

Abstract
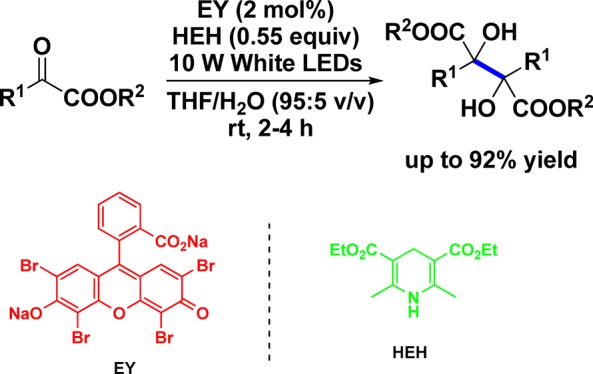
A mild transition-metal-free protocol to prepare 2,3-dialkylated tartaric acid esters has been developed by taking advantage of a visible light photoredox-catalyzed reductive dimerization of α-ketoesters with a combination of an organic dye photocatalyst and a Hantzsch-type 1,4-dihydropyridine hydrogen donor. A broad range of functional groups including cyclopropane, alkene, alkyne, 4-methoxybenzyl ether, acetal, silyl ether, carbamate, cyclic ether, cyclic thioether, bromoalkane, and N-alkoxyphthalimide are well-compatible. By employing the visible light photoredox-catalyzed reductive coupling and the subsequent optical resolution, both enantioenriched diastereomers of 2,3-dialkylated tartaric acid could be acquired conveniently.
Introduction
Since the first isolation by Carl Wilhelm Scheele in 1769, tartaric acid and its derivatives have been applied widely in organic synthesis as resolving agents,1 chiral auxiliaries,2 and natural chiral pools.3 Most importantly, an array of tartaric acid-derived compounds performed perfectly as privileged chiral ligands and catalysts in modern asymmetric synthesis, providing versatile opportunities to access optically active compounds in a catalytic fashion.4−6 These chiral ligands and catalysts were mainly generated by specific modifications on either the existing carboxylic acid or alcohol functionalities of the naturally occurring (2R,3R)-dimethyl tartaric acid, while the modification of the C2 and C3 positions of the backbone was kept to be inconvenient (Figure 1). In some cases, a few alkyl substituents could be introduced to the C2 and C3 positions of the tartaric acid through alkylation of the corresponding enolates, in which multiple group protections and usage of strong bases are always required and competitive side reactions might be caused.3a,3e,3f Therefore, the development of more convenient and general approaches to 2,3-disubstituted tartaric acids together with their derivatives still remains in high demand. Such methodologies will not only expand the ligand framework diversity but also be helpful to acquire new insightful understandings on stereoselective transformations.
Figure 1.

Tartaric acid and its derivatives.
Reductive dimerization of α-ketoesters makes 2,3-disubstituted tartaric acids available in a more straightforward manner, even though the bulky steric hindrance should be surmounted during the formation of two consecutive quaternary carbon centers. Actually, this kind of carbon–carbon bond formation based on the coupling of ketyl radicals has been carried out in a set of conditions in the past decades (Scheme 1), which involves the employment of stoichiometric amounts of one-electron-donating metal reagents [SmI2, TiCl3, and Mg(0)] and the utilization of high energetic UV light with coreductants.7,8 A recent Rueping’s work on visible light photoredox-catalyzed reductive coupling of ketones showed that two α-ketoester substrates were transformed to the corresponding 2,3-dialkylated tartaric acid esters in moderate yields under the catalysis of a polypyridyl–iridium complex.9 Almost at that time, we launched a project on scalable synthesis of 2,3-disubstituted tartaric acids, in which a protocol based on organic dye catalysis10 was envisioned with the consideration of obviation of transition metals and potential industrial applications. In this paper, we would like to report the developed transition-metal-free protocol, which features substantially mild condition and general functional group tolerance.
Scheme 1. Reductive Coupling of α-Ketoester under Different Conditions.
Results and Discussion
This study was initiated by choosing α-ketoester 1a as a model substrate, which contains a bulky isopropyl attached to the carbonyl group. An array of photocatalysts including rhodamine B (RhB), fluorescein (FL), rose bengal (RB), and Na2-Eosin Y (EY) were examined first by establishing a Hantzsch-type 1,4-dihydropyridine as a hydrogen donor and a 23 W compact fluorescent lamp (CFL) as a light source (Table 1, entries 1–4). It was demonstrated that Na2-EY exhibits the best catalytic efficiency in terms of yield and reaction time. Hence, in a degassed dimethylformamide (DMF) solution containing 10 mol % of Na2-EY and 0.6 equiv of diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (HEH), 1a was smoothly converted to dimethyl 2,3-diisopropyltartrate 2a in 64% yield at room temperature after 4 h of irradiation. However, when the light source was changed to 10 W white light-emitting diodes (LEDs), the reaction was further accelerated even though the yield was kept the same (Table 1, entry 5). Under identical irradiation, solvent screening suggested that a mixture of tetrahydrofuran (THF) and water (95:5 v/v) is more favorable than either DMF or THF, giving 2a in an increased isolated yield (Table 1, entries 6–7). The yield of product was improved to 87% when the amount of Na2-EY was decreased from 5% to 2 mol % (Table 1, entries 8–9), while further reduction of the photocatalyst resulted in a prolonged reaction time (Table 1, entry 10). Other two types of hydrogen donors, 2-phenyl-dihydrobenzothiazoline (BT) and N,N-diisopropylethylamine (DIPEA), were also attempted to replace HEH; inferior results were afforded (Table 1, entries 11–12). Further fine-tuning of the condition parameters indicated that 0.55 equiv of HEH are preferred to assure the complete conversion of 1a in a 0.05 M solution concentration (Table 1, entries 13–16). The control experiments also showed that no reaction occurred in the absence of either a photocatalyst or a light source (Table 1, entries 17–18). Accordingly, the optimal conditions for this metal-free reductive coupling were established as follows: 2 mol % of Na2-EY as the catalyst, 0.55 equiv of HEH as the hydrogen donor, THF/H2O (95:5 v/v) as the solvent, and 10 W white LEDs as the light source.
Table 1. Optimization Studies of Visible Light-Induced Reductive Couplinga.

| entry | light source | catalyst (mol %) | H donor (equiv)b | solventc | time (h) | yield (%)d |
|---|---|---|---|---|---|---|
| 1 | 23 W CFL | RhB (10%) | HEH (0.6) | DMF | 63 | 61 |
| 2 | 23 W CFL | FL (10%) | HEH (0.6) | DMF | 63 | 26 |
| 3 | 23 W CFL | RB (10%) | HEH (0.6) | DMF | 63 | 39 |
| 4 | 23 W CFL | EY (10%) | HEH (0.6) | DMF | 4 | 64 |
| 5 | 10 W white LEDs | EY (10%) | HEH (0.6) | DMF | 3 | 64 |
| 6 | 10 W white LEDs | EY (10%) | HEH (0.6) | THF | 6 | 70 |
| 7 | 10 W white LEDs | EY (10%) | HEH (0.6) | THF/H2O | 3 | 72 |
| 8 | 10 W white LEDs | EY (5%) | HEH (0.6) | THF/H2O | 4 | 78 |
| 9 | 10 W white LEDs | EY (2%) | HEH (0.6) | THF/H2O | 5 | 87 |
| 10 | 10 W white LEDs | EY (1%) | HEH (0.6) | THF/H2O | 12 | 83 |
| 11 | 10 W white LEDs | EY (2%) | BT (0.6) | THF/H2O | 12 | 51 |
| 12 | 10 W white LEDs | EY (2%) | DIPEA (0.6) | THF/H2O | 12 | 65 |
| 13 | 10 W white LEDs | EY (2%) | HEH (0.55) | THF/H2O | 5 | 87 |
| 14 | 10 W white LEDs | EY (2%) | HEH (0.5) | THF/H2O | 5 | 80 |
| 15e | 10 W white LEDs | EY (2%) | HEH (0.55) | THF/H2O | 4 | 81 |
| 16f | 10 W white LEDs | EY (2%) | HEH (0.55) | THF/H2O | 6 | 84 |
| 17 | 10 W white LEDs | HEH (0.55) | THF/H2O | 48 | n. r. | |
| 18 | EY (2%) | HEH (0.55) | THF/H2O | 48 | n. r. |
Concentration is 0.05 M unless otherwise mentioned.
HEH = diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate; BT = 2-phenyl-dihydrobenzothiazoline.
Ratio of THF/H2O is 95:5 v/v.
Mixture of dl-, meso-2a with a ratio of about 1:1.07 was isolated by column chromatography, and the ratios were determined by the 1H NMR method.
Concentration is 0.1 M.
Concentration is 0.025 M.
With the optimal reaction conditions in hand, the scope of the substrates was then explored (Table 2). Apart from 1a, α-ketoesters 1b–1e containing an acyclic primary alkyl substituent (Me, n-C6H13) or a cyclic secondary alkyl substituent (cyclopropyl, cyclohexyl) connected with a carbonyl group underwent the photoredox-catalyzed reductive coupling reaction smoothly, affording the corresponding dimethyl 2,3-dialkylated tartrates 2b–2e in a yield ranging from 80 to 92%. To differentiate the 1H NMR spectra of dl and meso forms of the products, we further confirmed the crystal structure of meso-2e diastereomer by using an X-ray diffraction technique (CCDC 1547831) (Figure 2). It is worthy to mention that the cyclopropyl moiety was kept intact in this type of radical process. However, for α-ketoester 1f, carbonyl reduction occurred instead of the expected coupling, suggesting that the bulky adamantyl substituent adjacent to the carbonyl group should inhibit the carbon–carbon bond formation of two ketyl radicals. For α-ketoesters 1g–1i with an appendant terminal alkene or a silylated internal alkyne group, all photoredox-catalyzed reductive coupling reactions were performed well to give 2g–2i in satisfactory yields (from 70 to 80%). The internal carbon–carbon π bonds remained untouched, and no cross-coupling between the carbon–carbon π bond and the carbonyl group was detected. A variety of α-ketoesters 1j–1q with different heteroatom-containing groups involving 4-methoxybenzyl ether, aldehyde acetal, silyl ether, secondary amine carbamate, cyclic ether, cyclic thioether, bromoalkane, and N-alkoxyphthalimide were converted to 2j–2q as expected in moderate to good yields, highlighting the general group tolerance of the conditions. Of note is the inertness of C–Br bond and N–O bond, both of which are prone to cleavage in the reductive conditions.11,12 For 1r bearing two electron-withdrawing groups (CO2Me) attached to the same carbonyl group, no reductive coupling was captured and only carbonyl reduction was observed as the major reaction. To further detect the effect of ester group, α-ketoesters 1s–1v with various alkoxy groups (PhO, BnO, cyclohexyloxy, and tert-butoxy) were then examined under the standard condition. However, 2s–2v were generated in a comparative yield with a similar dl/meso ratio, indicating that the effect of ester moiety is not distinguished. In addition, the reaction of the substrate 1w bearing a chiral moiety (R-1-phenylethyl ester) also afforded a mixture of two diastereomers 2w (dr = 1:1.1 by 1H NMR). This mentions that the stereogenic center of the substrate 1w did not have much impact on the behaviors of the ketyl radical coupling as well as the stereochemical selectivity.
Table 2. Visible Light Photoredox-Catalyzed Reductive Coupling of α-Ketoestersa.

Reaction conditions: α-ketoester (0.3 mmol), HEH (0.165 mmol), Na2-EY (2 mol %), THF/H2O (6.0 mL, 95:5 v/v), room temperature, N2, 10 W white LEDs, 2–4 h [monitored by thin-layer chromatography (TLC)].
dl and meso compounds can be separated by column chromatography.
Alcohol product was isolated.
Figure 2.

Thermal ellipsoid plot of meso-2e (30% probability levels).
A mechanism involving proton-coupled electron transfer was proposed to account for the visible light photoredox-catalyzed reductive coupling (Scheme 2).13 Initially, the excited-state EY* [*E1/2red = 0.83 V vs saturated calomel electrode (SCE)]14 is generated from Na2-EY by irradiation of visible light. A single electron transfer from Hantzsch-type 1,4-dihydropyridine (HEH) to the excited-state EY* would afford radical anion EY•– (E1/2 = −1.08 V vs SCE)15 and a radical cation HEH•+.16 After an electron transfer from radical anion EY•– and a proton transfer from either radical cation HEH•+ or cation HEH+, a neutral ketyl radical is formed from α-ketoester, which immediately undergoes dimerization to give 2,3-disubstituted tartaric acid ester. Owing to the high reduction potential of α-ketoester, as evidenced by 1a (E1/2red = −1.75 V vs SCE) (see Figure S1 in the Supporting Information for details), a transient hydrogen-bonded complex was supposed, which might facilitate the concurrent electron transfer from radical anion EY•– to the carbonyl group. To confirm the radical pathway, two radical-trapping experiments were performed using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 1,1-diphenylethylene as radical scavengers, respectively (Scheme 3). When 1 equiv of TEMPO was introduced to the standard reaction system, the visible light photoredox-catalyzed dimerization of 1b was completely inhibited and no dimerized product was detected. Instead, a high yield of TEMPOH was provided. In the other parallel experiment using 5 equiv of 1,1-diphenylethylene, a competitive ketyl–olefin–ketyl coupling product 3 was isolated in 35% yield, along with the dimeric product 2b (53% yield). These results mention that the ketyl radical species does play as the reactive intermediate in this type of visible light photoredox-catalyzed dimerization.
Scheme 2. Proposed Mechanism.

Scheme 3. Experiments of Trapping the Radical Intermediates.

To test the practicality of the transition-metal-free protocol, the visible light photoredox-catalyzed reductive coupling reactions were carried out on a 60 mmol scale. By prolonging the reaction times, 1b, 1e, and 1t were transformed to the desired products 2b, 2e, and 2t without erosion of the yield, demonstrating that the new protocol is valuable for scale synthesis (Table 3).
Table 3. Gram-Scale Reactionsa.

| entry | substrate | R1 | R2 | time (h) | 2, yield (%) |
|---|---|---|---|---|---|
| 1 | 1a (6.1 g) | Me | Me | 22 | 2a (5.2 g), 84 |
| 2 | 1e (10.2 g) | Cy | Me | 19 | 2e (9.2 g), 90 |
| 3 | 1t (10.7 g) | Me | Bn | 15 | 2t (8.9 g), 83 |
Reaction conditions: α-ketoester (60 mmol), HEH (33 mmol), Na2-EY (2 mol %), THF/H2O (1200 mL, 95:5 v/v), room temperature, N2, and 50 W white LEDs.
To get enantioenriched 2,3-dialkylated tartaric acids, two complementary optical resolution experiments were performed by using brucine and quinine as chiral-resolving reagents (Scheme 4).17 As reported, the racemic 2,3-dimethyl tartaric acid 4 can be separated from its meso diastereomer in barium salt forms. By taking advantage of the different solubility of the diastereomers in water or ethanol, (2S,3S)-4·brucine and (2R,3R)-4·quinine can be purified by repetitive recrystallizations. After acidic dissociation of the salts, (2S,3S)-dimethyl tartaric acid or (2R,3R)-dimethyl tartaric acid could be obtained with the optical purity almost identical to the literature values [[α]D20 = −13.2 (c = 4.0, H2O) and [α]D = +13.2 (c = 4.0, H2O)].17a
Scheme 4. Optical Resolution of rac-4.

Conclusions
A transition-metal-free protocol to prepare 2,3-dialkylated tartaric acid esters from α-ketoesters has been developed by means of a visible light photoredox-catalyzed reductive coupling, in which an organic dye Na2-EY was used as the photocatalyst and a Hantzsch-type 1,4-dihydropyridine was used as the hydrogen donor. A range of functional groups such as cyclopropane, alkene, alkyne, 4-methoxybenzyl ether, aldehyde acetal, silyl ether, secondary amine carbamate, cyclic ether, cyclic thioether, bromoalkane, and N-alkoxyphthalimide can be well-tolerated under the optimal conditions. Optically pure 2,3-dialkylated tartaric acid diastereomers can be obtained by combining the photoredox-catalyzed reductive coupling and optical resolution.
Experimental Section
General Information
All melting points were determined without correction. The 1H NMR spectra were obtained at 400 MHz, and the 13C NMR spectra were obtained at 100 MHz. The spectra were recorded in CDCl3 and dimethyl sulfoxide-d6 solution using the residual protonated solvent as the internal standard; J values were given in hertz. Mass spectra (ESI) were recorded on an LCQ Fleet spectrometer. High-resolution mass spectroscopy (HRMS) analyses were determined on a Q-TOF-MS spectrometer. α-Ketoester 1b was purchased directly.
General Procedure for the Preparation of α-Ketoesters 1a, 1c–1e, and 1g–1k(18)

To a stirred suspension of magnesium turnings (30 mmol, 1.5 equiv) in anhydrous THF (5 mL) was added a crystal of I2 as an activator. After stirring for 5 min, a solution of bromide (20 mmol, 1.0 equiv) in anhydrous THF (20 mL) was added dropwise (a water bath was used to control the temperature below 35 °C). The suspension was stirred for 1 h at room temperature, and the resulting Grignard reagent was used directly for the next step.
To a solution of dimethyl oxalate (20 mmol, 1.0 equiv) in anhydrous THF (30 mL) at −78 °C was added dropwise the Grignard reagents (commercially available or prepared as shown above) for more than 1 h. After stirring for 1 h at −78 °C, the mixture was warmed to room temperature, quenched with a saturated solution of NH4Cl (30 mL), and extracted with Et2O (3 × 20 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 30:1 v/v for 1c, 1e, 1g–1i; 15:1 v/v for 1j and 1k) or distillation under vacuum (1a, 1d) to afford pure α-ketoester.
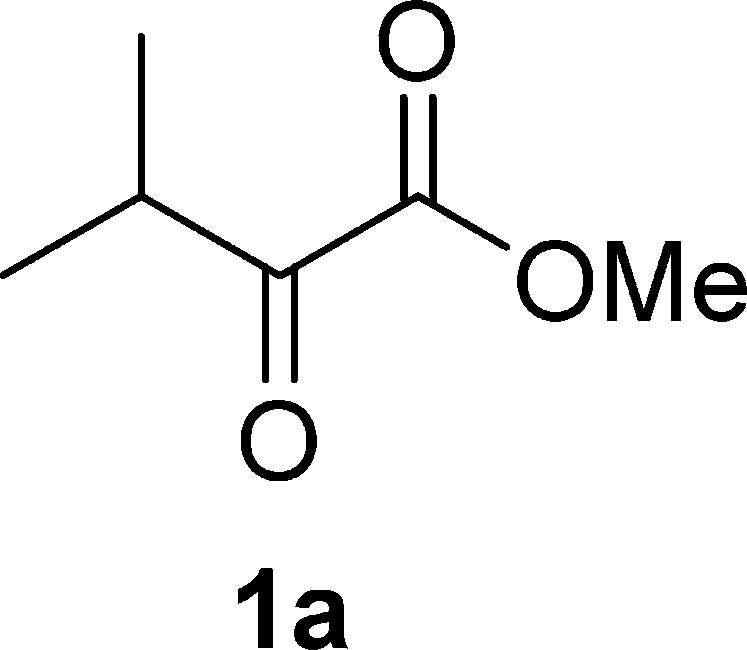
Methyl 3-Methyl-2-oxobutanoate (1a)19a
Pale yellow oil, 0.9 g, 35% yield; bp 71–73 °C/30 mm Hg; 1H NMR (400 MHz, CDCl3): δ 3.87 (s, 3H), 3.28 (hept, J = 7.0 Hz, 1H), 1.16 (d, J = 7.0 Hz, 6H); MS (ESI) m/z: 129 [M – 1]−.
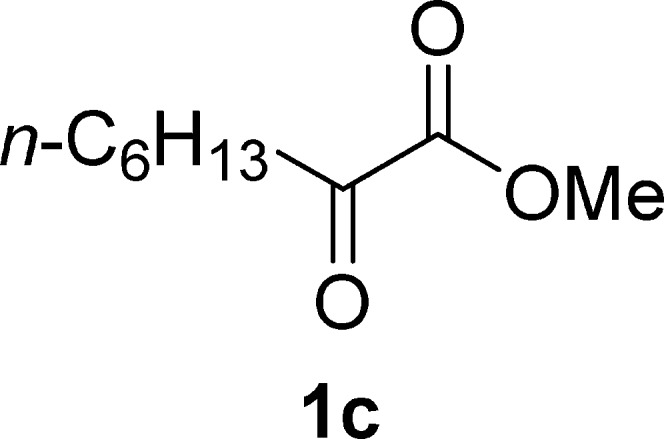
Methyl 2-Oxooctanoate (1c)19b
Pale yellow oil, 1.1 g, 31% yield; 1H NMR (400 MHz, CDCl3): δ 3.85 (s, 3H), 2.82 (t, J = 7.3 Hz, 2H), 1.62 (m, 2H), 1.34–1.24 (m, 6H), 0.87 (t, J = 6.8 Hz, 3H); MS (ESI) m/z: 171.15 [M – 1]−.
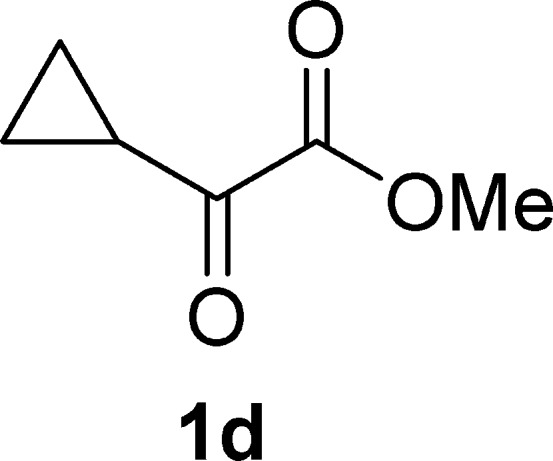
Methyl 2-Cyclopropyl-2-oxoacetate (1d)19c
Colorless oil, 0.95 g, 37% yield; bp 80–81 °C/30 mm Hg; 1H NMR (400 MHz, CDCl3): δ 3.89 (s, 3H), 2.80–2.68 (m, 1H), 1.28–1.22 (m, 2H), 1.15 (m, 2H); MS (ESI) m/z: 255 [2M – 1]−, 279 [2M + 23]+.
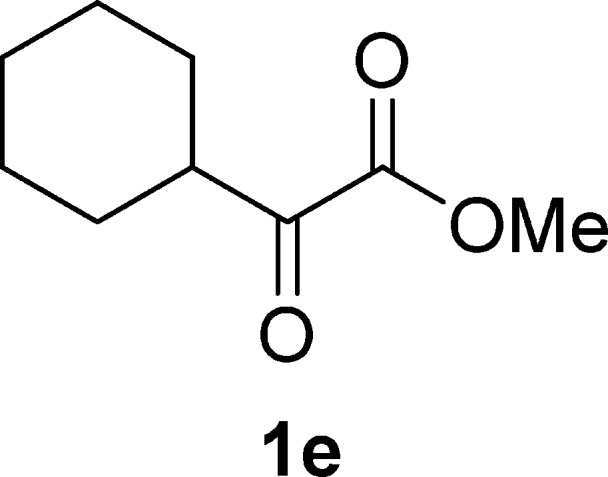
Methyl 2-Cyclohexyl-2-oxoacetate (1e)19d
Pale yellow oil, 1.1 g, 31% yield; 1H NMR (400 MHz, CDCl3): δ 3.85 (s, 3H), 3.07–3.00 (m, 1H), 1.94–1.84 (m, 2H), 1.84–1.74 (m, 2H), 1.72–1.64 (m, 1H), 1.42–1.27 (m, 4H), 1.27–1.15 (m, 1H); MS (ESI) m/z: 169 [M – 1]−.
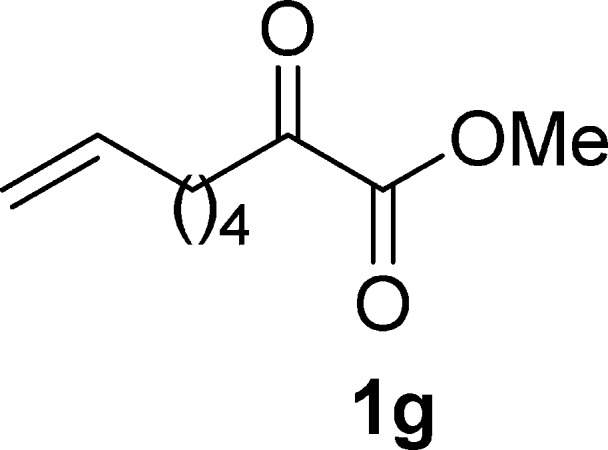
Methyl 2-Oxooct-7-enoate (1g)19e
Pale yellow oil, 1.2 g, 36% yield; 1H NMR (400 MHz, CDCl3): δ 5.81–5.70 (m, 1H), 5.01–4.95 (m, 1H), 4.95–4.91 (m, 1H), 3.84 (s, 3H), 2.82 (t, J = 7.2 Hz, 2H), 2.05 (dd, J1 = 14.3 Hz, J2 = 7.2 Hz, 2H), 1.70–1.56 (m, 2H), 1.45–1.34 (m, 2H); MS (ESI) m/z: 169 [M – 1]−.
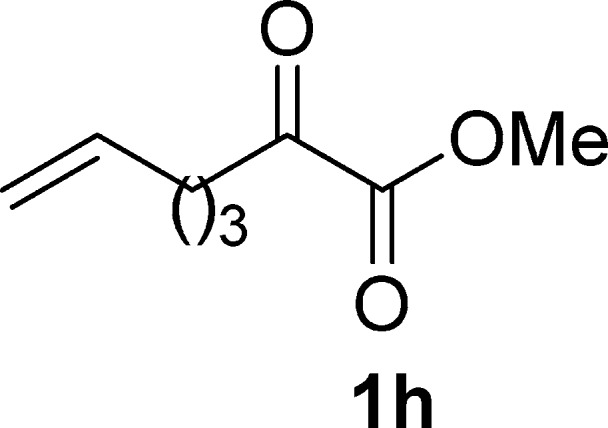
Methyl 2-Oxohept-6-enoate (1h)19e
Pale yellow oil, 1.3 g, 42% yield; 1H NMR (400 MHz, CDCl3): δ 5.80–5.67 (m, 1H), 5.03–4.95 (m, 2H), 3.84 (s, 3H), 2.82 (dd, J1 = 9.3 Hz, J2 = 5.3 Hz, 2H), 2.14–2.00 (m, 2H), 1.78–1.65 (m, 2H); MS (ESI) m/z: 155 [M – 1]−.

Methyl 2-Oxo-6-(trimethylsilyl)hex-5-enoate (1i)
Colorless oil, 0.6 g, 39% yield (7.5 mmol scale); 1H NMR (400 MHz, CDCl3): δ 3.86 (s, 3H), 3.08 (t, J = 7.2 Hz, 2H), 2.53 (t, J = 7.2 Hz, 2H), 0.11 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 191.9, 160.9, 104.4, 85.8, 53.1, 38.7, 13.9, 0.0; IR (KBr) νmax: 2960, 2178, 1736, 1252, 1081, 848 cm–1; HRMS (ESI) m/z: calcd for C10H16NaO3Si, 235.0761 [M + Na]+; found, 235.0753.

Methyl 5-((4-Methoxybenzyl)oxy)-2-oxopentanoate (1j)
Pale yellow oil, 2.8 g, 50% yield; 1H NMR (400 MHz, CDCl3): δ 7.25–7.17 (m, 2H), 6.92–6.79 (m, 2H), 4.37 (s, 2H), 3.79 (s, 6H), 3.46 (t, J = 5.9 Hz, 2H), 2.92 (t, J = 7.0 Hz, 2H), 2.00–1.90 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.8, 161.4, 159.2, 130.2, 129.3, 113.8, 72.5, 68.5, 55.3, 52.8, 36.4, 24.0; IR (KBr) νmax: 2949, 2859, 1733, 1614, 1514, 1448, 1249, 1178, 1101, 1040, 823 cm–1; HRMS (ESI) m/z: calcd for C14H18NaO5, 289.1064 [M + Na]+; found, 289.1064.

Methyl 4-(1,3-Dioxolan-2-yl)-2-oxobutanoate (1k)
Colorless oil, 1.0 g, 27% yield; 1H NMR (400 MHz, CDCl3): δ 4.91 (t, J = 3.7 Hz, 1H), 3.90–3.85 (m, 2H), 3.82 (s, 3H), 3.81–3.76 (m, 2H), 2.87 (t, J = 7.0 Hz, 2H), 2.06 (td, J1 = 7.0 Hz, J1 = 3.7 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 193.2, 161.2, 102.6, 65.0, 52.8, 33.0, 27.6; IR (KBr) νmax: 2954, 2887, 2537, 1730, 1546, 1401, 1247, 1134, 1088, 1031, 889 cm–1; HRMS (ESI) m/z: calcd for C8H12NaO5, 211.0577 [M + Na]+; found, 211.0579.
Procedure for the Preparation of α-Ketoester 1f(20,21)

To a solution of 2-(adamantan-1-yl)-2-oxoacetic acid (5 mmol, 1.0 equiv) in DMF (5 mL) was added dropwise 1,8-diazabicyclo[5.4.0]undec-7-ene (5 mmol, 1.0 equiv) at 0 °C. The mixture was stirred at the same temperature for an hour, and then iodomethane (10 mmol, 2.0 equiv) was added dropwise to the solution at the same temperature. After stirring for 0.5 h, the reaction mixture was stirred at room temperature until the completion of the reaction (monitored by TLC), quenched with a saturated solution of NH4Cl (5 mL), concentrated in a rotary evaporator under vacuum, and then extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 50:1 v/v) to give 1f.
Methyl 2-(Adamantan-1-yl)-2-oxoacetate (1f)21
White solid, 0.87 g, 78% yield; mp 63–65 °C; 1H NMR (400 MHz, CDCl3): δ 3.83 (s, 3H), 2.06 (s, 3H), 1.91 (d, J = 2.7 Hz, 6H), 1.73 (q, J = 12.3 Hz, 6H); MS (ESI) m/z: 245 [M + 23]+.
Procedure for the Preparation of α-Ketoester 1l(22)

To a solution of ((4-bromobut-3-yn-1-yl)oxy)(tert-butyl)dimethylsilane23 (5 mmol, 1.0 equiv) in MeOH (75 mL) was added a solution of NaHCO3 (3 mmol, 0.6 equiv) and MgSO4 (10 mmol, 2.0 equiv) in water (75 mL) at 0 °C. The mixture was stirred for 10 min before KMnO4 (12 mmol, 2.4 equiv) was added in portions. This mixture was then stirred at 0 °C until the completion of the reaction (monitored by TLC) and filtered through a Celite pad. The filter cake was washed with EtOAc (3 × 50 mL), and the filtrate was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 30:1 v/v) to give 1l.
Methyl 4-((tert-Butyldimethylsilyl)oxy)-2-oxobutanoate (1l)22
Colorless oil, 0.92 g, 73% yield; 1H NMR (400 MHz, CDCl3): δ 3.97 (t, J = 6.2 Hz, 2H), 3.87 (s, 3H), 3.04 (t, J = 6.2 Hz, 2H), 0.86 (s, 9H), 0.05 (s, 6H); MS (ESI) m/z: 247 [M + 1]+.
General Procedure for the Preparation of α-Ketoesters 1m–1o(24)

To a solution of methyl 2-((tert-butyldimethylsilyl)oxy)-2-(dimethoxyphosphoryl)acetate24a (11 mmol, 1.1 equiv) in THF (5 mL) was added dropwise a solution of LiHMDS (1 M solution in THF) (11 mmol, 1.1 equiv) at −78 °C under an N2 atmosphere. After stirring for 30 min, a solution of ketone (10 mmol, 1.0 equiv) in THF (5 mL) was added dropwise over 15 min. The reaction mixture was stirred at −78 °C for 0.5 h, allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction was quenched with a saturated solution of NH4Cl (20 mL) and extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over MgSO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 30:1 v/v) to give the corresponding silyl enol ether. To a solution of silyl enol ether (2 mmol, 1.0 equiv) and acetic acid (10 mmol, 5.0 equiv) in MeCN (20 mL) was added solid cesium fluoride (4 mmol, 2.0 equiv) in one portion at 0 °C. After stirring for 30 min, the mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction was quenched with a saturated solution of NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with a saturated solution of NaHCO3 (20 mL), brine (20 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 30:1 v/v) to give pure α-ketoester.
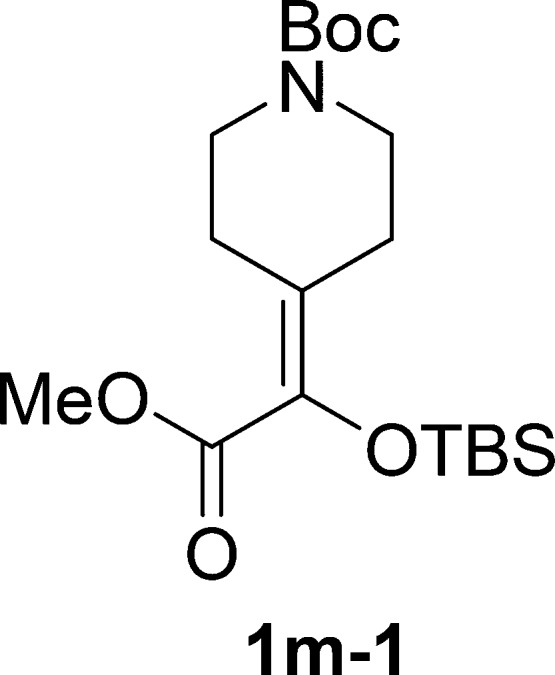
tert-Buty l4-(1-((tert-Butyldimethylsilyl)oxy)-2-methoxy-2-oxoethylidene)piperidine-1-carboxylate (1m-1)
Colorless oil, 3.4 g, 89% yield; 1H NMR (400 MHz, CDCl3): δ 3.73 (s, 3H), 3.49–3.32 (m, 4H), 2.69 (t, J = 6.1 Hz, 2H), 2.40 (t, J = 6.1 Hz, 2H), 1.45 (s, 9H), 0.93 (s, 9H), 0.07 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 165.4, 154.8, 135.0, 130.5, 79.6, 51.3, 43.7, 28.4, 27.9, 25.7, 18.4, −4.6; IR (KBr) νmax: 2938, 2860, 2361, 1700, 1634, 1411, 1284, 1225, 1163, 1112, 1024, 841, 781 cm–1; HRMS (ESI) m/z: calcd for C19H35NNaO5Si, 408.2177 [M + Na]+; found, 408.2177.
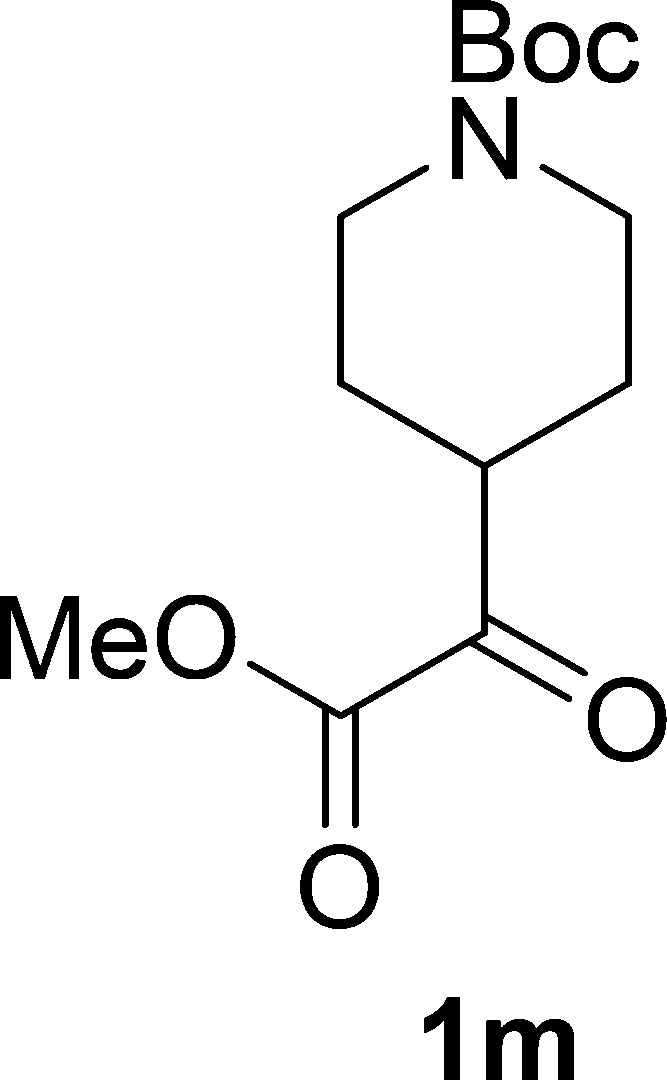
tert-Butyl 4-(2-Methoxy-2-oxoacetyl)piperidine-1-carboxylate (1m)
Colorless oil, 0.54 g, 98% yield; 1H NMR (400 MHz, CDCl3): δ 4.15–3.97 (m, 2H), 3.84 (s, 3H), 3.17 (tt, J1 = 11.2 Hz, J2 = 3.7 Hz, 1H), 2.83 (t, J = 12.0 Hz, 2H), 1.93–1.77 (m, 2H), 1.56–1.44 (m, 2H), 1.42 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 195.4, 161.5, 154.6, 79.8, 52.9, 44.4, 42.9, 28.4, 26.6; IR (KBr) νmax: 2961, 2860, 1691, 1419, 1271, 1168, 1124, 1069, 1011 cm–1; HRMS (ESI) m/z: calcd for C13H21NNaO5, 294.1312 [M + Na]+; found, 294.1313.
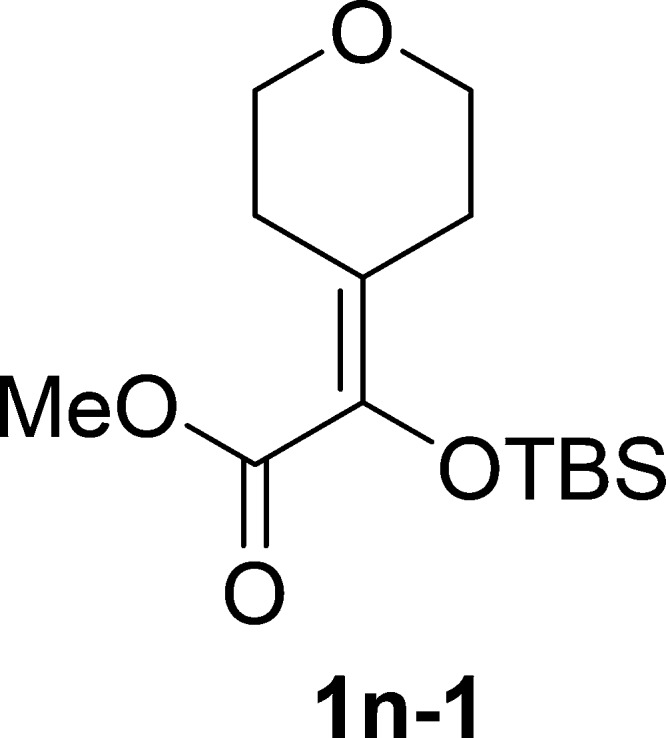
Methyl 2-((tert-Butyldimethylsilyl)oxy)-2-(tetrahydro-4H-pyran-4-ylidene)acetate (1n-1)
Colorless oil, 2.6 g, 92% yield; 1H NMR (400 MHz, CDCl3): δ 3.73 (s, 3H), 3.70 (t, J = 5.6 Hz, 2H), 3.67 (t, J = 5.5 Hz, 2H), 2.75 (t, J = 5.5 Hz, 2H), 2.44 (t, J = 5.6 Hz, 2H), 0.94 (s, 9H), 0.08 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 165.5, 134.5, 130.0, 68.5, 68.0, 51.3, 29.7, 29.2, 25.7, 18.4, −4.6; IR (KBr) νmax: 2950, 2853, 1721, 1633, 1437, 1290, 1232, 1146, 1100, 1028, 844 cm–1; HRMS (ESI) m/z: calcd for C14H26NaO4Si, 309.1493 [M + Na]+; found, 309.1487.
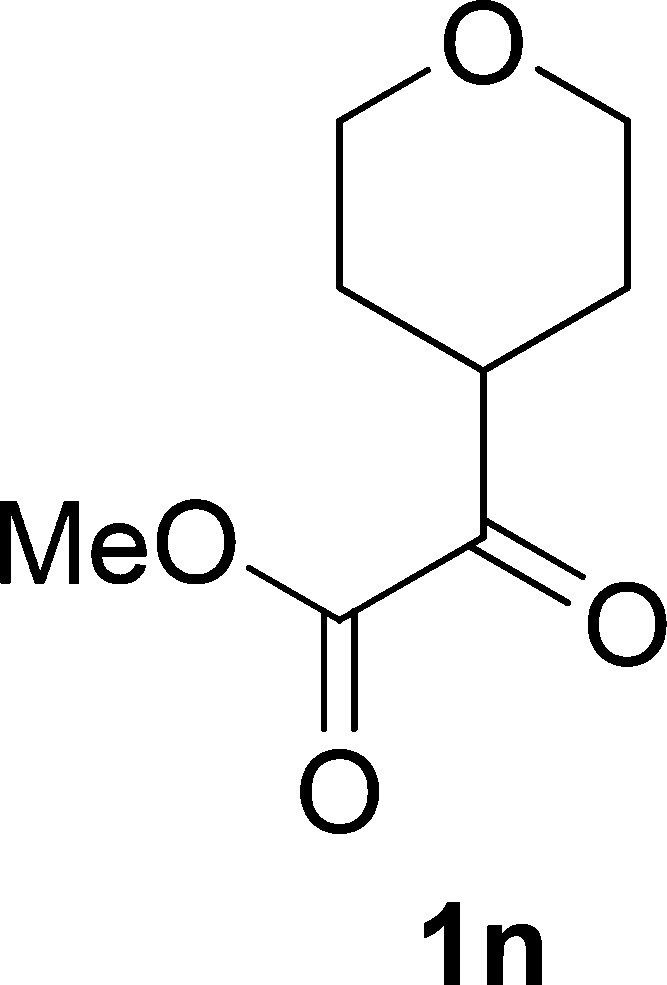
Methyl 2-Oxo-2-(tetrahydro-2H-pyran-4-yl)acetate (1n)
White solid, 0.29 g, 84% yield; mp 56–52 °C; 1H NMR (400 MHz, CDCl3): δ 3.99 (dd, J1 = 3.9 Hz, J2 = 3.1 Hz, 1H), 3.97 (dd, J1 = 3.9 Hz, J2 = 3.1 Hz, 1H), 3.86 (s, 3H), 3.47 (td, J1 = 11.5 Hz, J2 = 2.4 Hz, 2H), 3.27 (tt, J1 = 11.1 Hz, J2 = 3.9 Hz, 1H), 1.86–1.78 (m, 2H), 1.74–1.63 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 195.1, 161.6, 66.8, 52.9, 43.5, 27.2; IR (KBr) νmax: 2954, 2851, 1731, 1633, 1444, 1395, 1250, 1081, 1018 cm–1; HRMS (ESI) m/z: calcd for C8H12NaO4, 195.0628 [M + Na]+; found, 195.0639.
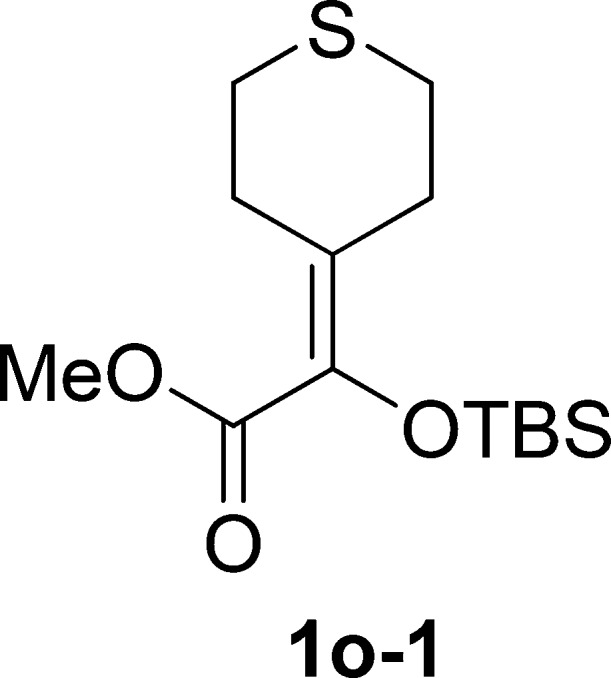
Methyl 2-((tert-Butyldimethylsilyl)oxy)-2-(tetrahydro-4H-thiopyran-4-ylidene)acetate (1o-1)
Colorless oil, 2.8 g, 94% yield; 1H NMR (400 MHz, CDCl3): δ 3.73 (s, 3H), 2.86–2.89 (m, 2H), 2.71–2.64 (m, 6H), 0.94 (s, 9H), 0.08 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 164.6, 134.1, 129.7, 50.4, 29.7, 29.3, 28.9, 28.5, 24.7, 17.4, 0.0, −5.6; IR (KBr) νmax: 2946, 2857, 2359, 1720, 1631, 1431, 1286, 1230, 1132, 1027, 835 cm–1; HRMS (ESI) m/z: calcd for C14H26NaO3SSi, 325.1264 [M + Na]+; found, 325.1257.
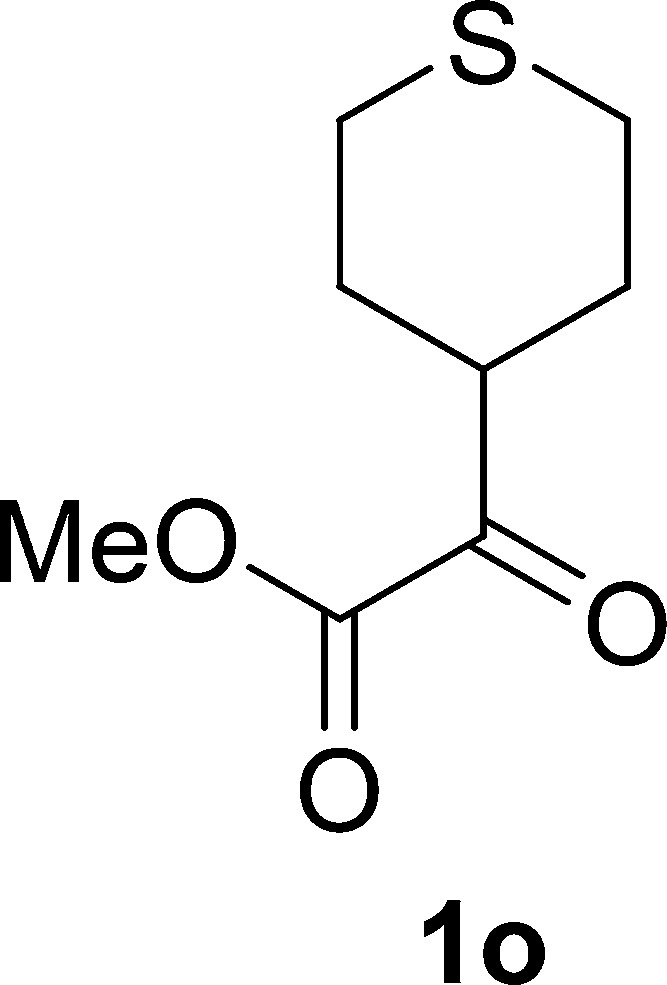
Methyl 2-Oxo-2-(tetrahydro-2H-thiopyran-4-yl)acetate (1o)
Colorless oil, 0.36 g, 96% yield; 1H NMR (400 MHz, CDCl3): δ 3.85 (s, 3H), 3.12 (tt, J1 = 11.1 Hz, J2 = 3.2 Hz, 1H), 2.75–2.65 (m, 4H), 2.26–2.13 (m, 2H), 1.81–1.66 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 195.5, 161.7, 53.0, 45.6, 28.3, 27.6; IR (KBr) νmax: 2914, 1728, 1433, 1261, 1190, 1071, 967 cm–1; HRMS (ESI) m/z: calcd for C8H11O3S, 187.0434 [M – H]−; found, 187.0420.
General Procedure for the Preparation of α-Ketoesters 1p–1q

Preparation of 1p-1
To a solution of methyl 2-((tert-butyldimethylsilyl)oxy)-6-hydroxyhex-2-enoate24c (5 mmol, 1.0 equiv) and CBr4 (7.5 mmol, 1.5 equiv) in DCM (15 mL) was added dropwise a solution of PPh3 (7.5 mmol, 1.5 equiv, in 5 mL DCM) at −30 °C under the N2 atmosphere. After stirring for 2 h, the reaction mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction was concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 50:1 v/v) to give 1p-1.
Preparation of 1q-1
To a solution of methyl 2-((tert-butyldimethylsilyl)oxy)-6-hydroxyhex-2-enoate20 (5 mmol, 1.0 equiv), O-phthalimide (6 mmol, 1.2 equiv), and PPh3 (6 mmol, 1.2 equiv) in THF (15 mL) was added dropwise diisopropyl azodiformate (6 mmol, 1.2 equiv) at 0 °C under the N2 atmosphere. After stirring for 0.5 h, the reaction mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction was concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 10:1 v/v) to give 1q-1.
Procedure for the Preparation of 1p–1q
To a solution of silyl enol ether (2 mmol, 1.0 equiv) and acetic acid (10 mmol, 5.0 equiv) in MeCN (20 mL) was added solid cesium fluoride (4 mmol, 2.0 equiv) in one portion at 0 °C. After stirring for 30 min, the mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction was quenched with a saturated solution of NaHCO3 (20 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with a saturated solution of NaHCO3 (20 mL), brine (20 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 30:1 v/v for 1p, 10:1 v/v for 1q) to give the corresponding α-ketoester.

Methyl 6-Bromo-2-((tert-butyldimethylsilyl)oxy)hex-2-enoate (1p-1)
Colorless oil, 1.1 g, 65% yield; 1H NMR (400 MHz, CDCl3): δ 5.46 (t, J = 8.1 Hz, 1H), 3.76 (s, 3H), 3.41 (t, J = 6.8 Hz, 2H), 2.61–2.56 (m, 2H), 2.03–1.90 (m, 2H), 0.93 (s, 9H), 0.12 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 164.0, 140.1, 122.1, 50.5, 32.0, 31.8, 24.8, 24.6, 17.2, −5.9; IR (KBr) νmax: 2946, 2860, 1789, 1731, 1356, 1253, 1164, 986, 839 cm–1; HRMS (ESI) m/z: calcd for C13H25NaBrO3Si, 359.0649 [M + Na]+; found, 359.0646.

Methyl 6-Bromo-2-oxohexanoate (1p)
Colorless oil, 0.27 g, 60% yield; 1H NMR (400 MHz, CDCl3): δ 3.86 (s, 3H), 3.41 (t, J = 6.5 Hz, 2H), 2.88 (t, J = 7.0 Hz, 2H), 1.95–1.85 (m, 2H), 1.84–1.74 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.5, 161.3, 53.0, 38.3, 32.9, 31.7, 21.5; IR (KBr) νmax: 1731, 1399, 1253, 1075, 739 cm–1; HRMS (ESI) m/z: calcd for C7H11BrNaO3, 244.9784 [M + Na]+; found, 244.9792.

Methyl 2-((tert-Butyldimethylsilyl)oxy)-6-((1,3-dioxoisoindolin-2-yl)oxy)hex-2-en-oate (1q-1)
Colorless oil, 2.0 g, 96% yield; 1H NMR (400 MHz, CDCl3): δ 7.83 (dd, J1 = 5.5 Hz, J2 = 3.0 Hz, 2H), 7.74 (dd, J1 = 5.5 Hz, J2 = 3.0 Hz, 2H), 5.56 (t, J = 8.1 Hz, 1H), 4.22 (t, J = 6.7 Hz, 2H), 3.75 (s, 3H), 2.69–2.62 (m, 2H), 2.07–1.80 (m, 2H), 0.93 (s, 9H), 0.12 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 165.0, 163.6, 140.9, 134.4, 129.0, 123.9, 123.5, 77.9, 51.5, 28.4, 25.6, 23.0, 18.2, −4.9; IR (KBr) νmax: 2947, 2886, 1784, 1650, 1462, 1394, 1184, 1125, 990 cm–1; HRMS (ESI) m/z: calcd for C21H29NNaO6Si, 442.1656 [M + Na]+; found, 442.1654.

Methyl 6-((1,3-Dioxoisoindolin-2-yl)oxy)-2-oxohexanoate (1q)
White solid, 0.45 g, mp 128–130 °C; 73% yield; 1H NMR (400 MHz, CDCl3): δ 7.85–7.81 (m, 2H), 7.77–7.73 (m, 2H), 4.22 (t, J = 6.0 Hz, 2H), 3.87 (s, 3H), 2.99 (t, J = 6.9 Hz, 2H), 1.94–1.80 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 193.8, 163.6, 161.4, 134.5, 128.9, 123.5, 77.9, 53.0, 38.7, 27.3, 19.3; IR (KBr) νmax: 2359, 1727, 1521, 1394, 1067, 696 cm–1; HRMS (ESI) m/z: calcd for C15H15NNaO6, 328.0792 [M + Na]+; found, 328.0792.
Procedure for the Preparation of α-Ketoester 1s(25)

To a solution of pyruvic acid (10 mmol, 1.0 equiv), phenol (10 mmol, 1.0 equiv), and pyridine (10 mmol, 1.0 equiv) in DCM (30 mL) was added dropwise a solution of dicyclohexylcarbodiimide (11 mmol, 1.1 equiv) in DCM (10 mL) at 0 °C. After stirring for 30 min, the reaction mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC). The reaction mixture was filtered, and the filter cake was washed with DCM (3 × 20 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The residue was purified by silica gel column chromatography (petroleum ether/ethyl acetate, 20:1 v/v) to give 1s.
Phenyl Pyruvate (1s)25
White solid, 1.2 g, 70% yield; mp 63–67 °C; 1H NMR (400 MHz, CDCl3): δ 7.47–7.39 (m, 2H), 7.33–7.27 (m, 1H), 7.21–7.14 (m, 2H), 2.60 (s, 3H); MS (ESI) m/z: 187 [M + 23]+.
General Procedure for the Preparation of α-Ketoesters 1t–1v(25)

To a solution of pyruvic acid (10 mmol, 1.0 equiv), alcohol (20 mmol, 2.0 equiv), and pyridine (25 mmol, 2.5 equiv) in THF (10 mL) was added dropwise mesyl chloride (MsCl) (12 mmol, 1.2 equiv) at 0 °C. After stirring for 30 min, the reaction mixture was allowed to be stirred at room temperature until the completion of the reaction (monitored by TLC), quenched with water (20 mL), and then extracted with Et2O (3 × 20 mL). The combined organic layers were then dried over Na2SO4, filtered, and concentrated in a rotary evaporator under vacuum. The resulting crude product was purified by silica gel flash column chromatography (petroleum ether/ethyl acetate, 20:1 v/v for 1t, 40:1 v/v for 1u, 1v) to afford the desired α-ketoester.
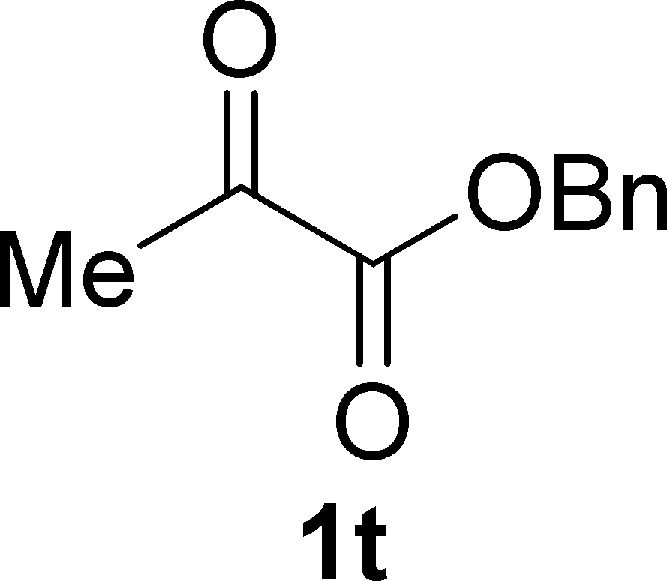
Benzyl Pyruvate (1t)25
Colorless oil, 1.1 g, 62% yield (20 mmol scale); 1H NMR (400 MHz, CDCl3): δ 7.46–7.33 (m, 5H), 5.28 (s, 2H), 2.47 (s, 3H); MS (ESI) m/z: 201 [M + 23]+.
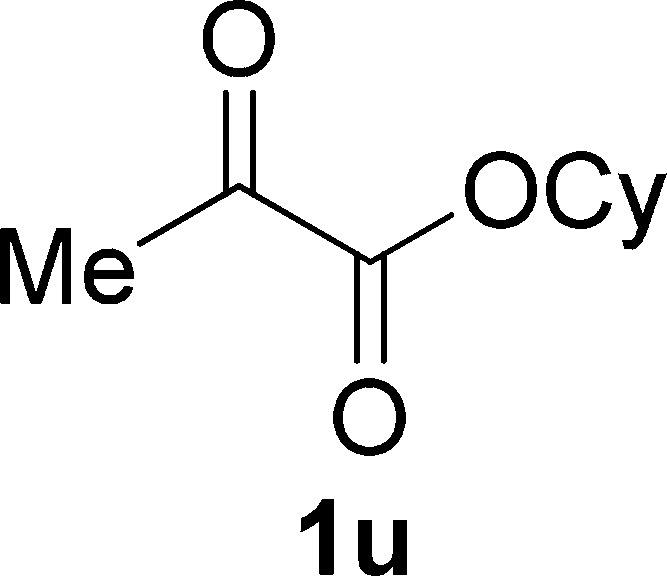
Cyclohexyl Pyruvate (1u)
Colorless oil, 0.9 g, 53% yield; 1H NMR (400 MHz, CDCl3): δ 4.93–4.83 (m, 1H), 2.45 (s, 3H), 1.94–1.87 (m, 2H), 1.80–1.72 (m, 2H), 1.59–1.50 (m, 3H), 1.44–1.28 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 192.5, 160.3, 75.5, 31.3, 26.7, 25.2, 23.7; IR (KBr) νmax: 2932, 1726, 1401, 1268, 1128, 1013 cm–1; HRMS (ESI) m/z: calcd for C9H14NaO3, 193.0835 [M + Na]+; found, 193.0830.
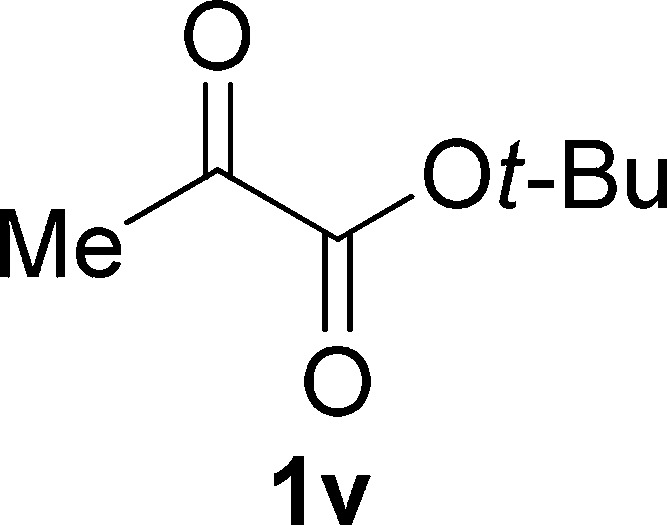
tert-Butyl Pyruvate (1v)
Colorless oil, 0.9 g, 62% yield; 1H NMR (400 MHz, CDCl3): δ 2.39 (s, 3H), 1.53 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 193.2, 160.3, 83.9, 27.8, 26.5; IR (KBr) νmax: 2926, 1633, 1399, 1119, 909, 737 cm–1; HRMS (ESI) m/z: calcd for C7H12NaO3, 167.0679 [M + Na]+; found, 167.0682.
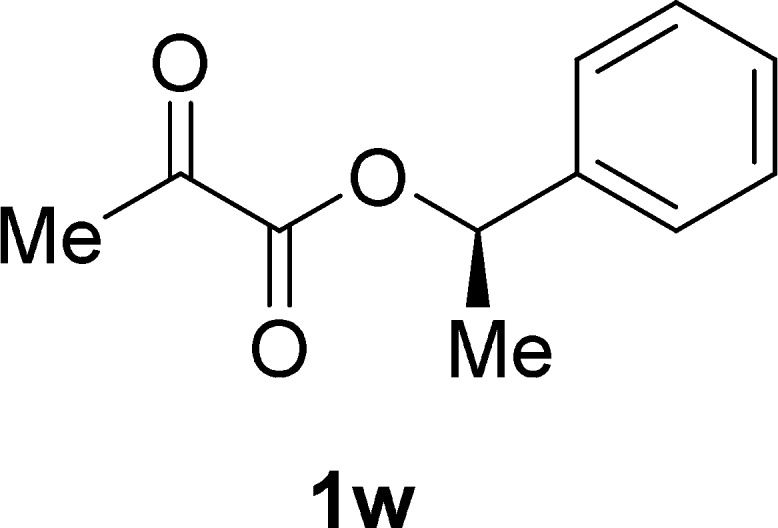
(R)-1-Phenylethyl Pyruvate (1w)
Colorless oil, 1.2 g, 62% yield; 1H NMR (400 MHz, CDCl3): δ 7.46–7.28 (m, 1H), 5.98 (q, J = 6.6 Hz, 1H), 2.45 (s, 1H), 1.65 (d, J = 6.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ 192.0, 160.1, 140.2, 128.7, 128.5, 126.3, 75.0, 26.7, 22.0; IR (KBr) νmax: 3416, 2985, 1728, 1452, 1291, 1142, 699 cm–1; HRMS (ESI) m/z: calcd for C11H12NaO3, 215.0679 [M + Na]+; found, 215.0682.
General Procedure for the Photoredox-Catalyzed Reductive Dimerization of α-Ketoesters
A solution of Na2-EY (0.006 mmol), Hantzsch-type 1,4-dihydropyridine (0.165 mmol), and α-ketoester (0.3 mmol) in THF/H2O (6.0 mL, 95:5 v/v, degassed) was stirred under the nitrogen atmosphere by irradiation of 10 W white LEDs at a distance of 5 cm at room temperature. The reaction mixture was concentrated in a rotary evaporator under vacuum upon the completion of the reaction (monitored by TLC). The residue was then purified by silica gel column chromatography (petroleum ether/ethyl acetate = 30:1 v/v for 2a–e, 2g–i, 2l–p, 2s–t; 20:1 v/v for 2j, 2l, 2u, 2v; 10:1 v/v for 2q) to give pure 2,3-dialkylated tartaric acid esters.
dl-Dimethyl 2,3-Diisopropyltartrate (dl-2a)
White solid, 16 mg, 42% yield; mp 46–51 °C; 1H NMR (400 MHz, CDCl3): δ 3.80 (s, 6H), 3.77 (s, 2H), 2.18 (m, J = 6.7 Hz, 2H), 1.02 (d, J = 6.7 Hz, 6H), 0.82 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 174.9, 83.4, 52.6, 33.8, 19.1, 17.2; IR (KBr) νmax: 2960, 1724, 1633, 1441, 1392, 1247, 1159, 1024, 763 cm–1; HRMS (ESI) m/z: calcd for C12H22NaO6, 285.1309 [M + Na]+; found, 285.1308.
meso-Dimethyl 2,3-Diisopropyltartrate (meso-2a)
White solid, 17.5 mg, 45% yield; mp 65–67 °C; 1H NMR (400 MHz, CDCl3): δ 3.82 (s, 2H), 3.79 (s, 6H), 2.37 (m, J = 6.7 Hz, 2H), 1.01 (d, J = 6.7 Hz, 6H), 0.86 (d, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 175.2, 83.6, 52.7, 34.0, 18.9, 17.4; IR (KBr) νmax: 2959, 1732, 1454, 1249, 1145, 1019 cm–1; HRMS (ESI) m/z: calcd for C12H22NaO6, 285.1309 [M + Na]+; found, 285.1310.
dl,meso-Dimethyl 2,3-Dimethyltartrate (2b)
Colorless oil, 25 mg, 80% yield (dl/meso = 1/1.05); 1H NMR (400 MHz, CDCl3): δ 3.84 (s, 1H), 3.81 (s, 2.94H), 3.77 (s, 3.08H), 3.59 (s, 1H), 1.51 (s, 2.94H), 1.49 (s, 3.08H); 13C NMR (100 MHz, CDCl3): δ 175.3, 174.7, 78.9, 78.8, 53.2, 53.0, 20.4, 20.3; IR (KBr) νmax: 2952, 1735, 1446, 1377, 1258, 1155, 1091, 976, 762 cm–1; HRMS (ESI) m/z: calcd for C8H14NaO6, 229.0683 [M + Na]+; found, 229.0681.
dl,meso-Dimethyl 2,3-Dihexyltartrate (2c)
Colorless oil, 45 mg, 87% yield (dl/meso = 1/1.1); 1H NMR (400 MHz, CDCl3): δ 3.80 (s, 2.83H), 3.76 (s, 3.14H), 3.72 (s, 1H), 3.51 (s, 1H), 1.96–1.89 (m, 3H), 1.80–1.71 (m, 1H), 1.54–1.37 (m, 2H), 1.35–1.15 (m, 12H), 0.98–0.78 (m, 8H); 13C NMR (100 MHz, CDCl3): δ 174.4, 174.3, 82.9, 82.4, 53.0, 52.8, 32.8, 32.7, 31.7, 31.7, 29.4, 29.4, 23.8, 23.6, 22.5, 14.0; IR (KBr) νmax: 2928, 2859, 1733, 1447, 1239, 1145, 1084, 762 cm–1; HRMS (ESI) m/z: calcd for C18H34NaO6, 369.2248 [M + Na]+; found, 369.2255.
dl-Dimethyl 2,3-Dicyclopropyltartrate (dl-2d)
White solid, 17 mg, 44% yield; mp 94–96 °C; 1H NMR (400 MHz, CDCl3): δ 3.81 (s, 6H), 3.32 (s, 2H), 1.66–1.59 (m, 2H), 0.71–0.64 (m, 2H), 0.53–0.41 (m, 2H), 0.37–0.23 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 173.5, 77.6, 51.6, 11.7, 0.0, −2.7; IR (KBr) νmax: 3008, 2949, 1728, 1435, 1255, 1166, 1017, 892 cm–1; HRMS (ESI) m/z: calcd for C12H18NaO6, 281.0996 [M + Na]+; found, 281.0993.
meso-Dimethyl 2,3-Dicyclopropyltartrate (meso-2d)
White solid, 19 mg, 48% yield; mp 95–99 °C; 1H NMR (400 MHz, CDCl3): δ 3.81 (s, 6H), 3.53 (s, 2H), 1.55–1.45 (m, 2H), 0.74–0.66 (m, 2H), 0.55–0.42 (m, 2H), 0.41–0.27 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 175.6, 79.4, 53.4, 13.0, 2.5, 0.0; IR (KBr) νmax: 3009, 2953, 1728, 1437, 1260, 1163, 1018, 887 cm–1; HRMS (ESI) m/z: calcd for C12H18NaO6, 281.0996 [M + Na]+; found, 281.0996.
dl-Dimethyl 2,3-Dicyclohexyltartrate (dl-2e)
White solid, 21.5 mg, 42% yield; mp 98–100 °C; 1H NMR (400 MHz, CDCl3): δ 3.79 (s, 2H), 3.79 (s, 6H), 2.10–1.97 (m, 2H), 1.90–1.79 (m, 2H), 1.79–1.66 (m, 4H), 1.60 (d, J = 10.5 Hz, 2H), 1.37 (d, J = 11.2 Hz, 2H), 1.28–1.05 (m, 10H); 13C NMR (100 MHz, CDCl3): δ 175.2, 83.7, 52.6, 43.9, 28.5, 26.8, 26.5, 26.5, 26.0; IR (KBr) νmax: 2926, 2853, 1726, 1633, 1445, 1236, 1149, 1110, 757 cm–1; HRMS (ESI) m/z: calcd for C18H30NaO6, 365.1935 [M + Na]+; found, 365.1938.
meso-Dimethyl 2,3-Dicyclohexyltartrate (meso-2e)
White solid, 23.5 mg, 46% yield; mp 106–108 °C; 1H NMR (400 MHz, CDCl3): δ 3.96 (s, 2H), 3.78 (s, 6H), 2.03–1.89 (m, 4H), 1.81–1.66 (m, 4H), 1.62–1.56 (m, 2H), 1.50–1.43 (m, 2H), 1.28–1.14 (m, 6H), 1.13–0.95 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 175.3, 83.6, 52.6, 44.9, 28.0, 26.9, 26.5, 26.4, 26.0; IR (KBr) νmax: 2926, 2853, 1730, 1637, 1444, 1239, 1114, 1022 cm–1; HRMS (ESI) m/z: calcd for C18H30NaO6, 365.1935 [M + Na]+; found, 365.1936.
dl,meso-Dimethyl 2,3-Di(hex-5-en-1-yl)tartrate (2g)
Colorless oil, 43 mg, 80% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 5.81–5.70 (m, 2H), 4.97 (dd, J1 = 17.1 Hz, J2 = 1.2 Hz, 2H), 4.92 (dd, J1 = 10.2 Hz, J2 = 0.9 Hz, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 3.74 (s, 1H), 3.53 (s, 1H), 2.09–1.92 (m, 5H), 1.92–1.86 (m, 2H), 1.82–1.71 (m, 1H), 1.55–1.42 (m, 2H), 1.42–1.32 (m, 4H), 1.00–0.80 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.3, 174.2, 138.6, 138.6, 114.5, 82.8, 82.3, 53.1, 52.9, 33.6, 32.6, 32.5, 28.9, 28.9, 23.3, 23.1; IR (KBr) νmax: 2925, 2856, 1731, 1637, 1440, 1238, 1167, 993, 906 cm–1; HRMS (ESI) m/z: calcd for C18H30NaO6, 365.1935 [M + Na]+; found, 365.1936.
dl,meso-Dimethyl 2,3-Di(pent-4-en-1-yl)tartrate (2h)
Colorless oil, 34 mg, 72% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 5.81–4.93 (m, 2H), 4.99 (d, J = 16.9 Hz, 2H), 4.94 (d, J = 10.2 Hz, 2H), 3.80 (s, 3H), 3.76 (s, 3H), 3.74 (s, 1H), 3.53 (d, J = 1.4 Hz, 1H), 2.10–1.97 (m, 5H), 1.96–1.90 (m, 2H), 1.82–1.71 (m, 1H), 1.65–1.52 (m, 2H), 1.11–0.90 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.2, 174.2, 138.2, 138.3, 114.8, 114.8, 82.8, 82.2, 53.1, 52.9, 33.7, 33.6, 32.2, 32.1, 23.1, 22.9; IR (KBr) νmax: 2937, 174, 1639, 1442, 1246, 1150, 1092, 995, 910 cm–1; HRMS (ESI) m/z: calcd for C16H26O6, 337.1622 [M + Na]+; found, 337.1622.
dl,meso-Dimethyl 2,3-Bis(4-(trimethylsilyl)but-3-yn-1-yl)tartrate (2i)
Colorless oil, 46 mg, 70% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 3.82 (s, 3H), 3.77 (s, 4H), 3.56 (s, 1H), 2.46–2.35 (m, 2H), 2.30–2.18 (m, 3H), 2.13–1.97 (m, 1H), 1.97–1.91 (m, 2H), 0.14 (s, 9H), 0.13 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 173.2, 173.2, 105.8, 105.8, 84.8, 84.8, 81.7, 81.0, 53.2, 53.0, 31.8, 31.6, 14.6, 14.5, 0.0, 0.0; IR (KBr) νmax: 2957, 2858, 2174, 1738, 1633, 1444, 1399, 1253, 1114, 844, 649 cm–1; HRMS (ESI) m/z: calcd for C20H34NaO6Si2, 449.1786 [M + Na]+; found, 449.1786.
dl,meso-Dimethyl 2,3-Bis(3-((4-methoxybenzyl)oxy)propyl)tartrate (2j)
Colorless oil, 66.4 mg, 83% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 7.25 (dd, J1 = 8.6 Hz, J2 = 1.2 Hz, 4H), 6.87 (d, J = 8.3 Hz, 4H), 4.42 (s, 4H), 3.90 (s, 1H), 3.79 (s, 6H), 3.78 (s, 3H), 3.74 (s, 4H), 3.48–3.37 (m, 4H), 2.20–1.95 (m, 3H), 1.84–1.72 (m, 3H), 1.36–1.24 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.1, 174.0, 159.2, 130.5, 130.5, 129.3, 129.3, 113.8, 82.7, 82.1, 72.4, 72.4, 69.8, 55.3, 53.1, 52.9, 29.8, 29.7, 24.2, 24.1; IR (KBr) νmax: 2944, 2855, 1735, 1512, 1450, 1246, 1090, 1031, 819 cm–1; HRMS (ESI) m/z: calcd for C28H38NaO10, 557.2357 [M + Na]+; found, 557.2363.
dl,meso-Dimethyl 2,3-Bis(2-(1,3-dioxolan-2-yl)ethyl)tartrate (2k)
Colorless oil, 44.5 mg, 78% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 4.83 (q, J = 4.8 Hz, 2H), 3.95–3.91 (m, 4H), 3.85 (s, 1H), 3.84–3.80 (m, 4H), 3.79 (s, 3H), 3.74 (s, 3H), 3.68 (d, J = 0.9 Hz, 1H), 2.14–2.05 (m, 3H), 1.98–1.88 (m, 1H), 1.84–1.78 (m, 2H), 1.38–1.23 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 173.9, 173.8, 104.0, 104.0, 82.4, 81.8, 64.9, 53.2, 53.0, 28.4, 28.1, 27.2, 27.0; IR (KBr) νmax: 2951, 1731, 1644, 1400, 1255, 1207, 1117, 1026, 803, 671 cm–1; HRMS (ESI) m/z: calcd for C16H26NaO10, 401.1418 [M + Na]+; found, 401.1421.
dl,meso-Dimethyl 2,3-Bis(2-((tert-butyldimethylsilyl)oxy)ethyl)tartrate (2l)
Colorless oil, 58 mg, 78% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 4.08 (s, 1H), 4.00 (s, 1H), 3.77 (s, 3H), 3.75–3.66 (m, 7H), 2.34–2.26 (m, 2H), 2.24–2.16 (m, 1H), 2.08–2.02 (m, 1H), 0.86 (s, 18H), 0.02–0.00 (m, 12H); 13C NMR (100 MHz, CDCl3): δ 173.8, 173.8, 81.2, 80.7, 59.4, 59.2, 52.7, 52.5, 34.9, 34.9, 25.9, 25.9, 18.4, 18.4, −5.5, −5.5, −5.6, −5.6; IR (KBr) νmax: 3856, 3736, 2945, 2865, 1740, 1640, 1459, 1256, 1095, 834, 670 cm–1; HRMS (ESI) m/z: calcd for C22H46NaO8Si2, 517.2623 [M + Na]+; found, 517.2629.
dl-Dimethyl 2,3-Bis(1-(tert-butoxycarbonyl)piperidin-4-yl)tartrate (dl-2m)
White solid, 33 mg, 40% yield; mp 73–77 °C; 1H NMR (400 MHz, CDCl3): δ 4.10 (s, 4H), 3.87 (s, 2H), 3.79 (s, 6H), 2.60 (s, 4H), 2.25–2.07 (m, 2H), 1.92 (d, J = 13.0 Hz, 2H), 1.77 (d, J = 11.0 Hz, 2H), 1.42 (s, 18H), 1.35–1.27 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.4, 154.6, 83.4, 79.5, 53.0, 43.7, 42.5, 28.4, 27.3, 26.3; IR (KBr) νmax: 2963, 2854, 1688, 1417, 1257, 1149, 1092, 1020, 798 cm–1; HRMS (ESI) m/z: calcd for C26H44N2NaO10, 567.2888 [M + Na]+; found, 567.2892.
meso-Dimethyl 2,3-Bis(1-(tert-butoxycarbonyl)piperidin-4-yl)tartrate (meso-2m)
White solid, 35 mg, 43% yield; mp 79–82 °C; 1H NMR (400 MHz, CDCl3): δ 4.09 (s, 4H), 3.80 (s, 2H), 3.77 (s, 6H), 2.60 (d, J = 8.7 Hz, 4H), 2.09–2.00 (m, 2H), 1.96 (d, J = 13.2 Hz, 2H), 1.44 (s, 2H), 1.42 (s, 18H), 1.38–1.28 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.6, 154.6, 83.0, 79.4, 52.9, 43.7, 42.0, 28.4, 27.9, 26.5; IR (KBr) νmax: 2961, 2855, 1735, 1688, 1414, 1251, 1093, 1022, 800 cm–1; HRMS (ESI) m/z: calcd for C26H44N2NaO10, 567.2888 [M + Na]+; found, 567.2893.
dl-Dimethyl 2,3-Bis(tetrahydro-2H-pyran-4-yl)tartrate (dl-2n)
White solid, 21 mg, 40% yield; mp 127–129 °C; 1H NMR (400 MHz, CDCl3): δ 3.98 (dd, J1 = 11.6 Hz, J2 = 3.9 Hz, 2H), 3.92 (dd, J1 = 11.6 Hz, J2 = 4.0 Hz, 2H), 3.81 (d, J = 5.8 Hz, 2H), 3.79 (s, 6H), 3.38–3.33 (m, 4H), 2.16 (tt, J1 = 11.8 Hz, J2 = 3.4 Hz, 2H), 1.87 (dd, J1 = 13.3 Hz, J2 = 2.4 Hz, 2H), 1.63–1.48 (m, 4H), 1.34 (dd, J1 = 10.5 Hz, J2 = 2.5 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 174.6, 82.9, 67.9, 67.7, 52.8, 41.0, 28.7, 27.4; IR (KBr) νmax: 2956, 2841, 2358, 1734, 1630, 1440, 1240, 1091, 805 cm–1; HRMS (ESI) m/z: calcd for C16H26NaO8, 369.1520 [M + Na]+; found, 369.1516.
meso-Dimethyl 2,3-Bis(tetrahydro-2H-pyran-4-yl)tartrate (meso-2n)
White solid, 22 mg, 43% yield; mp 184–188 °C; 1H NMR (400 MHz, CDCl3): δ 4.01–3.90 (m, 4H), 3.87 (s, 2H), 3.79 (s, 6H), 3.35–3.32 (m, 4H), 2.28 (tt, J1 = 11.9 Hz, J2 = 3.4 Hz, 2H), 1.83 (d, J = 13.2 Hz, 2H), 1.70–1.50 (m, 4H), 1.22–1.16 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.3, 83.4, 67.8, 67.7, 53.0, 41.4, 28.0, 27.3; IR (KBr) νmax: 2954, 2843, 1734, 1636, 1443, 1243, 1093, 1024, 875 cm–1; HRMS (ESI) m/z: calcd for C16H26NaO8, 369.1520 [M + Na]+; found, 369.1517.
dl-Dimethyl 2,3-Bis(tetrahydro-2H-thiopyran-4-yl)tartrate (dl-2o)
White solid, 22 mg, 39% yield; mp 138–140 °C; 1H NMR (400 MHz, CDCl3): δ 4.05 (s, 2H), 3.81 (s, 6H), 2.74–2.55 (m, 8H), 2.26 (d, J = 11.8 Hz, 2H), 1.97 (tt, J1 = 11.8 Hz, J2 = 2.6 Hz, 2H), 1.86 (d, J = 12.7 Hz, 2H), 1.70–1.57 (m, 2H), 1.41 (qd, J1 = 11.9 Hz, J2 = 4.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 174.5, 83.4, 53.0, 45.1, 29.4, 28.9, 28.3; IR (KBr) νmax: 2946, 1728, 1634, 1435, 1247, 1118, 1020, 802 cm–1; HRMS (ESI) m/z: calcd for C16H26NaO6S2, 401.1063 [M + Na]+; found, 401.1061.
meso-Dimethyl 2,3-Bis(tetrahydro-2H-thiopyran-4-yl)tartrate (meso-2o)
White solid, 24 mg, 42% yield; mp 144–146 °C; 1H NMR (400 MHz, CDCl3): δ 3.84 (s, 2H), 3.78 (s, 6H), 2.74–2.51 (m, 8H), 2.37 (d, J = 13.2 Hz, 2H), 1.98–1.83 (m, 4H), 1.61–1.45 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.7, 83.2, 52.9, 43.7, 30.2, 29.0, 28.9, 28.5; IR (KBr) νmax: 2947, 1731, 1633, 1434, 1246, 1116, 1019, 801 cm–1; HRMS (ESI) m/z: calcd for C16H26NaO6S2, 401.1063 [M + Na]+; found, 401.1060.
dl,meso-Dimethyl 2,3-Bis(4-bromobutyl)tartrate (2p)
Colorless oil, 57 mg, 85% yield (dl/meso = 1/1); 1H NMR (400 MHz, CDCl3): δ 3.82 (s, 3H), 3.78 (s, 3H), 3.77 (s, 1H), 3.56 (d, J = 1.4 Hz, 1H), 3.37 (t, J = 6.8 Hz, 4H), 2.07–1.74 (m, 8H), 1.64–1.53 (m, 2H), 1.18–1.00 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 173.9, 173.9, 82.6, 82.1, 53.3, 53.1, 33.3, 32.7, 32.6, 31.8, 31.7, 22.5, 22.4; IR (KBr) νmax: 2950, 1735, 1443, 1247, 1164, 1102, 810, 736 cm–1; HRMS (ESI) m/z: calcd for C14H24Br2NaO6, 470.9811 [M + Na]+; found, 470.9812.
dl,meso-Dimethyl 2,3-Bis(4-((1,3-dioxoisoindolin-2-yl)oxy)butyl)tartrate (2q)
Colorless oil, 37 mg, 40% yield (dl/meso = 1/1.05); 1H NMR (400 MHz, CDCl3): δ 7.82 (dd, J1 = 5.2 Hz, J2 = 3.1 Hz, 4H), 7.73 (dd, J1 = 5.2 Hz, J2 = 3.1 Hz, 4H), 4.22–4.14 (m, 4H), 3.85 (s, 2.91H), 3.80 (s, 3.07H), 3.77 (s, 1H), 3.58 (d, J = 0.9 Hz, 1H), 2.15–2.06 (m, 1H), 2.06–1.99 (m, 2H), 1.87 (dd, J1 = 12.1 Hz, J2 = 4.4 Hz, 1H), 1.84–1.74 (m, 4H), 1.69–1.60 (m, 2H), 1.23–1.10 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 174.1, 174.0, 163.6, 134.5, 128.9, 123.5, 82.7, 82.1, 78.1, 53.3, 53.1, 32.4, 32.2, 28.1, 20.0, 19.8; IR (KBr) νmax: 2950, 1730, 1458, 1385, 1251, 1123, 981, 703 cm–1; HRMS (ESI) m/z: calcd for C30H32N2NaO12, 635.1847 [M + Na]+; found, 635.1848.
dl-Diphenyl 2,3-Dimethyltartrate (dl-2s)
White solid, 20 mg, 40% yield; mp 107–109 °C; 1H NMR (400 MHz, CDCl3): δ 7.45–7.38 (m, 4H), 7.31–7.26 (m, 2H), 7.20–7.12 (m, 4H), 3.98 (s, 2H), 1.83 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 173.6, 150.4, 129.7, 126.5, 121.3, 79.5, 20.9; IR (KBr) νmax: 2930, 1753, 1592, 1487, 1382, 1185, 1082, 809, 743 cm–1; HRMS (ESI) m/z: calcd for C18H18NaO6, 353.0996 [M + Na]+; found, 353.0993.
meso-Diphenyl 2,3-Dimethyltartrate (meso-2s)
Colorless oil, 21 mg, 42% yield; 1H NMR (400 MHz, CDCl3): δ 7.40–7.34 (m, 4H), 7.25 (tt, J1 = 6.7 Hz, J2 = 1.1 Hz, 2H), 7.14–7.05 (m, 4H), 3.75 (s, 2H), 1.80 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 173.2, 150.3, 129.7, 126.5, 121.2, 79.0, 20.8; IR (KBr) νmax: 2932, 1752, 1632, 1595, 1487, 1238, 119, 1127, 1077, 745 cm–1; HRMS (ESI) m/z: calcd for C18H18NaO6, 353.0996 [M + Na]+; found, 353.0995.
dl-Dibenzyl 2,3-Dimethyltartrate (dl-2t)
Colorless oil, 21.5 mg, 40% yield; 1H NMR (400 MHz, CDCl3): δ 7.43–7.29 (m, 10H), 5.21 (d, J = 12.2 Hz, 2H), 5.17 (d, J = 12.2 Hz, 2H), 1.53 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 174.6, 134.9, 128.7, 128.6, 128.3, 79.0, 68.0, 20.6; IR (KBr) νmax: 2359, 1730, 1635, 1452, 1394, 1248, 1119, 740 cm–1; HRMS (ESI) m/z: calcd for C20H22NaO6, 381.1309 [M + Na]+; found, 381.1310.
meso-Dibenzyl 2,3-Dimethyltartrate (meso-2t)
White solid, 21.5 mg, 40% yield; mp 61–63 °C; 1H NMR (400 MHz, CDCl3): δ 7.38–7.30 (m, 6H), 7.30–7.22 (m, 4H), 5.10 (d, J = 12.1 Hz, 2H), 4.92 (d, J = 12.1 Hz, 2H), 3.64 (s, 2H), 1.49 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 174.1, 134.8, 128.6, 128.5, 78.7, 68.1, 20.4; IR (KBr) νmax: 2358, 171, 1635, 1450, 1389, 1254, 1148, 954, 742 cm–1; HRMS (ESI) m/z: calcd for C20H22NaO6, 381.1309 [M + Na]+; found, 381.1308.
dl-Dicyclohexyl 2,3-Dimethyltartrate (dl-2u)
White solid, 20.5 mg, 40% yield; mp 38–41 °C; 1H NMR (400 MHz, CDCl3): δ 4.94–4.81 (m, 2H), 3.82 (s, 2H), 1.93–1.82 (m, 4H), 1.76–1.68 (m, 4H), 1.59–1.54 (m, 2H), 1.52 (s, 6H), 1.52–1.50 (m, 2H), 1.50–1.46 (m, 2H), 1.46–1.32 (m, 6H); 13C NMR (100 MHz, CDCl3): δ 174.2, 78.6, 75.0, 31.3, 31.3, 25.3, 23.5, 20.7; IR (KBr) νmax: 2935, 2859, 1727, 1633, 1450, 1250, 1162, 1089, 1020, 802 cm–1; HRMS (ESI) m/z: calcd for C18H30NaO6, 365.1935 [M + Na]+; found, 365.1936.
meso-Dicyclohexyl 2,3-Dimethyltartrate (meso-2u)
White solid, 22.5 mg, 44% yield; mp 56–58 °C; 1H NMR (400 MHz, CDCl3): δ 4.90–4.76 (m, 2H), 3.62 (s, 2H), 1.90–1.82 (m, 4H), 1.75–1.68 (m, 4H), 1.59–1.50 (m, 3H), 1.49 (s, 6H), 1.47–1.42 (m, 3H), 1.42–1.36 (m, 4H), 1.35–1.28 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 173.9, 78.4, 75.1, 31.4, 31.2, 25.2, 23.6, 23.5, 20.7; IR (KBr) νmax: 2933, 2858, 1726, 1635, 1450, 1391, 1258, 1155, 1111, 1019, 804 cm–1; HRMS (ESI) m/z: calcd for C18H30NaO6, 365.1935 [M + Na]+; found, 365.1938.
dl-Di-tert-butyl 2,3-Dimethyltartrate (dl-2v)
White solid, 16 mg, 38% yield; mp 76–78 °C; 1H NMR (400 MHz, CDCl3): δ 3.79 (s, 2H), 1.51 (s, 18H), 1.48 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 174.0, 83.2, 78.6, 27.9, 21.1; IR (KBr) νmax: 2970, 2360, 1721, 1633, 1397, 1264, 1155, 1113, 802 cm–1; HRMS (ESI) m/z: calcd for C14H26NaO6, 313.1622 [M + Na]+; found, 313.1624.
meso-Di-tert-butyl 2,3-Dimethyltartrate (meso-2v)
White solid, 17 mg, 40% yield; mp 101–102 °C; 1H NMR (400 MHz, CDCl3): δ 3.63 (s, 2H), 1.49 (s, 18H), 1.44 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 173.6, 83.2, 78.5, 28.0, 21.1; IR (KBr) νmax: 2968, 2358, 1725, 1635, 1395, 1265, 1150, 1110, 1025, 803 cm–1; HRMS (ESI) m/z: calcd for C14H26NaO6, 313.1622 [M + Na]+; found, 313.1621.
dl-Di-((R)-1-phenylethyl) 2,3-Dimethyltartrate (dl-2w)
Colorless oil, 26 mg, 41% yield; 1H NMR (400 MHz, CDCl3): δ 7.41–7.31 (m, 5H), 7.30–7.25 (m, 3H), 7.18–7.09 (m, 2H), 5.93 (q, J = 6.5 Hz, 1H), 5.65 (q, J = 6.6 Hz, 1H), 3.63 (s, 1H), 3.59 (s, 1H), 1.60 (d, J = 6.5 Hz, 3H), 1.57 (s, 3H), 1.42 (s, 3H), 1.41 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 173.7, 173.5, 140.9, 140.2, 128.6, 128.5, 128.4, 128.1, 126.6, 125.8, 78.6, 78.3, 75.1, 74.78, 21.8, 21.8, 20.7, 20.4; IR (KBr) νmax: 3167, 1732, 1399, 1257, 1062, 698 cm–1; HRMS (ESI) m/z: calcd for C22H26NaO6, 409.1622 [M + Na]+; found, 409.1622.
meso-Di-((R)-1-phenylethyl) 2,3-Dimethyltartrate (meso-2w)
Colorless oil, 24 mg, 45% yield; 1H NMR (400 MHz, CDCl3): δ 7.53–7.27 (m, 6H), 5.93 (p, J = 6.5 Hz, 1H), 3.81 (d, J = 7.4 Hz, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.57 (s, 2H), 1.50 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 174.3, 173.8, 140.8, 140.5, 128.6, 128.6, 128.2, 128.2, 126.1, 126.0, 78.8, 78.7, 74.9, 74.7, 22.1, 21.9, 20.8, 20.4; IR (KBr) νmax: 2984, 1732, 1209, 1158, 1060, 698 cm–1; HRMS (ESI) m/z: calcd for C22H26NaO6, 409.1622 [M + Na]+; found, 409.1624.
Methyl 2-(Adamantan-1-yl)-2-hydroxyacetate (2f-ol)
White solid, 21.5 mg, 32% yield; mp 39–41 °C; 1H NMR (400 MHz, CDCl3): δ 3.78 (s, 3H), 3.66 (d, J = 8.1 Hz, 1H), 2.60 (d, J = 8.1 Hz, 1H), 1.99 (s, 3H), 1.70 (d, J = 12.2 Hz, 3H), 1.65–1.58 (m, 9H); 13C NMR (100 MHz, CDCl3): δ 174.4, 79.1, 52.0, 37.9, 37.0, 36.9, 28.2; IR (KBr) νmax: 2906, 2852, 173, 1633, 1447, 1253, 1211, 1706, 988, 733, 609 cm–1; HRMS (ESI) m/z: calcd for C13H20NaO3, 247.1305 [M + Na]+; found, 247.1304.
Diethyl 2-Hydroxymalonate (2r-ol)26
Colorless oil, 27 mg, 46% yield; 1H NMR (400 MHz, CDCl3): δ 4.68 (d, J = 8.3 Hz, 1H), 4.37–4.19 (m, 4H), 3.54 (d, J = 8.3 Hz, 1H), 1.29 (t, J = 7.1 Hz, 6H); MS (ESI) m/z: 177 [M + 1]+, 199 [M + 23]+.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (21572098, 21532002, and 21472087) and the Natural Science Foundation of Jiangsu Province (BK 20141313).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00749.
The authors declare no competing financial interest.
Supplementary Material
References
- a Schanz H.-J.; Linseis M. A.; Gilheany D. G. Tetrahedron: Asymmetry 2003, 14, 2763–2769. 10.1016/s0957-4166(03)00586-x. [DOI] [Google Scholar]; b Hu Y.; Yuan J.-J.; Sun X.-X.; Liu X.-M.; Wei Z.-H.; Tuo X.; Guo X. Tetrahedron: Asymmetry 2015, 26, 791–796. 10.1016/j.tetasy.2015.04.007. [DOI] [Google Scholar]; c Bagi P.; Fekete A.; Kállay M.; Hessz D.; Kubinyi M.; Holczbauer T.; Czugler M.; Fogassy E.; Keglevich G. Heteroat. Chem. 2015, 26, 79–90. 10.1002/hc.21218. [DOI] [PubMed] [Google Scholar]; d Bagi P.; Ujj V.; Czugler M.; Fogassy E.; Keglevich G. Dalton Trans. 2016, 45, 1823–1842. 10.1039/c5dt02999f. [DOI] [PubMed] [Google Scholar]; e Schmitt M.; Schollmeyer D.; Waldvogel S. R. Eur. J. Org. Chem. 2014, 1007–1012. 10.1002/ejoc.201301566. [DOI] [Google Scholar]
- a Takinami M.; Ukaji Y.; Inomata K. Tetrahedron: Asymmetry 2006, 17, 1554–1560. 10.1016/j.tetasy.2006.05.025. [DOI] [Google Scholar]; b Ding X.; Ukaji Y.; Fujinami S.; Inomata K. Chem. Lett. 2003, 32, 582–583. 10.1246/cl.2003.582. [DOI] [Google Scholar]; c Wei W.; Kobayashi M.; Ukaji Y.; Inomata K. Chem. Lett. 2006, 35, 176–177. 10.1246/cl.2006.176. [DOI] [Google Scholar]; d Kim Y.; Park K. J.; Park Y. S. Bull. Korean Chem. Soc. 2012, 33, 3853–3856. 10.5012/bkcs.2012.33.11.3853. [DOI] [Google Scholar]; e Castaldi G.; Cavicchioli S.; Giordano C.; Uggeri F. J. Org. Chem. 1987, 52, 3018–3027. 10.1021/jo00390a013. [DOI] [Google Scholar]
- a Miranda V.; Maycock C. D.; Ventura M. R. Eur. J. Org. Chem. 2015, 7529–7533. 10.1002/ejoc.201501002. [DOI] [Google Scholar]; b Tao D. J.; Slutskyy Y.; Overman L. E. J. Am. Chem. Soc. 2016, 138, 2186–2189. 10.1021/jacs.6b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ananthan B.; Chang W.-C.; Lin J.-S.; Li P.-H.; Yan T.-H. J. Org. Chem. 2014, 79, 2898–2905. 10.1021/jo402764v. [DOI] [PubMed] [Google Scholar]; d Lo H.-J.; Chang Y.-K.; Yan T.-H. Org. Lett. 2012, 14, 5896–5899. 10.1021/ol3028237. [DOI] [PubMed] [Google Scholar]; e Kelly T. R.; Cai X.; Tu B.; Elliott E. L.; Grossmann G.; Laurent P. Org. Lett. 2004, 6, 4953–4956. 10.1021/ol047922h. [DOI] [PubMed] [Google Scholar]; f Barros M. T.; Maycock C. D.; Ventura M. R. Org. Lett. 2003, 5, 4097–4099. 10.1021/ol0354932. [DOI] [PubMed] [Google Scholar]
- a Yoon T. P.; Jacobsen E. N. Science 2003, 299, 1691–1693. 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]; b Doyle A. G.; Jacobsen E. N. Chem. Rev. 2007, 107, 5713–5743. 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]; c Taylor M. S.; Jacobsen E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. 10.1002/anie.200503132. [DOI] [PubMed] [Google Scholar]
- a Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2024–2032. . [DOI] [PubMed] [Google Scholar]; b Hoye T. R.; Ye Z. J. Am. Chem. Soc. 1996, 118, 1801–1802. 10.1021/ja953781q. [DOI] [Google Scholar]; c Paterson I.; De Savi C.; Tudge M. Org. Lett. 2001, 3, 3149–3152. 10.1021/ol010150u. [DOI] [PubMed] [Google Scholar]; d Sunazuka T.; Hirose T.; Shirahata T.; Harigaya Y.; Hayashi M.; Komiyama K.; O̅mura S.; Smith A. B. J. Am. Chem. Soc. 2000, 122, 2122–2123. 10.1021/ja9938074. [DOI] [Google Scholar]
- a Gratzer K.; Gururaja G. N.; Waser M. Eur. J. Org. Chem. 2013, 4471–4482. 10.1002/ejoc.201201675. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seebach D.; Beck A. K.; Heckel A. Angew. Chem., Int. Ed. 2001, 40, 92–138. . [DOI] [PubMed] [Google Scholar]; c Pellissier H. Tetrahedron 2008, 64, 10279–10317. 10.1016/j.tet.2008.08.029. [DOI] [Google Scholar]
- a Fang J.-M.; Chen M.-Y.; Shiue J.-S.; Lu L.; Hsu J.-L. Tetrahedron Lett. 2000, 41, 4633–4636. 10.1016/s0040-4039(00)00680-8. [DOI] [Google Scholar]; b Kim S. M.; Byun I. S.; Kim Y. H. Angew. Chem., Int. Ed. 2000, 39, 728–731. . [DOI] [PubMed] [Google Scholar]; c Clerici A.; Porta O. J. Org. Chem. 1982, 47, 2852–2856. 10.1021/jo00136a006. [DOI] [Google Scholar]; d Clerici A.; Pastori N.; Porta O. Tetrahedron Lett. 2004, 45, 1825–1827. 10.1016/j.tetlet.2004.01.025. [DOI] [Google Scholar]; e Clerici A.; Pastori N.; Porta O. J. Org. Chem. 2005, 70, 4174–4176. 10.1021/jo048279f. [DOI] [PubMed] [Google Scholar]; f Yamashita H.; Reddy N. P.; Tanaka M. Chem. Lett. 1993, 315–318. 10.1246/cl.1993.315. [DOI] [Google Scholar]; g Takikawa G.; Katagiri T.; Uneyama K. J. Org. Chem. 2005, 70, 8811–8816. 10.1021/jo051242q. [DOI] [PubMed] [Google Scholar]; h Xu K.; Fang Y.; Yan Z.; Zha Z.; Wang Z. Org. Lett. 2013, 15, 2148–2151. 10.1021/ol4006344. [DOI] [PubMed] [Google Scholar]
- a Hu S.; Neckers D. C. Tetrahedron 1997, 53, 7165–7180. 10.1016/s0040-4020(97)00420-1. [DOI] [Google Scholar]; b Hu S.; Neckers D. C. J. Chem. Soc., Perkin Trans. 2 1997, 1751–1754. 10.1039/a700948h. [DOI] [Google Scholar]; c Pappas S. P.; Pappas B. C.; Okamoto Y.; Sakamoto H. J. Org. Chem. 1988, 53, 4404. 10.1021/jo00253a042. [DOI] [Google Scholar]
- Nakajima M.; Fava E.; Loescher S.; Jiang Z.; Rueping M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. 10.1002/anie.201501556. [DOI] [PubMed] [Google Scholar]
- a Romero N. A.; Nicewicz D. A. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; b Hari D. P.; König B. Chem. Commun. 2014, 50, 6688–6699. 10.1039/c4cc00751d. [DOI] [PubMed] [Google Scholar]; c Ravelli D.; Fagnoni M.; Albini A. Chem. Soc. Rev. 2013, 42, 97–113. 10.1039/c2cs35250h. [DOI] [PubMed] [Google Scholar]
- a Duan Z.; Li W.; Lei A. Org. Lett. 2016, 18, 4012–4015. 10.1021/acs.orglett.6b01837. [DOI] [PubMed] [Google Scholar]; b Pratsch G.; Overman L. E. J. Org. Chem. 2015, 80, 11388–11397. 10.1021/acs.joc.5b01962. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Nicewicz D. A.; MacMillan D. W. C. Science 2008, 322, 77–80. 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shih H.-W.; Vander Wal M. N.; Grange R. L.; MacMillan D. W. C. J. Am. Chem. Soc. 2010, 132, 13600–13603. 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Wallentin C.-J.; Nguyen J. D.; Finkbeiner P.; Stephenson C. R. J. J. Am. Chem. Soc. 2012, 134, 8875–8884. 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]
- a Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342–15345. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Allen L. J.; Cabrera P. J.; Lee M.; Sanford M. S. J. Am. Chem. Soc. 2014, 136, 5607–5610. 10.1021/ja501906x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Schnermann M. J.; Overman L. E. Angew. Chem., Int. Ed. 2012, 51, 9576–9580. 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Huynh M. H. V.; Meyer T. J. Chem. Rev. 2007, 107, 5004–5064. 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gentry E. C.; Knowles R. R. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T.; Zhao Z.-G.; Yu Q.; Xu H.-J. J. Photochem. Photobiol., A 1989, 47, 203–212. 10.1016/1010-6030(89)87066-2. [DOI] [Google Scholar]
- a Zhu X.-Q.; Li H.-R.; Li Q.; Ai T.; Lu J.-Y.; Yang Y.; Cheng J.-P. Chem.—Eur. J. 2003, 9, 871–880. 10.1002/chem.200390108. [DOI] [PubMed] [Google Scholar]; b Fukuzumi S.; Koumitsu S.; Hironaka K.; Tanaka T. J. Am. Chem. Soc. 1987, 109, 305–316. 10.1021/ja00236a003. [DOI] [Google Scholar]
- a Qi L.; Chen Y. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. 10.1002/anie.201607813. [DOI] [PubMed] [Google Scholar]; b Lee K. N.; Lei Z.; Ngai M.-Y. J. Am. Chem. Soc. 2017, 139, 5003–5006. 10.1021/jacs.7b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lackner G. L.; Quasdorf K. W.; Overman L. E. J. Am. Chem. Soc. 2013, 135, 15342–15345. 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tarantino K. T.; Liu P.; Knowles R. R. J. Am. Chem. Soc. 2013, 135, 10022–10025. 10.1021/ja404342j. [DOI] [PubMed] [Google Scholar]; e Rono L. J.; Yayla H. G.; Wang D. Y.; Armstrong M. F.; Knowles R. R. J. Am. Chem. Soc. 2013, 135, 17735–17738. 10.1021/ja4100595. [DOI] [PubMed] [Google Scholar]
- a Tatsumi S.; Izumi Y.; Imaida M.; Fukuda Y.; Akabori S. Bull. Chem. Soc. Jpn. 1966, 39, 602–604. 10.1246/bcsj.39.602. [DOI] [PubMed] [Google Scholar]; b Smith A. C.; Williams R. M. Angew. Chem., Int. Ed. 2008, 47, 1736–1740. 10.1002/anie.200705421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C.; Loh T.-P. Angew. Chem., Int. Ed. 2013, 52, 12414–12417. 10.1002/anie.201307245. [DOI] [PubMed] [Google Scholar]
- a Bassignani L.; Brandt A.; Caciagli V.; Re L. J. Org. Chem. 1978, 43, 4245–4247. 10.1021/jo00415a059. [DOI] [Google Scholar]; b Page P. C. B.; Rosenthal S. Tetrahedron 1990, 46, 2573–2586. 10.1016/s0040-4020(01)82037-8. [DOI] [Google Scholar]; c Prokopenko V. V.; Okonnishnikova G. P.; Klimenko I. P.; Shulishov E. V.; Tomilov Y. V. Russ. Chem. Bull. 2007, 56, 1515–1521. 10.1007/s11172-007-0234-6. [DOI] [Google Scholar]; d Kurono N.; Uemura M.; Ohkuma T. Eur. J. Org. Chem. 2010, 1455–1459. 10.1002/ejoc.200901501. [DOI] [Google Scholar]; e Horne D.; Gaudino J.; Thompson W. J. Tetrahedron Lett. 1984, 25, 3529–3532. 10.1016/s0040-4039(01)91067-6. [DOI] [Google Scholar]
- Cheng W.-M.; Shang R.; Yu H.-Z.; Fu Y. Chem.—Eur. J. 2015, 21, 13191–13195. 10.1002/chem.201502286. [DOI] [PubMed] [Google Scholar]
- Reznikov A. N.; Martynova N. A.; Sibiryakova A. E.; Klimochkin Y. N. Russ. Chem. Bull. 2015, 85, 2024–2029. 10.1134/s1070363215090029. [DOI] [Google Scholar]
- Li L.-S.; Wu Y.-L. Tetrahedron Lett. 2002, 43, 2427–2430. 10.1016/s0040-4039(02)00290-3. [DOI] [Google Scholar]
- Fujino D.; Yorimitsu H.; Osuka A. J. Am. Chem. Soc. 2014, 136, 6255–6258. 10.1021/ja5029028. [DOI] [PubMed] [Google Scholar]
- a Feng Y.; Dong J.; Xu F.; Liu A.; Wang L.; Zhang Q.; Chai Y. Org. Lett. 2015, 17, 2388–2391. 10.1021/acs.orglett.5b00901. [DOI] [PubMed] [Google Scholar]; b Su C.; Xie Y.; Pan H.; Liu M.; Tian H.; Shi Y. Org. Biomol. Chem. 2014, 12, 5856–5860. 10.1039/c4ob00684d. [DOI] [PubMed] [Google Scholar]; c Gergely J.; Morgan J. B.; Overman L. E. J. Org. Chem. 2006, 71, 9144–9152. 10.1021/jo061537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y.-S.; Jiang Y.-J.; An D.; Sha D.; Antilla J. C.; Zhang S. Org. Lett. 2014, 16, 6112–6115. 10.1021/ol502965p. [DOI] [PubMed] [Google Scholar]
- Companyó X.; Mazzanti A.; Moyano A.; Janecka A.; Rios R. Chem. Commun. 2013, 49, 1184–1186. 10.1039/c2cc38659c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.