 3’ UTR of IL-6 mRNA [65]. The region it binds contains several AREs and overlaps with the sequence that Regnase-1 binds to, which has led to speculation that the local Regnase-1/Arid5a ratio governs the fate of IL-6 mRNA [64]. Understanding the role of AREs in IL-6 mRNA stability will be important for understanding the progression of inflammatory diseases.
While we focused the discussion of AREs on TNF-α and IL-6 due to their importance in cardiovascular inflammation, it is important to keep in mind that AREs are widespread and regulated in a large variety of ways. This regulation is complicated by the observations that microRNA can also mediate ARE-dependent mRNA decay [66]. Thus, it will be important to determine the extent to which microRNA and other noncoding RNA species regulate ARE-BP activity and ARE-containing transcript stability. Despite the complex and diverse nature of ARE-mediated mRNA regulation, the general cellular strategy for regulation of ARE-containing transcripts seems to be broadly applicable. A given ARE-containing transcript is kept in default on or off state, but different stresses alter the cellular distribution of ARE regulators (as seen in HuR, BEX1, and Arid5a described above) and rapidly inactivate or activate the gene (Fig. 1). This keeps the cell poised to rapidly upregulate inflammatory genes or downregulate anti-inflammatory genes when necessary.
Fig. 1.
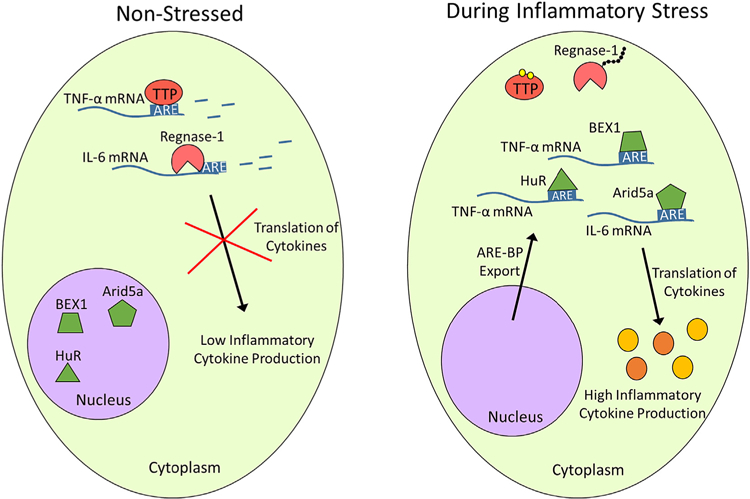
AU-rich Elements Regulate Inflammatory Protein Production. AU-rich elements within pro-inflammatory mRNAs typically destabilize the mRNA in unstressed states. TPP and Regnase-1 destabilize TNF-α and IL-6 mRNA, respectively. Following inflammatory stress, TTP is inactivated via phosphorylation, Regnase-1 becomes ubiquitinated and degraded, and nuclear ARE-BPs are exported. BEX1 and HuR stabilize TNF-α mRNA, while Arid5a stabilizes IL-6 mRNA. This ultimately leads to enhanced production of pro-inflammatory cytokines, which will be released to signal to the body that damage has occurred.
2.2. Long non-coding RNAs
In addition to mRNA-based mechanisms of regulating the inflammatory response, many non-coding RNAs (ncRNAs) also regulate inflammation. One broad class of ncRNA is long non-coding RNA (lncRNA). Long non-coding RNAs are transcripts greater than 200 nucleotides in length that have little or no coding potential. While they were once thought to be functionless evolutionary leftovers whose production represented transcriptional noise, the lncRNA field has exploded over the past few decades and revealed hundreds of functionally significant lncRNAs. Not surprisingly, lncRNAs exhibit extreme functional diversity. They can act as mediators between protein and DNA to regulate transcription, interact with mRNAs or miRNAs to regulate translation, and act as molecular scaffolds to regulate the spatial distribution of different factors [67]. Several recent reviews [68,69] have discussed the roles of lncRNA in cardiovascular diseases, and here we will highlight several lncRNAs involved in cardiovascular inflammation.
One of the most well-studied lncRNAs is MALAT1 (metastasis-associated lung adenocarcinoma transcript 1). MALAT1 resides in the nucleus, where it serves as a regulator of transcription and splicing [70]. While most of the study of MALAT1 has focused on its role in cancer progression, it is becoming clear that MALAT1 also plays an important role in regulating cardiovascular inflammation. Injection of mice with LPS causes an increase in MALAT1 expression in the heart, and in vitro studies suggest that MALAT1 mediates LPS-induced TNF-α production and apoptosis in cardiomyocytes [71]. Additionally, knockdown of MALAT1 in a rat model of systemic inflammation reduced the cardiac impairment and amount of circulating TNF-α and IL-6 that resulted from inflammatory stress [72]. MALAT1 was reported to increase NF-κB protein levels in this study, potentially suggesting that MALAT1 is involved in transcriptional upregulation of pro-inflammatory genes. MALAT1 has also been shown to regulate T-cell and macrophage activation, suggesting that its pro-inflammatory role could be in part due to regulation of immune cells [73,74]. Interestingly, diabetic MALAT1 KO mice have decreased expression of inflammatory cytokines in the heart and kidneys compared to their diabetic WT counterparts, suggesting that MALAT1 contributes to the well-established relationship between hyperglycemia and cardiac inflammation [75]. While these studies support a pro-inflammatory role for MALAT1 in cardiac inflammation, it has been shown that MALAT1 KO mice subjected to pressure overload cardiac stress do not develop more severe cardiac inflammation than WT mice, suggesting that the effect of MALAT1 on cardiac inflammation is dependent on the type of inflammatory stimulus [76].
MALAT1 has also been shown to regulate vascular inflammation. Consistent with the above observation that MALAT1 is involved in metabolic stress-induced inflammation, hyperglycemia causes a MALAT1-dependent increase in reactive oxygen species (ROS), TNF-α, and IL-6 production in endothelial cells [77]. Hyperglycemia also causes endothelial cell death in a MALAT1-dependent manner [78]. In contrast to the pro-inflammatory role observed in hyperglycemia, MALAT1 actually appears to serve an anti-inflammatory role in conditions of dyslipidemia. For example, MALAT1 has been observed to protect human coronary artery endothelial cells from oxidized LDL-induced damage [79]. This again suggests that MALAT1 mediates inflammation in response to metabolic stress. Interestingly, this anti-inflammatory role was suggested to be a consequence of MALAT1 sponging pro-inflammatory miR-155 (discussed below) [79]. Additional studies have investigated the role of MALAT1 in atherosclerosis using an apolipoprotein E (ApoE) deficient mouse model [80,81]. ApoE loss of function (ApoE−/−) mice have much higher levels of circulating LDL, which predisposes them to atherosclerosis and makes them a common atherosclerosis model. ApoE−/− mice that are also heterozygous for MALAT1 have been shown to develop significantly more severe atherosclerosis and vascular inflammation than ApoE−/− mice alone [80]. A subsequent study found that loss of MALAT1 in hematopoietic cells appears to be responsible for the increased atherosclerosis observed in MALAT1 deficient ApoE−/− mice. Replacement of ApoE−/− mouse bone marrow (and the associated hematopoietic cells) with that of MALAT1−/− ApoE−/− mice was sufficient to drive increased atherosclerosis [81]. This suggests that MALAT1 expression in inflammatory cells plays a vasculoprotective role. These studies have revealed that MALAT1 is an important regulator of cardiovascular inflammation, but inconsistencies with respect to a pro or anti-inflammatory role for MALAT1 warrants future research to clarify the effect of MALAT1 in different conditions.
Another lncRNA involved in cardiovascular inflammation is ANRIL (antisense RNA in the INK4 locus). The ANRIL locus is a major hotspot for disease associated mutations (including coronary artery disease), and ANRIL’s involvement in cardiovascular inflammation is becoming increasingly apparent [82]. In vitro studies using endothelial cells have shown that ANRIL expression is induced by TNF-α in a manner that involves the NF-κB pathway [83]. Moreover, ANRIL increases the expression of inflammatory proteins, including IL-6 as well as the cell adhesion molecules ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 (vascular cell adhesion molecule-1) [84]. Apoptosis of human coronary endothelial cells was also increased upon transfection with ANRIL and attenuated when transfected with siRNAs targeting ANRIL, suggesting a pro-inflammatory role for ANRIL in the vasculature [84]. Interestingly, a circular isoform of ANRIL has been reported to drive vascular inflammation when injected into rats, while injection of ANRIL inhibitors reduced vascular inflammation [85]. This further supports the idea that ANRIL plays a pro-inflammatory role. ANRIL has been found to interact with a pro-inflammatory transcription factor called Yin Yang 1 (YY1), which is known to be a downstream modulator of NF-κB signaling [83,86]. This has led to speculation that ANRIL exerts its pro-inflammatory effect in part by interacting with YY1 and subsequently binding to the promoters of inflammatory genes and driving their transcription [83]. Circular ANRIL has also been found to drive apoptosis in endothelial cells and macrophages by binding and inactivating a protein necessary for ribosomal RNA synthesis called pescadillo homologue 1 (PES1) [87]. Thus, ANRIL appears to regulate expression of pro-inflammatory cytokines and endothelial apoptosis. Nevertheless, due to significant differences between mouse and human ANRIL, the in vivo data is showing a causal role of ANRIL in human atherosclerosis are sparse and future studies will be necessary to evaluate that claim. Future studies will also have to investigate the role of ANRIL in immune cells, as immune cells are key regulators of cardiovascular inflammation and lncRNAs often exhibit cell type-specific functions. Moreover, it will be important to consider differences in function between different ANRIL isoforms.
The lncRNA HOTAIR (HOX transcript antisense RNA) has also been implicated in cardiovascular inflammation. Injection of mice with LPS leads to cardiac dysfunction and an increase in HOTAIR expression, TNF-α production, and NF-κB activity [88]. Knockdown of HOTAIR in cardiomyocytes leads to a reduction in LPS-induced TNF-α production and NF-κB activity, whereas HOTAIR overexpression increases both. This suggests that HOTAIR serves a pro-inflammatory role in cardiomyocytes in response to LPS. Inhibition of NF-κB impaired HOTAIR’s ability to stimulate TNF-α production, indicating that HOTAIR operates via regulation of the NF-κB pathway. Interestingly, simultaneous injection of mice with LPS and siRNA against HOTAIR lead to a reduction in LPS-induced cardiac dysfunction, agreeing with the pro-inflammatory role of HOTAIR implied by the in vitro studies [88]. HOTAIR has also been shown to regulate inflammation in macrophages. Treatment of macrophages with oxidized LDL leads to a HOTAIR-dependent increase in ROS and expression of TNF-α and IL-6 [89]. HOTAIR was found to operate by sponging microRNA-330–5p; a known regulator of macrophage activity [89]. While these results indicate that HOTAIR plays a pro-inflammatory role in cardiomyocytes and macrophages, studies have suggested that HOTAIR actually plays a protective role in endothelial cells. Knockdown of HOTAIR in human aortic endothelial cells lead to increased apoptosis, while HOTAIR overexpression reduced apoptosis [90]. HOTAIR was also shown to protect endothelial cells from oxidized LDL stress, further supporting the idea that it protects from vascular inflammation. The conflicting inflammatory roles of HOTAIR could be due to tissue-specific or stress-specific differences in HOTAIR function. Future studies will clarify the inflammatory activity of HOTAIR.
Only a fraction of lncRNAs have been characterized, yet they have revolutionized our understanding of how cells regulate complicated processes. Their condition-specific expression and frequent misregulation in disease makes them of great interest to the inflammation field. The examples discussed above (summarized in Table 1) are likely just the tip of the iceberg of lncRNAs involved in cardiovascular inflammation. With time and the development of higher sensitivity methods of detection, we can expect to see many more examples of lncRNAs regulating cardiovascular inflammation. A key future challenge will be to unravel the mechanisms by which lncRNAs regulate cardiovascular inflammation. Much of the work so far has been correlative, and identifying which proteins, RNA molecules, and signaling cascades are regulated by lncRNAs will be necessary to make sense of observed relationships between the abundance of a given lncRNA and the magnitude of the inflammatory response. Additionally, it will be important to determine the basis for the observations that certain lncRNAs can serve either pro or anti-inflammatory roles depending on the cell type or condition.
Table 1.
Relevant functions and potential effectors of the discussed lncRNA species.
| lncRNA | Functions | Potential effectors | References |
|---|---|---|---|
| MALAT1 | LPS-induced cardiac inflammation (+) | SAA3, NF-κB | [71,72,75,77,79–81] |
| Hyperglycemia-associated cardiac and vascular inflammation (+) | TNF-α, IL-6 | ||
| LDL-associated vascular inflammation (−) | mir-155 | ||
| ANRIL | Endothelial cytokine production (+) | YY1 | [83,84,86,87] |
| Endothelial apoptosis (+) | PES1 | ||
| HOTAIR | LPS-induced cardiac inflammation (+) | NF-κB | [88–90] |
| LDL-induced macrophage inflammation (+) | miR-330–5p | ||
| Endothelial apoptosis (−) |
; The process is upregulated by that lncRNA.
; The process is downregulated by that lnRNA.
2.3. MicroRNA
MicroRNAs (miRNAs) are a class of ncRNAs around 22 nucleotides in length. They regulate diverse biological processes, primarily by binding to the 3′UTR of mRNAs and directly reducing their translation or facilitating their degradation. Not surprisingly, miRNAs are heavily involved in regulating cardiovascular inflammation. In fact, miRNAs regulate essentially every aspect of atherogenesis; from maintaining lipid/cholesterol homeostasis to regulating immune cell recruitment and differentiation to regulating cytokine production [91]. We will briefly discuss key miRNAs involved in regulating each of these steps of atherogenesis. We will then discuss the roles of miRNAs in cardiac inflammation.
One key regulator of lipid homeostasis is miR-122. It regulates fatty acid and cholesterol synthesis in the liver (in part by targeting Sirtuin 6 (SIRT6) and 1-acylglycerol-3-phosphate O-acyltransferase (AGPAT) mRNAs), and its circulating levels correlate with both hyperlipidemia and coronary artery disease in humans [92–95]. MiR-122 knock out mice also have severe inflammatory phenotypes, consistent with the association between dyslipidemia and inflammation [94]. MiR-33 also regulates lipid homeostasis. It downregulates genes (including peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α), pyruvate dehydrogenase kinase isozyme 4 (PDK4), ATP binding cassette transporter A1 (ABCA1), ATP binding cassette transporter G1 (ABCG1), and Niemann-Pick disease, type C1 (NPC1)) required for the biosynthesis and trafficking of HDL, and thus leads to reduced levels of circulating HDL [96–98]. Inhibition of miR-33 leads to a reduction in atherosclerosis in mice, which is not surprising given the atheroprotective activity of HDL [99]. Lastly, miR-223 is also involved in maintaining cholesterol homeostasis as its transcription is induced by cholesterol and it downregulates expression of cholesterol biosynthesis proteins such as methylsterol monooxygenase 1 (SC4MOL) and hydroxymethylglutaryl-CoA synthase (HMGCS1) [100]. Interestingly, miR-223 appears to be involved in a number of other steps in the progression of atherosclerosis as will be discussed below, potentially explaining (in part) the known connection between dyslipidemia and vascular inflammation.
Following aggregation of LDL at the endothelial membrane, atherogenesis progresses as endothelial cells express cell adhesion molecules to bind circulating inflammatory cells. MiRNAs also regulate this process. For example, endothelial cell expression of ICAM-1 is repressed by miR-223 [101]. Interestingly, it has been shown that HDL can carry and deliver miR-223 to endothelial cells and subsequently reduce their expression of ICAM-1 [101]. This could be one mechanism by which HDL protects the vasculature from atherosclerosis. Expression of VCAM-1 is also regulated by miRNAs. VCAM-1 is normally repressed by miR-126, but adverse conditions (i.e. hypoxic or hyperglycemic stress) can reduce endothelial miR-126 levels and drive VCAM-1 expression [102–104]. Interestingly, vasculoprotective miR-126 can be transferred to endothelial cells via exosomes from endothelial progenitor cells or from apoptotic bodies of other endothelial cells [105,106]. MiR-126 expression is also sensitive to environmental conditions, as its expression is reduced in EPCs in diabetic conditions and circulating levels increase with physical activity [104,107,108]. This suggests that altered expression of miR-126 could underlie vascular inflammation associated with sedentary lifestyle.
Cell adhesion molecule expression also regulated by miR-181b. miR-181b represses the expression importin-α3, which is a nuclear membrane protein that mediates translocation of NF-κB to the nucleus in response to inflammatory stress [109]. With reduced NF-κB nuclear translocation, the cell cannot properly activate VCAM-1 and ICAM-1 (and other inflammatory genes) under stress. Not surprisingly given its role in downregulating NF-κB signaling, miR-181b has been shown to have atheroprotective effects. More specifically, ApoE−/− mice injected with miR-181b have reduced atherosclerosis compared to controls [110]. Interestingly, miR-181b levels are higher in endothelial cells with ANRIL knocked down, potentially suggesting that ANRIL’s pro-inflammatory activity operates via a reduction in the anti-inflammatory miR-181b [84]. This example also highlights the fact that there is cross-talk between inflammatory miRNAs and lncRNAs.
MicroRNAs also regulate the steps of atherogenesis that follow recruitment of inflammatory cells to the vascular wall. For example, miR-223 regulates the activation and differentiation of monocytes. More specifically, addition of miR-223 inhibitors have been shown to prevent their differentiation in vitro [111]. Interestingly, activated macrophages were shown to release microvesicles rich in miR-223 that can then be taken up by monocytes and facilitate their differentiation [111]. MiR-223 targets multiple key regulators of inflammation, including NLRP3 (NACHT, LRR And PYD Domains-Containing Protein 3), IKK-α (I-kappa-B kinase subunit alpha), and STAT3 (signal transducer and activator of transcription 3) [112–116]. Thus, the mechanism by which miR-223 drives monocyte differentiation is likely complex and multifaceted. Taken together, these examples highlight how miRNAs are involved in regulating the key steps of atherogensis.
MicroRNAs also regulate cardiac inflammation. For example, miR-155 appears to serve a pro-inflammatory role in the heart. It is upregulated following myocardial infarction in mice, and mice lacking miR-155 have reduced expression of inflammatory cytokines in the heart following myocardial infarction [117]. It appears to operate in part by targeting an anti-inflammatory protein called suppressor of cytokine signaling-1 (SOCS-1). Interestingly, the authors found that miR-155 is primarily produced in cardiac macrophages following myocardial infarction and that miR-155 can be transferred to fibroblasts (where it is thought to exert its effects) via macrophage-derived exosomes [117]. MiR-155 has also been shown to play a pro-inflammatory role during viral myocarditis. Mice infected with cardiotropic viruses have increased expression of miR-155 in the heart, and injection of miR-155 inhibitors reduced cardiac damage associated with viral infection [118]. This study revealed that miR-155 positively regulates both the production of inflammatory cytokines and the activation of immune cells in the heart during infection [118]. While these studies suggest that miR-155 plays a pro-inflammatory role in the heart, miR-155 has also been shown to play an anti-inflammatory and cardioprotective role during sepsis [119]. This suggests that miR-155 has context-specific effects on cardiac inflammation.
MiR-223 is also involved in cardiac inflammation in response to injury. Ischemia-reperfusion injury in the heart causes an increase in both miR-223 expression and cardiac inflammation [120]. The hearts of miR-223 KO mice exhibited increased expression of TNF-α and IL-1β production in response to ischemia-reperfusion injury. They also experienced more cardiomyocyte necrosis, suggesting that miR-223 plays a cardioprotective role. This anti-inflammatory role of miR-223 appears to result from downregulation of the target genes TNFR1 and NLRP3 [120]. MiR-21 also regulates cardiomyocyte death following ischemia-reperfusion injury. Inhibiting miR-21 during reperfusion leads to increased cardiomyocyte apoptosis, suggesting that miR-21 is cardio-protective [121]. Interestingly, miR-21 has also been shown to prevent apoptosis following myocardial infarction. It serves an anti-inflammatory role by negatively regulating NF-κB signaling by targeting a protein called KBTBD7 (Kelch repeat and BTB (POZ) domain containing 7) [122]. Lastly, the miRNA Let-7i is also involved in cardiac inflammation. It is downregulated in response to angiotensin II-based cardiac inflammatory stress [123]. Addition of Let-7i mimics or Let-7i inhibitors revealed that Let-7i reduces cardiac fibrosis in angiotensin II-stressed mice. It does so by targeting pro-inflammatory and pro-fibrotic genes such as IL-6 and collagen type 1 alpha 2 (Col1α2) in cardiac fibroblasts [123].
These examples (summarized in Table 2) highlight the diverse roles of miRNAs in cardiovascular inflammation. They regulate essentially every step of the inflammatory response and often operate in a stress and cell type-specific manner. MicroRNAs have been recognized as potential therapeutic targets, as they represent a relatively easy way to hijack cellular processes to reduce protein levels. Moreover, increasing evidence that they can transfer from cell to cell via exosomes makes them even more attractive for therapeutic uses. Nevertheless, the fact that a given miRNA can target multiple mRNAs suggests that off-target effects could be a challenge for miRNA-based therapies. Additionally, reports of cross-talk between miRNAs and lncRNAs suggest that miRNA biology is much more complicated than previously thought [124]. It is important to keep this complexity in mind when trying to design therapeutics.
Table 2.
Relevant functions and targets of the discussed MicroRNA species.
| miRNA | Function | Relevant Targets | References |
|---|---|---|---|
| miR-122 | Fatty acid/cholesterol synthesis (+) | AGPAT1, SIRT6 | [94,95] |
| miR-33 | Biosynthesis/transport of HDL (−) | ABCA1, ABCG1, NPC1, PGC1-α, PDK4 | [96,98] |
| miR-223 | Cholesterol biosynthesis (−), | HMGCS1, SC4MOL ICAM-1 | [100,101,112–115] |
| Cell adhesion molecule production (−), | NLRP3, IKK-α, STAT3 | ||
| Inflammatory cell activity | |||
| miR-126 | Cell adhesion molecule expression (−), | VCAM-1 | [102,104,107] |
| Endothelial progenitor cell activity (+) | Spred-1 | ||
| miR-181b | Anti-inflammatory | Importin-α3 | [109] |
| miR-155 | Myocardial inflammation (+/−) | SOCS1 | [117] |
| miR-21 | Myocardial inflammation (−) | KBTBD7 | [122] |
| Let-7i | Cardiac Fibrosis (−) | IL-6, Col1α2 | [123] |
; The process is upregulated by that miRNA.
; The process is downregulated by that miRNA.
2.4. Viral RNA
In addition to host RNA, cardiovascular inflammation is also regulated by exogenous RNA from pathogens. RNA viruses are the most common cause of viral myocarditis, and thus it is not surprising that the heart is equipped with mechanisms to recognize and respond to foreign RNA. Coxsackievirus B3 (CVB3) is the dominant cardiotropic virus in humans and mice, and its mechanisms of action are the most well-studied. It has a single stranded RNA (ssRNA) genome with a double stranded RNA (dsRNA) replication intermediate, both of which can be recognized by host factors [125]. Among the factors that appear to be involved in sensing CVB3 RNA are the toll-like receptors TLR3, TLR4, and TLR8 [126–129]. After encountering the foreign RNA, these receptors recruit signal adaptors like MyD88 (myeloid differentiation primary response gene 88) or TRIF (TIR domain-containing adaptor-inducing interferon-β), both of which have been shown to mediate the heart’s response to CVB3 [130–132]. MyD88 then instigates a signaling cascade that ultimately results in degradation of I-kappa-B-alpha (IκBα), which is a cytoplasmic factor that binds to NF-κB under normal conditions. Following IκBα degradation, NF-κB is free to translocate to the nucleus where it can drive the transcription of pro-inflammatory cytokines [133]. The TRIF signaling cascade results in activation of NF-κB and another transcription factor called interferon regulatory factor 3 (IRF3), which then translocates to the nucleus and drives expression of interferons [130]. Immune cells are then recruited to destroy infectious agents and infected cells.
Toll-like receptors are not the only viral RNA sensors involved in mediating pathogen-induced cardiovascular inflammation. A group of RNA helicases called RIG-I-like receptors (RLRs) can also recognize viral RNA and stimulate the immune response [134]. Members of this group include RIG-I (retinoic acid-inducible gene 1) and MDA-5 (melanoma differentiation-associated protein 5). They recognize viral RNA structures and subsequently interact with mitochondrial antiviral sensing protein (MAVS) [135]. This interaction is mediated by caspase recruitment domains (CARDs), which are found in both MAVS and the two RLRs. Following RLR binding, MAVS proteins aggregate with one another and ultimately activate NF-κB and IRF3, which leads to the expression of inflammatory genes [135].
Recent studies have investigated the role of NOD-like receptors (NLRs) in responding to CVB infection. Like TLRs and RLRs, NLRs sense pathogen or damage-associated molecular patterns and facilitate the inflammatory response. NLRs drive pyroptosis, a specialized form of cell death characterized by cell bursting and release of pro-inflammatory cytokines [136]. CVB infection resuwlts in activation of the NLR and inflammasome protein NLRP3 [137]. Active NLRP3 then recruits and activates caspase-1, which processes several pro-inflammatory cytokines (namely pro-IL-1β and pro-IL-18) and causes cell bursting [136]. Release of IL-1β and IL-18 from pyroptotic cells is thought to contribute to the cardiac inflammation seen during viral infection. In fact, IL-1β plays a general role in cardiac inflammatory pathologies, as the CANTOS (Canakinumab anti-inflammatory thrombosis outcome study) clinical trial has shown that the IL-1β-nullifying antibody Canakinumab can improve outcomes for those with cardiovascular inflammation [18]. Importantly, Canakinumab has also recently been shown to reduce cardiac damage during murine CVB infection [138]. Thus, it will be interesting to see if IL-1β-nullifying antibodies can be used to improve outcomes for viral myocarditis patients. The viral RNA sensing mechanisms described above are depicted in Fig. 2.
Fig. 2.
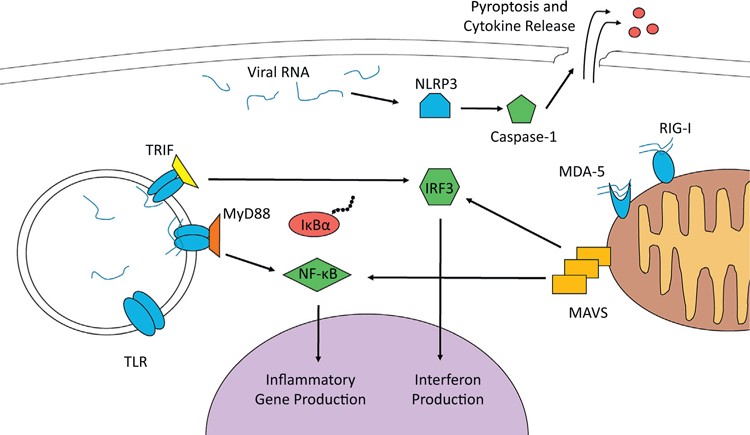
Viral RNA Drives Cardiovascular Inflammation. RNA viruses are sensed by multiple pattern recognition receptors within the host cell. These include TLRs, RIG-I-like receptors (MDA-5 and RIG-I), and NLRs. Activated TLRs interact with signal transduction adaptor proteins (such as TRIF and MyD88) which the drive the activation of transcription factors NF-κB and IRF3. These factors move into the nucleus, where they activate pro-inflammatory genes. The RIG-I-like receptors also can lead to the activation of NF-κB and IRF3. They do so by associating with and activating the signal transducer MAVS at the mitochondrial membrane. The NLRs can exacerbate inflammation and drive pyroptosis as shown for NLRP3 above. It activates caspase-1, which disrupts the cell membrane and allows the release of inflammatory cytokines.
RNA viruses can also cause vascular inflammation. For example, hepatitis C virus (HCV) appears to drive vascular inflammation and atherosclerosis. Studies have shown an association between chronic HCV infection and atherosclerosis, and HCV RNA has been detected within atherosclerotic plaques [139,140]. In vitro experiments have shown that HCV RNA drives expression of inflammatory genes in endothelial cells, supporting the idea that HCV plays a causal role in vascular inflammation and atherosclerosis [141]. Ultimately, the fact that HCV infection is restricted to humans and chimpanzees has hampered the investigation of the pro-atherosclerotic role of HCV in vivo [142]. Other RNA viruses that have been shown to cause damage to endothelial cells include human immunodeficiency virus and hepatitis B virus, although the precise mechanisms underlying their role in vascular inflammation are poorly understood [143]. Most of the work on viral RNAs in vascular inflammation has been correlational, and thus demonstrating a causal role for viruses in vascular inflammation and atherosclerosis will be an essential next step. This review focused on the role of RNA viruses, but it is important to note that DNA viruses (such as cytomegalovirus and parvovirus B19) can also cause cardiovascular inflammation [144,145].
3. Conclusion
Chronic cardiovascular inflammation constitutes a major societal problem, as the physiological damage that results is severe, costly, and widespread. Unfortunately, the more we learn about cardiovascular inflammation, the more complicated (both in terms of its causes and how the body regulates the inflammatory response) the phenomenon is shown to be. This complexity demands highly specific treatment strategies. The use of antibodies reduces the activity of specific inflammatory proteins is a promising approach. Among the factors that have been targeted in an attempt to reduce cardiovascular inflammation are TNF-α, IL-6, and IL-1β [18,146]. Antibody-based therapies have had some success, but there are often unintended side effects that can harm the individual (see reference [146] for a review). Lifestyle changes are still the most effective way to avoid and reduce cardiovascular inflammation.
The study of inflammation has historically been focused on protein factors involved in the inflammatory response, but it has become clear in recent decades that proteins are only a fraction of the story and that a diverse array of RNA molecules fine-tune the process. The mRNA of inflammatory genes are densely packed with regulatory elements that allow environment-sensitive translation, which is necessary for inflammatory responses given that they must be rapidly induced by a wide variety of stimuli. AU-rich regulatory elements are widespread in inflammatory genes and characterization of AREs and their corresponding ARE-BPs will be necessary for the advancement of the cardiovascular inflammation field. Additionally, the expanding knowledge of the roles of lncRNAs and miRNAs in cardiovascular inflammation warrants further investigation of the inflammatory function of these and other ncRNAs. Moreover, cross-talk between inflammatory lncRNAs and miRNAs suggest an additional layer of complexity of the inflammatory response, and understanding the extent and effects of such cross-talk remains as an important future step in the cardiovascular inflammation field. Importantly, the study of miRNAs in inflammation could potentially uncover new therapeutic targets, as adjusting miRNA levels has been recognized as a potential mechanism of disease treatment. Finally, RNA viruses are the most common cardiac pathogen, and thus understanding how viral RNA drives cardiovascular inflammation and subsequent tissue damage will be essential for prevention or treatment of cardiac infections. Additionally, while the evidence for a causal role of RNA viruses in vascular inflammation is relatively weak, the fact that RNA viruses are often found within atherosclerotic plaques coupled with the tight relationship between pathogens and the inflammatory response suggests that RNA viruses may play an underappreciated role in vascular inflammation. Additional studies are needed to evaluate the role of pathogens in chronic cardiovascular inflammation, as it could change the way we think about the resulting diseases.
Acknowledgments
Funding source
This work was supported by grants from NIH (HL121284, HL136951 to F.A.; and R00 HL132123 S.S.B.); from the American Heart Association AHA 17IRG33460198 (to F.A.) and from the United States-Israel Binational Science Foundation (BSF) 2017094 to F.A.
Footnotes
Disclosures
None.
References
- [1].Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, Libby P, Weissleder R, Pittet MJ, The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions, J. Exp. Med 204 (12) (2007) 3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chen GY, Nuñez G, Sterile inflammation: sensing and reacting to damage, Nat. Rev. Immunol 10 (12) (2010) 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Prabhu SD, Frangogiannis NG, The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis, Circ. Res 119 (1) (2016) 91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, von Schlippenbach J, Skurk C, Steendijk P, Riad A, Poller W, Schultheiss HP, Tschope C, Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction, Circ. Heart Fail 4 (1) (2011) 44–52. [DOI] [PubMed] [Google Scholar]
- [5].Ismahil Mohamed A, Hamid T, Bansal Shyam S, Patel B, Kingery Justin R, Prabhu Sumanth D, Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure, Circ. Res 114 (2) (2014) 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL, Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart, Proc. Natl. Acad. Sci. U.S.A 111 (1091–6490 (Electronic)) (2014) 16026–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Van der Borght K, Scott CL, Martens L, Sichien D, Van Isterdael G, Nindl V, Saeys Y, Boon L, Ludewig B, Gillebert TC, Lambrecht BN, Myocarditis elicits dendritic cell and monocyte infiltration in the heart and self-antigen presentation by conventional type 2 dendritic cells, Front. Immunol 9 (2018) 2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD, Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure, Circulation 10 (1941–3297 (Electronic)) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sakakura K, Nakano M, Otsuka F, Ladich E, Kolodgie FD, Virmani R, Pathophysiology of atherosclerosis plaque progression, Heart Lung Circ 22 (6) (2013) 399–411. [DOI] [PubMed] [Google Scholar]
- [10].Björkerud S, Björkerud B, Apoptosis is abundant in human atherosclerotic lesions, especially in inflammatory cells (macrophages and T cells), and may contribute to the accumulation of gruel and plaque instability, Am. J. Pathol 149 (2) (1996) 367–380. [PMC free article] [PubMed] [Google Scholar]
- [11].Ross R, Atherosclerosis — an inflammatory disease, N. Engl. J. Med 340 (2) (1999) 115–126. [DOI] [PubMed] [Google Scholar]
- [12].Raggi P, Genest J, Giles JT, Rayner KJ, Dwivedi G, Beanlands RS, Gupta M, Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions, Atherosclerosis 276 (2018) 98–108. [DOI] [PubMed] [Google Scholar]
- [13].Boesen ME, Singh D, Menon BK, Frayne R, A systematic literature review of the effect of carotid atherosclerosis on local vessel stiffness and elasticity, Atherosclerosis 243 (1) (2015) 211–222. [DOI] [PubMed] [Google Scholar]
- [14].van Oijen M, Jan de Jong F, Witteman JCM, Hofman A, Koudstaal PJ, Breteler MMB, Atherosclerosis and risk for dementia, Ann. Neurol 61 (5) (2007) 403–410. [DOI] [PubMed] [Google Scholar]
- [15].Goraya TY, Leibson CL, Palumbo PJ, Weston SA, Killian JM, Pfeifer EA, Jacobsen SJ, Frye RL, Roger VL, Coronary atherosclerosis in diabetes mellitus, J. Am. Coll. Cardiol 40 (5) (2002) 946. [DOI] [PubMed] [Google Scholar]
- [16].Davies MJ, Thomas AC, Plaque fissuring–the cause of acute myocardial infarction, sudden ischaemic death, and crescendo angina, Br. Heart J 53 (4) (1985) 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Spagnoli L, Mauriello A, Sangiorgi G, et al. , Extracranial thrombotically active carotid plaque as a risk factor for ischemic stroke, JAMA 292 (15) (2004) 1845–1852. [DOI] [PubMed] [Google Scholar]
- [18].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Antiinflammatory therapy with canakinumab for atherosclerotic disease, N. Engl. J. Med 377 (12) (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [19].Barreau C, Paillard L, Osborne HB, AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res 33 (22) (2005) 7138–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H, Involvement of KSRP in the post-transcriptional regulation of human iNOS expression–complex interplay of KSRP with TTP and HuR, Nucleic Acids Res 33 (15) (2005) 4813–4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Xu N, Chen C-YA, Shyu A-B, Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover, Mol. Cell. Biol 21 (20) (2001) 6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW, MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14–3-3 binding, J. Biol. Chem 279 (11) (2004) 10176–10184. [DOI] [PubMed] [Google Scholar]
- [23].Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WFC, Blackwell TK, Anderson P, MK2-induced tristetraprolin:14–3-3 complexes prevent stress granule association and ARE-mRNA decay, EMBO J 23 (6) (2004) 1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kubota T, McTiernan CF, Frye CS, Demetris AJ, Feldman AM, Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice, J. Card. Fail 3 (2) (1997) 117–124. [DOI] [PubMed] [Google Scholar]
- [25].Meldrum DR, Tumor necrosis factor in the heart, Am. J. Phys. Regul. Integr. Comp. Phys 274 (3) (1998) R577–R595. [DOI] [PubMed] [Google Scholar]
- [26].Popa C, Netea MG, van Riel PLCM, van der Meer JWM, Stalenhoef AFH, The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk, J. Lipid Res 48 (4) (2007) 751–762. [DOI] [PubMed] [Google Scholar]
- [27].Szekanecz Z, Shah MR, Pearce WH, Koch AE, Human atherosclerotic abdominal aortic aneurysms produce interleukin (IL)-6 and interferon-gamma but not IL-2 and IL-4: the possible role for IL-6 and interferon-gamma in vascular inflammation, Agents Act 42 (3) (1994) 159–162. [DOI] [PubMed] [Google Scholar]
- [28].Tieu BC, Lee C, Sun H, LeJeune W, Recinos A 3rd, Ju X, Spratt H, Guo D-C, Milewicz D, Tilton RG, Brasier AR, An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice, J. Clin. Invest 119 (12) (2009) 3637–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sabio G, Davis RJ, TNF and MAP kinase signalling pathways, Semin. Immunol 26 (3) (2014) 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dember LM, Kim ND, Liu K-Q, Anderson P, Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities, J. Biol. Chem 271 (5) (1996) 2783–2788. [DOI] [PubMed] [Google Scholar]
- [31].Chen C-Y, Gherzi R, Ong S-E, Chan EL, Raijmakers R, Pruijn GJM, Stoecklin G, Moroni C, Mann M, Karin M, AU binding proteins recruit the exosome to degrade ARE-containing mRNAs, Cell 107 (4) (2001) 451–464. [DOI] [PubMed] [Google Scholar]
- [32].Dean JLE, Wait R, Mahtani KR, Sully G, Clark AR, Saklatvala J, The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR, Mol. Cell. Biol 21 (3) (2001) 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Garcin ED, GAPDH as a model non-canonical AU-rich RNA binding protein, Semin. Cell Dev. Biol 86 (2018) 162–173. [DOI] [PubMed] [Google Scholar]
- [34].Carballo E, Lai WS, Blackshear PJ, Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin, Science 281 (5379) (1998) 1001. [DOI] [PubMed] [Google Scholar]
- [35].Lykke-Andersen J, Wagner E, Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1, Genes Dev 19 (3) (2005) 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ, A pathogenetic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency, Immunity 4 (5) (1996) 445–454. [DOI] [PubMed] [Google Scholar]
- [37].Ghosh S, Hoenerhoff MJ, Clayton N, Myers P, Stumpo DJ, Maronpot RR, Blackshear PJ, Left-sided cardiac valvulitis in tristetraprolin-deficient mice: the role of tumor necrosis factor alpha, Am. J. Pathol 176 (3) (2010) 1484–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Carballo E, Blackshear PJ, Roles of tumor necrosis factor-α receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome, Blood 98 (8) (2001) 2389. [DOI] [PubMed] [Google Scholar]
- [39].Carballo E, Gilkeson GS, Blackshear PJ, Bone marrow transplantation reproduces the tristetraprolin-deficiency syndrome in recombination activating gene-2 (−/−) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha overproduction, J. Clin. Investig 100 (5) (1997) 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ebner F, Sedlyarov V, Tasciyan S, Ivin M, Kratochvill F, Gratz N, Kenner L, Villunger A, Sixt M, Kovarik P, The RNA-binding protein tristetraprolin schedules apoptosis of pathogen-engaged neutrophils during bacterial infection, J. Clin. Invest 127 (6) (2017) 2051–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang Q, Ning H, Peng H, Wei L, Hou R, Hoft DF, Liu J, Tristetraprolin inhibits macrophage IL-27-induced activation of antitumour cytotoxic T cell responses, Nat. Commun 8 (1) (2017) 867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang H, Taylor WR, Joseph G, Caracciolo V, Gonzales Donna M, Sidell N, Seli E, Blackshear Perry J, Kallen Caleb B, mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells, Arterioscler. Thromb. Vasc. Biol 33 (6) (2013) 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bollmann F, Wu Z, Oelze M, Siuda D, Xia N, Henke J, Daiber A, Li H, Stumpo DJ, Blackshear PJ, Kleinert H, Pautz A, Endothelial dysfunction in tristetraprolin-deficient mice is not caused by enhanced tumor necrosis factor-alpha expression, J. Biol. Chem 289 (22) (2014) 15653–15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fan XC, Steitz JA, Overexpression of HuR, a nuclear–cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs, EMBO J 17 (12) (1998) 3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Doller A, Pfeilschifter J, Eberhardt W, Signalling pathways regulating nucleocytoplasmic shuttling of the mRNA-binding protein HuR, Cell. Signal 20 (12) 2008) 2165–2173. [DOI] [PubMed] [Google Scholar]
- [46].Tiedje C, Ronkina N, Tehrani M, Dhamija S, Laass K, Holtmann H, Kotlyarov A, Gaestel M, The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU–rich element–dependent translation, PLoS Genet 8 (9) (2012) e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J, Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing Deadenylase recruitment, Mol. Cell. Biol 31 (2) (2011) 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL, Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis, J. Clin. Invest 122 (1) (2012) 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R, IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR, Circ. Res 104 (2) (2009) e9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Misquitta CM, Iyer VR, Werstiuk ES, Grover AK, The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology, Mol. Cell. Biochem 224 (1) (2001) 53–67. [DOI] [PubMed] [Google Scholar]
- [51].Srikantan S, Gorospe M, HuR function in disease, Front Biosci. (Landmark Ed) 17 (2012) 189–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rhee WJ, Ni C-W, Zheng Z, Chang K, Jo H, Bao G, HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells, Proc. Natl. Acad. Sci 107 (15) (2010) 6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Green LC, Anthony SR, Slone S, Lanzillotta L, Nieman ML, Wu X, Robbins N, Jones SM, Roy S, Owens III AP, Aube J, Xu L, Lorenz JN, Blaxall BC, Rubinstein J, Benoit JB, Tranter M, Human antigen R as a therapeutic target in pathological cardiac hypertrophy, JCI Insight 4 (4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhou A, Xie A, Kim TY, Liu H, Shi G, Kang G-J, Jiang N, Liu M, Jeong E-M, Choi B-R, Dudley SC, HuR-mediated SCN5A messenger RNA stability reduces arrhythmic risk in heart failure, Heart Rhythm 15 (7) (2018) 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Accornero F, Schips TG, Petrosino JM, Gu S-Q, Kanisicak O, van Berlo JH, Molkentin JD, BEX1 is an RNA-dependent mediator of cardiomyopathy, Nat. Commun 8 (1) (2017) 1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Heinrich PC, Behrmann I, MÜLler-Newen G, Schaper F, Graeve L, Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway, Biochem. J 334 (2) (1998) 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhao W, Liu M, D’Silva NJ, Kirkwood KL, Tristetraprolin regulates interleukin-6 expression through p38 MAPK-dependent affinity changes with mRNA 3′ untranslated region, J. Interf. Cytokine Res 31 (8) (2011) 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Griseri P, Pages G, Control of pro-angiogenic cytokine mRNA half-life in cancer: the role of AU-rich elements and associated proteins, J. Interf. Cytokine Res 34 (4) (2014) 242–254. [DOI] [PubMed] [Google Scholar]
- [59].Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, Satoh T, Kato H, Tsujimura T, Nakamura H, Akira S, Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay, Nature 458 (2009) 1185. [DOI] [PubMed] [Google Scholar]
- [60].Iwasaki H, Takeuchi O, Teraguchi S, Matsushita K, Uehata T, Kuniyoshi K, Satoh T, Saitoh T, Matsushita M, Standley DM, Akira S, The IkappaB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1, Nat. Immunol 12 (12) (2011) 1167–1175. [DOI] [PubMed] [Google Scholar]
- [61].Cui X, Mino T, Yoshinaga M, Nakatsuka Y, Hia F, Yamasoba D, Tsujimura T, Tomonaga K, Suzuki Y, Uehata T, Takeuchi O, Regnase-1 and Roquin non-redundantly regulate Th1 differentiation causing cardiac inflammation and fibrosis, J. Immunol 199 (12) (2017) 4066–4077. [DOI] [PubMed] [Google Scholar]
- [62].Mao R, Yang R, Chen X, Harhaj EW, Wang X, Fan Y, Regnase-1, a rapid response ribonuclease regulating inflammation and stress responses, Cell. Mol. Immunol 14 (2017) 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhu T, Yao Q, Hu X, Chen C, Yao H, Chao J, The role of MCPIP1 in ischemia/reperfusion injury-induced HUVEC migration and apoptosis, Cell. Physiol. Biochem 37 (2) (2015) 577–591. [DOI] [PubMed] [Google Scholar]
- [64].Masuda K, Ripley B, Nishimura R, Mino T, Takeuchi O, Shioi G, Kiyonari H, Kishimoto T, Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo, Proc. Natl. Acad. Sci 110 (23) (2013) 9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Higa M, Oka M, Fujihara Y, Masuda K, Yoneda Y, Kishimoto T, Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a, Proc. Natl. Acad. Sci. U. S. A 115 (6) (2018) E1214–e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, Di Padova F, Lin S-C, Gram H, Han J, Involvement of MicroRNA in AU-rich element-mediated mRNA instability, Cell 120 (5) (2005) 623–634. [DOI] [PubMed] [Google Scholar]
- [67].Chew CL, Conos SA, Unal B, Tergaonkar V, Noncoding RNAs: master regulators of inflammatory signaling, Trends Mol. Med 24 (1) (2018) 66–84. [DOI] [PubMed] [Google Scholar]
- [68].Greco S, Salgado Somoza A, Devaux Y, Martelli F, Long noncoding RNAs and cardiac disease, Antioxid. Redox Signal 29 (9) (2017) 880–901. [DOI] [PubMed] [Google Scholar]
- [69].Salamon I, Saccani Jotti G, Condorelli G, The long noncoding RNA landscape in cardiovascular disease: a brief update, Curr. Opin. Cardiol 33 (3) (2018) 282–289. [DOI] [PubMed] [Google Scholar]
- [70].Gutschner T, Hämmerle M, Diederichs S, MALAT1 — a paradigm for long noncoding RNA function in cancer, J. Mol. Med 91 (7) (2013) 791–801. [DOI] [PubMed] [Google Scholar]
- [71].Zhuang YT, Xu DY, Wang GY, Sun JL, Huang Y, Wang SZ, IL-6 induced lncRNA MALAT1 enhances TNF-alpha expression in LPS-induced septic cardiomyocytes via activation of SAA3, Eur. Rev. Med. Pharmacol. Sci 21 (2) (2017) 302–309. [PubMed] [Google Scholar]
- [72].Chen H, Wang X, Yan X, Cheng X, He X, Zheng W, LncRNA MALAT1 regulates sepsis-induced cardiac inflammation and dysfunction via interaction with miR-125b and p38 MAPK/NFκB, Int. Immunopharmacol 55 (2018) 69–76. [DOI] [PubMed] [Google Scholar]
- [73].Cui H, Banerjee S, Guo S, Xie N, Ge J, Jiang D, Zörnig M, Thannickal VJ, Liu G, Long noncoding RNA Malat1 regulates differential activation of macrophage and response to lung injury, JCI Insight 4 (2019) e124522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Masoumi F, Ghorbani S, Talebi F, Branton WG, Rajaei S, Power C, Noorbakhsh F, Malat1 long noncoding RNA regulates inflammation and leukocyte differentiation in experimental autoimmune encephalomyelitis, J. Neuroimmunol 328 (2019) 50–59. [DOI] [PubMed] [Google Scholar]
- [75].Gordon AD, Biswas S, Feng B, Chakrabarti SCE, MALAT1: a regulator of inflammatory cytokines in diabetic complications, Endocrinol. Diabet. Metab 1 (2) (2018) e00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Peters T, Hermans-Beijnsberger S, Beqqali A, Bitsch N, Nakagawa S, Prasanth KV, de Windt LJ, van Oort RJ, Heymans S, Schroen B, Long noncoding RNA Malat-1 is dispensable during pressure overload-induced cardiac Remodeling and failure in mice, PLoS One 11 (2) (2016) e0150236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Puthanveetil P, Chen S, Feng B, Gautam A, Chakrabarti S, Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells, J. Cell. Mol. Med 19 (6) (2015) 1418–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Song Y, Yang L, Guo R, Lu N, Shi Y, Wang X, Long noncoding RNA MALAT1 promotes high glucose-induced human endothelial cells pyroptosis by affecting NLRP3 expression through competitively binding miR-22, Biochem. Biophys. Res. Commun 509 (2) (2019) 359–366. [DOI] [PubMed] [Google Scholar]
- [79].Li S, Sun Y, Zhong L, Xiao Z, Yang M, Chen M, Wang C, Xie X, Chen X, The suppression of ox-LDL-induced inflammatory cytokine release and apoptosis of HCAECs by long non-coding RNA-MALAT1 via regulating microRNA-155/SOCS1 pathway, Nutr. Metab. Cardiovasc. Dis 28 (11) (2018) 1175–1187. [DOI] [PubMed] [Google Scholar]
- [80].Gast M, Rauch BH, Nakagawa S, Haghikia A, Jasina A, Haas J, Nath N, Jensen L, Stroux A, Böhm A, Friebel J, Rauch U, Skurk C, Blankenberg S, Zeller T, Prasanth KV, Meder B, Kuss A, Landmesser U, Poller W, Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE−/−mice, Cardiovasc. Res 115 (2018) 302–314 (cvy202-cvy202). [DOI] [PubMed] [Google Scholar]
- [81].Cremer S, Michalik Katharina M, Fischer A, Pfisterer L, Jaé N, Winter C, Boon Reinier A, Muhly-Reinholz M, John D, Uchida S, Weber C, Poller W, Günther S, Braun T, Li Daniel Y, Maegdefessel L, Matic Perisic L, Hedin U, Soehnlein O, Zeiher A, Dimmeler S, Hematopoietic deficiency of the long noncoding RNA MALAT1 promotes atherosclerosis and plaque inflammation, Circulation 0 (0) (2018). [DOI] [PubMed] [Google Scholar]
- [82].Pasmant E, Sabbagh A, Vidaud M, Bieche I, ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS, FASEB J 25 (2) (2011) 444–448. [DOI] [PubMed] [Google Scholar]
- [83].Zhou X, Han X, Wittfeldt A, Sun J, Liu C, Wang X, Gan LM, Cao H, Liang Z, Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway, RNA Biol 13 (1) (2016) 98–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Guo F, Tang C, Li Y, Liu Y, Lv P, Wang W, Mu Y, The interplay of LncRNA ANRIL and miR-181b on the inflammation-relevant coronary artery disease through mediating NF-kappaB signalling pathway, J. Cell. Mol. Med 22 (2018) 5062–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Song CL, Wang JP, Xue X, Liu N, Zhang XH, Zhao Z, Liu JG, Zhang CP, Piao ZH, Liu Y, Yang YB, Effect of circular ANRIL on the inflammatory response of vascular endothelial cells in a rat model of coronary atherosclerosis, Cell. Physiol. Biochem 42 (3) (2017) 1202–1212. [DOI] [PubMed] [Google Scholar]
- [86].Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gäbel G, Bergert H, Bechmann I, Stadler PF, Thiery J, Teupser D, Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks, PLoS Genet 9 (7) (2013) e1003588–e1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, Gäbel G, Beutner F, Scholz M, Thiery J, Musunuru K, Krohn K, Mann M, Teupser D, Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans, Nat. Commun 7 (2016) 12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wu H, Liu J, Li W, Liu G, Li Z, LncRNA-HOTAIR promotes TNF-alpha production in cardiomyocytes of LPS-induced sepsis mice by activating NF-kappaB pathway, Biochem. Biophys. Res. Commun 471 (1) (2016) 240–246. [DOI] [PubMed] [Google Scholar]
- [89].Liu J, Huang G-Q, Ke Z-P, Silence of long intergenic noncoding RNA HOTAIR ameliorates oxidative stress and inflammation response in ox-LDL-treated human macrophages by upregulating miR-330–5p, J. Cell. Physiol 234 (4) (2019) 5134–5142. [DOI] [PubMed] [Google Scholar]
- [90].Peng Y, Meng K, Jiang L, Zhong Y, Yang Y, Lan Y, Zeng Q, Cheng L, Thymic stromal lymphopoietin-induced HOTAIR activation promotes endothelial cell proliferation and migration in atherosclerosis, Biosci. Rep 37 (4) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Madrigal-Matute J, Rotllan N, Aranda JF, Fernández-Hernando C, MicroRNAs and atherosclerosis, Curr. Atheroscler. Rep 15 (5) (2013) 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP, miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting, Cell Metabol 3 (2) (2006) 87–98. [DOI] [PubMed] [Google Scholar]
- [93].Gao W, He HW, Wang ZM, Zhao H, Lian XQ, Wang YS, Zhu J, Yan JJ, Zhang DG, Yang ZJ, Wang LS, Plasma levels of lipometabolism-related miR-122 and miR-370 are increased in patients with hyperlipidemia and associated with coronary artery disease, Lipids Health Dis 11 (2012) 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Hsu S.-h., Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, Mao H, Wei M, Clark KR, Mendell JR, Caligiuri MA, Jacob ST, Mendell JT, Ghoshal K, Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver, J. Clin. Invest 122 (8) (2012) 2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Elhanati S, Ben-Hamo R, Kanfi Y, Varvak A, Glazz R, Lerrer B, Efroni S, Cohen HY, Reciprocal regulation between SIRT6 and miR-122 controls liver metabolism and predicts hepatocarcinoma prognosis, Cell Rep 14 (2) (2016) 234–242. [DOI] [PubMed] [Google Scholar]
- [96].Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C, MiR-33 contributes to the regulation of cholesterol homeostasis, Science 328 (5985) (2010) 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM, MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis, Science 328 (5985) (2010) 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Karunakaran D, Thrush AB, Nguyen M-A, Richards L, Geoffrion M, Singaravelu R, Ramphos E, Shangari P, Ouimet M, Pezacki JP, Moore KJ, Perisic L, Maegdefessel L, Hedin U, Harper M-E, Rayner KJ, Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-miR33 in atherosclerosis, Circ. Res 117 (3) (2015) 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ, Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis, J. Clin. Invest 121 (7) (2011) 2921–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Vickers KC, Landstreet SR, Levin MG, Shoucri BM, Toth CL, Taylor RC, Palmisano BT, Tabet F, Cui HL, Rye KA, Sethupathy P, Remaley AT, MicroRNA-223 coordinates cholesterol homeostasis, Proc. Natl. Acad. Sci. U. S. A 111 (40) (2014) 14518–14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Tabet F, Vickers KC, Cuesta Torres LF, Wiese CB, Shoucri BM, Lambert G, Catherinet C, Prado-Lourenco L, Levin MG, Thacker S, Sethupathy P, Barter PJ, Remaley AT, Rye K-A, HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells, Nat. Commun 5 (2014) 3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ, MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1, Proc. Natl. Acad. Sci. U. S. A 105 (5) (2008) 1516–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Ye P, Liu J, He F, Xu W, Yao K, Hypoxia-induced deregulation of miR-126 and its regulative effect on VEGF and MMP-9 expression, Int. J. Med. Sci 11 (1) (2014) 17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN, The endothelial-specific MicroRNA miR-126 governs vascular integrity and angiogenesis, Dev. Cell 15 (2) (2008) 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, Fan H, Exosomes from endothelial progenitor cells improve the outcome of a murine model of Sepsis, Mol. Ther 26 (5) (2018) 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Köppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C, Delivery of MicroRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection, Sci. Signal 2 (100) (2009) ra81. [DOI] [PubMed] [Google Scholar]
- [107].Meng S, Cao JT, Zhang B, Zhou Q, Shen CX, Wang CQ, Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1, J. Mol. Cell. Cardiol 53 (1) (2012) 64–72. [DOI] [PubMed] [Google Scholar]
- [108].Wang S, Liao J, Huang J, Yin H, Yang W, Hu M, miR-214 and miR-126 were associated with restoration of endothelial function in obesity after exercise and dietary intervention, J. Appl. Biomed 16 (1) (2018) 34–39. [Google Scholar]
- [109].Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Blackwell TS, Baron RM, Feinberg MW, MicroRNA-181b regulates NF-κB–mediated vascular inflammation, J. Clin. Invest 122 (6) (2012) 1973–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sun X, He S, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW, Systemic delivery of microRNA-181b inhibits nuclear factor-κB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice, Circ. Res 114 (1) (2014) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, Shah P, Wisler J, Eubank TD, Tridandapani S, Paulaitis ME, Piper MG, Marsh CB, Macrophage microvesicles induce macrophage differentiation and < em > miR-223 < /em > transfer, Blood 121 (6) (2013) 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Haneklaus M, Gerlic M, Kurowska-Stolarska M, Rainey A-A, Pich D, McInnes IB, Hammerschmidt W, O’Neill LAJ, Masters SL, Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1β production, J. Immunol 189 (8) (2012) 3795. [DOI] [PubMed] [Google Scholar]
- [113].Bauernfeind F, Rieger A, Schildberg FA, Knolle PA, Schmid-Burgk JL, Hornung V, NLRP3 inflammasome activity is negatively controlled by miR-223, J. Immunol 189 (8) (2012) 4175. [DOI] [PubMed] [Google Scholar]
- [114].Li T, Morgan MJ, Choksi S, Zhang Y, Kim Y-S, Liu Z.-g., MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation, Nat. Immunol 11 (9) (2010) 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, Wang Q, Inducible MicroRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1β production in macrophages by targeting STAT3, PLoS One 7 (8) (2012) e42971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Haneklaus M, Gerlic M, O’Neill LA, Masters SL, miR-223: infection, inflammation and cancer, J. Intern. Med 274 (3) (2013) 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Wang C, Zhang C, Liu L, Chen XAB, Li Y, Du J, Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury, Mol. Ther 25 (1) (2017) 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Corsten MF, Papageorgiou A, Verhesen W, Carai P, Lindow M, Obad S, Summer G, Coort SL, Hazebroek M, van Leeuwen R, Gijbels MJ, Wijnands E, Biessen EA, De Winther MP, Stassen FR, Carmeliet P, Kauppinen S, Schroen B, Heymans S, MicroRNA profiling identifies microRNA-155 as an adverse mediator of cardiac injury and dysfunction during acute viral myocarditis, Circ. Res 111 (4) (2012) 415–425. [DOI] [PubMed] [Google Scholar]
- [119].Zhou Y, Song Y, Shaikh Z, Li H, Zhang H, Caudle Y, Zheng S, Yan H, Hu D, Stuart C, Yin D, MicroRNA-155 attenuates late sepsis-induced cardiac dysfunction through JNK and β-arrestin 2, Oncotarget 8 (29) (2017) 47317–47329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Qin D, Wang X, Li Y, Yang L, Wang R, Peng J, Essandoh K, Mu X, Peng T, Han Q, Yu K-J, Fan G-C, MiR-223–5p and −3p cooperatively suppress necroptosis in ischemic/reperfused hearts, J. Biol. Chem 291 (2016) 20247–20259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tu Y, Wan L, Fan Y, Wang K, Bu L, Huang T, Cheng Z, Shen B, Ischemic postconditioning-mediated miRNA-21 protects against cardiac ischemia/reperfusion injury via PTEN/Akt pathway, PLoS One 8 (10) (2013) e75872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Yang L, Wang B, Zhou Q, Wang Y, Liu X, Liu Z, Zhan Z, MicroRNA-21 prevents excessive inflammation and cardiac dysfunction after myocardial infarction through targeting KBTBD7, Cell Death Dis 9 (7) (2018) 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang X, Wang HX, Li YL, Zhang CC, Zhou CY, Wang L, Xia YL, Du J, Li HH, MicroRNA let-7i negatively regulates cardiac inflammation and fibrosis, Hypertension 66 (4) (2015) 776–785. [DOI] [PubMed] [Google Scholar]
- [124].Bayoumi SA, Sayed A, Broskova Z, Teoh J-P, Wilson J, Su H, Tang Y-L, Kim I.-m., Crosstalk between long noncoding RNAs and MicroRNAs in health and disease, Int. J. Mol. Sci 17 (3) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Yajima T, Viral myocarditis: potential defense mechanisms within the cardiomyocyte against virus infection, Fut. Microbiol 6 (5) (2011) 551–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Henke A, Huber S, Stelzner A, Whitton JL, The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis, J. Virol 69 (11) (1995) 6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, Rose NR, IL-12 receptor β1 and toll-like receptor 4 increase IL-1β- and IL-18-associated myocarditis and coxsackievirus replication, J. Immunol 170 (9) (2003) 4731. [DOI] [PubMed] [Google Scholar]
- [128].Triantafilou K, Orthopoulos G, Vakakis E, Ahmed MA, Golenbock DT, Lepper PM, Triantafilou M, Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly toll-like receptor (TLR) 8-dependent, Cell. Microbiol 7 (8) (2005) 1117–1126. [DOI] [PubMed] [Google Scholar]
- [129].Weinzierl AO, Szalay G, Wolburg H, Sauter M, Rammensee H-G, Kandolf R, Stevanović S, Klingel K, Effective chemokine secretion by dendritic cells and expansion of cross-presenting CD4−/CD8+ dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis, J. Virol 82 (16) (2008) 8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kawasaki T, Kawai T, Toll-like receptor signaling pathways, Front. Immunol 5 (2014) 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Fuse K, Chan G, Liu Y, Gudgeon P, Husain M, Chen M, Yeh W-C, Akira S, Liu Peter P, Myeloid differentiation Factor-88 plays a crucial role in the pathogenesis of coxsackievirus B3–induced myocarditis and influences type I interferon production, Circulation 112 (15) (2005) 2276–2285. [DOI] [PubMed] [Google Scholar]
- [132].Riad A, Westermann D, Zietsch C, Savvatis K, Becher PM, Bereswill S, Heimesaat MM, Lettau O, Lassner D, Dörner A, Poller W, Busch M, Felix SB, Schultheiss HP, Tschöpe C, TRIF is a critical survival factor in viral cardiomyopathy, J. Immunol 186 (2011) 2561–2570. [DOI] [PubMed] [Google Scholar]
- [133].Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y, Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway, Proc. Natl. Acad. Sci 92 (23) (1995) 10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Loo Y-M, Gale M, Immune signaling by RIG-I-like receptors, Immunity 34 (5) (2011) 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Jacobs JL, Coyne CB, Mechanisms of MAVS regulation at the mitochondrial membrane, J. Mol. Biol 425 (24) (2013) 5009–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Bergsbaken T, Fink SL, Cookson BT, Pyroptosis: host cell death and inflammation, Nat. Rev. Microbiol 7 (2009) 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Wang Y, Gao B, Xiong S, Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis, Am. J. Phys. Heart Circ. Phys 307 (10) (2014) H1438–H1447. [DOI] [PubMed] [Google Scholar]
- [138].Kraft L, Erdenesukh T, Sauter M, Tschöpe C, Klingel K, Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling, Basic Res. Cardiol 114 (2) (2019) 11. [DOI] [PubMed] [Google Scholar]
- [139].Boddi M, Abbate R, Chellini B, Giusti B, Solazzo V, Sofi F, Pratesi G, Pratesi C, Gensini G, Zignego AL, HCV infection facilitates asymptomatic carotid atherosclerosis: preliminary report of HCV RNA localization in human carotid plaques, Dig. Liver Dis 39 (2007) S55–S60. [DOI] [PubMed] [Google Scholar]
- [140].Boddi M, Abbate R, Chellini B, Giusti B, Giannini C, Pratesi G, Rossi L, Pratesi C, Gensini GF, Paperetti L, Zignego AL, Hepatitis C virus RNA localization in human carotid plaques, J. Clin. Virol 47 (1) (2010) 72–75. [DOI] [PubMed] [Google Scholar]
- [141].Pircher J, Czermak T, Merkle M, Mannell H, Krötz F, Ribeiro A, Vielhauer V, Nadjiri J, Gaitzsch E, Niemeyer M, Porubsky S, Gröne H-J, Wörnle M, Hepatitis C virus induced endothelial inflammatory response depends on the functional expression of TNFα receptor subtype 2, PLoS One 9 (11) (2014) e113351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tawar RG, Mailly L, Baumert TF, A new HCV mouse model on the block, Cell Res 24 (10) (2014) 1153–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Chetty R, Vasculitides associated with HIV infection, J. Clin. Pathol 54 (4) (2001) 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Zhu J, Quyyumi AA, Norman JE, Csako G, Epstein SE, Cytomegalovirus in the pathogenesis of atherosclerosis, J. Am. Coll. Cardiol 34 (6) (1999) 1738. [DOI] [PubMed] [Google Scholar]
- [145].Bock CT, Klingel K, Aberle S, Duechting A, Lupescu A, Lang F, Kandolf R, Human parvovirus B19: a new emerging pathogen of inflammatory cardiomyopathy, J. Veterinary Med. Ser. B 52 (7–8) (2005) 340–343. [DOI] [PubMed] [Google Scholar]
- [146].Van Taunay JS, Biologics and cardiovascular disease, J. Cardiovasc. Pharmacol 72 (2) (2018). [DOI] [PubMed] [Google Scholar]