

Abstract
Introduction:
Various phenotypes have been identified for MYH7 gene mutation-related myopathy. Here, we describe a patient with severe muscular weakness and skeletal deformity with de novo heterozygous MYH7 gene mutation.
Patient concerns:
A 33-year-old woman presented with early onset of muscular weakness, with delayed motor development during infancy. At age 8 years, she was unable to walk, with signs of skeletal deformity, including the progression of kyphoscoliosis. At age 31 years, she developed dyspnea.
Diagnosis:
She diagnosed with esophageal hiatal hernia with abdominal CT. In electromyography, short duration, small amplitude motor unit action potential (MUAP), and early recruitment patterns were observed in the involved proximal muscles, suggesting myopathy. Muscle histopathology showed fiber-type disproportion.
Interventions:
Next-generation sequencing study revealed a heterozygous in-frame deletion variation in the exon 14 of the MYH7 gene (c.1498_1500del/p.Glu500del), which is a novel variation confirmed by conventional Sanger sequencing. Compared with the parental test, this variant was concluded as de novo.
Outcomes:
She received laparoscopic hiatal hernia repair and Nissen fundoplication for esophageal hiatal hernia. After surgery, her postural dyspnea improved. As there is no fundamental treatment for MYH7-related myopathies, she continued conservative treatment for her symptoms.
Conclusion:
Here, we presented a rare case of de novo mutation of the myosin head domain in the MYH7 gene. This report broadens both the phenotypic and genotypic spectra of MYH7-related myopathies.
Keywords: distal myopathies, high-throughput nucleotide sequencing, human, MYH7 protein
1. Introduction
MYHCI (slow skeletal muscle / b-cardiac myosin heavy chain), a type of myosin heavy chain, is expressed by the MYH7 gene and presented primarily in the slow twitch type I skeletal muscle and heart muscle of the ventricle.[1] MyHCI, found in the skeletal muscle, is known to be important for maintaining normal posture.[2]
Various phenotypes have been reported to be associated with MYH7 gene mutation-related myopathy, including familial hypertrophic / dilated cardiomyopathy (CMH1; MIM # 192600), Laing distal myopathy (MPD1; MIM # 160500), myosin storage myopathy (MSM; MIM # 608358), and Congenital fiber-type disproportion (CFTD; MIM # 255310).[3–5]
Here, we describe a patient with severe muscular weakness and skeletal deformity with de novo heterozygous MYH7 gene mutation.
2. Case reports
A 33-year-old female patient visited the outpatient department of Rehabilitation Clinic for further evaluation of severe scoliosis and weakness. There were no significant abnormal findings during the perinatal period and at the time of birth; however, the development was delayed from the time of birth. She was referred to a neurologist at age 12 months. At that time, she presented muscle weakness and hypotonic posture; she was clinically diagnosed as muscular dystrophy without gene study. At age 2 years, she was able to walk alone; at age 8 years, she was unable to walk, completely dependent on a wheelchair due to gradual worsening of weakness. At the same time, there was progression of skeletal deformity and kyphoscoliosis.
At age 31 years, she developed dyspnea. she visited another neurologist for evaluation, and SMN 1del/dup test was performed under suspicion of motor neuron disease, SMA; however, no specific findings were observed.
At age 33 years, she visited our rehabilitation clinic for the treatment of dyspnea. After admission, a comprehensive reevaluation was done for systemic review. At first, physical exam, serologic test, and neurophysiologic test, including nerve conduction study (NCS) and electromyogram (EMG), were performed. On manual muscle test (MMT), she received a grade of 4 for wrist flexor, extensor muscle, finger flexor, and extensor muscle, and a grade of 1 for all other upper and lower limb muscles. Moreover, the contracture associated with weakness further progressed to the upper and lower limbs. The patient was in a state of immobilization in shoulder flexed, elbow flexed, hip/knee flexed, and ankle plantar flexed posture, except for the wrist and finger. Kyphoscoliosis progressed severely, and the Cobb angle was 126° at the time of measurement (Fig. 1A, B). We did not observe any abnormal signs on the upper motor neuron. Cognitive function was normal, and her family history was uneventful. She was diagnosed with esophageal hiatal hernia with abdominal CT.
Figure 1.
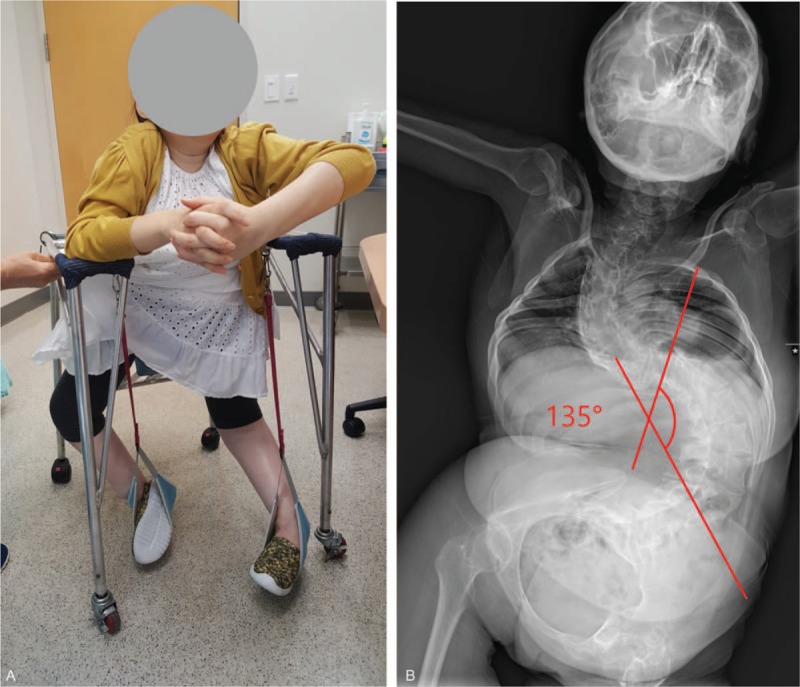
These figures show the multiple contractures and kyphoscoliosis. The patient was in a state of immobilization in shoulder flexed, elbow flexed, hip / knee flexed, and ankle plantar flexed posture, except for the wrist and finger. Kyphoscoliosis progressed severely, and the Cobb angle was 126° at the time of measurement (A, B).
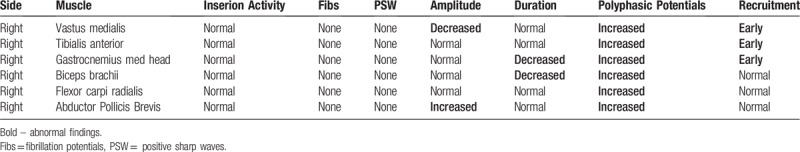
Nerve conduction studies showed normal findings (Table 1). In electromyography, short duration, small amplitude motor unit action potential (MUAP), and early recruitment patterns were observed in the involved proximal muscles, suggesting myopathy (Table 2).
Table 1.
Electrodiagnostic testing analysis and summary.
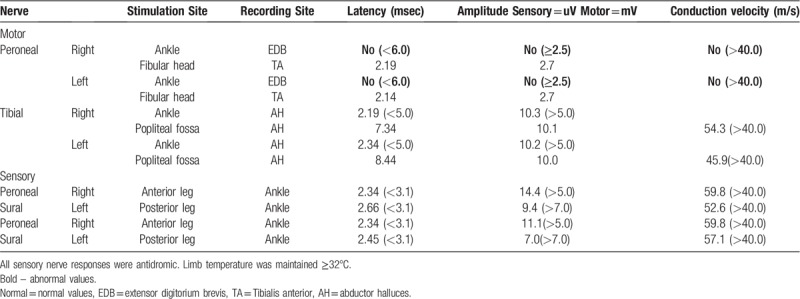
Table 2.
Concentric needle EMG analysis and summary.
The chromosomal microarray showed no abnormal findings. We performed the Next-generation sequencing (NGS) study on the patient. The patient was analyzed via genetic testing, using a targeted gene sequencing panel, analyzing 293 genes—5609 exons—associated with genetic neuromuscular diseases. Genomic DNA was extracted from the peripheral blood of the patient. Library preparation and target enrichment were performed using the hybridization capture method. Custom oligo design and synthesis were done by Celemics (Korea). Massively parallel sequencing was performed using 2 × 150 bp in the paired-end mode of MiSeq platform (Illumina, San Diego, CA). Sequence reads were aligned with the Burrow–Wheeler Aligner (version 0.7.12, MEM algorithm, MEM algorithm). After duplicated reads were removed with Picard, local realignment and recalibration were performed using the Genome Analysis Tool Kit (GATK, version 3.5). Variant calling was conducted with GATK. Variants were annotated by Variant Effect Predictor and dbNSFP. Common variants with minor allele frequency ≥1% were filtered using public databases (1000 Genomes Project, Exome Variant Server, Exome Aggregation Consortium). Average coverage of depth was 100X, and 99% of target bases were covered by 10X sequence reads.
We found a heterozygous in-frame deletion variation in the patient's MYH7 gene in exon 14 (c.1498_1500del/p.Glu500del), which is a novel variation confirmed by conventional Sanger sequencing. This variant was concluded as de novo compared with the parental conventional Sanger sequencing (Fig. 2). This variation has not been reported in the control databases, such as the 1000 Genomes Project, Exome Aggregation Consortium, gnomAD, and the dbSNP Database.
Figure 2.
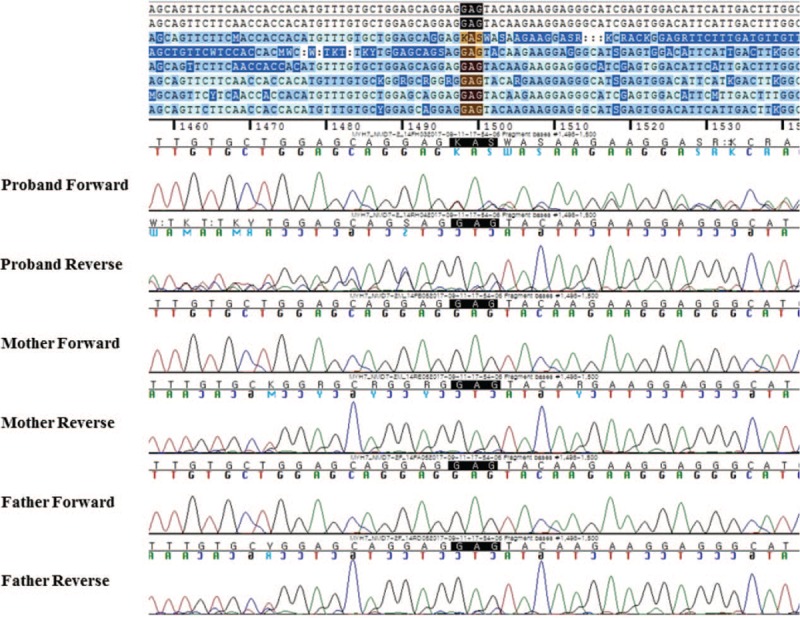
These figures show a heterozygous in-frame deletion variation in the patient's MYH7 gene in exon 14 (c.1498_1500del/p.Glu500del), which is a novel variation confirmed by conventional Sanger sequencing. This variant was concluded as de novo compared with the parental conventional Sanger sequencing.
Muscle biopsy samples were taken from the vastus lateralis muscle for confirmation. Immunohistochemical and histological studies, including the electron microscopic exam, were performed. Histochemical evidence showed specific atrophy, with increase fiber size variability and increased nuclear internalization of type I fiber on muscle biopsy, consistent with congenital myopathy. On electron microscopy, we did not observe any nemaline body, centronuclear core or multi-minicore.
One month later, she received laparoscopic hiatal hernia repair and Nissen fundoplication for esophageal hiatal hernia. After the surgical treatment, dyspnea symptom disappeared.
This study received an approval from the institutional review board and the signed consent form was obtained from the patient to publish this report.
3. Discussion
The evidence presented above suggests that this in-frame deletion variant is the pathogenic element, based on the American College of Medical Genetics and Genomics guidelines regarding the interpretation of sequence variations.[6]MYH7 mutation was de novo (PS2), and the muscle biopsy findings were compatible with Laing muscular dystrophy (PS3). Moreover, the mutation was absent in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium (PM2). Therefore, our patient is compatible to “pathogenic”.[6]
According to previous studies that reported various phenotypes of MYH7 gene-related myopathy, skeletal myopathies occurred in the LMM domain of the tail domain and cardiomyopathy occurred in the head domain.[7] One interesting thing in our case was that the location of the MYH7 gene mutation was in the myosin head domain (c.1498_1500del: p.Glu500del), unlike previously reported skeletal myopathies. Moreover, 2D-echocardiography and electrocardiogram (ECG) showed no specific findings other than grade I diastolic dysfunction. These tests showed that there was no occurrence of cardiomyopathy. A recent study, like in our case, reported skeletal myopathies occurring in the head domain and cardiomyopathies in the tail domain.[8] Thus, the phenotypic variance of MYH7 gene-related myopathy is often not explained by monogenic mutation consequences.[9,10]
In limitation, only a few patients were reported regarding the phenotypes of the MYH7 gene-related myopathy. Therefore, additional reports are required to understand the diverse phenotypes of MYH7 gene-related skeletal myopathy.
In conclusion, MYH7 gene-related skeletal myopathy has been observed in a variety of spectra, and the factors determining these phenotypic variances can be the primary pathognomic gene mutation and the mechanical overloading causing myofibrillar injury. Here, we presented a rare case of de novo mutation of the myosin head domain (c.1498_1500del: p.Glu500del) in the MYH7 gene. Practitioners should consider all the various phenotypes of the MYH7 gene-related myopathy.
Acknowledgments
We thank the family and the patient for their cooperation.
Author contributions
Conceptualization: Jin Young Ko, Minyong Lee, Dae-Hyun Jang, Ju Seok Ryu.
Data curation: Jin Young Ko, Minyong Lee, Ja-Hyun Jang, Ju Seok Ryu.
Formal analysis: Ja-Hyun Jang, Dae-Hyun Jang.
Funding acquisition: Dae-Hyun Jang.
Investigation: Jin Young Ko, Ja-Hyun Jang, Ju Seok Ryu.
Methodology: Jin Young Ko, Ju Seok Ryu.
Project administration: Dae-Hyun Jang.
Resources: Dae-Hyun Jang.
Supervision: Minyong Lee.
Writing – original draft: Jin Young Ko, Ju Seok Ryu.
Writing – review & editing: Jin Young Ko, Dae-Hyun Jang, Ju Seok Ryu.
Footnotes
Abbreviations: MUAP = motor unit action potential, MYHCI = slow skeletal muscle / b-cardiac myosin heavy chain, NCS = nerve conduction study, EMG = electromyogram, MMT = manual muscle test, NGS = next generation sequencing, ECG = electrocardiogram.
JSR and D-HJ contributed equally to this paper and should, therefore, be regarded as equivalent corresponding authors.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2017R1C1B5014840).
The authors have no conflicts of interests to disclose.
References
- [1].Jandreski MA, Sole MJ, Liew CC. Two different forms of beta myosin heavy chain are expressed in human striated muscle. Hum Genet 1987;77:127–31. [DOI] [PubMed] [Google Scholar]
- [2].Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord 2007;17:355–67. [DOI] [PubMed] [Google Scholar]
- [3].Walsh R, Rutland C, Thomas R, et al. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010;115:49–60. [DOI] [PubMed] [Google Scholar]
- [4].Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 2004;75:703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Darin N, Tajsharghi H, Ostman-Smith I, et al. New skeletal myopathy and cardiomyopathy associated with a missense mutation in MYH7. Neurology 2007;68:2041–2. [DOI] [PubMed] [Google Scholar]
- [6].Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fiorillo C, Astrea G, Savarese M, et al. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis 2016;11:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Homayoun H, Khavandgar S, Hoover JM, et al. Novel mutation in MYH7 gene associated with distal myopathy and cardiomyopathy. Neuromuscul Disord 2011;21:219–22. [DOI] [PubMed] [Google Scholar]
- [9].Muelas N, Hackman P, Luque H, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology 2010;75:732–41. [DOI] [PubMed] [Google Scholar]
- [10].Udd B. 165th ENMC International Workshop: distal myopathies 6–8th February 2009 Naarden, The Netherlands. Neuromuscul Disord 2009;19:429–38. [DOI] [PubMed] [Google Scholar]