

Abstract
Many natural organic compounds with pharmaceutical applications, including antibiotics (chlortetracycline and vancomycin), antifungal compounds (pyrrolnitrin), and chemotherapeutics (salinosporamide A and rebeccamycin) are chlorinated. Halogenating enzymes like tryptophan 7-halogenase (PrnA) and tryptophan 5-halogenase (PyrH) perform regioselective halogenation of tryptophan. In this study, the conformational dynamics of two flavin-dependent tryptophan halogenases—PrnA and PyrH—was investigated through molecular dynamics simulations, which are in agreement with the crystallographic and kinetic experimental studies of both enzymes and provide further explanation of the experimental data at an atomistic level of accuracy. They show that the binding sites of the cofactor-flavin adenine dinucleotide and the substrate do not come into close proximity during the simulations, thus supporting an enzymatic mechanism without a direct contact between them. Two catalytically important active site residues, glutamate (E346/E354) and lysine (K79/K75) in PrnA and PyrH, respectively, were found to play a key role in positioning the proposed chlorinating agent, hypochlorous acid. The changes in the regioselectivity between PrnA and PyrH arise as a consequence of differences in the orientation of substrate in its binding site.
Introduction
Many pharmaceutically important natural organic compounds (including antibiotics, such as chlortetracycline1 and vancomycin,2 the antifungal compound pyrrolnitrin3 and chemotherapeutics, such as salinosporamide A4 and rebeccamycin5) are chlorinated. Halogenating enzymes perform regioselective halogenation of aromatic compounds efficiently in a solution using only chloride ions at physiological temperatures and atmospheric pressure. However, selective nonenzymatic chlorination of the C–H bonds is a chemical synthesis challenge.6 For example, the halogenation of tryptophan in the solution lacks regioselectivity and produces a mixture of products with chlorine added at the 1st, 5th, and 7th carbon of the indole ring.7 From an industrial point of view, this is unacceptable, as the desired isomer is produced with a lower yield and is expensive to separate from the other isomers. Interestingly, many natural products with pharmaceutical relevance contain halogen atoms at a range of different positions. These would be difficult to synthesize chemocatalytically and rely on the use of protecting groups and metal-based catalysts. Such strategies introduce extra reaction steps to the synthesis, increasing financial costs and lowering yields.8 Hence, a detailed understanding of the enzymatic mechanism of regioselective chlorination/halogenation of natural organic compounds and knowledge of the origin of the regioselectivity is of importance to organic chemical synthesis. Halogenating enzymes are attractive as biocatalysts because they can be engineered to suit different synthetic purposes,9 not only adjusting their regioselectivity but also their ability to accept a range of different substrates, such as indoles and other aryl-based substrates.10
The indole ring of tryptophan gets chlorinated at different positions of the 5th, 6th, or 7th carbon atom by distinct flavin-dependent halogenases, these include tryptophan 5-halogenase (PyrH11), tryptophan 6-halogenases (Thal12 and SttH13), and tryptophan 7-halogenases (RebH14 and PrnA15), respectively. All of these enzymes exhibit high levels of regio- and stereoselectivity. For example, chlorination of the indole ring of tryptophan at its sixth carbon atom by tryptophan 6-halogenase (Thal12) has been suggested to be the first step of the biosynthesis of the indole alkaloid thienodolin—a natural compound that exhibits plant growth-regulating activity. Our study focuses on the structural analysis through extensive molecular dynamics (MD) simulations of two flavin-dependent halogenases, namely, PrnA (tryptophan 7-halogenase) and PyrH (tryptophan 5-halogenase). PrnA catalyzes the chlorination of free tryptophan to 7-chlorotryptophan as a first step in the biosynthesis of the antibiotic and antifungal compound pyrrolnitrin.3 PyrH catalyzes the chlorination of free tryptophan to 5-chlorotryptophan as a part of the biosynthesis of the antibiotic pyrroindomycin B.11 It is important to understand the reasons for regioselectivity, with a focus on the structural differences at the active sites of these structurally similar enzymes.
X-ray crystallographic structures of the two halogenases are available from the Protein Data Bank (PDB).15,16 The structure of PrnA is shown in Figure 1.
Figure 1.
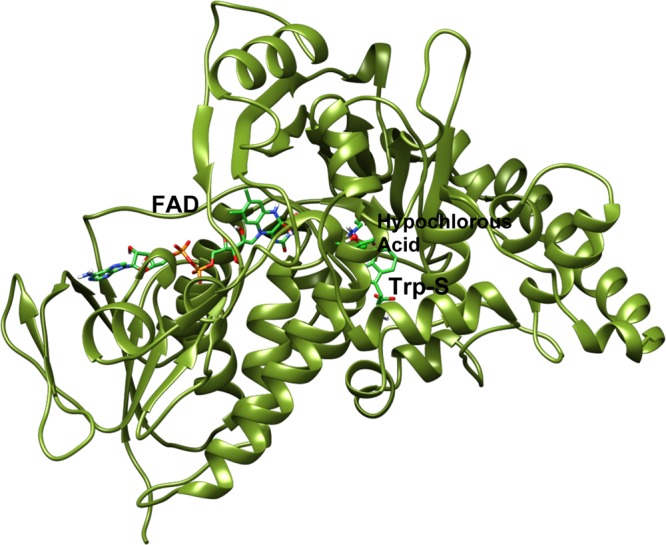
X-ray PrnA structure drawn using PrnA displayed in silhouette round ribbon. Substrate (Trp-S), cofactor FAD, chlorination agent, hypochlorous acid, and the side chains of the catalytically important K79 and E346 are shown in tube representation. Hypochlorous acid and the to-be halogenated carbon of Trp-S are shown in spherical representation. Carbons are green, nitrogens are dark blue, oxygens are red, and hydrogens are white.
Several reaction mechanisms were proposed for the enzymatic chlorination of tryptophan performed by tryptophan 7-halogenase.9 In the van Pée mechanism, the aryl ring of tryptophan reacts directly with hydroperoxy-FAD to produce a hydroxylated tryptophan intermediate.17 The positively charged intermediate is then attacked by a chloride ion to produce a chlorinated hydroxyl-tryptophan product. This product undergoes an elimination of water to produce the chlorinated tryptophan product.17 In the Walsh mechanism, chloride attacks hydroperoxy-FAD to produce a FAD-OCl intermediate. The tryptophan substrate (Trp-S) can then attack the chlorine of the FAD-OCl intermediate in a classical electrophilic aromatic substitution reaction.18 The van Pée and Walsh mechanisms rely on the possibility that the FAD and tryptophan-binding sites of the enzyme can be brought within a suitable proximity for direct contact between the FAD and Trp-S.17,18 Inspection of the crystal structures of the known FAD-dependent halogenase enzymes shows a >10 Å distance between the FAD and tryptophan-binding sites (Figure 2).11−15 The separation between the ligands would be too distant for direct interaction. However, close contact between the cofactor and the substrate, though not observable in the crystal structure, is a possibility that cannot be entirely excluded. A large conformational change could take place in the protein structure, bringing the two binding sites into close proximity and allowing direct reaction between the two ligands.
Figure 2.
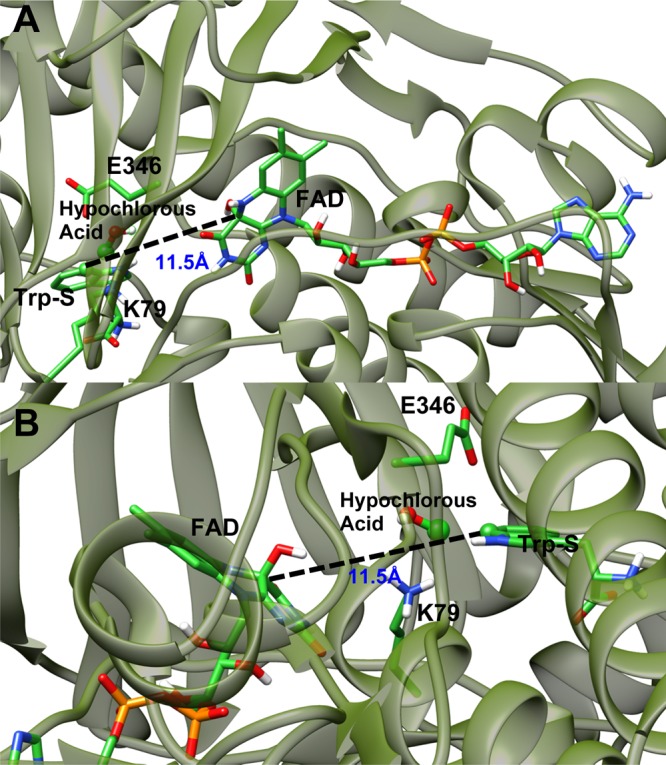
Two different views A and B of the ribbon representation of the PrnA crystal structure, with Trp-S, FAD, hypochlorous acid, K79, and E346 rendered as tubes and labeled. Hypochlorous acid and the to-be halogenated carbon of Trp-S are represented with a sphere rendering. Carbons are bright green and additional element colors are as follows: nitrogen is dark blue, oxygen is red, and hydrogen is white. In addition, the distance between the FAD C4A carbon and Trp-S C7 atom is drawn with a dashed line and the distance is labeled in blue.
A third mechanism put forward by Naismith et al. suggests that hypochlorous acid is produced at the FAD-binding site by the reaction of a chloride ion and hydroperoxy-FAD. Hypochlorous acid then travels through a channel between the FAD and tryptophan-binding sites.15,19 Once in proximity to tryptophan, the active site lysine and glutamate residues facilitate a reaction between hypochlorous acid and tryptophan to produce the chlorinated tryptophan product.15,20 Because the FAD- and tryptophan-binding sites are distant, the chlorinating agent, hypochlorous acid, is thought to travel through a channel in the protein.15 Two amino acids K79 and E346 in PrnA (analogous to K75 and E354 in PyrH) are positioned in close proximity to the reactive carbon of tryptophan’s indole ring. They are thought to be involved in the activation of the hypochlorous acid for the halogenation step of the reaction.19 The role of K79 and E346 in PrnA is supported by an experimental mutagenesis, showing that the K79A mutant had no detectable activity, and in the E346Q mutant, the Kcat value for the halogenation is decreased by 2 orders of magnitude.15
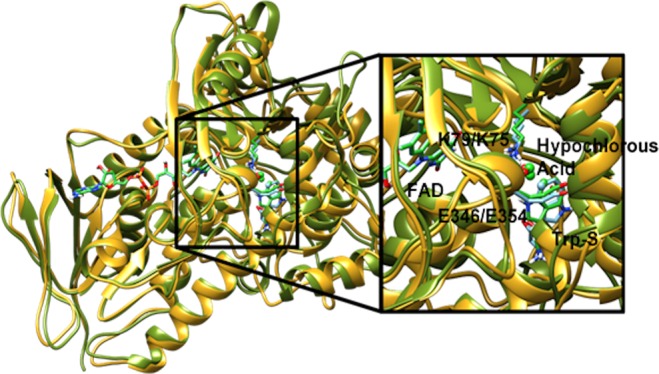
PyrH and PrnA share a 40% sequence identity and a 58% sequence similarity, making their structures similar.21 Despite this similarity, the catalytic turnovers of tryptophan 7-halogenase and tryptophan 5-halogenase differ. For example, PyrH was found to convert 100% of its Trp-S, whereas PrnA converted only 59% of its substrate and the origin of this difference has not yet been elucidated.10 In the FAD-binding site of the crystal structure of PyrH, a “strap” region was identified and hypothesized as a structural feature that allows for “communication” between the two binding sites that are involved in the regulation of FAD binding. The overlaid crystal structures of PyrH and PrnA reveal that the FAD-binding sites are almost identical (Figure 3). Structural analysis showed that PyrH possesses a structurally different tryptophan binding site to that of PrnA. Trp-S in the PyrH crystal structure is bound in a way that is upside down with respect to tryptophan in the PrnA crystal structure (Figure 3). However, the to-be halogenated carbon of Trp-S in PrnA (C7) and PyrH (C5) superimpose well when the two protein structures are aligned. The positioning of the reactive carbon is located between the active site lysine and glutamate residues, which show similar orientation across the two enzymes. In a recent study investigating the reaction mechanism for the chlorination of tryptophan in PrnA, quantum mechanics (QM)/molecular mechanics (MM) methods were applied to study the potential energy and free energy surfaces of the chlorination reaction.22 Key atomistic interactions in the stationary points and energetic changes along the reaction path were explored. They reported that E346 fulfills the role of a proton acceptor and hydrogen-bonding residue for Trp-S, whereas K79 acts as a proton donor and hydrogen-bonding residue for the hypochlorous acid. The structural data suggest that the reason for different regioselectivity of the two enzymes would be related to the binding interactions of Trp-S in the active sites.22
Figure 3.
View of the aligned crystal structures of PrnA and PyrH rendered with transparent protein ribbons, the FAD-binding straps are rendered as solid ribbons to highlight them. FAD, Trp-S, hypochlorous acid, and the active lysine and glutamate residues are rendered as tubes with carbon atoms colored according to the protein: PrnA in bright green and PyrH in light blue.
Enzymes are large, flexible, and dynamic molecules that naturally undergo a wide range of conformational changes and molecular motions ranging from femtoseconds to hours.23 Many of these motions are functionally important and relate enzyme structure to function.24 Experimentally determined protein structures (e.g., by X-ray crystallography) provide valuable structural information, however, limited to a static structure, averaged over the number of molecules in the crystal lattice, and the duration of the experiment.25 In addition, steric effects can also arise due to the compactness of the crystal environment.26 Enzyme conformational flexibility plays a substantial role in stabilizing the protein interactions vital in facilitating ligand binding and unbinding events.27 Molecular plasticity is involved in assisting the migration of ligands to the binding site, as well as the diffusion of gases and small molecules through the protein.28 Mutations of key residues, involved in catalysis and binding, can not only influence locally the structure but also exercise a long-range structural effect on the protein conformation as a whole. Exploration of the dynamic events in proteins, using experimental methods, can be a challenge; thus, computer-based experiments, for example, MD simulations can be applied to study this.23−25,29 Long-range atomistic MD simulations were performed to elucidate structure–function relationships and mechanistic implications related to the origin of regioselectivity in both enzymes.
Methods
An initial structure for the MD simulations of the wild-type full complex PrnA was created from the pdb structure of the enzyme (PDBID: 2AR8).15 The product 7-chlorotryptophan was separated to create tryptophan and hypochlorous acid; in addition, the chloride ion bound at the FAD-binding site was removed and FAD was modified to create hydroxy-FAD (from this point forward, FAD will refer to hydroxy-FAD). These changes were made with the aim to create the active full complex before the halogenation of Trp-S. Modification of the atomic coordinates was performed using Maestro 9.9.013.30 Structures of the mutant forms K79A and E346Q were prepared by changing the respective residues in the wild-type full complex structure using Maestro.30 The initial structure of PyrH (PDBID: 2WET) for MD simulations was prepared by superimposing the pdb structure with that of the wild-type full complex PrnA. The coordinates of hypochlorous acid from this were then added to 2WET, as they were not present in the crystal structure.16 In addition to this, the sulfate and chloride ions from the crystal structure were also removed. The parameters for FAD and hypochlorous acid were generated by the PRODRG web server31 for the GROMOS96 43a1 forcefield32 with atomic partial charges for hypochlorous acid supplemented from QM calculations performed by the Automated Topology Builder web server.33 The missing coordinates of the two loop regions in the 2WET structure were modeled using the Modeller34 plug-in for Chimera 1.10.2.35 The setup for PyrH then followed the same protocol as the one for PrnA. In total, the PrnA full complex had 94 114 atoms and PyrH 97 090 atoms. The hydrogen atoms missing from the X-ray crystal structure were added using Gromacs 4.5.5.36 To remove unfavorable steric clashes in the starting structure, in vacuo energy minimization was performed using the steepest descent algorithm until the maximum force was less than 100 kJ mol–1 nm–1, the protein was then placed in a box with periodic boundary conditions. The energy-minimized protein structure was then solvated using the single point charge37 model for water. The total charge of the system was neutralized by adding the correct number of Na+ or Cl– ions to make the overall charge of the system zero. Another energy minimization (using the same conditions as described for in vacuo energy minimization) was then performed to reduce close contacts between the solvent molecules or the ions that may be unfavorably close to the protein structure. The energy-minimized structure was then subject to position-restrained MD for 50 ps at 300 K, during that, the protein structure was restrained and the water was allowed to equilibrate. The position-restrained dynamics simulations were performed in NVT ensemble, a constant number of particles, volume, and temperature with a time step of 2 fs. The productive MD was then carried out with the output structure from the position-restrained MD providing the initial structure for 1 μs as in NPT ensemble at a temperature of 300 K. The MD trajectories were analyzed over the time period of 100–1000 ns, after equilibration phase was reached, using tools provided in Gromacs. Visualization and inspection of the trajectories were performed with visual molecular dynamics.38 Dynamic cross correlation analysis (DCCA) was performed using the Bio3D package39 for Rstudio.40 The DCCA is used to visualize which residues play a role in correlated motions that occur between different components of the protein structure.41 The level of correlation between each Cα atom can be quantified and visualized on a plot, with correlations ranging from +1 to −1, indicating a strong positive to a negative correlation. This allows the identification of regions in the protein, showing correlated motion in the simulation.
Results and Discussion
Conformational Dynamics of Full Complex Wild-Type PrnA
In total, five 1 μs MD simulations were performed: the full complex of wild-type PrnA, apoenzyme PrnA, two single point mutant forms, K79A and E346Q, of PrnA, as well as the full complex wild-type PyrH.
The root mean square deviation (RMSD) profile for all Cα atoms for the 1 μs MD simulation of the wild-type full complex PrnA is 3.5 Å. The RMSD profiles of all five of the 1 μs simulations (Figure 4) indicated that the initial equilibration phase was completed after 100 ns. In addition to the 1 μs simulation, three additional 200 ns MD simulations of the wild-type full complex of PrnA were performed. These used the same initial structure but different initial velocities (Supporting Information (SI) Figure S1). These simulations were created to evaluate the effect of statistical error on the quality of the simulations. The RMSDs for each of these trajectories was consistent with the 1 μs wild-type full complex PrnA simulation indicating good quality of the simulations. The radius of gyration of all four 1 μs simulations (SI Figure S2) was 23 Å, showing that the protein remains relatively compact during the simulation time scale.
Figure 4.

RMSD plot of all five 1 μs MD simulations: the PrnA full complex, apoenzyme PrnA, PyrH full complex, and the K79A and E346Q mutant forms of PrnA.
The average RMSD of the full complex wild-type PrnA is 3.5 Å and in the apoenzyme PrnA is 3.6 Å, indicating a slight trend of increased flexibility of the apoenzyme form possibly due to the absence of bound ligands. The average RMSD of PyrH was 2.8 Å, reflecting its relatively lower flexibility compared with PrnA. The solvent accessible area (SAS) of the apoenzyme PrnA is lower than the SAS of the full complex wild-type PrnA (Figure 5). The radius of gyration also indicates a more compact structure of the apoenzyme form of PrnA with respect to the full complex PrnA (SI Figure S2). These observations are consistent with the differences in the RMSDs of the full complex PrnA and apoenzyme PrnA, and are indicative of conformational changes associated with ligand binding and an opening of the PrnA structure upon ligand binding. PyrH is characterized as having overall lower levels of flexibility and a more compact structure than the full complex PrnA (Figure 4 and SI Figure S2).
Figure 5.

Solvent accessible area graph of the PrnA full complex and PrnA apoenzyme simulations.
The root mean square fluctuation (RMSF) profiles of the full complex PrnA and apoenzyme PrnA are presented in Figure 6. For the full complex PrnA, the peak centered on residue P93 exhibits a high RMSF, reflecting its position at a particularly flexible portion of the loop that precedes key tryptophan interacting residues: H101, F103, G104, and N105. These residues are involved in the binding of Trp-S. It is therefore possible that the flexibility of this loop is related to the binding and orientation of Trp-S.
Figure 6.

RMSF plot of the PrnA full complex and PrnA apoenzyme simulations.
A flexible loop consisting of residues 147–159 has a maximum RMSF value of 6.7 Å (centered on residue G152) in the full complex PrnA (Figure 6). The same region has an RMSF value of 1.8 Å in the apoenzyme form of PrnA. This loop is located on the exterior of the protein and is solvent exposed, forming several intraloop hydrogen bonds. The loop immediately precedes S157, a hydrogen bond stabilizing residue of Trp-S. The RMSD plot of the loop region 147–159 (Figure 7) shows that the loop is adopting a stable orientation after 300 ns. In this conformation, S157 forms a hydrogen bond with the carboxylate of Trp-S. The dynamics of the loop differ greatly between the full complex wild-type PrnA and the apoenzyme PrnA, suggesting that a conformational change occurs in the loop upon binding of Trp-S. In the apoenzyme, the S157 side chain forms hydrogen bonds with the neighboring residues A80, M156, and Y443 instead. These intraloop protein–protein hydrogen bonds stabilize the 149–159 loop of the apoenzyme and maintain a more compact conformation, which is reflected in the lower RMSD of the loop in the apoenzyme (Figure 7).
Figure 7.

Plot showing the relationship between the RMSD of the flexible loop region in both the full complex PrnA and the apoenzyme PrnA spanning residues 147–170 (left y-axis), and the hydrogen-bonding interaction distance between S157 side chain oxygen and Trp-S (right y-axis).
Conformational changes in enzymes are complex and involve collective motions between different regions of the protein molecule.42 To analyze the collective correlated motions in the studied tryptophan halogenase enzymes, we performed DCCA. In the full complex PrnA, a correlated motion between the portion of the FAD strap region closest to Trp-S (residues 50–54) and the important catalytic residue E346 was found, and supports the idea of the strap region being an important link between the two binding sites (Figure 8).
Figure 8.

DCCA plot of the PrnA full complex simulation. Areas of strongly positive correlation are in red and areas of strongly negative correlation are in blue.
In the DCCA plot of the full complex wild-type PrnA (Figure 8), a region of positive correlations corresponds to two areas of protein from residues 205 to 255 and 305 to 350 (Figure 9). The area makes up a large part of the FAD-binding site and contains many important FAD-binding residues. It also contains important residues from the tryptophan-binding site, such as E346 and S347.
Figure 9.
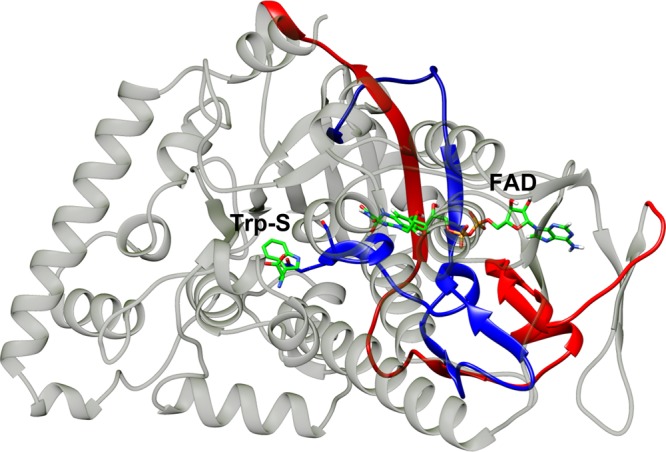
Two DCCA correlated regions described as spanning residues 205–255 in red and the region spanning from residues 305 to 350 in blue. The important residues E346 and S347 are displayed along with FAD and the substrate tryptophan in green carbon tubes.
The region of residues from 355 to 380 that shows fluctuation in both the RMSF plot (Figure 6) of the full complex wild-type PrnA and apoenzyme PrnA correspond to a long α-helix that intersects the FAD- and tryptophan-binding sites. It contains the tryptophan hydrogen-bonding residue, Y351. In the DCCA plot of the full complex wild-type PrnA (Figure 8), this region of residues, 355–380, shows correlation with an important tryptophan-binding residue W455. A relatively large span of residues 396–456 directly precedes the important tryptophan binding residues (Y443, Y444, W455, E450, F454, and N459) and shows more fluctuation in the full complex than the apoenzyme (Figure 6). These residues form several helices joined by short loops. Interactions between the helices create a compact and less flexible hydrophobic cluster.
Tryptophan-Binding Site Interactions of Wild-Type PrnA
The high level of regioselectivity of FAD-dependent halogenases is thought to depend on the proper orientation of Trp-S.43 Tryptophan positioning and orientation allows for the respective carbon atom from the indole ring (C7 in PrnA and C5 in PyrH) to be favorably oriented for the reaction. To accomplish stable binding of tryptophan, an extensive network of hydrogen bonds (Figure 10), electrostatic interactions (SI Figure S3), and van der Waals interactions (Figure 11) were found. The measured distances of interactions of Trp-S observed in the X-ray crystal structure and wild-type full complex PrnA MD simulation are recorded in Tables 1 and 2.15 K79 and E346, thought to be important for hypochlorous acid activation, are also involved in a network of hydrogen-bonding and electrostatic interactions that maintain their orientations in the active site relative to Trp-S and the chlorinating agent, hypochlorous acid.19,22
Figure 10.
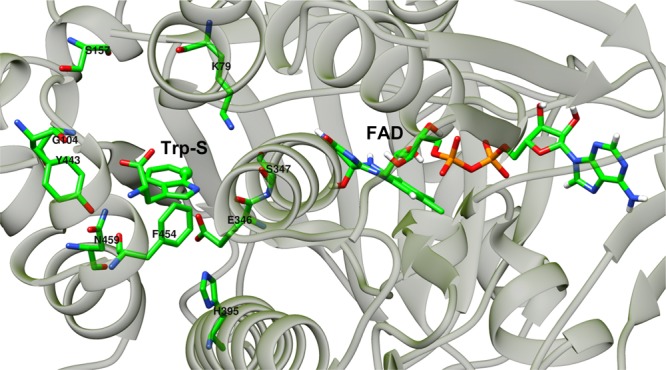
Hydrogen-bonding interactions surrounding Trp-S in PrnA. The distances between the donors and acceptors are shown in Table 1.
Figure 11.
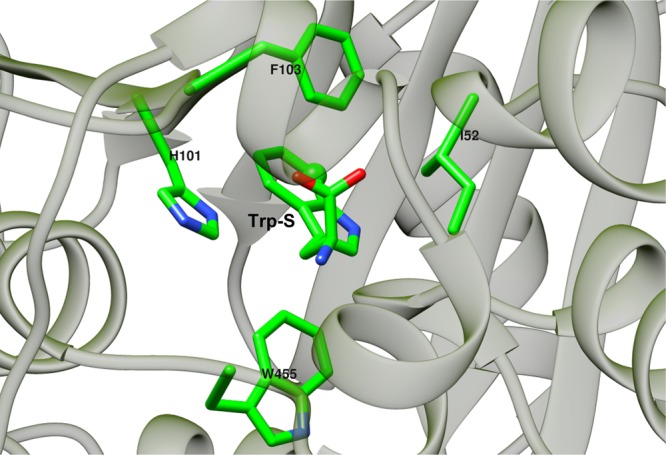
Hydrophobic contacts surrounding Trp-S in PrnA. The distances between the centers of mass of the Trp-S indole ring and the hydrophobic amino acid side chains are shown in Table 2.
Table 1. Hydrogen-Bonding and Electrostatic Interactions for the Tryptophan-Binding Site in the Wild-Type Full Complex PrnA.
| residue 1 | atom 1 | residue 2 | atom 2 | % of the simulation time <3.5 Å | average distance in MD (Å) | distance in crystal structure (Å) |
|---|---|---|---|---|---|---|
| H395 | NE2 | E346 | OE2 | 53 | 3.5 | 2.5 |
| E346 | OE1 | HYP | O | 91 | 3.0 | 3.5 |
| E346 | OE2 | HYP | O | 90 | 3.1 | 5.2 |
| G104 | N | Trp-S | O | 82 | 3.0 | 6.0 |
| G104 | N | Trp-S | carboxylate | 81 | 3.1 | 8.0 |
| Y443 | OH | Trp-S | amino | 95 | 3.1 | 3.1 |
| F454 | O | Trp-S | amino | 69 | 3.4 | 2.8 |
| H395 | NE2 | E346 | carboxylate | n/a | 3.7 | 4.5 |
| E346 | carboxylate | HYP | H | n/a | 2.5 | 4.6 |
| K79 | NZ | HYP | O1 | n/a | 6.7 | 3.2 |
| E450 | carboxylate | Trp-S | N | n/a | 4.2 | 3.8 |
| K57 | NZ | E450 | CD | n/a | 5.8 | 10.3 |
Table 2. Distances between the Centers of Mass between the Hydrophobic Side Chains and the Indole Ring of Trp-S.
| residue name and number | average distance (Å) | distance in crystal structure (Å) |
|---|---|---|
| I52 | 6.2 | 5.0 |
| H101 | 6.3 | 5.4 |
| F103 | 4.7 | 5.6 |
| W455 | 5.1 | 5.9 |
Measurements were made between the donor and acceptor atoms for hydrogen bonds. Measurements for electrostatic interactions (highlighted in gray) were measured between the centers of the charged groups.
Trp-S (Table 1) can act as both hydrogen bond donor and acceptor with its amino nitrogen, carboxylate oxygen, and indole ring nitrogen atoms. The backbone nitrogen of G104 participates in a hydrogen bond with Trp-S’s carboxylate. This interaction does not exist in the crystal structure but is stable during the MD trajectory. The amino group of tryptophan is hydrogen bonded to the side chain phenolic oxygen of Y443, and the backbone carbonyl oxygen of F454. The amino group of tryptophan can also make electrostatic interactions (Table 1) with the carboxylate of E450 with an average distance of 4.2 Å. E450 in turn interacts with the side chain amino group of K57 (distance 5.8 Å), which would help more efficient binding of Trp-S.
Hypochlorous acid can participate in hydrogen bonds and interactions with charged residues in the enzyme active site. The hydrogen atom of hypochlorous acid has a partial positive charge (0.455e) and forms a strong hydrogen bond with the side chain of E346 (Table 1). In the initial structure of the PrnA full complex, K79 is in close proximity to hypochlorous acid and seems a likely candidate for hydrogen bonding; however, during the MD simulation, hypochlorous acid moves away from K79, reflected in the average distance of 6.7 Å in the MD (Table 1). The carboxylate side chain of E346 has two oxygen atoms, OE1 and OE2, with which it is possible to form hydrogen bonds (Table 1). The E346 carboxylate forms an electrostatic interaction with the NE2 nitrogen atom of the protonated H395 side chain. Hypochlorous acid makes strong hydrogen-bonding interactions with the carboxylate side chain of E346. The E346 carboxylate side chain also interacts with the positively charged doubly protonated H395 (Table 1). In the crystal structure, the indole nitrogen atom of tryptophan forms a hydrogen bond with the backbone carbonyl oxygen of E346. However, in the MD simulation, the backbone carbonyl of E346 moves away from tryptophan to make other hydrogen-bonding interactions with T348 and the hydroxyl oxygen of hydroperoxyflavin moiety of FAD.10
F103, W455, and H101 form π–π stacking interactions with tryptophan (respective average distances of 4.7, 5.1, and 6.3 Å). Throughout the MD simulation, W455 remains close to Trp-S participating in a stable π–π stacking interaction with the substrate (Table 2). E346 and hypochlorous acid are also located in a close proximity to the side chain of W455; however, W455 does not become halogenated. K79 is not found in proximity to W455, indicating that the proximity to both K79 and E346 is needed for the halogenation reaction to occur.
A positive correlation between residues 100–130 and residues 475–505 is found in the DCCA of the full complex wild-type PrnA (Figure 8). The region of residues 475–505 forms a long α-helix running perpendicular to the H101/F103 region. The correlation is likely to be caused by hydrophobic interactions between the two regions of the loop portions of the H101/F103 (SI Figure S4). These two regions are interwoven and any movement in one will affect the other region as well.
FAD-Binding Site in PrnA
A structural feature previously observed in the crystal structure of PyrH is the FAD-binding strap. This strap region is thought to control the binding of FAD and also hypothesized to act as a line of communication between the FAD-binding and tryptophan-binding sites in PyrH.16 In the crystallographic study of PyrH, the electron density of the strap region is relatively low, implying that it is a particularly flexible region of PyrH.16 Through inspection of the crystal structure of PrnA, we found that a similar strap region superimposes with that of the PyrH crystal structure and would also exists in PrnA (Figure 3). The high flexibility of the strap region shown in SI Figure S5, together with its probable influence on both FAD and tryptophan (Figure 3), points toward the strap region fulfilling a similar role to the one hypothesized to perform in PyrH. In both enzymes, the strap region consists of a long straight section of residues running parallel to FAD without secondary structure elements (Figure 3). The region of the strap that is in close contact with FAD forms several hydrogen bonds (SI Figure S6) and hydrophobic and cation−π interactions (SI Figure S7) with FAD. These are evident in the crystal structure as well as the MD simulations of the PrnA wild-type full complex (SI Tables S1 and S2).20
FAD is a relatively large molecule, and in the crystal structure, it adopts a linear extended conformation, whereas in the MD, we see it undergoing a structural transition around 200 ns to adopt a bent conformation (SI Figure S8). We also see this structural transition of FAD in the MD simulations of PrnA and PyrH. In the PrnA full complex simulation, the change in the conformation of FAD happens simultaneously with a structural transition seen in the FAD strap region (SI Figure S5). After 200 ns, the strap adopts a conformation that shows reduced structural flexibility. In comparison, the RMSD of the strap region in the apoenzyme form of PrnA shows a higher flexibility as well as larger fluctuations (SI Figure S5). The increased flexibility of the strap region in the apoenzyme PrnA MD in contrast to the full complex PrnA MD suggests that the strap region is involved in the binding of FAD and becomes more stable in its presence, which can be seen by comparing hydrogen-bonding interactions of FAD in the X-ray crystal structure to those from the MD (SI Table S2).
The RMSF profile of the strap region (SI Figure S9) shows that although the strap region in the apoenzyme form of PrnA possesses a higher flexibility than the strap region in the full complex PrnA form, this is mainly due to the high flexibility of residues 45–49. These residues are located in close proximity to the flavin moiety of FAD and form stable hydrogen-bonding interactions with it (SI Table S2). The region of residues from 50 to 53 connects the FAD-binding residues to those of the tryptophan-binding site. S54 hydrogen bonds E450, which is a key residue for the binding of the amino group of tryptophan. The equilibration of the strap region and FAD after 150 ns causes a conformational change that brings the side chain of S54 into the proximity of E450 to form a hydrogen bond (SI Figure S10). This movement brings E450 into the proximity of the tryptophan amino group, where it forms an electrostatic interaction (Table 1). This conformational change provides an atomistic basis for the predicted communication between FAD and tryptophan-binding sites. The binding of FAD can influence the binding of tryptophan by means of the strap region running through both domains.
The hydrophobic interactions between FAD and the protein in the MD simulations and X-ray crystal structure are predominantly with the flavin moiety of FAD (SI Table S1). The adenine moiety of FAD also has the potential to form a cation−π interaction with the side chain of R221 (SI Table S1). Most hydrogen bonds with the adenine moiety of FAD are formed with the backbone carbonyl oxygen and nitrogen atoms of the surrounding residues (SI Table S2). A50, S347, T348, and I350 are the key residues found to form stable hydrogen-bonding interactions with the flavin ring moiety of FAD. The backbone carbonyl groups of E346 and P344 interact with the hydroxyl group of FAD. The interaction between hydroxy-FAD and E346 could influence the communication between FAD and Trp-S.
Possibility of Direct Contact between FAD- and Tryptophan-Binding Site/Module
The 1 μs MD simulations show that close contact between FAD and tryptophan does not occur at this time scale. The distance between the FAD-binding site and the substrate-binding site remains relatively high during MD. The average distance between the centers of mass of the FAD- and tryptophan-binding sites was found to be 12.1, 13.5, and 14.2 Å, respectively, for PrnA full complex, apoenzyme PrnA, and PyrH full complex MD simulations (Figure 12). The distance between C4A atom of FAD and the to-be halogenated carbon of tryptophan (C7/C5) remains high throughout the MD simulations of PrnA and PyrH (Table 3). The side chains of residues K79 and E346 remain distant from flavin at the 1 μs time scale of the MD simulations (Table 3). Some hydrogen bonding is found between the hydroxyl group of flavin and the backbone carbonyl oxygen of E346. This would not, however, allow for direct halogenation of the E346 or K79 side chains (SI Table S1). These observations support the main catalytic mechanism in which the intermediary halogenating agent is created at the FAD-binding site and travels through a channel between the FAD- and tryptophan-binding sites.15,22
Figure 12.

Distance between the center of mass of the binding sites of the substrate tryptophan and the flavin ring moiety of FAD in the PrnA full complex, PrnA apoenzyme, and PyrH full complex MD simulations.
Table 3. Average Distances between the Proposed Reactive Atoms in the Tryptophan-Binding Site.
| average distance PrnA full complex MD (Å) | average distance PyrH full complex MD (Å) | |
|---|---|---|
| flavin C4A—substrate tryptophan C7/C5 | 11.6 | 11.0 |
| flavin C4X—active lysine NZ | 7.0 | 6.9 |
| flavin C4X—active glutamate CD | 8.3 | 10.8 |
Distances were measured between the lysine (K79/K75 of PrnA and PyrH, respectively)–NZ, glutamate (E346/E354 of PrnA and PyrH, respectively), CD, and substrate tryptophan C7/C5 atoms and the proposed reactive atom of FAD-C4A in the MD simulations.
Conformational Effects of the Mutations of K79 and E346
Mutational studies show that both K79 and E346 in PrnA and K75 and E354 in PyrH play a vital role in the reaction of chlorination of tryptophan. The residues are conserved across the known FAD-dependent halogenases: PyrH11 (tryptophan 5-halogenase), Thal12 and SttH13 (tryptophan 6-halogenases) and RebH,14 and PrnA15 (trypthophan 7-halogenases), indicating their key roles in the catalysis.44 In PrnA, the mutation K79A leads to a complete loss of activity and the mutation E346Q shows activity that is reduced by 2 orders of magnitude to a level where it is barely detectable.15 Although possessing no formal charge, hypochlorous acid has a strong dipole moment (oxygen −0.456 D and hydrogen 0.445 D calculated by the automated force field topology builder33) and interactions with K79 and E346 will have an influence on its position and orientation. To test the stabilizing effect of the two charged residues and to explain the experimental effects of the mutations, we performed the MD simulations on in silico mutated forms of PrnA K79A and E346Q. Without the electrostatic environment created by both K79 and E346, hypochlorous acid moves away from tryptophan and back along the channel toward FAD. In this position, hypochlorous acid is too distant from tryptophan and would likely be unable to participate in the halogenation reaction (SI Figure S11). In the MD simulation of K79A mutant, the hypochlorous acid remains closer to the flavin ring and forms hydrogen bonds with the O4 atom of FAD. In the E346Q mutant form MD simulation, the hypochlorous acid moves away from the tryptophan-binding site along the channel toward FAD, where it forms a hydrogen bond with T263. This residue, although close to K79, is separated by internal protein structure and not accessible for interaction with hypochlorous acid. The simulations of the two mutant forms show increased hypochlorous acid Cl to Trp-C7 distances relative to the wild-type PrnA. This indicates to us that both residues are of key importance for the positioning of hypochlorous acid in proximity to tryptophan.
Comparison of PyrH to PrnA
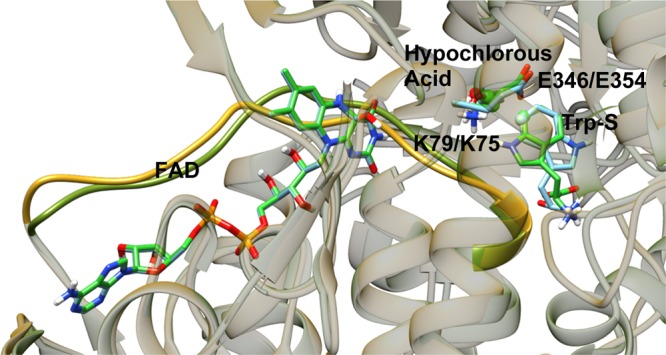
PyrH and PrnA are structurally similar enzymes that carry out similar reactions but exhibit different kinetics. PyrH is a more efficient enzyme at chlorinating tryptophan, it achieves 100% conversion to 5-chlorotryptophan, whereas under the same conditions, PrnA converts only 59% of tryptophan to 7-chlorotryptophan, with the remaining tryptophan unreacted.10 The binding mode of FAD in both enzymes is almost identical, and the FAD molecules overlay almost perfectly when the two proteins are aligned (Figures 3 and 13). The differences in their regioselectivity and kinetics probably originate in the tryptophan-binding domain. The way these two enzymes bind Trp-S in the active site is quite different, Trp-S in PyrH is oriented in an upside-down position relative to its binding orientation in PrnA. The benzene moieties of the indole rings for both PrnA and PyrH are nearly superimposable in the crystal structure (Figure 14). This indicates that the C5 atom in PyrH is in an almost identical place to that of the C7 atom in PrnA. During the MD simulation of PyrH, we observe a rotation of Trp-S to a slightly different orientation, and after equilibration, this orientation remains relatively stable (SI Figure S12). Despite this movement, the relative orientation of the C5 atom of the indole ring of tryptophan toward K75 and E354 remains the same. The position of hypochlorous acid in the PyrH MD simulation also remains more stable relative to the PrnA MD, which is indicated by the lower levels of fluctuation in the RMSD plot of hypochlorous acid in SI Figure S13.
Figure 13.

Ribbon structure of PrnA (dark green) and PyrH (gold) structurally aligned with one another. Stick representations of the bound ligands FAD and tryptophan as well as the active glutamate and lysine residues. Hypochlorous acid and the to-be halogenated carbon of tryptophan are represented as spheres. Light blue carbons represent PrnA and green carbons represent PyrH. Additional element colors are as follows: nitrogen is dark blue, oxygen is red, and hydrogen is white.
Figure 14.
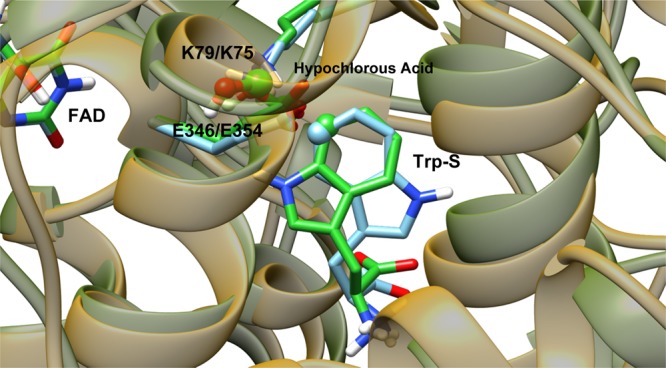
View of the aligned crystal structures of PrnA and PyrH rendered with transparent protein ribbons, PrnA with yellow ribbons, and PyrH with green ribbons. The substrate tryptophan, hypochlorous acid, and the active lysine and glutamate residues are rendered as tubes with carbon atoms colored according to the protein: PrnA in bright green and PyrH in light blue. The to-be halogenated carbon (C7/C5) of the substrate tryptophan is rendered as a sphere.
The higher efficiency of PyrH as an enzyme may correlate with the reduced flexibility of the PyrH tryptophan-binding site. A more rigid binding site can make PyrH a more efficient enzyme for halogenating tryptophan.10 The averaged distances from the MD trajectories between K79/K75-NZ, hypochlorous acid Cl, and Trp (C7 in PrnA and C5 in PyrH) are shown in SI Figures S14 and S15. K79/K75-NZ to hypochlorous acid Cl and tryptophan C7/C5 in PyrH show that hypochlorous acid makes more stable interactions with relatively lower levels of fluctuations between the active lysine and glutamate residues. The stability of these interactions could mean that more energetically favorable interactions take place in PyrH as opposed to PrnA. This could be one of the contributing factors to the experimentally observed greater catalytic turnover of PyrH compared to PrnA.
The average RMSD of PyrH was 2.8 Å, which is significantly lower than the PrnA full complex and PrnA apoenzyme simulations and means that PyrH is less flexible than PrnA (Figure 4). In the RMSF plot comparing the PyrH full complex and PrnA full complex MD simulations (SI Figure S16), the region around G37, which immediately precedes the FAD binding strap that runs from residues 37–50, we see a similar feature, the FAD strap region, seen in the RMSF plot of PrnA. However, in PyrH, this region shows lower levels of flexibility. In PyrH, both the FAD strap region (SI Figure S17) and FAD show more conformational fluctuations than those in the PrnA full complex simulation (SI Figure S18). Due to the differences in the tryptophan-binding sites between PyrH and PrnA, we found no analogous interaction between S50 and E452 in PrnA. Instead, a direct hydrogen-bonding interaction between the side chain of S50 and the carboxylate moiety of Trp-S is found (SI Table S3). This suggests that despite the differences in tryptophan-binding between PyrH and PrnA, the role of the strap region remains the same in the two enzymes.
In PrnA, the flexible loop region, which spans residues 147–159, might be important for tryptophan binding. In the MD simulation, we identified a hydrogen bond between the side chain of S157 and the carboxylate moiety of tryptophan, which was affected by the dynamics of the 147–159 loop. We propose that this relationship is important for tryptophan binding (Figure 7). In PyrH, there is a similarly positioned flexible loop that had to be modeled due to its lack of coordinates in the crystal structure.16 This loop of residues from 148 to 165 in PyrH is similar to the loop 147–159 in PrnA in that G153 acts as a hinge residue with several internal hydrogen-bonding interactions forming within the loop during the MD simulation, such as between T156 and D149, S151 and D149, and R154 and E150. This loop could play a similar role in PyrH as the equivalent loop 157–159 does in PrnA, acting as a structural link between the FAD-binding strap and Trp-S. However, the residue S157 from PrnA has no analogous residue in PyrH; instead, F164 is found in a similar spatial position and probably fulfills a similar role in hydrogen-bonding Trp-S. After equilibration, F164 forms stable hydrogen-bonding interactions with Trp-S amino and carboxylate moieties (SI Figure S19). DCCA of the PyrH full complex (SI Figure S20) shows similar correlations to PrnA (Figure 8, SI Figure S4).
In general, most of the binding interactions between the protein, Trp-S, and FAD in PrnA are similar to those in PyrH. For example, tryptophan is bound in a similar way, i.e., positioned between hydrophobic side chains, making π–π stacking interactions with the indole ring (shown in SI Figure S21, Table 2 and SI Table S4). In PrnA, W455 is replaced by a similarly positioned F451 in PyrH, although the distance is greater (average distance 7.0 Å), therefore making the interaction weaker and less significant for tryptophan binding. F49 in PyrH occupies a similar position to F454 in PrnA, with an average distance of 5.3 Å to Trp-S (SI Table S4). H101 and F103 from PrnA are conserved in PyrH as H92 and F94 and fulfill a similar role as hydrophobic residues in proximity to Trp-S (SI Table S4).
In PyrH, the amino group of Trp-S interacts electrostatically with the side chain of E452 (distance of 3.9 Å) (SI Table S5 and Figure S22). In the crystal structure of PyrH, a similar electrostatic interaction supports E452 and appears to be created by R96; however, the average distance of this interaction during the MD simulation is much greater and therefore weaker than the analogous interaction between K57 and E450 observed in the MD simulation of PrnA. In PyrH, the important catalytic residue E354 interacts with the protonated H40 with a similar distance to that of H395 to E346 seen in PrnA (Table 1 and SI Table S5). Hypochlorous acid forms strong interactions with both the active site K75 and E354. Low average distances indicate that these interactions are stronger and more stable than in PrnA (SI Figures S14 and S15). This may be another contributing factor to the increased efficiency of PyrH as an enzyme.
The greatest differences in tryptophan binding between PyrH and PrnA observed in our MD simulations were found in the hydrogen-bonding interactions between the protein and Trp-S. The different orientations of the substrate between PrnA and PyrH lead to very different hydrogen bonding. In PyrH, the relative upside-down positioning of tryptophan means that the tryptophan NE1 atom no longer points toward hypochlorous acid channel, meaning it is more able to make hydrogen bonds with the surface residues of the tryptophan binding pocket (Figures 14 and 15). In PyrH, S345 forms a hydrogen bond with the Trp-S NE1 atom, S50 is the main residue responsible for the hydrogen bonding of Trp-S carboxylate moiety and F164 forms hydrogen bonds with both amino and carboxylate moiety of tryptophan (SI Table S3).
Figure 15.
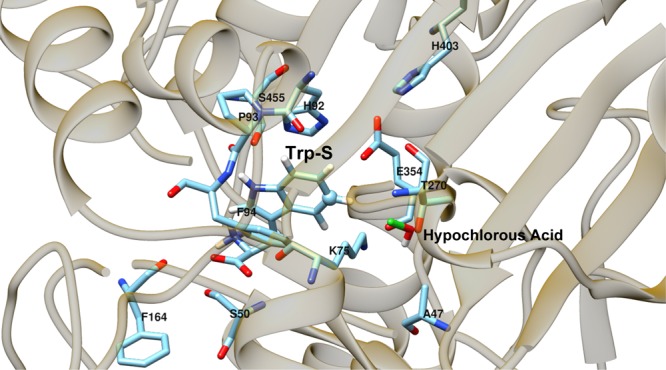
Hydrogen-bonding interactions of Trp-S in the PyrH enzyme.
Similarly, in the PrnA MD simulation, FAD undergoes a structural transition from a linear to a bent form (SI Figure S8). The transition occurs much more rapidly in PyrH than in PrnA (SI Figure S18). This is seen in the loss of several hydrogen bonds in the crystal structure as compared to the MD simulation (SI Table S6). The binding mode of FAD in PyrH shows a high level of similarity to that of PrnA, in that it is mainly bound by backbone hydrogen bonds (SI Figure S23). Some of the interactions between hydrophobic side chains and the adenine and flavin moieties are conserved by similar residues to those in PrnA in both the crystal structure and MD simulations (SI Table S7 and Figure S24).
Correlations to Experimental Studies
The simulations of PrnA and PyrH agree with the experimental crystallographic studies of both enzymes on the structural organization of these enzymes and the structural basis of their functions. The simulations are consistent with the crystallographic data for the majority of the interactions that stabilize the binding of Trp-S and FAD in their respective binding sites.15,16
Important justification concerning the nature of the reaction mechanism arises from the observation of the crystal structure of PrnA and especially by the fact that the FAD-binding site and tryptophan-binding site are separated by 10 Å.15 Although experimental studies assert that there is a lack of a potential large conformational transition that brings into close proximity the two binding sites, they cannot completely exclude such a possibility. Instead, the MD simulations for 1 μs of the full complex, apoenzyme form of PrnA, and full complex PyrH (Figure 12) demonstrate that there is no such kind of conformational transition that would bring the two binding sites together and disfavor the reaction mechanism that involves direct contact between FAD and Trp-S.
The computational results of the PrnA and PyrH simulations agree with the experimental studies on the key interactions in the tryptophan-binding site. The experiments show no activity in PrnA for the K79A mutant and a 2 order of magnitude reduction of activity for the E346Q mutant, and our computational studies provide the atomistic details about these experimental findings. Furthermore, the present simulations agree with the overall profile of stabilizing interactions in the FAD-binding site and further explain the role of the strap region in both enzymes. Furthermore, the present MD studies complement and agree with previous QM/MM studies on the reaction mechanism of PrnA.22
Conclusions
Applying MD simulations, atomistic insights into the structure–function relationships of two halogenases—PrnA and PyrH—were gained and the origin of their regioselectivity was found to be related to structural features. The MD study showed that during the 1 μs time scale, no major conformational change occurs that can bring the cofactor FAD and the substrate tryptophan binding sites together. This confirms the feasibility of the reaction mechanism that involves first formation of hypochlorous acid in the FAD-binding site and then its transfer to the tryptophan binding site, where the halogenation reaction takes place. Key residues involved in positioning the substrate tryptophan in the enzyme active sites were identified. Specific active site orientation of tryptophan is likely a key factor in the regioselectivity of the two enzymes. The MD simulations identified several flexible regions that have implications for substrate binding. A possible function was proposed for the strap region. Atomistic details about the communication that links the tryptophan and FAD binding sites were provided. The study of the two mutant forms of PrnA confirmed the experimental mutagenesis results and provided a better understanding of the structural basis for reduced activities observed for these mutants. Analysis of PyrH and PrnA MD simulations showed that although the two enzymes share very similar structures, they exhibit fine differences in interactions in the respective tryptophan-binding sites. These observations suggest a structural basis for PyrH as a more efficient halogenating enzyme.
Acknowledgments
The authors acknowledge Jimmy Gibson, Northumbria University, for the provided technical IT support. J.A. acknowledges the technical support of Dr. Warispreet Singh.
Glossary
Abbreviations
- PrnA
tryptophan 7-halogenase from Pseudomonas fluorescens
- PyrH
tryptophan 5-halogenase from Streptomyces rugosporus
- MD
molecular dynamics
- FAD
flavin adenine dinucleotide
- Trp-S
the substrate tryptophan
- Thal
tryptophan 6-halogenase from Streptomyces albogriseolus
- SttH
tryptophan 6-halogenase from Streptomyces toxytricini
- RebH
tryptophan 7-halogenase from Lechevalieria aerocolonigenes
- DCCA
dynamic cross-correlation analysis
- RMSD
root mean square deviation
- SAS
solvent accessible area
- RMSF
root mean square fluctuation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00385.
Supporting figures and tables for structural insights from molecular dynamics simulations of tryptophan 7-halogenase and tryptophan 5-halogenase (PDF)
Author Contributions
J.A. conducted the calculations, analyzed the results, and wrote the manuscript. A.J.M., G.W.B., and O.S. analyzed and discussed the results and revised the manuscript. T.G.K.-C. designed the study and T.G.K.-C. and C.Z.C. analyzed and discussed the results and wrote and revised the manuscript. The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
T.G.K.-C. and C.Z.C. acknowledge Marie Curie International Outgoing Career Development Fellowships, NSCCS grants, HEC-Biosim grants, and Michigan Tech start-up grants. T.G.K.-C. is grateful to the University of Bristol and the U.K. Overseas Postgraduate Research Scholarships. J.A. acknowledges Northumbria University PhD Scholarship. The authors acknowledge the High-Performance Computing Cluster “Pasteur” and GPU System “Newton” at the Department of Applied Sciences at Northumbria University.
The authors declare no competing financial interest.
Supplementary Material
References
- Jukes T. H.Some Historical Notes on Chlortetracycline. In Reviews of Infectious Diseases; Oxford University Press: Oxford, 1985; Vol. 7, pp 702–707. [DOI] [PubMed] [Google Scholar]
- Moellering R. C.Vancomycin: A 50-year Reassessment. In Clinical Infectious Diseases; Oxford University Press: Oxford, 2006; Vol. 42, pp S3–S4. [DOI] [PubMed] [Google Scholar]
- Hammer P. E.; Hill D. S.; Lam S. T.; Van Pée K.-H.; Ligon J. M. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl. Environ. Microbiol. 1997, 63, 2147–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feling R. H.; Buchanan G. O.; Mincer T. J.; Kauffman C. A.; Jensen P. R.; Fenical W. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem., Int. Ed. 2003, 42, 355–357. 10.1002/anie.200390115. [DOI] [PubMed] [Google Scholar]
- a Neumann C. S.; Fujimori D. G.; Walsh C. T. Halogenation strategies in natural product biosynthesis. Chem. Biol. 2008, 15, 99–109. 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]; b Long B. H.; Rose W. C.; Vyas D. M.; Matson J. A.; Forenza S. Discovery of antitumor indolocarbazoles: rebeccamycin, NSC 655649, and fluoroindolocarbazoles. Curr. Med. Chem.: Anti-Cancer Agents 2002, 2, 255–266. 10.2174/1568011023354218. [DOI] [PubMed] [Google Scholar]
- Frese M.; Schnepel C.; Minges H.; Voß H.; Feiner R.; Sewald N. Modular Combination of Enzymatic Halogenation of Tryptophan with Suzuki-Miyaura Cross-Coupling Reactions. ChemCatChem 2016, 8, 1799–1803. 10.1002/cctc.201600317. [DOI] [Google Scholar]
- Somei M.A Frontier in Indole Chemistry: 1-Hydroxyindoles, 1-Hydroxytryptamines, and 1-Hydroxytryptophans. In Bioactive Heterocycles I; Eguchi S., Ed.; Springer, 2006; pp 77–111. [Google Scholar]
- a Young I. S.; Baran P. S. Protecting-group-free synthesis as an opportunity for invention. Nat. Chem. 2009, 1, 193–205. 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]; b Wang X.; Lane B. S.; Sames D. Direct C-arylation of free (NH)-indoles and pyrroles catalyzed by Ar-Rh (III) complexes assembled in situ. J. Am. Chem. Soc. 2005, 127, 4996–4997. 10.1021/ja050279p. [DOI] [PubMed] [Google Scholar]
- Anderson J. L. R.; Chapman S. K. Molecular mechanisms of enzyme-catalysed halogenation. Mol. BioSyst. 2006, 2, 350–357. 10.1039/b607813c. [DOI] [PubMed] [Google Scholar]
- Shepherd S. A.; Karthikeyan C.; Latham J.; Struck A. W.; Thompson M. L.; Menon B. R.; Styles M. Q.; Levy C.; Leys D.; Micklefield J. Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem. Sci. 2015, 6, 3454–3460. 10.1039/C5SC00913H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehner S.; Kotzsch A.; Bister B.; Süssmuth R. D.; Méndez C.; Salas J. A.; van Pée K.-H. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol. 2005, 12, 445–452. 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Seibold C.; Schnerr H.; Rumpf J.; Kunzendorf A.; Hatscher C.; Wage T.; Ernyei A. J.; Dong C.; Naismith J. H.; Van Pée K.-H. A flavin-dependent tryptophan 6-halogenase and its use in modification of pyrrolnitrin biosynthesis. Biocatal. Biotransform. 2006, 24, 401–408. 10.1080/10242420601033738. [DOI] [Google Scholar]
- Shepherd S. A.; Menon B. R.; Fisk H.; Struck A. W.; Levy C.; Leys D.; Micklefield J. A Structure-Guided Switch in the Regioselectivity of a Tryptophan Halogenase. ChemBioChem 2016, 17, 821–824. 10.1002/cbic.201600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitto E.; Huang Y.; Bingman C. A.; Singh S.; Thorson J. S.; Phillips G. N. The structure of flavin-dependent tryptophan 7-halogenase RebH. Proteins 2008, 70, 289–293. 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- Dong C.; Flecks S.; Unversucht S.; Haupt C.; Van Pee Ligon K.-H.; Naismith J. H. Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309, 2216–2219. 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu X.; De Laurentis W.; Leang K.; Herrmann J.; Ihlefeld K.; van Pée K.-H.; Naismith J. H. Structural insights into regioselectivity in the enzymatic chlorination of tryptophan. J. Mol. Biol. 2009, 391, 74–85. 10.1016/j.jmb.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pée K. H. Biosynthesis of halogenated metabolites by bacteria. Annu. Rev. Microbiol. 1996, 50, 375–399. 10.1146/annurev.micro.50.1.375. [DOI] [PubMed] [Google Scholar]
- Yeh E.; Garneau S.; Walsh C. T. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3960–3965. 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flecks S.; Patallo E. P.; Zhu X.; Ernyei A. J.; Seifert G.; Schneider A.; Dong C.; Naismith J. H.; van Pée K. H. New insights into the mechanism of enzymatic chlorination of tryptophan. Angew. Chem., Int. Ed. 2008, 47, 9533–9536. 10.1002/anie.200802466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.; Kotzsch A.; Dorward M.; van Pée K.-H.; Naismith J. H. Crystallization and X-ray diffraction of a halogenating enzyme, tryptophan 7-halogenase, from Pseudomonas fluorescens. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1438–1440. 10.1107/S0907444904012521. [DOI] [PubMed] [Google Scholar]
- a Prlić A.; Bliven S.; Rose P. W.; Bluhm W. F.; Bizon C.; Godzik A.; Bourne P. E. Pre-calculated protein structure alignments at the RCSB PDB website. Bioinformatics 2010, 26, 2983–2985. 10.1093/bioinformatics/btq572. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith T. F.; Waterman M. S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Karabencheva-Christova T. G.; Torras J.; Mulholland A. J.; Lodola A.; Christov C. Z. Mechanistic Insights into the Reaction of Chlorination of Tryptophan Catalyzed by Tryptophan 7-Halogenase. Sci. Rep. 2017, 7, 17395 10.1038/s41598-017-17789-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Karplus M.; McCammon J. A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]; b Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- a Karplus M.; Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 6679–6685. 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Garcia-Viloca M.; Gao J.; Karplus M.; Truhlar D. G. How enzymes work: analysis by modern rate theory and computer simulations. Science 2004, 303, 186–195. 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- Kruschel D.; Zagrovic B. Conformational averaging in structural biology: issues, challenges and computational solutions. Mol. BioSyst. 2009, 5, 1606–1616. 10.1039/b917186j. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Thai V.; Lei M.; Ott M.; Wolf-Watz M.; Fenn T.; Pozharski E.; Wilson M. A.; Petsko G. A.; Karplus M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838–844. 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- Csermely P.; Palotai R.; Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco M.; Luque F. J. Theoretical methods for the description of the solvent effect in biomolecular systems. Chem. Rev. 2000, 100, 4187–4226. 10.1021/cr990052a. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Gorfe A. A.; McCammon J. A. Large conformational changes in proteins: signaling and other functions. Curr. Opin. Struct. Biol. 2010, 20, 142–147. 10.1016/j.sbi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Release S. d.3: Maestro, version 9.9; Schrödinger, LLC: New York, 2014.
- Schüttelkopf A. W.; Van Aalten D. M. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Schuler L. D.; Daura X.; Van Gunsteren W. F. An improved GROMOS96 force field for aliphatic hydrocarbons in the condensed phase. J. Comput. Chem. 2001, 22, 1205–1218. 10.1002/jcc.1078. [DOI] [Google Scholar]
- Malde A. K.; Zuo L.; Breeze M.; Stroet M.; Poger D.; Nair P. C.; Oostenbrink C.; Mark A. E. An automated force field topology builder (ATB) and repository: version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. 10.1021/ct200196m. [DOI] [PubMed] [Google Scholar]
- Fiser A.; Šali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pronk S.; Páll S.; Schulz R.; Larsson P.; Bjelkmar P.; Apostolov R.; Shirts M. R.; Smith J. C.; Kasson P. M.; van der Spoel D.; et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 845–854. 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J.; Postma J. P.; van Gunsteren W. F.; Hermans J.. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular forces; Springer: Dordrecht, 1981; pp 331–342. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P.; ElSawy K. M.; McCammon J. A.; Caves L. S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Studio R.RStudio: Integrated Development Environment for R; RStudio Inc: Boston, MA, 2012. [Google Scholar]
- Singh W.; Fields G. B.; Christov C. Z.; Karabencheva-Christova T. G. Effects of Mutations on Structure-Function Relationships of Matrix Metalloproteinase-1. Int. J. Mol. Sci. 2016, 17, 1727 10.3390/ijms17101727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hünenberger P. H.; Mark A.; Van Gunsteren W. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol. 1995, 252, 492–503. 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- Yeh E.; Blasiak L. C.; Koglin A.; Drennan C. L.; Walsh C. T. Chlorination by a Long-Lived Intermediate in the Mechanism of Flavin-Dependent Halogenases. Biochemistry 2007, 46, 1284–1292. 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- Andorfer M. C.; Park H. J.; Vergara-Coll J.; Lewis J. C. Directed evolution of RebH for catalyst-controlled halogenation of indole C-H bonds. Chem. Sci. 2016, 7, 3720–3729. 10.1039/C5SC04680G. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.