

Abstract
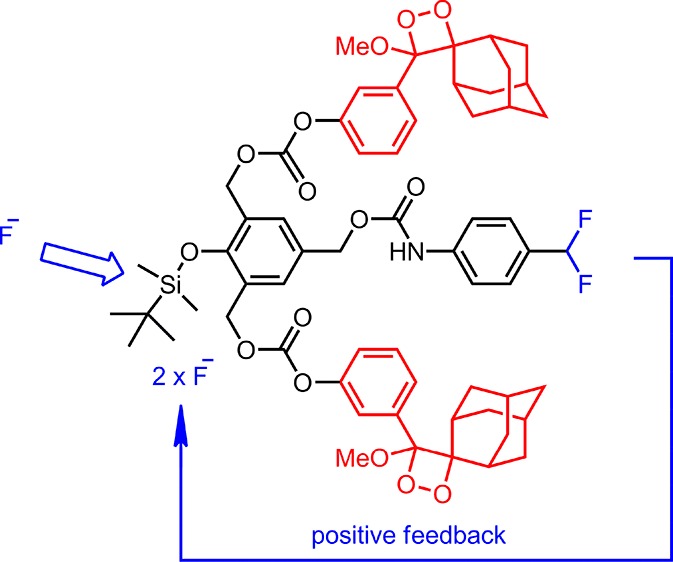
Bringing together the concepts of self-immolative linkers and chemiluminogen dioxetane modules, a chemiluminescence-based sensor for fluoride with signal amplification is presented. Signal amplification is obtained by triggering two chemiluminescence events for each reacting fluoride ion that in turn releases two fluoride ions for each ion. As expected, the chemiluminescence signal starts to rise following an induction period. In addition to the analytical potential, this chemical system is also of interest as a demonstration of positive feedback loop character.
Introduction
Molecular agents that respond to chemical species of interest, with externally observable/detectable responses proved to be immensely useful. In addition, they served as a proving ground for testing a number of photophysical concepts. Fluoride ion is an important species as an analytical target as it is implicated in certain disease states.1 As a chemosensing target, it is challenging owing to its very strong hydrogen bonding capability in aqueous solution, which deactivates it toward additional interactions.
There are a number of strategies used for sensing fluoride ions in polar solvents.2 Most of them are reaction-based. High affinity of fluoride ions for silicon centers in silyl groups is often exploited in such chemosensor designs. tert-Butyldimethylsilyl is such a group, which would leave alkoxy or phenoxy units following the reaction with fluoride ions.3 In fact, the smooth reactivity under very mild conditions makes these groups very useful as protecting groups for phenols, alcohols, and amines.4
On the other hand, self-immolative linkers were shown to be very valuable in drug release.5 Typically, a quinone methide rearrangement leads to the cleavage of a critical bond, releasing an active agent or initiating further breakdown in the molecular structure.6 Single trigger may initiate one (1,4) or multiple (1,4; 1,2; and 1,6) quinone methide reactions.
In order to make use of fluoride-initiated self-immolative breakdown of the linker for chemiluminescence signaling, we made use of sterically stabilized adamantyl dioxetane units as the chemiluminogen unit.7 Chemical triggering of chemiluminescence using this particular module is well-documented in the literature.8 Final addition to our design (Scheme 1) was the strategic placement of the protected 4-difluoromethylaniline module: the attachment was made in the form of a carbamate at the para-position to the silyloxy substituent.
Scheme 1. Synthesis of a Chemiluminescent Signal Amplifier 10.

As an external source of fluoride is introduced, a number of chemical reactions are expected to be triggered in accord with this design: the initial removal of the silyl group, which leads to 1,2- and 1,6-quinone methide reactions that would be then coupled to carbonate and carbamate decompositions, generating chemiluminescence as a result of dioxetane ring opening, and the 1,4-quinone methide reaction results in the release of unstable p-difluoromethylaniline-derived species, which eventually ejects two fluoride ions. Thus, signal amplification can be achieved by a positive feedback loop or autoinduction.
Results and Discussion
The synthesis scheme is shown in Scheme 1. p-Hydroxybenzoate ester 1 can be converted into bishydroxymethyl-substituted compound 2. All three hydroxyl functionalities were then protected with tert-butyldimethylsilyl chloride (TBDMSCl). Diisobutyl aluminum hydride (DIBAL-H) reduction of the ester yields benzylic alcohol 4. Curtius rearrangement9 of acyl azide obtained from 4-(difluoromethyl)-benzoic acid, by the reaction with SOCl2, followed by NaN3, yields an isocyanate that generates the carbamate 5 on reaction with the benzylic alcohol 4. Mild deprotection conditions with p-toluenesulfonic acid (TsOH) remove benzylic protections while keeping the silylated phenol group untouched (6). p-Nitrophenylchloroformate activation yields a reactive carbonate 7. This compound was then reacted with the adamantyl derivative 8(2d) to yield the penultimate compound 9 that was then converted into stable dioxetane that is the target compound 10, by singlet oxygen addition to the electron-rich double bond.
Following spectroscopic characterization, our first investigation was the titration of the chemiluminescence response (Figure 1) and the demonstration of selectivity of the response (Figure 2). It is clear that micromolar concentrations of fluoride ions lead to clear luminescence emission with a peak at 470 nm.
Figure 1.
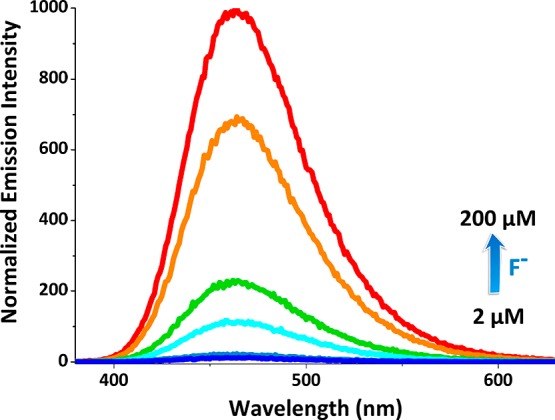
Normalized chemiluminescence spectra of compound 10 in the presence of increasing F– concentrations. Probe concentration is 200 μM in dimethyl sulfoxide (DMSO). [Chemiluminescence intensities were recorded 10 s after the addition of tetra-n-butylammonium fluoride (TBAF)].
Figure 2.
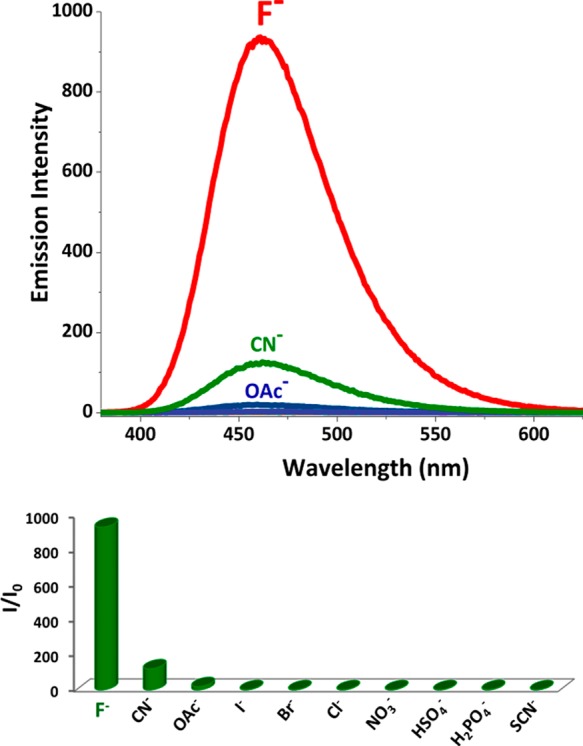
Top: Chemiluminescence spectra of compound 10 upon the addition of 1.0 equiv of I–, Br–, Cl–, SCN–, CN–, AcO–, H2PO4–, HSO4–, NO3–, and F–, and the probe concentration is 200 μM in DMSO. (Chemiluminescence spectra were recorded 10 s after the addition of TBAF). Bottom: The bar graph representing intensity enhancement over the background signal.
As expected, the chemiluminescence signal is highly selective. Only the other response at the same concentration of the anions was observed with CN– ions, which was considerably weaker compared to the fluoride response. The design of the probe (Figure 3) is such that for every fluoride ion that reacts to remove the silyl protecting group will lead to a cascade of events that would result in triggering two chemiluminescence events and generate two fluoride ions as a result of 4-(difluoromethyl)aniline hydrolysis.
Figure 3.
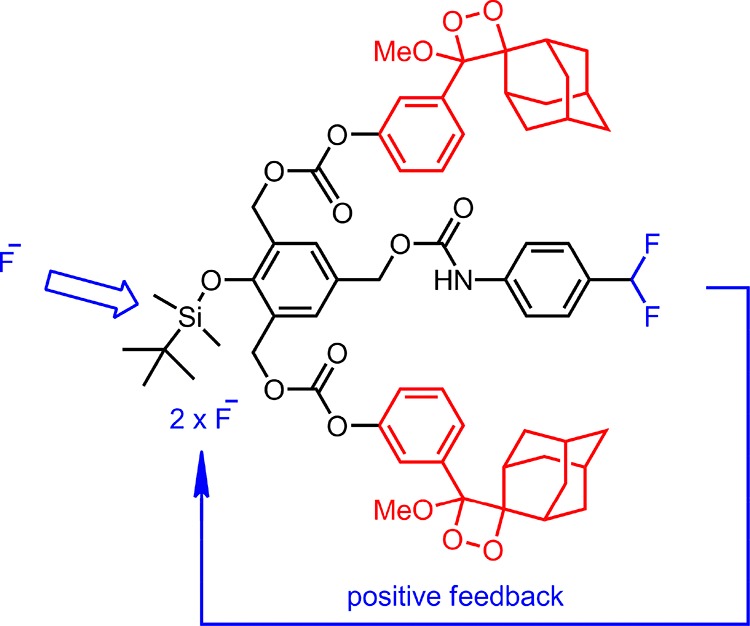
Amplification of fluoride signal, resulting in faster enhanced chemiluminescence signal.
Considering the number of independent steps involved in signal generation, actual kinetics of the process can be complicated.
Depending on the initial concentration of the fluoride ions, the chemiluminescence signal at all initial concentrations starts to rise only after an initial lag period. It is also interesting to note that at high fluoride concentrations there is a clear dip in the signal intensity for the first few seconds, followed by an increase, most likely indicating faster dioxetane opening compared with other processes involved. Small oscillations in the signal intensity as shown in the figure above (Figure 4) are also to be expected, as autoinductive (self-amplified) processes with essential steps of different rates result in nonlinear behavior.
Figure 4.

Normalized chemiluminescence emission data for compound 10 as a function of time in the presence of increasing F– concentrations. The probe concentration is 200 μM in DMSO. Chemiluminescence data were recorded 10 s after the addition of TBAF.
Conclusions
In conclusion, we were able to demonstrate that ion signals can be amplified and coupled to a chemiluminescence output. Also, it appears that the rational design of oscillating reactions based on similar systems would be possible by studying the rates of the different processes involved.
Our work in that direction is in progress.
Experimental Section
Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker Spectrospin Avance DPX 400 spectrometer using CDCl3 as the solvent. Chemical shift values were reported in ppm from tetramethylsilane as internal standard. Spin multiplicities were reported as follows: s (singlet), d (doublet), and m (multiplet). High-resolution mass spectrometry (HRMS) data were acquired on an Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS. Chemiluminescence measurements were taken on a Varian Eclipse spectrofluorometer. For other experimental details, please refer to an earlier publication.10
Synthesis of Compound 2
To a cold 12% NaOH (70.0 mL, 0.21 mol), commercially available ethyl-4-hydroxybenzoate 1 (15.0 g, 0.09 mol) was added while being cooled to 0 °C. Formaldehyde (37% in water, 60.0 mL, 4.0 mol) was added. The reaction mixture was stirred at 55 °C for 3 days and was monitored by thin-layer chromatography (TLC) (EtOAc/hexane, 3:2, v/v). After completion, the reaction mixture was diluted with EtOAc and was washed with NH4Cl. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by recrystallization with EtOAc to give compound 2 (11.07 g, 54%). 1H NMR (200 MHz, CDCl3): δ 9.39 (s, 1H), 7.84 (s, 2H), 5.37 (s, 2H), 4.56 (s, 4H), 4.27 (q, J = 8.0, 4.0 Hz, 2H), 1.30 (t, J = 8.0 Hz, 3H). MS (TOF-ESI) m/z: calcd: 226.08412 [M – H]−; found, 226.08568 [M – H]−, Δ = −6.88 ppm.
Synthesis of Compound 3
Compound 2 (1.0 g, 4.42 mmol) was dissolved in dimethylformamide (DMF) (5.0 mL) and cooled to 0 °C. Imidazole (0.993 g, 14.59 mmol) and TBDMSCl (2.19 g, 14.59 mmol) were added. The reaction mixture was allowed to warm to room temperature and was stirred for an additional 2 h. The reaction was then monitored by TLC. Upon completion, the reaction mixture was diluted with diethyl ether and washed with a saturated NH4Cl solution followed by brine. The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel dichloromethane (DCM)/hexane (1:1, v/v). Compound 3 was obtained as colorless oil (2.19 g, 87%). 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 2H), 4.74 (s, 4H), 4.36 (q, J = 8.0, 4.0 Hz, 2H), 1.39 (t, J = 8.0 Hz, 3H), 1.04 (s, 9H), 0.98 (s, 18H), 0.22 (s, 6H), 0.12 (s, 12H). 13C NMR (100 MHz, CDCl3): δ 131.9, 127.9, 124.0, 60.5, 60.4, 26.0, 25.9, 18.3, 14.2, −3.2, −5.2 ppm. MS (TOF-ESI) m/z: calcd: 591.33278 [M + Na]+; found, 591.33578 [M + Na]+, Δ = −5.08 ppm.
Synthesis of Compound 4
Compound 3 (1.0 g, 1.76 mmol) was dissolved in dry THF (10.0 mL) under an argon atmosphere and cooled to −78 °C. DIBAL-H (1 M in toluene, 8.50 mL, 8.50 mmol) was added dropwise. The reaction mixture was stirred for 30 min and was monitored by TLC. Upon completion, the reaction mixture was quenched with a saturated NH4Cl solution (5.0 mL) and diluted with diethyl ether. Celite was added, and the reaction mixture was stirred at room temperature for 15 min. After filtration, the organic layer was dried over magnesium sulfate, and the solvent was removed under reduced pressure. The crude product was purified by flash column chromatography on silica gel DCM/methanol (98:2, v/v). Compound 4 was obtained as colorless oil (0.742 g, 80%). 1H NMR (400 MHz, CDCl3): δ 7.40 (s, 2H), 4.74 (s, 4H), 4.66 (d, J = 4.0 Hz, 2H), 1.06 (s, 9H), 0.97 (s, 18H), 0.20 (s, 6H), 0.12 (s, 12H). 13C NMR (100 MHz, CDCl3): δ 147.6, 134.1, 131.8, 125.1, 65.6, 60.6, 26.0, 25.9, 18.4, −3.3, −5.2 ppm. MS (TOF-ESI) m/z: calcd: 549.32221 [M + Na]+; found, 549.32564 [M + Na]+, Δ = −6.24 ppm.
Synthesis of Compound 5
p-Difluoromethylbenzoic acid (0.140 mg, 0.81 mmol) was dissolved in 1,2-dichloroethane (2.0 mL), and the resulting solution was cooled to 0 °C. Thionyl chloride (0.148 mL, 2.03 mmol) was added dropwise to the ice-cold solution, followed by 4 drops of N,N-DMF. The reaction mixture was heated to 100 °C and was stirred for 1 h. The resultant solution was allowed to cool to room temperature and was concentrated under reduced pressure. The resulting residue was redissolved in acetone (1.0 mL) and was cooled to 0 °C. A solution of sodium azide (0.159 g, 2.44 mmol; dissolved in 1.0 mL water) was added dropwise, and the reaction mixture was stirred at 0 °C for 1 h. The solution was diluted with ethyl acetate (10.0 mL), and the layers were separated. The organic layer was dried over sodium sulfate, the solids were filtered through a fritted Büchner funnel, and the remaining liquid was concentrated under reduced pressure. The residue was dissolved in toluene (3.0 mL), and this solution was heated to 100 °C for 1 h. The reaction mixture was allowed to cool to room temperature, and compound 4 (0.514 g, 0.976 mmol) dissolved in toluene was added in one portion. The reaction mixture was heated to 100 °C and stirred for 3 h. The reaction mixture was cooled to room temperature, and the solution was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel DCM/hexane (1:1, v/v). Compound 5 was obtained as colorless oil (0.450 g, 80%). 1H NMR (400 MHz, CDCl3): δ 7.5 (d, J = 12.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.44 (s, 2H), 6.63 (t, J = 56.0 Hz, 1H), 5.21 (s, 2H), 4.74 (s, 4H), 1.06 (s, 9H), 0.96 (s, 18H), 0.20 (s, 6H), 0.11 (s, 12H). 13C NMR (100 MHz, CDCl3): δ 148.2, 132.0, 128.8, 126.6, 126.5, 126.5, 126.1, 118.2, 114.6, 67.6, 60.5, 31.5, 26.0, 25.9, 14.1, −3.3, −5.2 ppm. MS (TOF-ESI) m/z: calcd: 718.35613 [M + Na]+; found, 718.36340 [M + Na]+, Δ = −10.12 ppm.
Synthesis of Compound 6
Compound 5 (0.450 g, 0.65 mmol) was dissolved in 2.0 mL of methanol. After adding a catalytic amount of TsOH, the reaction mixture was stirred at room temperature. The progress of the reaction was monitored by TLC. When TLC showed no starting material, the reaction mixture was diluted with EtOAc and extracted first with the saturated solution of NaHCO3 and brine. Combined organic phases were dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by silica gel flash column chromatography using EtOAc/hexane (1:5, v/v) as the eluant. Compound 6 was obtained as pale yellow solid (0.292 g, 96%). 1H NMR (400 MHz, CDCl3): δ 7.60 (br, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.30 (s, 2H), 6.59 (t, J = 56.0 Hz, 1H), 5.02 (s, 2H), 4.63 (s, 4H), 2.84 (br, 2H), 1.02 (s, 9H), 0.17 (s, 6H). MS (TOF-ESI) m/z: calcd: 490.18318 [M + Na]+; found, 490.18673 [M + Na]+, Δ = −7.25 ppm.
Synthesis of Compound 7
A solution of compound 6 (0.290 g, 0.62 mmol) was dissolved in dry DCM (5.0 mL), and then the reaction mixture was cooled to 0 °C. Triethyl amine (0.346 mL, 2.48 mmol) and a catalytic amount of 4-dimethylaminopyridine (DMAP) were added. Then, 4-nitrophenyl chloroformate (0.500 g, 2.48 mmol) dissolved in dry DCM was added dropwise to the reaction mixture at 0 °C. The reaction mixture was allowed to be stirred at room temperature. The progress of the reaction was monitored by TLC. When TLC showed no starting material, the reaction mixture was diluted with EtOAc and extracted first with the saturated solution of NH4Cl and brine. Combined organic phases were dried over anhydrous Na2SO4. After removal of the solvent, the residue was purified by silica gel flash column chromatography using EtOAc/hexane (1:5, v/v) as the eluant. Compound 7 was obtained as pale yellow solid (0.242 g, 55%). 1H NMR (400 MHz, CDCl3): δ 8.29 (d, J = 8.0 Hz, 4H), 7.55 (s, 2H), 7.51–7.46 (m, 4H), 7.4 (d, J = 8.0 Hz, 4H), 6.62 (t, J = 56.0 Hz, 1H), 5.36 (s, 4H), 5.23 (s, 2H), 1.09 (s, 9H), 0.29 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 155.4, 152.4, 151.7, 145.5, 131.2, 129.9, 126.7, 126.6, 126.6, 126.2, 126.1, 125.3, 121.5, 118.4, 115.6, 66.1, 66.0, 25.8, 18.7, −3.6 ppm. MS (TOF-ESI) m/z: calcd: 820.19559 [M + Na]+; found, 820.20374 [M + Na]+, Δ = −9.93 ppm.
Synthesis of Compound 9
Compound 7 (0.050 g, 0.064 mmol) was dissolved in DCM. After the addition of compound 8(2d) (0.0415 g, 0.15 mmol) and DMAP (0.019 g, 0.15 mmol), the reaction mixture was stirred at room temperature overnight. When the starting material was consumed, the mixture was concentrated under vacuo and the residue was subjected to the silica gel flash column chromatography using EtOAc/hexane (1:5, v/v) as the eluant. Compound 9 was obtained as yellow oil (0.054 g, 80%). 1H NMR (400 MHz, CDCl3): δ 7.54 (s, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.0 Hz, 2H), 7.37 (t, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 7.17 (s, 2H), 7.13 (d, J = 8.0 Hz, 2H), 6.95 (s, 1H), 6.62 (t, J = 56.0 Hz, 1H), 5.33 (s, 4H), 5.21 (s, 2H), 3.33 (s, 6H), 3.27 (s, 2H), 2.69 (s, 2H), 1.78–1.89 (m, 24H), 1.09 (s, 9H), 0.29 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 153.5, 151.5, 150.9, 142.5, 137.2, 132.9, 130.8, 129.6, 128.9, 127.0, 126.7, 126.6, 126.5, 121.7, 119.9, 118.4, 114.5, 114.0, 66.5, 66.5, 57.9, 39.1, 39.0, 37.1, 32.1, 30.3, 28.2, 25.9, −3.6 ppm. MS (TOF-ESI) m/z: calcd: 1082.46567 [M + Na]+; found, 1082.47612 [M + Na]+, Δ = −9.66 ppm.
Synthesis of Compound 10
Compound 9 (0.054 g, 0.051 mmol) was dissolved in DCM. Methylene blue (5.0 mg) was added to the reaction mixture at −78 °C, which was irradiated while oxygen gas was passed through it. The progress of the reaction was monitored by TLC. When TLC showed no starting material, the mixture was concentrated under vacuo and the residue was subjected to the silica gel flash column chromatography by using DCM as the eluant. Compound 10 was obtained as white solid (0.055 g, 96%). 1H NMR (400 MHz, CDCl3): δ 7.46–7.55 (m, 14H), 6.93 (s, 1H), 6.62 (t, J = 56.0 Hz, 1H), 5.34 (s, 4H), 5.22 (s, 2H), 3.25 (s, 6H), 3.06 (s, 2H), 2.16 (s, 2H), 1.61–1.91 (m, 24H), 1.10 (s, 9H), 0.29 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 153.4, 151.1, 136.7, 131.0, 129.7, 129.3, 126.7, 126.6, 126.5, 126.4, 122.04, 118.4, 114.5, 111.5, 95.4, 66.4, 65.6, 50.0, 36.3, 34.7, 33.1, 32.8, 32.2, 31.7, 31.5, 29.6, 26.0, 25.9, 25.8, −3.6 ppm.
Acknowledgments
Support from the Bilkent University in the form of a graduate student scholarship to S.A. and a postdoctoral scholarship to O.S. (TUBITAK-2218) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00537.
Additional spectral and characterization data, including 1H, 13C NMR, and HRMS data (PDF)
Author Contributions
The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Farley J. R.; Wergedal J. E.; Baylink D. J. Science 1983, 222, 330–332. 10.1126/science.6623079. [DOI] [PubMed] [Google Scholar]; b Horowitz H. S. J. Publ. Health Dent. 2003, 63, 3–8. 10.1111/j.1752-7325.2003.tb03467.x. [DOI] [PubMed] [Google Scholar]; c Kleerekoper M. Endocrinol. Metab. Clin. North Am. 1998, 27, 441–452. 10.1016/s0889-8529(05)70015-3. [DOI] [PubMed] [Google Scholar]; d Lennon M. A. Bull. W. H. O. 2006, 84, 759–760. 10.2471/blt.05.028209. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wimalawansa S. J. Environ. Geochem. Health 2016, 38, 639–678. 10.1007/s10653-015-9768-y. [DOI] [PubMed] [Google Scholar]; f Yang Z.; Zhang K.; Gong F.; Li S.; Chen J.; Ma J. S.; Sobenina L. N.; Mikhaleva A. I.; Yang G.; Trofimov B. A. Beilstein J. Org. Chem. 2011, 7, 46–52. 10.3762/bjoc.7.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baker M. S.; Phillips S. T. Org. Biomol. Chem. 2012, 10, 3595–3599. 10.1039/c2ob25363a. [DOI] [PubMed] [Google Scholar]; b Black C. B.; Andrioletti B.; Try A. C.; Ruiperez C.; Sessler J. L. J. Am. Chem. Soc. 1999, 121, 10438–10439. 10.1021/ja992579a. [DOI] [Google Scholar]; c Cao X.; Lin W.; Yu Q.; Wang J. Org. Lett. 2011, 13, 6098–6101. 10.1021/ol202595t. [DOI] [PubMed] [Google Scholar]; d Turan I. S.; Akkaya E. U. Org. Lett. 2014, 16, 1680. 10.1021/ol5003412. [DOI] [PubMed] [Google Scholar]; e Zhang J. F.; Lim C. S.; Bhuniya S.; Cho B. R.; Kim J. S. Org. Lett. 2011, 13, 1190–1193. 10.1021/ol200072e. [DOI] [PubMed] [Google Scholar]
- a Adam W.; Trofimov A. V. J. Org. Chem. 2000, 65, 6474–6478. 10.1021/jo000495a. [DOI] [PubMed] [Google Scholar]; b Baker M. S.; Phillips S. T. J. Am. Chem. Soc. 2011, 133, 5170–5173. 10.1021/ja108347d. [DOI] [PubMed] [Google Scholar]; c Matsumoto M.; Mizuno T.; Watanabe N. Chem. Commun. 2003, 482–483. 10.1039/b210857g. [DOI] [PubMed] [Google Scholar]; d Nery A. L. P.; Röpke S.; Catalani L. H.; Baader W. J. Tetrahedron Lett. 1999, 40, 2443–2446. 10.1016/s0040-4039(99)00236-1. [DOI] [Google Scholar]; e Schaap A. P.; Chen T.-S.; Handley R. S.; DeSilva R.; Giri B. P. Tetrahedron Lett. 1987, 28, 1155–1158. 10.1016/s0040-4039(00)95313-9. [DOI] [Google Scholar]; f Watanabe N.; Nagashima Y.; Yamazaki T.; Matsumoto M. Tetrahedron 2003, 59, 4811–4819. 10.1016/s0040-4020(03)00727-0. [DOI] [Google Scholar]
- a Corey E. J.; Venkateswarlu A. J. Am. Chem. Soc. 1972, 94, 6190–6191. 10.1021/ja00772a043. [DOI] [Google Scholar]; b Djuric S. W. J. Org. Chem. 1984, 49, 1311–1312. 10.1021/jo00181a045. [DOI] [Google Scholar]; c Sabitha G.; Syamala M.; Yadav J. S. Org. Lett. 1999, 1, 1701–1703. 10.1021/ol990140h. [DOI] [Google Scholar]
- a Avital-Shmilovici M.; Shabat D. Soft Matter 2010, 6, 1073–1080. 10.1039/b922341j. [DOI] [Google Scholar]; b Lee C. C.; MacKay J. A.; Fréchet J. M. J.; Szoka F. C. Nat. Biotechnol. 2005, 23, 1517–1526. 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]; c Sagi A.; Weinstain R.; Karton N.; Shabat D. J. Am. Chem. Soc. 2008, 130, 5434–5435. 10.1021/ja801065d. [DOI] [PubMed] [Google Scholar]; d Wang W.; Alexander C. Angew. Chem., Int. Ed. 2008, 47, 7804–7806. 10.1002/anie.200802474. [DOI] [PubMed] [Google Scholar]
- a Amir R. J.; Pessah N.; Shamis M.; Shabat D. Angew. Chem. 2003, 115, 4632–4637. 10.1002/ange.200351962. [DOI] [PubMed] [Google Scholar]; b Gabrielli L.; Mancin F. J. Org. Chem. 2016, 81, 10715–10720. 10.1021/acs.joc.6b01787. [DOI] [PubMed] [Google Scholar]
- a Gu J.-A.; Mani V.; Huang S.-T. Analyst 2015, 140, 346–352. 10.1039/c4an01723d. [DOI] [PubMed] [Google Scholar]; b Kim T.-H.; Swager T. M. Angew. Chem., Int. Ed. 2003, 42, 4803–4806. 10.1002/anie.200352075. [DOI] [PubMed] [Google Scholar]; c Mahoney K. M.; Goswami P. P.; Winter A. H. J. Org. Chem. 2012, 78, 702–705. 10.1021/jo301698v. [DOI] [PubMed] [Google Scholar]; d Perry-Feigenbaum R.; Sella E.; Shabat D. Chem.—Eur. J. 2011, 17, 12123–12128. 10.1002/chem.201101796. [DOI] [PubMed] [Google Scholar]
- a Adam W.; Bronstein I.; Trofimov A. V. J. Phys. Chem. A 1998, 102, 5406–5414. 10.1021/jp973175c. [DOI] [Google Scholar]; b Adam W.; Fell R.; Schulz M. H. Tetrahedron 1993, 49, 2227–2238. 10.1016/s0040-4020(01)80366-5. [DOI] [Google Scholar]; c Adam W.; Matsumoto M.; Trofimov A. V. J. Org. Chem. 2000, 65, 2078–2082. 10.1021/jo991665q. [DOI] [PubMed] [Google Scholar]; d Adam W.; Reinhardt D. J. Chem. Soc., Perkin Trans. 2 1997, 1453–1464. 10.1039/a701189j. [DOI] [Google Scholar]; e Adam W.; Reinhardt D.; Saha-Möller C. R. Analyst 1996, 121, 1527–1531. 10.1039/an9962101527. [DOI] [Google Scholar]; f Hananya N.; Boock A. E.; Bauer C. R.; Satchi-Fainaro R.; Shabat D. J. Am. Chem. Soc. 2016, 138, 13438–13446. 10.1021/jacs.6b09173. [DOI] [PubMed] [Google Scholar]; g MacManus-Spencer L. A.; Latch D. E.; Kroncke K. M.; McNeill K. Anal. Chem. 2005, 77, 1200–1205. 10.1021/ac048293s. [DOI] [PubMed] [Google Scholar]; h Richard J.-A.; Jean L.; Schenkels C.; Massonneau M.; Romieu A.; Renard P.-Y. Org. Biomol. Chem. 2009, 7, 2941–2957. 10.1039/b905725k. [DOI] [PubMed] [Google Scholar]; i Roda A.Chemiluminescence and Bioluminescence; RSC Publishing: Cambridge, 2010. [Google Scholar]; j Turan I. S.; Yilmaz O.; Karatas B.; Akkaya E. U. RSC Adv. 2015, 5, 34535–34540. 10.1039/c5ra01551k. [DOI] [Google Scholar]; k Seven O.; Sozmen F.; Simsek-Turan I. Sens. Actuators, B 2017, 239, 1318–1324. 10.1016/j.snb.2016.09.120. [DOI] [Google Scholar]
- Shioiri T.; Ninomiya K.; Yamada S. J. Am. Chem. Soc. 1972, 94, 6203–6205. 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- Turan I. S.; Cakmak F. P.; Sozmen F. Tetrahedron Lett. 2014, 55, 456–459. 10.1016/j.tetlet.2013.11.059. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.