

Cross-linked polymer films synthesized via vapor deposition improve cycling behavior of silicon anodes for lithium ion batteries.
Abstract
Electrochemical reduction of lithium ion battery electrolyte on Si anodes was mitigated by synthesizing nanoscale, conformal polymer films as artificial solid electrolyte interface (SEI) layers. Initiated chemical vapor deposition (iCVD) was used to deposit poly(1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane) (pV4D4) onto silicon thin film electrodes. pV4D4 films (25 nm) on Si electrodes improved initial coulombic efficiency by 12.9% and capacity retention over 100 cycles by 64.9% relative to untreated electrodes. pV4D4 coatings improved rate capabilities, enabling higher lithiation capacity at all current densities. Impedance spectroscopy showed that SEI resistance grew from 50 to 191 ohms in untreated Si and only 34 to 90 ohms in pV4D4-coated Si over 30 cycles. Post-cycling Fourier transform infrared and x-ray photoelectron spectroscopy showed that pV4D4 moderated electrolyte reduction and altered SEI composition, with LiF formation being favored. This work will guide further development of polymeric artificial SEIs to mitigate electrolyte reduction and enhance capacity retention in Si electrodes.
INTRODUCTION
Si anodes promise breakthroughs in energy densities and lower costs for lithium ion batteries, but several roadblocks preclude practical realization of their key advantages. Graphite remains the dominant anode material in commercial lithium ion battery anodes despite its relatively low specific capacity of 372 mAh g−1 because of its unique ability to form a stable, self-limiting solid electrolyte interface (SEI) in alkyl carbonate electrolytes, e.g., 1 M LiPF6 in a mixture of ethylene carbonate (EC) and linear carbonates. The SEI eliminates continual reduction of the electrolyte as the anode operates at potentials below the electrochemical stability window of the electrolyte. In contrast to graphite, silicon (Li15Si4) has a theoretical specific capacity of 3579 mAh g−1 at room temperature, making it an attractive candidate material for next-generation anodes (1). However, silicon undergoes 300% volume dilation during cycling, resulting in physical damage to the SEI and low coulombic efficiencies as electrolyte is consumed to reform the SEI and through other side reactions (2).
A common approach to addressing the unstable SEIs on silicon anodes is the fabrication of artificial SEIs or composites where the silicon is embedded within another material, often carbon (3). These layers are intended to prepassivate the silicon surface, minimizing irreversible capacity in the critical first charge of the cell and ideally over extended cycling. Many materials and fabrication approaches have been developed, and several recent review articles detail these strategies (2, 4). One approach is physical or chemical vapor deposition of nanoscale glass or metal oxide coatings onto silicon surfaces via magnetron sputtering or atomic layer deposition (ALD) (2, 4, 5). A challenge with this approach is that the coatings must be mechanically compliant to accommodate the volume dilation of Si; otherwise, they will fracture and expose new surface area that can further reduce electrolytes (6). For example, He et al. showed that ALD-synthesized Al2O3 enables good capacity utilization in silicon electrodes (~2750 mAh g−1) but had insufficient mechanical toughness to enable long-term cycling. Cracks formed in the surface layer after 40 cycles of deep lithiation (0.005 to 2 V) (7, 8). Other techniques include synthesis of fixed volume shells that accommodate expansion of smaller-diameter silicon nanoparticles within the shell. One such example is carbon shells surrounding silicon nanoparticles. These materials yielded anodes with good capacity and longevity (1160 mAh g−1 after 1000 cycles at C/2); however, these techniques require many fabrication steps, including the carbonization of a polymer coating and the dissolution of a sacrificial SiO2 layer to form the shells (9). Thus, although performance is relatively good, electrode synthesis is process intensive. Another example is silicon particle confinement in TiO2 shells. This yielded good capacity (804 mAh g−1 after 100 cycles), which is similar to the previous study, but again multistep synthesis is complex and the energy density is sacrificed because of the inactive volume of the shell (8). Last, electrolyte additives are being extensively explored, as they have been shown to alter the SEI formation and composition leading to enhanced surface passivation. The additives fluoroethylene carbonate (FEC) and vinylene carbonate (VC) have been shown to promote the formation of a thin SEI of varying compositions and to efficiently passivate the anode surface; however, passivation by FEC is incomplete and will eventually grow a thick SEI. Passivation by VC yields an ionically resistive layer, limiting high rate capability (10).
Thin synthetic polymer layers are promising materials as artificial SEIs because they can be designed for mechanical flexibility to maintain the passivation of silicon electrode in spite of volume dilation (3, 11). In addition, polymer film synthesis can be relatively simple and rapid compared with other deposition techniques, such as ALD or sputtering, and can be executed at low temperatures. Unlike glass or ceramic surface layers, which are susceptible to fracture because of silicon volume changes during (de)lithiation, polymer thin films have been shown to accommodate silicon expansion while simultaneously exhibiting chemical stability, maintaining structural integrity during cycling and enhancing silicon cycling performance (8). A previous study showed that surface-tethered poly(methyl methacrylate) (PMMA) brush films partially exclude electrolyte from the Si electrochemical interface, resulting in enhanced specific capacity, higher coulombic efficiency, and improved cycling stability (12). However, the brushes do not fully exclude electrolyte solvent, and some irreversible reduction still occurs. Using this approach, thick films (>150 nm) are required to substantially enhance coulombic efficiency, but the drawback is greater ohmic resistance and lower specific capacities during galvanostatic cycling. Films that can effectively exclude the electrolyte but are also ultrathin and ionically conductive should provide both enhanced specific capacities and coulombic efficiencies. In this work, we exploit initiated chemical vapor deposition (iCVD) to synthesize ultrathin, cross-linked polymer thin films on silicon thin film electrodes. iCVD is a thin film processing tool that integrates polymer synthesis with processing (film formation) as it directly converts monomer vapor to polymer film on a substrate. Heterogeneous polymerization is achieved by adsorbing monomer molecules to the surface and generating primary radicals via thermolysis of peroxide initiators at a heated filament array positioned above the substrate. The radicals collide with the adsorbed monomer, initiating a free radical polymerization. Several recent review articles further highlight the mechanism and capabilities of iCVD, which include exquisite control of polymer chemistry and thin film structure on both planar and highly complex, nanostructured substrates (13–16). In short, iCVD can be used to conformally coat battery electrodes with ultrathin polymer films without extensive processing steps. We used iCVD to deposit thin films of poly(1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane) (pV4D4) on silicon electrodes (17). The polymer, pV4D4, was chosen for its adhesion to silicon surfaces, which eliminates the need for covalent surface-tethering agents, and for its potential as an ionically conductive and electronically resistive polymer layer (17–20). Recent studies show that the siloxane ring in the 1,3,5,7-tetravinyl-1,3,5,7-tetramethylcyclotetrasiloxane (V4D4) monomer can interact and coordinate with lithium cations. These overlaid rings in an ultrathin pV4D4 film provide a path for lithium transport, and appreciable lithium conductivities are obtained in ultrathin layers that are less than 30 nm in thickness. Furthermore, the tetrafunctionality of the V4D4 monomer ensures the formation of highly cross-linked films, which should exclude the electrolyte and enhance surface passivation (21). Last, the films are thermally stable to at least 400°C in N2, showing negligible mass loss over a 2-hour soak duration (22). The end result is a stable nanoscale, electrically resistive, and ionically conductive polymeric artificial SEI that mitigates capacity loss from surface side reactions and electrolyte reduction. In this work, we explore the effects of pV4D4 coatings on silicon thin film anode cycling performance. We demonstrate improved first-cycle coulombic efficiency and average reversible capacity of pV4D4-coated silicon thin film electrodes, and long-term passivation of the silicon is confirmed using electrochemical impedance spectroscopy (EIS). This work provides the groundwork for developing polymeric artificial SEIs for silicon anodes using iCVD.
RESULTS
Material characterization
pV4D4 films were deposited to target thicknesses of 15, 25, 35, and 45 nm using laser interferometry for real-time thickness control. The deposition rate was 10 nm/min, which is consistent with other reports of pV4D4 by iCVD (23). The thickness of the deposited films was later confirmed via spectral reflectivity, and measurements at multiple spots across the sample showed good uniformity in thickness and optical constants. Visual observations of thicker films (>100 nm) that were deposited but not used for this study found that the films are homogeneous and smooth. Reflection haze, which can be indicative of microscale nonuniformities, was not observed.
The set of specific thicknesses was chosen on the basis of the expectation that an effective artificial SEI should have a thickness on the order of 20 nm (24). Moreover, previous studies of organosilicon polymer electrolytes showed that 25-nm layers of pV4D4 can absorb lithium salts, obtaining a conductivity of 6 × 10−8 S cm−1 in the dry state at room temperature (20). However, there was an upper threshold on the extent of salt absorption. Reeja-Jayan et al. (25) demonstrated that Li content reaches a maximum of 1 weight % (1 Li+ per ring) at a pV4D4 film thickness of 25 nm; beyond 25 nm, the film generally exhibits dielectric behavior. In addition, density functional theory suggested that the Si─O─Si bonds of the monomer interact favorably with lithium ions and can facilitate transport through the film via ion hoping (25). Therefore, overlaid pV4D4 can act as channels for lithium ions to transport to and from the underlying silicon anode. Another important consideration is the stability and thickness retention of the films in solvents. Ideally, the pV4D4 films should adhere to the Si without the aid of an organosilane coupling agent, which influences the chemical and electrochemical behavior of the electrode interface. Physical stability of the films was assessed using 100-nm-thick coatings, with the expectation that swelling stresses will have lower magnitudes and be less likely to delaminate in thinner films. Pre- and post-characterization of the 100-nm films soaked in deionized water and dimethyl carbonate (DMC) for 5 min revealed negligible changes in film thicknesses, confirming the good adhesion of these highly cross-linked films. Without the use of organosilane adhesion promoters, the adhesion of iCVD films is controlled by physical adsorption. The affinity of organosilicon polymers to silicon surfaces and the highly cross-linked nature of the pV4D4 contribute to the strong adhesion (18, 26). Moreover, film swelling due to solvent uptake was measured using spectral reflectivity (table S1). Thicknesses remained constant despite soaking, indicating that the films undergo minimal swelling in the presence of these solvents, which is expected on the basis of previous studies of pV4D4 (24).
The composition of the pV4D4 films was qualitatively characterized using Fourier transform infrared (FTIR)–attenuated total reflectance (ATR). Spectra of the V4D4 monomer and pV4D4 films are presented in Fig. 1, A and B. The two spectra are very similar, suggesting that the monomer chemical structure is largely preserved in the polymer film. The structure of the film has been reported previously (27). SiO2 peaks are observed at 1235 cm−1 from native oxide on the silicon thin film electrode or siloxane bonds within the pV4D4 bulk. Prominent absorbance bands associated with Si─O modes are 1071 cm−1 (Si─O─Si bending, network), 1065 cm−1 (Si─O stretching, ring), and 1009 cm−1 (─Si─O stretching, linear). Peaks associated with Si─C vibrational modes are 1408 cm−1 (CH3 bending in Si-Mex), 1261 cm−1 (CH3 bending in Me2SiO2), 791 cm−1 (Si─C asymmetric rocking), and 749 cm−1 (Si─C rocking) (28).
Fig. 1. Surface characterization of pV4D4-coated and uncoated silicon electrodes.
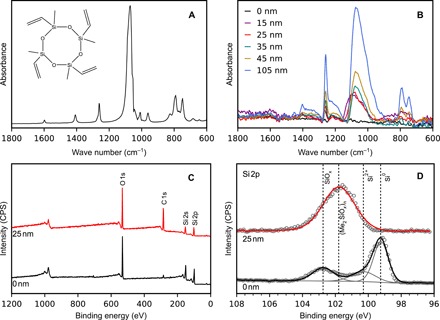
FTIR-ATR spectra of (A) the V4D4 monomer and (B) the pV4D4-coated thin film silicon electrodes. (C) Normalized XPS survey spectra and (D) normalized high-resolution spectra of uncoated and 25-nm pV4D4–coated silicon thin films. CPS, counts per second.
The most distinct differences between the two spectra are the absence of the following two absorbance bands in the pV4D4 spectra: 961 cm−1 attributed to CH2 bending in vinyl bonds and 1598 cm−1 associated with the C═C stretch mode of the vinyl. The absence of these peaks is an indication of polymerization of the V4D4 monomer and/or film cross-linking, as the vinyl species react to form large networks (17). Polymerization is further supported by the C─H vibrational bands found in the complete mid-infrared spectra of pV4D4 films. Spectra for the V4D4 monomer and 200-nm-thick pV4D4 film are presented in fig. S1. In the pV4D4 spectrum, strong absorbances at 2915 and 2870 cm−1 appear and are assigned to the asymmetric and symmetric C─H stretches, respectively, of the saturated aliphatic backbone resulting from chain polymerization of the vinyl moieties of V4D4 monomer (17, 18). In total, the coated silicon electrode spectra, which closely match the spectrum of the V4D4 monomer, strongly support that the Si thin films have been successfully coated with pV4D4.
Polymer film composition was quantitatively characterized using x-ray photoelectron spectroscopy (XPS). Survey spectra for uncoated and 25-nm pV4D4–coated silicon electrodes are shown in Fig. 1C. The elemental composition of the uncoated Si electrode was determined to be 8.4% C, 35.8% O, and 55.8% Si. The carbon component can be attributed to adventitious carbon, yet it is likely that some organosilicon components (Si─C and Si─O─C) are integrated into the sputtered Si thin films. The elemental Si/O ratio is 1.6, which is far higher than the expected Si/O ratio of ~0.50 for native SiO2, indicating that the native oxide layer on the sputtered Si is less than 10 nm in thickness or has a substoichiometric composition. Typically, we do not achieve a perfect native oxide in our sputtered films (12). The elemental composition of the 25-nm pV4D4–coated electrode was determined to be 50.6% C, 26.8% O, and 22.6% Si. The Si/O ratio is 0.85, which is reasonably close to the expected monomer Si/O ratio of 1.0, further supporting the claim that pV4D4 has been prepared on the surface of the Si. This Si/O ratio is also consistent with as-deposited pV4D4 films previously explored by Trujillo et al. (19).
The Si 2p high-resolution spectra, shown in Fig. 1D, further support that the Si thin film electrode has been coated with pV4D4. The uncoated electrode spectrum contains three deconvoluted component peaks, attributed to native SiOx (102.7 eV) and elemental silicon (Si 2p1/2 at 99.25 eV and Si 2p3/2 at 100.3 eV) (29, 30). These peaks are not observed in the 25-nm pV4D4–coated silicon spectrum, which suggests that the film not only covers the native oxide and/or the bulk silicon but also is greater than 10 nm in thickness, the scanning depth of the XPS instrument. Instead, a new peak is observed at 101.8 eV, which is assigned to disubstituted siloxane (31). While the V4D4 monomer is cyclic, the chemical composition is close to that of polydimethylsiloxane (PDMS), that is, Si─O─Si bonding is present in pV4D4. The high-resolution Si 2p spectrum of pV4D4 closely matches that of PDMS in previous literature (31). In summary, both the coated silicon electrode survey and high-resolution spectra confirm that a pV4D4 film coats the electrode surface.
Electrochemical cycling
The effect of the pV4D4 coatings on the electrochemical behavior of Si thin film was initially assessed through a comparison of galvanostatic cycling performance, shown in Fig. 2, A and B. Coated and uncoated silicon thin film electrodes were cycled at 25°C against lithium metal in a coin cell format using 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC as electrolyte. All cells were initially cycled twice at a C/10 rate to establish an SEI on the uncoated silicon surface and then 1C thereafter.
Fig. 2. Electrochemical characterization of pV4D4-coated silicon electrodes.
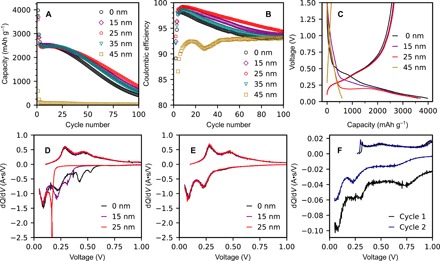
(A) Lithiation capacity and (B) coulombic efficiency of pV4D4-coated silicon films cycled against lithium metal from 0.05 to 1.5 V at 25°C at a rate of C/10 for the first two cycles and then at 1C after. Electrolyte was 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC. (C) First cycle voltage profiles and (D) first and (E) second cycle differential capacity curves of pV4D4-coated silicon electrodes. (F) Differential capacity curves of 45-nm pV4D4–coated silicon electrodes.
The untreated silicon electrodes show cycling behavior typical of thin film electrodes (29, 32). Initial lithiation and delithiation capacities were 3980 and 2560 mAh g−1, respectively, and reversible capacity decayed to 404 mAh g−1 over 100 cycles. Initial coulombic efficiency was 64.4%, reaching a maximum of 98.1% at cycle 7 and then decaying consistently over 100 cycles (Fig. 2B). The addition of pV4D4 thin films up to a thickness of 25 nm improved overall cycling performance. Films thinner than 25 nm provided only a modest improvement. The optimum thickness appears to be 25 nm, providing the highest initial (2657 mAh g−1) and final (789 mAh g−1) reversible capacities. In addition to reversible capacity, the 25-nm film improved the initial coulombic efficiency to 73.2% and reached a maximum coulombic efficiency of 99.2% at cycle 8. This is an important consideration, as commercial cells are assembled in the discharged state and any lithium lost during the SEI formation is detrimental to the capacity matching provided by the cathodes, which tend to be capacity limiting and the most expensive material component (33). The pV4D4 reduces irreversible capacity by prepassivating the silicon surface, reducing the extent of the electrolyte reduction (21). This should also manifest as a lower film resistance, leading to better power performance. A comprehensive summary of cycling data is provided in Table 1.
Table 1. Summary of initial and final cycling data for pV4D4-coated silicon electrodes.
Average (de)lithiation capacity and average coulombic efficiency at cycles 1 and 100 for (un)coated silicon electrodes.
| Coating thickness (nm) | Cycle 1 | Cycle 100 | ||||
|
Charge capacity (mAh g−1) |
Discharge capacity (mAh g−1) |
Coulombic efficiency |
Charge capacity (mAh g−1) |
Discharge capacity (mAh g−1) |
Coulombic efficiency | |
| 0 | 2705 ± 204 | 4174 ± 278 | 64.8 ± .6 | 442 ± 92 | 472 ± 97 | 93.5 ± .2 |
| 15 | 2491 ± 107 | 3428 ± 226 | 72.8 ± 1.7 | 565 ± 12 | 605 ± 13 | 93.5 ± .1 |
| 25 | 2685 ± 105 | 3670 ± 139 | 73.2 ± .8 | 729 ± 46 | 776 ± 47 | 93.9 ± .4 |
| 35 | 2632 ± 28 | 3720 ± 28 | 70.7 ± .2 | 482 ± 24 | 516 ± 27 | 93.4 ± .2 |
| 45 | 168 | 598 | 28 | 33 | 35 | 93.2 |
As film thickness exceeds 25 nm, cycling performance begins to suffer, which is most likely an effect of increased ionic resistance contributed by the pV4D4 film, limiting lithium ion transport to the silicon surface (32). The first cycle and average cycling performance of the 35-nm pV4D4–coated electrodes is observed to be lower than those of the 25-nm coated electrode and is more comparable to the performance of the 15-nm pV4D4. As the polymer thickness increases further to 45 nm, impractical lithiation capacity is observed, which is a function of the limited penetration of charge carriers throughout the pV4D4 film thickness (20).
For further insight into the electrochemical effect of the pV4D4 films, the voltage profiles of the first cycle were analyzed (Fig. 2C). It is important to consider that, at 25°C, lithium begins to alloy with silicon from 300 to 250 mV versus Li/Li+ to form LixSi, where x = 0.0 to 2.0 (34). Thus, the specific capacity observed above 300 mV is due to electrochemical reactions not associated with lithiation and is potentially irreversible. The formation of the surface layer and the reduction of SiOx layers are examples of irreversible electrochemical processes. First cycle lithiation of the uncoated silicon electrode shows a significant lithiation plateau due to the electrolyte reduction and the SEI formation at 0.53 V. Capacity observed below 0.3 V is 2709 mAh g−1, which is fairly close to the initial delithiation capacity observed (2561 mAh g−1). The addition of thin films of pV4D4 (15 and 25 nm) reduces the irreversible capacity above 0.3 V while increasing the reversible capacity. The uncoated and coated (15- and 25-nm pV4D4) electrodes have nearly identical delithiation profiles. The primary difference is the irreversible processes observed during the first lithiation. The voltage profiles are different for the thicker pV4D4 films (45-nm pV4D4). These films appear to limit lithiation capacity because of a large overpotential from the high ionic resistance of the thicker film. Another indirect indication of this overpotential is the rapid relaxation of the electrode to 0.27 V during the rest step (open circuit) in the cycling protocol.
Differential capacity curves for the first and second cycles shown in Fig. 2 (D and F) further support this analysis. In the untreated Si, a peak is observed at 0.53 V, which can be attributed to the SEI formation and the electrolyte reduction, and an additional peak is observed at 0.42 V, which can be attributed to lithium reaction with surface oxides (35). Lithiation peaks are observed at 0.06 V (the formation of crystalline Li3.75Si) and 0.22 V (previously discussed). The addition of a 15-nm pV4D4 film slightly changes the differential capacity curve. The electrolyte reduction peak is shifted to a lower potential at 0.31 V. This behavior has been observed previously with LiPON and PMMA thin film coatings on Si (12, 32). However, the reduction peak area from 0.30 to 0.39 V is smaller than that of the processes in the untreated silicon from 0.41 to 0.59 V, indicating that less total charge is associated with irreversible processes. Moreover, the lithiation peak at 0.21 V is larger than in the untreated electrode, improving first cycle reversible capacity and coulombic efficiency. In conjunction with previous cycling data, the presence of this peak indicates that, while the 15-nm film improves the initial cycling performance, it is insufficient in passivating the silicon surface.
Differential capacity curves of the 25-nm pV4D4–coated silicon electrode, previously observed as the optimum thickness, show little current for electrochemical processes above 0.3 V, suggesting good surface passivation. The first lithiation peak occurs at a much lower potential at 0.18 V, which may be a combination of the silicon lithiation and the SEI formation, which is difficult to fully deconvolute. However, the delithiation/oxidation plateaus observed at 0.28 and 0.49 V remain similar in electrodes coated with 0- to 25-nm V4D4, indicating that the 15- and 25-nm pV4D4 films do not kinetically limit lithiation to the extent that cycling is adversely affected.
Differential capacity curves in the second cycle (Fig. 2E) are largely the same. Significant capacity associated with processes above 0.3 V is not observed for any of the samples, suggesting that the electrodes are largely passivated by the second cycle. However, the untreated silicon electrode underwent far more undesirable electrochemical processes to achieve this passivation, indicating a higher consumption of electrolyte required to passivate the silicon surface. pV4D4, on the other hand, limits the electrolyte reduction in the first cycle. From the potential shifts observed in the differential capacity curves, it is expected that the pV4D4 coating not only limits the electrolyte reduction in the first cycle but also alters the composition of the SEI. Therefore, the addition of pV4D4 is responsible for the improved coulombic efficiency observed in the pV4D4-coated electrode cycling data, as less capacity is lost to irreversible reactions and electrolyte reduction.
With an excessively thick polymer film (45 nm; Fig. 2F), lithium transport to the silicon surface is hindered, as demonstrated by the relatively shallow lithiation peaks, with an order of magnitude less lithiation capacity. While the complete exclusion of the electrolyte is desirable to prevent electrolyte reductions, the kinetic limitations imposed by the film lead to the poor and incomplete lithiation of the silicon. The limited ionic conductivity in pV4D4 imposes limitations on the thicknesses of these passivation layers.
To further understand the effect of the pV4D4 film on the electrochemical stability of silicon electrodes, we performed a rate capability test. Specific lithiation capacity as a function of rate is shown in Fig. 3. The lithiation capacity of both the uncoated and pV4D4-coated electrodes decreases as the cycling rate increases from C/10 to 1C but recovers as the rate is returned to C/10. The lithiation capacity of the pV4D4-coated silicon electrode is observed to be higher than that of the uncoated electrode at every cycling rate during the test. Final lithiation capacities for the pV4D4-coated and uncoated silicon electrodes are 3107 and 2481 mAh g−1, respectively. A calculation of the recovery (the ratio of capacity at cycle 20 to capacity at cycle 4) shows that the pV4D4-coated silicon electrode retains 98.7% of its capacity, whereas the uncoated silicon electrode retains only 88.3%, although this is likely a function of the ability of the pV4D4 to stabilize the lithiation capacity for the duration of the test. Regardless, the higher capacity observed at all C rates in pV4D4-coated films suggests that the film improves the rate performance of the silicon electrode. This may be attributed to the film’s ability to passivate the silicon surface despite large volume expansion during lithiation. For cycling rates exceeding 1C, we expect both coated and bare electrodes to have further reduced capacities because of kinetic limitations; however, the coated electrodes should have better capacity retention over extended cycling because of the protective nature of the pV4D4.
Fig. 3. Rate capability of pV4D4-coated and uncoated silicon electrodes.
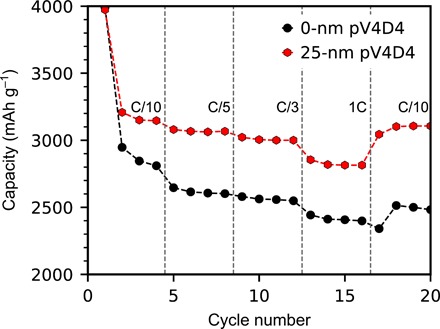
Lithiation capacity of pV4D4-coated silicon electrodes during a variable rate test. Cells were cycled four times at C/10, C/5, C/3, 1C, and lastly at C/10 each.
Electrochemical impedance spectroscopy
EIS was performed as a function of cycling to compare longer-term behavior of the SEI in pV4D4-coated and uncoated electrodes; the data are provided in Fig. 4. Three electrode split cells were assembled, and silicon electrodes [working electrode (WE)] were cycled against lithium metal [counter electrode (CE)/reference electrode (RE)] from 1.5 to 0.05 V at 25°C. Impedance measurements were collected every five cycles at a potential of 0.05 V. In all spectra, the frequency-dependent data make a single semicircle spanning the medium frequencies (100 kHz to 10 Hz) and a line at 45° then nearly vertical relative to the x axis at low frequencies (<10 Hz). The impedance spectra can be described as Relectrolyte − [R − CPE]surface − Wdiff, where the bracketed elements are in parallel (36). A constant phase element (CPE) was used instead of an ideal capacitor to describe the dispersion in capacitance caused by the rough morphology of the silicon/copper substrate. Relectrolyte is the resistance of the liquid electrolyte. [R − CPE]surface is assigned to a slightly depressed, difficult-to-deconvolute semicircle, which is attributed to a combination of charge transfer and ion transport through the surface film (SEI), and Wdiff corresponds to solid-state diffusion of Li+ into the Si thin film bulk (37).
Fig. 4. EIS of pV4D4-coated and uncoated silicon electrodes.
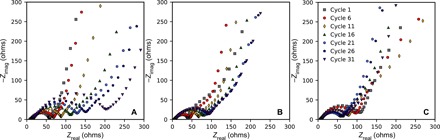
EIS spectra of silicon electrodes coated with (A) 0-nm, (B) 25-nm, and (C) 45-nm pV4D4 films. Three electrode potentiostatic EIS measurements were performed after the silicon electrodes had been lithiated to 50 mV. Silicon electrodes acted as the WE, while lithium metal foil acted as the CE and the RE. Electrolyte used was 1 M LiPF6 in 1:1:1 EC:EMC:DMC. Cell temperature was maintained at 25°C.
Impedance spectra of the uncoated electrodes (Fig. 4A) show that the semicircle continually increases over the 30 cycles. After the SEI formation in the first lithiation, the surface resistance determined from fitting was 50 ohms. Over the next 30 cycles, the resistance of the semicircle grows to 191 ohms. This rise in the surface film resistance is consistent with development of a nonpassivating SEI on an uncoated silicon surface and has been observed in other studies of Si thin film anodes (38). Subsequent cycling of silicon electrodes results in a thicker SEI and continual capacity loss as an additional electrolyte is consumed to reform the SEI.
The addition of a 25-nm pV4D4 film reduces growth in the surface film resistance over these cycles, shown in Fig. 4B. The resistance of the surface processes was determined to be 34 ohms after the first lithiation. The lower resistance compared to the uncoated Si anode suggests that pV4D4 reduces the SEI formation and/or alters the SEI composition to improve surface film conductivity, which is consistent with the galvanostatic cycling data. The ability of the pV4D4 to limit electrolyte reduction or decomposition is also responsible for the improved initial lithiation capacity and coulombic efficiency. Over the 30 cycles, the semicircle resistance only increases to 90 ohms, which is less than half of the untreated electrodes, providing further evidence that the improved passivation from the pV4D4 is also sustained over long cycling. The reduction in the surface film resistance growth is consistent with previous cycling, as average lithiation capacity and cycling stability improved with the addition of a 25-nm pV4D4 film (Fig. 2B).
The general trend in impedance growth observed for the bare and pV4D4-coated silicon electrodes is consistent with the capacity decay observed during galvanostatic cycling (Fig. 2B). The interfacial resistance continues to grow in both treated and untreated electrodes likely because of the continued SEI formation, leading to a reduced accessible capacity. However, the 25-nm pV4D4 film passivates the electrode and slows the growth in interfacial resistance. While the coating is not 100% effective, the general trend in impedance over the first 30 cycles and in capacity over 100 cycles shows that the effect of the pV4D4 coating is significant.
In contrast, coating of an excessively thick film (45 nm; Fig. 4C) substantially alters the passivation behavior. After the first lithiation, the surface resistance is 102 ohms, much larger than the uncoated and 25-nm pV4D4–coated silicon electrodes. This larger resistance is due to the greater ohmic resistance of the film and due to charge transfer resistance (convoluted with the surface layer) being potential dependent. (37). From the cycling and dQ/dV data, we suspect that Si has not fully lithiated to 0.05 V even after the potentiostatic hold step. Thus, the true potential is higher in the first impedance measurement, contributing to the larger resistance. As cycling continues, the semicircle decreases in diameter, reaching a minimum resistance of 52 ohms. Over the next 10 cycles (to the 31st lithiation), the semicircle continually increases in diameter to a final resistance of 68.4 ohms. This trend in impedance is consistent with anomalous cycling results in fig. S2 that were obtained for several films (45 nm and thicker). The behavior suggests that the 45-nm films start off being highly resistive, hence the limited capacities. Through repeated cycling, the gradually increasing capacity is attributed to more lithium salt being incorporated into the passivation films. The chemical or physical conditioning of the film that is occurring to enable the salt incorporation results in greater ion diffusivity in the film. Then, as the film begins to reach lower lithiation potentials, potential-dependent irreversible processes can occur (the passivation layers are imperfect), leading to an increased interfacial impedance and less capacity.
Post-cycling electrode characterization
To understand the effect of pV4D4 on the SEI composition, we performed post-mortem characterization of cycled electrodes. Electrodes were cycled 30 times against lithium metal from 0.05 to 1.5 V at 1C at 25°C using 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC. The cells were disassembled, and the anodes were washed three times in DMC and dried at 80°C overnight. The FTIR spectra of the pV4D4-coated and uncoated electrodes, shown in Fig. 5A, are quite different. First, FTIR shows that the overall absorbance of the pV4D4-coated electrode is generally lower than that of the uncoated electrode, which suggests that the pV4D4 coating on the silicon electrode limits the electrolyte reduction and the SEI formation. This observation is consistent with previous cycling and impedance data. Second, a comparison of the two spectra of cycled silicon electrodes reveals that the pV4D4 film also alters the composition of the SEI. Post-cycling FTIR suggests that the pV4D4 polymer remains on the surface throughout the cycling. The vibrational mode at 1264 cm−1 associated with the Si─CH3 bend in pV4D4 is well resolved from absorption bands associated with electrolyte reduction products. This absorption band can be found in the spectrum of pV4D4-coated electrodes before and after cycling. However, it is absent in the spectra of uncoated electrodes, further confirming that the 1264 cm−1 peak can be attributed to pV4D4. The features spanning from 1250 to 900 cm−1 are similar in both spectra and can be attributed to an overlap of multiple species, including (CH2OCO2Li)2 (LEDC), Si─O─Si, and Li-alkoxy species (29, 39). The notable difference in the cycled silicon is the peaks associated with Li2CO3 at 1510 to 1410 cm−1 (C─O stretching), 1416 cm−1 (C─O stretching), and 864 cm−1 (40). Relative to untreated Si, the intensities of these vibrational modes in Si coated with 25-nm pV4D4 are much weaker, indicating that less Li2CO3 is present in the reduction products.
Fig. 5. Post-cycling surface characterization of pV4D4-coated and uncoated silicon electrodes.
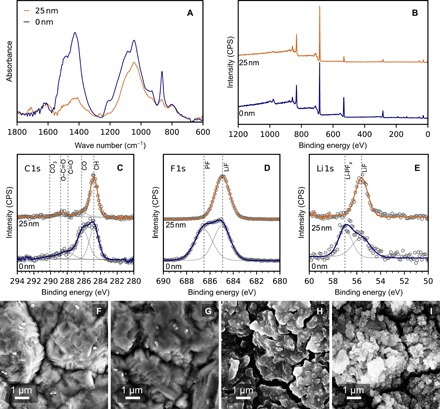
(A) FTIR-ATR and (B) XPS survey spectra of uncoated and 25-nm pV4D4–coated silicon electrodes after 30 cycles against lithium metal from 0.05 to 1.5 V at 25°C at 1C using 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC as electrolyte. Electrodes were rinsed in DMC and dried at 80°C for 48 hours. (C) Carbon, (D) fluorine, and (E) lithium high-resolution scans of the cycled electrodes are shown. Scanning electron micrographs are shown for (F) as-deposited Si electrodes, (G) as-deposited 25-nm pV4D4–coated Si electrodes, (H) Si electrodes after 30 cycles, and (I) 25-nm pV4D4–coated Si electrodes after 30 cycles.
The lower concentration of Li2CO3 in pV4D4-coated silicon electrodes suggests that the polymer film inhibits the formation of insoluble electrolyte reduction products. The electrical resistivity of the film may be responsible for the diminution of electrolyte reduction products observed near the film surface. As a result, less cycling capacity would be lost to irreversible and undesired side reactions, which agrees with previous cycling data. Future work will be required to elucidate the exact mechanism responsible for the effect of pV4D4 on the SEI composition.
XPS spectra for both uncoated and pV4D4-coated electrodes after 30 cycles are provided in Fig. 5, B to E. The survey spectra in Fig. 5B show the obvious incorporation of F and Li into the SEIs of both electrodes. As these elements were absent before coating, it can be concluded that LiPF6 salt decomposition products are incorporated into the SEI during cycling. The compositions of the SEI layers of (un)coated silicon electrodes are summarized in table S2. The concentrations of C, O, and Si decrease in silicon electrodes after cycling, while the concentrations of Li and F increase. The composition of the SEI formed on the uncoated Si electrode is consistent with previous XPS studies, which show appreciable salt and solvent decomposition (41, 42). For the uncoated Si electrode, the high-resolution C 1s spectra in Fig. 5C are composed of multiple component peaks. Spectral deconvolution revealed contributions of aliphatic carbon (285 eV) and species containing ─CO3 (290.1 eV), C═O (288.1 eV), and C─O (286.3 eV). The oxygen-containing moieties are the result of the electrolyte decomposition and are consistently observed in Si SEIs (41–43). The concentration of the ─CO3 component is relatively low, suggesting that the Li2CO3 observed in the FTIR is concentrated deeper within the SEI layer near the Si interface. Moreover, the reduced concentration of C and O in the survey scan is further evidence of a graded concentration of Li2CO3. This has also been observed by Philippe et al. (44), who showed that Li2CO3 is concentrated at the buried Si/SEI interface. The F 1s spectra for the uncoated Si (Fig. 5D) contain peaks that are characteristic of LiF (285 eV) and LiPFx/LixPFyOz (286.6 eV) (45). These assignments are internally consistent with the two component peaks in the Li 1s spectra (Fig. 5E) associated with LiF (55.7 eV) and LixPFy/LixPFyOz (57 eV) (43). These XPS data suggest that appreciable salt decomposition is occurring on the uncoated Si electrode.
XPS spectra for the Si electrode coated with 25-nm pV4D4 are quite distinct. The SEI composition quantified from the survey spectrum shows further enrichment in lithium and fluorine content and diminishment of the carbon and oxygen content in the film. The carbon and oxygen concentrations are only 6.9 and 4.6 atomic %, suggesting that less solvent degradation products are present. This is further supported by the high-resolution spectra. The C 1s spectra are described almost entirely by a hydrocarbon component, which can be attributed to the aliphatic backbone and methyl groups in the pV4D4 and/or adventitious carbon. Integrated areas for the higher binding energy components associated with oxygen-containing functional groups are much lower relative to the uncoated Si SEI, indicating that fewer solvent degradation products are on the surface. Moreover, the F 1s and Li 1s spectra are dominated primarily by LiF. Note that LiF is FTIR transparent, which explains why this difference in SEI compositions was not detected by our FTIR analysis.
Coating the Si electrode with 25-nm pV4D4 has a similar effect on the SEI composition as adding large concentrations of FEC (>10%) to the electrolyte solution, much higher than the typical 3% commonly used in electrolyte formulations. Previous studies show that the large concentrations of FEC favor the formation of LiF over other salt decomposition products; in addition, LiF is thought to effectively passivate Si anode surfaces (45, 46). While the exact role of FEC is still under debate, it is generally acknowledged that FEC improves reversible capacity and coulombic efficiencies. Thus, from XPS data, it can be concluded that pV4D4 improves electrochemical performance in a similar fashion to FEC additives by limiting the formation of organic reduction products and favoring the formation of a LiF-enriched SEI layer.
The mechanism of LiF enrichment in pV4D4 has not been fully characterized in this report and will be the subject of future studies. We do not expect the mechanism of FEC and pV4D4 to be analogous, as fluorine enrichment in SEIs of FEC-containing electrolytes is thought to be associated with FEC reduction, whereas the pV4D4 does not contain fluorine. We can conclude that the uncoated Si is subject to the typical electrolyte degradation processes observed for EC-based electrolytes (without FEC additive). During cycling, both electrolyte solvent and salt react on the electrode surface. The organic SEI components can further degrade, ultimately forming Li2CO3 (47). Parimalam et al. (48) have also proposed that these organic species and Li2CO3 can react with LiPF6 salt to form LiF, CO2, and several PF–bearing species, as shown in the reactions below
| (1) |
| (2) |
| (3) |
These processes explain the multiple component peaks observed in the high-resolution Li 1s and F 1s spectra for the uncoated electrode. While fewer organic reaction products are observed in the pV4D4-coated electrode, the surface is enriched in LiF. One possibility is that the source of LiF is homogenous salt decomposition in the electrolyte (47)
| (4) |
PV4D4 coatings are also expected to limit the electrochemical reduction pathways because of its high electrical resistivity (19, 25). An analysis of the voltage profiles (Fig. 2) shows that the electrolyte reduction does occur, but the reaction pathways are presumably altered, given the inhibited electron transfer from the Si through the polymer layer. These new reaction pathways may lead to soluble organic products, which would explain the enrichment in LiF (insoluble in carbonate solvents) and the slower yet consistent growth in interfacial impedance observed in Fig. 4B. Alternatively, these products could also be soluble in the DMC solvent and would have been washed away during the rinsing step before XPS analysis.
Micrographs of as-deposited and pV4D4-coated electrodes, before and after cycling, are shown in Fig. 5, F to I. The as-deposited 100-nm silicon micrograph (Fig. 5F) closely matches that of the underlying copper foil, which is provided fig. S3. The large morphological features observed are from the copper foil rather than the silicon material. Likewise, micrographs of the as-deposited 25-nm V4D4 on the 100-nm silicon (Fig. 5G) suggest good conformality during the deposition of the polymer film via iCVD, as the topography of both samples closely matches. After 30 cycles, the micrographs of the uncoated silicon (fig. 5H) and pV4D4-coated silicon (Fig. 5I) show completely different morphologies. The surface of the cycled uncoated silicon is smoother, which is consistent with an SEI composed of oligomeric species such as EC reduction products. In contrast, the cycled pV4D4-coated silicon surface appears more faceted, with more distinct deposits. This morphology can be attributed to increased LiF coverage on the electrode surface. Future work using smoother electrode surfaces is required to better understand the significance of this morphology and how it develops.
DISCUSSION
In this work, we demonstrate that pV4D4 films deposited by iCVD are effective artificial SEIs, passivating silicon electrode surfaces at high cycling rates. Cycling shows that 25-nm pV4D4 films on silicon electrodes increased the average initial coulombic efficiency by 12.9% and improved the silicon delithiation capacity at cycle 100 by 64.9%. The pV4D4 films also limit the SEI growth; impedance measurements show that the surface film resistance of an untreated silicon electrode rose from 50 to 191 ohms. In contrast, the surface layer resistance of a pV4D4-coated silicon electrode rose from 34 to 90 ohms over the same number of cycles. Post-mortem FTIR and XPS characterizations of cycled, uncoated silicon suggest that the SEI formed is primarily composed of salt and solvent decomposition products. LiPF6 salt irreversibly reacts with these products to form a mixture of LiF and insoluble P-F–containing species. In contrast, post-mortem FTIR and XPS characterizations indicate that the pV4D4 polymer film improves cycling stability by primarily limiting the electrolyte reduction and the formation of SEI products, such as LEDC or Li2CO3, on the electrode surface. Because the film limits irreversible reactions between LiPF6 salt and these SEI products, the electrode ultimately forms an SEI primarily consisting of LiF, which has been shown to passivate silicon surfaces. Thus, improved silicon passivation and enhanced electrochemical performance are achieved through pV4D4 films synthesized using a relatively straightforward polymer thin film coating technique.
This study also reveals key material properties required for effective polymeric artificial SEIs. The polymer film should be electrically resistive and exclude the electrolyte to limit solvent reduction or salt decomposition near the electrode surface. It must also allow transport of Li cations. These properties are found in the pV4D4. The polymer is electrically insulating, and the tetrafunctionality of the V4D4 monomer ensures a high cross-linking density, making it an effective barrier layer to liquid permeation (17). However, the siloxane ring of the V4D4 monomer can accommodate lithium cations and facilitate their transport. Film thickness is a critical parameter, as the pV4D4 films rely on limited uptake of lithium salts into the film bulk (20, 24). The optimal SEI thickness was determined to be 25 nm, as shown in Fig. 2, which is remarkably similar to thicknesses specified for typical SEI layers that develop through normal electrolyte reduction processes. Beyond a thickness of 25 nm, lithium salt is not fully incorporated within the film bulk, leading to higher resistances and consequently lower charge capacities obtained in galvanostatic cycling, as shown in Figs. 2 and 4.
The improvement in electrochemical performance achieved with the iCVD pV4D4 coating is similar to previous studies using ALD, sputtering, and other thin film deposition techniques (6–9). Improvements are also similar to studies using electrolyte additives to engineer the interfacial chemistry of Si anodes (10). However, a distinguishing feature of the iCVD process used in this study is the ability to finely control the composition of dense, conformal, cross-linked, ultrathin films over large areas. pV4D4 homopolymer was synthesized to establish baseline performance for these highly cross-linked films. While this study shows that pV4D4 is able to successfully exclude the electrolyte and limit the electrolyte reduction, ultrathin films are required to limit ohmic resistances and realize the full utilization of electrode capacity. To address this limitation and potentially further improve performance, copolymerization of V4D4 will be an important strategy to tune film composition and properties. Copolymerization of V4D4 with various classes of comonomers is a straightforward, well-established exercise in iCVD; several copolymers incorporating V4D4 have been reported in the literature (49, 50). For future SEI studies, anionic comonomers can be incorporated to coordinate Li+, perfluorinated comonomers can be used to lower the surface energy of the film and modulate the SEI composition, or enumerable other monomers can be used to tune the network microstructure and physical properties of the film. More complex structure such as compositional grading normal to the surface can also be engineered using iCVD.
This study was unable to fully elucidate the underlying mechanism responsible for an SEI composition primarily consisting of LiF in pV4D4-coated electrodes. In addition, while the pV4D4 films in this study limited the growth of interfacial resistance relative to the uncoated case, they were not perfect and some resistance growth still occurred. It does not appear that the pV4D4 disintegrates or completely decomposes during cycling because the post-mortem FTIR in Fig. 5 shows vibrational modes characteristic of the pV4D4. Considering the high cross-linking density of the pV4D4 coating, it is possible that the pV4D4 fractured or delaminated because of silicon volume changes during extended cycling. The mechanical properties of the polymer films are expected to be critical, especially when coated on nominally spherical nanoparticles, further suggesting that copolymerization is needed to tune copolymer chemistries. Future work will develop iCVD capabilities to deposit ultrathin, conformal, electrically resistive, ionically conductive, and mechanically compliant polymeric artificial SEIs on silicon electrodes. The design of an optimal SEI is a challenging task, given the coupled chemical and physical phenomena that must be considered, but the pV4D4 deposited in this study is a strong proof of concept, providing a foundation for future efforts.
MATERIALS AND METHODS
Material synthesis
Thin films (100 nm) of amorphous silicon were deposited onto battery-grade copper foil (99.8% purity; Oak-Mitsui) by radio frequency magnetron sputtering of a 2-inch Si target (undoped, 99.999% purity; Kurt J. Lesker Company) in a custom-built deposition chamber. Key deposition parameters were a forward power of 90 W, an argon gas flow rate of 20 scm3, an operating pressure of 20 mTorr, and a target-substrate distance of 7 cm.
After Si deposition, polymer thin films were then deposited onto the Si films via iCVD in a custom-built deposition system. The deposition system was similar in design to previously published examples (51). The V4D4 monomer (>99%; Gelest Inc.) was introduced into the chamber using a bubbler. Nitrogen was sparged through the V4D4 at a flow rate of 5 scm3 to generate near-saturated vapor. Di-tert-butyl peroxide (TBPO) initiator (98%; Sigma-Aldrich) was introduced at 1 scm3 using a mass flow controller. All precursors were degassed before the deposition but otherwise were used as received. Chamber pressure was maintained at 3 torr using a downstream feedback-controlled throttling valve (model T3Bia, MKS Instruments). The substrate temperature was maintained at 50°C using a recirculating chiller (Polysciences). The filament was resistively heated at approximately 250°C to thermolyze the TBPO initiator using a dc power supply. Film thickness was monitored in real time using in situ laser interferometry on a silicon monitor wafer, and the deposition was terminated by turning off the filament after achieving the desired thickness.
Material characterization
The thickness and optical constants of the polymer coatings deposited on the Si monitor wafers were determined by measuring spectral reflectivity and fitting to an appropriate optical model (F20-UV, Filmetrics). The composition of the polymer coatings on the Si electrodes was characterized using FTIR spectroscopy (Tensor 27, Bruker) with an ATR accessory having a monolithic diamond crystal with a 45° incident angle (Specac Golden Gate). Measurements were collected from 600 to 4000 cm−1 with a resolution of 4 cm−1. A total of 128 scans were integrated for an improved signal-to-noise ratio. The spectra were baseline-corrected in the Bruker OPUS software.
XPS was performed with a monochromatized Al Kα source operated at 15 kV and 10 mA (AXIS Ultra spectrometer, Kratos Analytical). Samples were loaded onto a sample holder using a double-sided copper tape. Survey scans were performed from 1200 to −5 eV with a 1-eV step. Survey scan pass energy was set at 160 eV. Spectra binding energies were calibrated to the aliphatic C 1s peak position at 284.8 eV. Ten scans were integrated for high-resolution spectra for C 1s, O 1s, Li 1s, and Si 2p with a pass energy of 20 eV. CasaXPS software was used to analyze and deconvolute peaks using a Gaussian-Lorentzian mix with a baseline modeled using the Shirley method. Uncoated and pV4D4-coated silicon thin film electrodes were imaged using a Zeiss Auriga scanning electron microscope with variable pressure operated between 10 and 15 kV.
Silicon electrodes were cycled against lithium metal in a coin cell format, assembled in an Ar-filled glovebox. Before battery assembly, the silicon electrodes were dried at 100°C in nitrogen gas. Aromatic polyamide (aramid) fiber–based separators (Gold 40, Dreamweaver) were used. These separators were infiltrated with 100 μl of 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC electrolyte (Gotion). The cells were cycled galvanostatically from 0.05 to 1.5 V using a battery cycler (BT-2043, Arbin Instruments). The first two cycles were performed at a C/10 rate to establish the SEI, and the rate was then increased to 1C for long-term cycling. Cell temperature was maintained at 25°C.
Potentiostatic EIS was performed using a three-electrode split cell configuration (EQ-3ESTC15, MTI Corporation) to isolate behaviors on the silicon electrode. The silicon electrode (diameter, 7/16″) was the WE. Lithium metal (diameter, 9/16″) was the CE. A small piece of foil (~1.5 mm by 1.5 mm) at the end of a copper foil strip (~1 mm by 12 mm) served as the RE. The three electrodes were separated by two separators (diameter, 3/4″; Gold 40, Dreamweaver). Electrolyte used was 200 μl of 1.0 M LiPF6 in 1:1:1 EC:EMC:DMC (Gotion). For impedance as a function of cycling, cells were cycled from 0.05 to 1.5 V at 1C (SP-200, Bio-Logic Science Instruments). Before each impedance measurement every five cycles, the silicon electrode was lithiated to 0.05 V and potentiostatically held for 2 hours to achieve equilibrium. Impedance spectroscopy was performed with a frequency sweep from 3 to 25 mHz with an amplitude of 10 mV.
Supplementary Material
Acknowledgments
Funding: This work was supported in part by the National Science Foundation under CBET award no. CBET-1604471. Author contributions: The manuscript was written through the contribution of all authors. B.H.S. synthesized the samples, performed the FTIR/XPS surface characterization, and conducted the electrochemical characterization. S.W. performed the SEM characterization. W.E.T. contributed to the planning of the research study and supervision of the experiments. All authors have given approval to the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from W.E.T. (wyatt.tenhaeff@rochester.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw4856/DC1
Fig. S1. FTIR of the V4D4 monomer and the 200-nm pV4D4 film.
Fig. S2. Anomalous cycling behavior observed with thick (45 nm) pV4D4 films.
Fig. S3. Micrograph of the uncoated copper foil.
Table S1. Spectral reflectivity measurements of the pV4D4-coated silicon in various media.
Table S2. XPS data for (un)cycled (un)coated silicon electrodes.
REFERENCES AND NOTES
- 1.Obrovac M. N., Christensen L., Structural changes in silicon anodes during lithium insertion/extraction. Electrochem. Solid State Lett. 7, A93–A96 (2004). [Google Scholar]
- 2.Su X., Wu Q., Li J., Xiao X., Lott A., Lu W., Sheldon B. W., Wu J., Silicon-based nanomaterials for lithium-ion batteries: A review. Adv. Energy Mater. 4, 1300882 (2014). [Google Scholar]
- 3.Franco Gonzalez A., Yang N.-H., Liu R.-S., Silicon anode design for lithium-ion batteries: Progress and perspectives. J. Phys. Chem. C 121, 27775–27787 (2017). [Google Scholar]
- 4.Feng K., Li M., Liu W., Kashkooli A. G., Xiao X., Cai M., Chen Z., Silicon-based anodes for lithium-ion batteries: From fundamentals to practical applications. Small 14, 1702737 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Li J., Xiao X., Cheng Y.-T., Verbrugge M. W., Atomic layered coating enabling ultrafast surface kinetics at silicon electrodes in lithium ion batteries. J. Phys. Chem. Lett. 4, 3387–3391 (2013). [Google Scholar]
- 6.Zhu C., Han K., Geng D., Ye H., Meng X., Achieving high-performance silicon anodes of lithium-ion batteries via atomic and molecular layer deposited surface coatings: An overview. Electrochim. Acta 251, 710–728 (2017). [Google Scholar]
- 7.He Y., Yu X., Wang Y., Li H., Huang X., Alumina-coated patterned amorphous silicon as the anode for a lithium-ion battery with high coulombic efficiency. Adv. Mater. 23, 4938–4941 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Luo W., Chen X., Xia Y., Chen M., Wang L., Wang Q., Li W., Yang J., Surface and interface engineering of silicon-based anode materials for lithium-ion batteries. Adv. Energy Mater. 7, 1701083 (2017). [Google Scholar]
- 9.Liu N., Lu Z., Zhao J., McDowell M. T., Lee H.-W., Zhao W., Cui Y., A pomegranate-inspired nanoscale design for large-volume-change lithium battery anodes. Nat. Nanotechnol. 9, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Jaumann T., Balach J., Langklotz U., Sauchuk V., Fritsch M., Michaelis A., Teltevskij V., Mikhailova D., Oswald S., Klose M., Stephani G., Hauser R., Eckert J., Giebeler L., Lifetime vs. rate capability: Understanding the role of FEC and VC in high-energy Li-ion batteries with nano-silicon anodes. Energy Storage Mater. 6, 26–35 (2017). [Google Scholar]
- 11.Assresahegn B. D., Bélanger D., Synthesis of binder-like molecules covalently linked to silicon nanoparticles and application as anode material for lithium-ion batteries without the use of electrolyte additives. J. Power Sources 345, 190–201 (2017). [Google Scholar]
- 12.Shen B. H., Veith G. M., Tenhaeff W. E., Silicon surface tethered polymer as artificial solid electrolyte interface. Sci. Rep. 8, 11549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen N., Kim D. H., Kovacik P., Sojoudi H., Wang M., Gleason K. K., Polymer thin films and surface modification by chemical vapor deposition: Recent progress. Annu. Rev.Chem. Biomol. Eng. 7, 373–393 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Tenhaeff W. E., Gleason K. K., Initiated and oxidative chemical vapor deposition of polymeric thin films: iCVD and oCVD. Adv. Funct. Mater. 18, 979–992 (2008). [Google Scholar]
- 15.Yu S. J., Pak K., Kwak M. J., Joo M., Kim B. J., Oh M. S., Baek J., Park H., Choi G., Kim D. H., Choi J., Choi Y., Shin J., Moon H., Lee E., Im S. G., Initiated chemical vapor deposition: A versatile tool for various device applications. Adv. Eng. Mater. 20, 1700622 (2018). [Google Scholar]
- 16.Moni P., Al-Obeidi A., Gleason K. K., Vapor deposition routes to conformal polymer thin films. Beilstein J. Nanotechnol. 8, 723–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Shaughnessy W. S., Gao M., Gleason K. K., Initiated chemical vapor deposition of trivinyltrimethylcyclotrisiloxane for biomaterial coatings. Langmuir 22, 7021–7026 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Coclite A. M., Ozaydin-Ince G., d’Agostino R., Gleason K. K., Flexible cross-linked organosilicon thin films by initiated chemical vapor deposition. Macromolecules 42, 8138–8145 (2009). [Google Scholar]
- 19.Trujillo N. J., Wu Q., Gleason K. K., Ultralow dielectric constant tetravinyltetramethylcyclotetrasiloxane films deposited by initiated chemical vapor deposition (iCVD). Adv. Funct. Mater. 20, 607–616 (2010). [Google Scholar]
- 20.Chen N., Reeja-Jayan B., Liu A., Lau J., Dunn B., Gleason K. K., iCVD cyclic polysiloxane and polysilazane as nanoscale thin-film electrolyte: Synthesis and properties. Macromol. Rapid Commun. 37, 446–452 (2016). [DOI] [PubMed] [Google Scholar]
- 21.R. Carter, J. W. Long, J. F. Parker, M. B. Sassin, C. T. Love, in 48th Power Sources Conference, Denver, CO, 11 to 14 June 2018. [Google Scholar]
- 22.Burkey D. D., Gleason K. K., Structure and mechanical properties of thin films deposited from 1,3,5-trimethyl-1,3,5-trivinylcyclotrisiloxane and water. J. Appl. Phys. 93, 5143–5150 (2003). [Google Scholar]
- 23.Baxamusa S. H., Gleason K. K., Thin polymer films with high step coverage in microtrenches by initiated CVD. Chem. Vap. Deposition 14, 313–318 (2008). [Google Scholar]
- 24.Chen N., Reeja-Jayan B., Lau J., Moni P., Liu A., Dunn B., Gleason K. K., Nanoscale, conformal polysiloxane thin film electrolytes for three-dimensional battery architectures. Mater. Horiz. 2, 309–314 (2015). [Google Scholar]
- 25.Reeja-Jayan B., Chen N., Lau J., Kattirtzi J. A., Moni P., Liu A., Miller I. G., Kayser R., Willard A. P., Dunn B., Gleason K. K., A group of cyclic siloxane and silazane polymer films as nanoscale electrolytes for microbattery architectures. Macromolecules 48, 5222–5229 (2015). [Google Scholar]
- 26.O'Shaughnessy W. S., Edell D. J., Gleason K. K., Initiated chemical vapor deposition of biopassivation coatings. Thin Solid Films 516, 684–686 (2008). [Google Scholar]
- 27.Lubguban J. A. Jr., Rajagopalan T., Mehta N., Lahlouh B. I., Simon S. L., Gangopadhyay S., Low-k organosilicate films prepared by tetravinyltetramethylcyclotetrasiloxane. J. Appl. Phys. 92, 1033–1038 (2002). [Google Scholar]
- 28.Gates S. M., Neumayer D. A., Sherwood M. H., Grill A., Wang X., Sankarapandian M., Preparation and structure of porous dielectrics by plasma enhanced chemical vapor deposition. J. Appl. Phys. 101, 094103 (2007). [Google Scholar]
- 29.Veith G. M., Doucet M., Sacci R. L., Vacaliuc B., Baldwin J. K., Browning J. F., Determination of the solid electrolyte interphase structure grown on a silicon electrode using a fluoroethylene carbonate additive. Sci. Rep. 7, 6326 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pereira-Nabais C., Światowska J., Chagnes A., Ozanam F., Gohier A., Tran-van P., Cojocaru C.-S., Cassir M., Marcus P., Interphase chemistry of Si electrodes used as anodes in Li-ion batteries. Appl. Surf. Sci. 266, 5–16 (2013). [Google Scholar]
- 31.Mundry T., Surmann P., Schurreit T., Surface characterization of polydimethylsiloxane treated pharmaceutical glass containers by x-ray-excited photo- and Auger electron spectroscopy. Fresenius J. Anal. Chem. 368, 820–831 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Li J., Dudney N. J., Nanda J., Liang C., Artificial solid electrolyte interphase to address the electrochemical degradation of silicon electrodes. ACS Appl. Mater. Interfaces 6, 10083–10088 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Kwade A., Haselrieder W., Leithoff R., Modlinger A., Dietrich F., Droeder K., Current status and challenges for automotive battery production technologies. Nat. Energy 3, 290–300 (2018). [Google Scholar]
- 34.Ogata K., Salager E., Kerr C. J., Fraser A. E., Ducati C., Morris A. J., Hofmann S., Grey C. P., Revealing lithium–silicide phase transformations in nano-structured silicon-based lithium ion batteries via in situ NMR spectroscopy. Nat. Commun. 5, 3217 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Fears T. M., Doucet M., Browning J. F., Baldwin J. K. S., Winiarz J. G., Kaiser H., Taub H., Sacci R. L., Veith G. M., Evaluating the solid electrolyte interphase formed on silicon electrodes: A comparison of ex situ x-ray photoelectron spectroscopy and in situ neutron reflectometry. Phys. Chem. Chem. Phys. 18, 13927–13940 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Ruffo R., Hong S. S., Chan C. K., Huggins R. A., Cui Y., Impedance analysis of silicon nanowire lithium ion battery anodes. J. Phys. Chem. C 113, 11390–11398 (2009). [Google Scholar]
- 37.Kulova T. L., Skundin A. M., Pleskov Y. V., Terukov E. I., Kon'kov O. I., Lithium insertion into amorphous silicon thin-film electrodes. J. Electroanal. Chem. 600, 217–225 (2007). [Google Scholar]
- 38.Huang Q., Loveridge M. J., Genieser R., Lain M. J., Bhagat R., Electrochemical evaluation and phase-related impedance studies on silicon–few layer graphene (FLG) composite electrode systems. Sci. Rep. 8, 1386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Etacheri V., Haik O., Goffer Y., Roberts G. A., Stefan I. C., Fasching R., Aurbach D., Effect of fluoroethylene carbonate (FEC) on the performance and surface chemistry of Si-nanowire Li-ion battery anodes. Langmuir 28, 965–976 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Pasierb P., Komornicki S., Rokita M., Rȩkas M., Structural properties of Li2CO3–BaCO3 system derived from IR and Raman spectroscopy. J. Mol. Struct. 596, 151–156 (2001). [Google Scholar]
- 41.Dalavi S., Guduru P., Lucht B. L., Performance enhancing electrolyte additives for lithium ion batteries with silicon anodes. J. Electrochem. Soc. 159, A642–A646 (2012). [Google Scholar]
- 42.Chan C. K., Ruffo R., Hong S. S., Cui Y., Surface chemistry and morphology of the solid electrolyte interphase on silicon nanowire lithium-ion battery anodes. J. Power Sources 189, 1132–1140 (2009). [Google Scholar]
- 43.Nie M., Abraham D. P., Chen Y., Bose A., Lucht B. L., Silicon solid electrolyte interphase (SEI) of lithium ion battery characterized by microscopy and spectroscopy. J. Phys. Chem. C 117, 13403–13412 (2013). [Google Scholar]
- 44.Philippe B., Dedryvère R., Gorgoi M., Rensmo H., Gonbeau D., Edström K., Role of the LiPF6 salt for the long-term stability of silicon electrodes in Li-ion batteries—A photoelectron spectroscopy study. Chem. Mater. 25, 394–404 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Nguyen C. C., Lucht B. L., Comparative study of fluoroethylene carbonate and vinylene carbonate for silicon anodes in lithium ion batteries. J. Electrochem. Soc. 161, A1933–A1938 (2014). [Google Scholar]
- 46.Markevich E., Salitra G., Aurbach D., Fluoroethylene carbonate as an important component for the formation of an effective solid electrolyte interphase on anodes and cathodes for advanced Li-ion batteries. ACS Energy Lett. 2, 1337–1345 (2017). [Google Scholar]
- 47.Michan A. L., Leskes M., Grey C. P., Voltage dependent solid electrolyte interphase formation in silicon electrodes: Monitoring the formation of organic decomposition products. Chem. Mater. 28, 385–398 (2016). [Google Scholar]
- 48.Parimalam B. S., MacIntosh A. D., Kadam R., Lucht B. L., Decomposition reactions of anode solid electrolyte interphase (SEI) components with LiPF6. J. Phys. Chem. C 121, 22733–22738 (2017). [Google Scholar]
- 49.Yoo Y., You J. B., Choi W., Im S. G., A stacked polymer film for robust superhydrophobic fabrics. Polym. Chem. 4, 1664–1671 (2013). [Google Scholar]
- 50.Seok J.-H., Kim S. H., Cho S. M., Yi G.-R., Lee J. Y., Crosslinked organosilicon-acrylate copolymer moisture barrier thin film fabricated by initiated chemical vapor deposition (iCVD). Macromol. Res. 26, 1257–1264 (2018). [Google Scholar]
- 51.Lau K. K. S., Gleason K. K., Initiated chemical vapor deposition (iCVD) of poly(alkyl acrylates): An experimental study. Macromolecules 39, 3688–3694 (2006). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw4856/DC1
Fig. S1. FTIR of the V4D4 monomer and the 200-nm pV4D4 film.
Fig. S2. Anomalous cycling behavior observed with thick (45 nm) pV4D4 films.
Fig. S3. Micrograph of the uncoated copper foil.
Table S1. Spectral reflectivity measurements of the pV4D4-coated silicon in various media.
Table S2. XPS data for (un)cycled (un)coated silicon electrodes.