

Abstract
Objective:
To determine the frequency and characteristics of clinical diagnostic change in frontotemporal dementia (FTD)-spectrum syndromes and Alzheimer’s disease (AD)-type dementia.
Methods:
We reviewed records and categorized diagnostic changes in patients seen ≥2 times with behavioral variant FTD (bvFTD, n=99), nonfluent and semantic variant primary progressive aphasia (nfvPPA, n=32; svPPA, n=59), corticobasal syndrome (CBS, n=40), progressive supranuclear palsy Richardson syndrome (PSP-RS, n=34), and AD-type dementia (n=49). For bvFTD we compared patients with and without diagnostic change, and assessed predictors of diagnostic change by logistic regression.
Results:
Initial diagnoses changed infrequently at subsequent visits in svPPA (6.8%), PSP-RS (8.8%), and nfvPPA (12.5%), with rare changes largely involving clinicopathological overlap or diagnostic ambiguity. Changes in AD-type dementia (30.6%) and CBS (37.5%) were more common, but reflected greater specificity, predicted co-pathology, or overlapping syndromes. Diagnostic change in bvFTD was also common (32.3%), but more diverse, including motor neuron disease development, alternative neurodegenerative syndromes, and non-neurodegenerative diseases. Diagnostic change occurred more often in those who met possible rather than probable bvFTD criteria (70.6% vs 15.3%, p<.001). Patients with stable diagnoses showed greater overall impairment, bvFTD behavioral severity, and atrophy in core right hemisphere bvFTD regions. Patients with diagnostic change had more severe depression (p<.05) and more frequent contributing, secondary diagnoses (p=.01), such as cerebrovascular disease. By logistic regression the accuracy of predicting stable bvFTD diagnoses using first visit data was 80%.
Conclusion:
bvFTD displays more diverse diagnostic change than other neurodegenerative syndromes. First visit bvFTD diagnoses may waver if based on meeting possible criteria only.
Keywords: Frontotemporal dementia, corticobasal degeneration, progressive supranuclear palsy, Alzheimer’s disease
INTRODUCTION
Clinical care and research related to neurodegenerative diseases depend upon the accuracy of clinical diagnoses. Patients given an incorrect diagnosis may receive inappropriate treatment and have an erroneous impression of prognosis. Observational studies that include misdiagnosed patients give skewed descriptions of syndrome characteristics. Clinical trials that enroll patients who do not have the targeted disease lose power to detect an illness-specific effect. For example, recent Alzheimer’s disease (AD) trials found that 20–39% of enrolled patients who met AD-type dementia criteria were amyloid biomarker negative, indicating that AD is unlikely to be the cause of their syndrome [1, 2]. Well-validated biomarkers are not yet available for other neurodegenerative diseases, and diagnosis still depends on clinical and neuroimaging features.
Clinical diagnostic criteria exist for many neurodegenerative syndromes. These are often derived from cross-sectional assessment or by retrospectively tabulating symptoms over a patient’s entire disease course, rather than only including information available at first presentation. In some cases, the sensitivity and specificity analyses for diagnostic criteria are based on autopsy diagnoses. Autopsy analyses are valuable, but can underestimate certain groups of patients, in particular those who are less likely to die in the expected time frame. For example, patients may be diagnosed with behavioral variant frontotemporal dementia (bvFTD), yet subsequently either not progress, a scenario termed bvFTD phenocopy [3], or have the diagnosis switched to a psychiatric illness [4]. In either case longer follow-up is needed to include these patients in clinicopathological series to measure diagnostic accuracy.
It can be difficult to judge how often patients receive different diagnoses over time. Only the most recent diagnoses are generally reported, and patients with unstable diagnoses may be excluded from published cohorts entirely. Diagnostic stability through longitudinal follow-up provides another measurement of diagnostic accuracy. While longer follow-up may improve diagnostic accuracy, the extent to which this is true and whether this differs among diagnoses is unclear. The duration of follow-up in observational studies is limited by patient factors, study logistics, and resources. Information regarding the optimal duration of follow-up needed to have diagnostic certainty would influence study design.
Our objective was to determine the frequency of diagnostic change within a large neurodegenerative disease cohort across longitudinal follow-up in six common syndromes: bvFTD, AD-type dementia, nonfluent and semantic variant primary progressive aphasia (nfvPPA, svPPA), corticobasal syndrome (CBS), and progressive supranuclear palsy-Richardson syndrome (PSP-RS). We sought to identify factors that predict diagnostic stability. Based on the high rate of misdiagnoses in bvFTD [4], which can resemble both psychiatric and other neurodegenerative diseases, we hypothesized that bvFTD would have the greatest diagnostic instability.
METHODS
Subjects
We retrospectively identified all patients who had been seen in the UCSF FTD program project grant on at least two time points. This project targets yearly visits, though the interval sometimes varies due to programmatic or patient factors. We included patients who received first visit diagnoses of bvFTD, AD-type dementia, svPPA, nfvPPA, CBS, or PSP-RS. As part of their assessment all patients underwent a multidisciplinary evaluation including neurological examination, neuropsychological assessment, and caregiver interview. Functional status was assessed by the Clinical Dementia Rating scale (CDR) [5] including modifications for frontotemporal lobar degeneration (FTLD) [6]. Behavioral assessment included the Neuropsychiatric Inventory (NPI) [7]. Neuropsychological assessment included a previously-described battery of tests of memory, language, visuospatial ability, and executive function [8]. After reviewing all available information gathered during the research evaluation clinicians coded each visit with the best-fit clinical syndrome, any additional syndromes considered, contributing factors, and whether the patient met criteria used at the time for each syndromic diagnosis [9–14]. AD biomarkers and genetic results were typically not available at the time of first visit diagnoses. Our search captured patients who did not meet research criteria for their best-fit syndrome to be able to assess how meeting criteria influenced longitudinal diagnostic stability. While UCSF’s status as a tertiary referral center may influence the mix of patients seen, this project recruits all patients thought to have FTD-spectrum illnesses, without focusing on diagnostically challenging patients.
Assessment of Diagnostic Stability
For most patients the diagnoses were stable across visits. For others the diagnoses changed in light of the most recent findings. As not all diagnostic changes are equal in clinical importance we coded changes in four categories: 1) those that reflected either greater diagnostic specificity (e.g., frontal variant AD rather than unspecified AD-type dementia) or common developments (e.g., AD-type dementia evolving to AD plus vascular disease), 2) those that reflected a change to another neurodegenerative diagnosis (e.g., bvFTD changed to AD-type dementia), 3) those that reflected change to non-neurodegenerative diagnoses (e.g., bvFTD to bipolar disorder), and 4) those that changed as noted in categories 2 and 3, but ultimately reverted back to the original diagnosis. We compared the frequency of any change and of the 4 categories of change between the diagnostic groups.
Because we sought to determine the extent that additional years of follow-up led to greater diagnostic stability, and whether this differed by diagnosis, we also compared rates of diagnostic change between time points 1 and 2, or if there were 3+ or 4+ time points. The FTD Program Project Grant explicitly targets two time points for each patient, with additional time points scheduled when possible. The number of time points for each patient is non-random and reflects rate of clinical decline (i.e., whether a patient is still able to come in) and patient willingness to return.
Characteristics of patients with diagnostic change in bvFTD
Because we hypothesized that bvFTD would be the most diagnostically challenging and therefore least stable over time we reviewed the records for all patients with bvFTD to identify the cause for and factors associated with their change in diagnosis. To increase clinical applicability, we limited these analyses to patients who met either FTDC possible or probable bvFTD criteria [10] at first visit. Patients with a behavioral syndrome who do not meet accepted criteria cannot be considered to have a settled bvFTD diagnosis regardless of which features are present or whether the same uncertain diagnosis is given in follow-up. For patients seen before the publication of the FTDC criteria they were applied retrospectively by a behavioral neurologist who reviewed all available records. For purposes of these analyses we grouped diagnostic change category 1 (progression or refinement of terminology) with the stable diagnoses, since these patients retained their bvFTD diagnosis in spite of developing additional features. For example, a patient with bvFTD who developed motor neuron disease (MND) might receive a primary diagnosis of FTD-MND, but would continue to have stable bvFTD syndromic features. Using features from year 1 we compared the following variables between patients with and without diagnostic change: age at presentation, sex, degree of functional impairment, the presence of the 6 core FTDC diagnostic features, whether patients had secondary (contributing or alternative) diagnoses, the 12 NPI subscale scores, and cognitive test results. Cognitive tests were selected to represent the executive (modified Trails B, D-word fluency, design fluency), memory (California Verbal Learning Test – short form), and visuospatial (Benson figure copy) domains that the FTDC criteria employ to determine a bvFTD neuropsychological profile.
To determine whether degree or pattern of atrophy differed between those with and without diagnostic change we derived group-level atrophy maps by voxel-based morphometry (VBM) using SPM12 (http://www.fil.ion.ucl.ac.uk/spm/). We included all who had useable neuroimaging within 6 months of first diagnosis. Based on the scanner in use at the time, MRI images were acquired on 1.5T, 4T, or one of two 3T scanners, with previously published acquisition parameters [15–17] and pre-processing as previously described [18]. Multiple regression was performed to assess for regions of gray matter volume associated with diagnostic stability. Sex, age, total intracranial volume, scanner type, and MMSE were included as covariates. The threshold for significance was set at p < .05, Family Wise Error (FWE) corrected for multiple comparisons. Statistical maps were examined at p < .001, uncorrected for multiple comparisons.
To assess whether bvFTD diagnostic changes improved prediction of the pathological diagnosis, we compared clinical and pathological diagnoses in all autopsied patients. Neuropathological assessments followed previously described procedures [19, 20]. Patients were classified into major molecular classes and subtypes of FTLD [21, 22] or AD [23, 24].
Prediction of change in bvFTD diagnosis
To determine which first-visit features allow optimal prediction of diagnostic stability we entered the variables that were available on the greatest number of patients (demographics, MMSE, CDR, CDR sum of boxes (CDR-SB), FTDC features, and NPI subscale scores) into a stepwise logistic regression with α to enter and α to leave = 0.15. This yielded the most strongly predictive variables, which were then entered into leave-one-out, cross-validated logistic regression to determine their classification accuracy. Analyses were performed in R [25].
RESULTS
Diagnostic stability
Stable diagnoses
For svPPA, nfvPPA, and PSP-RS, diagnostic change was uncommon (Table 1). Many of these changes involved progression or switching to a syndrome with known clinicopathological overlap or similarity. Of the four patients with svPPA who had diagnostic change, three had right temporal-predominant atrophy, with two of those patients receiving a subsequent bvFTD diagnosis. The fourth patient was diagnosed at the second visit with unspecified PPA. Of the four patients with nfvPPA and diagnostic change, one developed MND, two received primary CBS diagnoses (which had been supportive diagnoses at the first visit), and one was diagnosed with AD-type dementia. Of the three patients with PSP-RS and diagnostic change, one reverted back to PSP-RS after receiving a CBS diagnosis at time point 2, and the other two, subsequently diagnosed with AD or MCI, either did not meet PSP diagnostic criteria or only met possible PSP criteria [13] at first visit.
Table 1 –
Frequency of any change in diagnosis after the first time point
| 1st visit diagnosis | Total | # changed | % changed |
|---|---|---|---|
| bvFTD | 99 | 32 | 32.3 |
| svPPA | 59 | 4 | 6.8 |
| AD-type dementia | 49 | 15 | 30.6 |
| CBS | 40 | 15 | 37.5 |
| PSP-RS | 34 | 3 | 8.8 |
| nfvPPA | 32 | 4 | 12.5 |
Less stable diagnoses
For patients initially diagnosed with bvFTD (n=99), AD-type dementia (n=49), or CBS (n=40), the diagnosis changed in follow-up more than 30% of the time (Table 1). Though change in diagnosis was common in all three, the types of changes significantly differed among the diagnoses (X2(8,191) = 23.4, p < .001, Figure 1). Nearly 75% of the diagnostic changes among those with AD-type dementia reflected development of common pathological comorbidities (e.g., prediction of vascular or Lewy body co-pathology) or greater specificity (e.g., focal cortical AD syndromes). Diagnostic changes among those initially diagnosed with CBS typically centered on closely related neurodegenerative syndromes (e.g., PSP-RS or nfvPPA). Diagnostic changes observed in patients with first visit bvFTD diagnoses ranged from other FTD-spectrum conditions to AD-type dementia to psychiatric illness.
Figure 1.
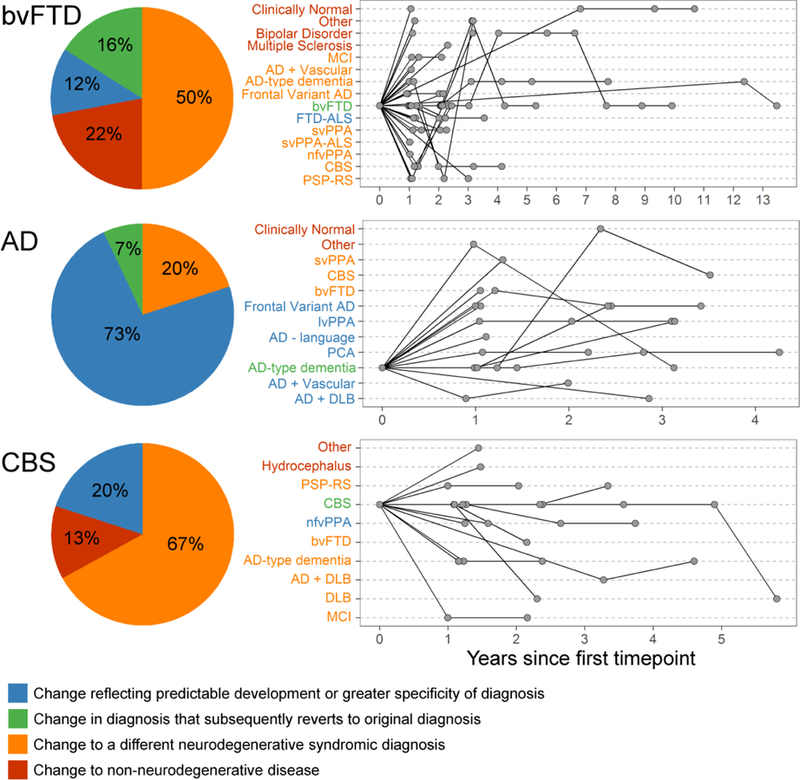
Type and timing of diagnostic change in bvFTD (n=99), AD-type dementia (n=49), and CBS (n=40). (left) The proportion of each category of diagnostic change among those who had more than one primary diagnosis throughout follow-up. (right) For individual patients who had a change in diagnosis line graphs depict all primary diagnoses received over the course of follow-up. lvPPA – logopenic variant primary progressive aphasia, DLB – dementia with Lewy bodies, MCI – mild cognitive impairment, PCA – posterior cortical atrophy
Number of time points and diagnostic change in less stable diagnoses
In bvFTD, AD-type dementia, and CBS, the majority of diagnostic changes occurred at time point 2, though a small number of changes occurred at time points 3 or beyond, even among those who had stable diagnoses in their first 2–3 time points (Table 2, Figure 1).
Table 2.
Frequency of change in diagnosis in relation to number of time points seen
| Diagnostic changes from time point 1 to 2 | ||||||
|---|---|---|---|---|---|---|
| Diagnosis at time point 1 | Change to other neurodegenerative syndrome | Change to non-neurodegenerative disease | Change reflecting development or specificity | |||
| n | % | n | % | n | % | |
| bvFTD, n=99 | 18 | 18.1% | 5 | 5.1% | 2 | 6.3% |
| AD-type dementia, n=49 | 3 | 6.1% | 1 | 2% | 7 | 14.3% |
| CBS, n=40 | 6 | 15% | 2 | 5% | 2 | 5% |
| Diagnostic changes in those seen at 3+ time points | ||||||||
|---|---|---|---|---|---|---|---|---|
| Diagnosis at time point 1 | No change in diagnosis | Changed at time point 2, then stable | Changed at time point 2 and again later | Same at time point 2, then changed | ||||
| n | % | n | % | n | % | n | % | |
| bvFTD, n=43 | 27 | 62.8% | 4 | 9.3% | 5 | 11.6% | 7 | 16.3% |
| AD-type dementia, n=19 | 10 | 52.6% | 2 | 10.5% | 3 | 15.8% | 4 | 21.1% |
| CBS, n=16 | 7 | 43.8% | 3 | 18.8% | 1 | 6.3% | 5 | 31.3% |
| Diagnostic changes in those seen at 4+ time points | ||||||||
| Diagnosis at time point 1 | No change in diagnosis | Changed time point 2 or 3, then stable | Changed at time point 2 or 3 and again later | Same at time point 2 or 3, then changed | ||||
|---|---|---|---|---|---|---|---|---|
| n | % | n | % | n | % | n | % | |
| bvFTD, n=19 | 11 | 57.9% | 2 | 10.5% | 3 | 15.8% | 3 | 15.8% |
| AD-type dementia, n=7 | 3 | 42.9% | 3 | 42.9% | 1 | 14.3% | 0 | 0% |
| CBS, n=2 | 0 | 0% | 1 | 50% | 0 | 0% | 1 | 50% |
Characteristics of patients with bvFTD with and without diagnostic change
For patients with bvFTD common themes emerged underlying the cause for their diagnostic changes (n=28 with diagnostic change, Figure 2). Symptom progression over time led to diagnosis of another neurodegenerative syndrome among three (e.g., two patients with behavioral symptoms emphasized at the initial visit had cognitive trajectories that were more consistent with AD-type dementia). For five patients the diagnostic change was influenced by amyloid PET biomarkers. Two patients lacked typical neurodegenerative progression and had signs of other neurological disease (e.g., multiple sclerosis). Another seven lacked expected neurodegenerative progression and had features of psychiatric illness. None of these met FTDC criteria on follow-up, either due to a shift in the perception of their symptoms or meeting FTDC exclusion criteria. The features of these patients are described in Table 3. The remaining six patients had diagnostic changes that revolved around confusion or disagreement over the appropriate primary diagnosis when FTD-spectrum syndromes overlap (e.g., a patient with an initial behavioral syndrome re-classified as PSP-RS when increasingly prominent motor features clarified prediction of the pathological diagnosis). These continued to meet the same or a higher level of FTDC criteria in spite of being given a different best-fit syndromic diagnosis.
Figure 2.
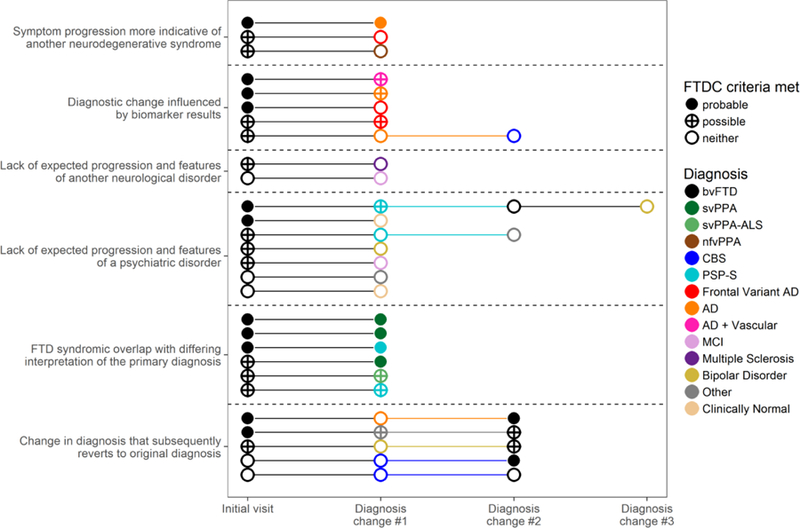
Causes of diagnostic change in bvFTD and diagnostic criteria met at each point when patients received a different primary diagnosis.
Table 3 –
Features of patients who changed from bvFTD diagnoses due to lack of progression and psychiatric symptomatology
| Age at first diagnosis | Sex | Follow-up (yrs) | Cognitive testing | Clinician’s neuroimaging interpretation | Family history | Psychiatric features | Behavior comments | |
|---|---|---|---|---|---|---|---|---|
| Patient 1 | 47 | M | 10 | Low-average verbal memory that improved through follow-up | Differing interpretations given – mild frontal and insular atrophy or normal | Autosomal dominant FTD | Depression (onset in 30s, moderate to severe on initial presentation), one prior possible hypomanic episode | Marital discord; spouse reported decrease in empathy, pornography viewing initially reported as compulsive, later deemed longstanding |
| Patient 2 | 66 | M | 4 | Mild executive dysfunction, but no clear progression over time. | Dorsal frontal atrophy, but no progression over follow-up | Late-life dementia in both parents | Depression/anxiety (onset in 40s), possible auditory hallucinations in childhood, episodes suggestive of mania throughout disease course. | At first visit, spouse reported loss of empathy, rigidity, impulsivity, obsessions, sweet craving. Separated from spouse after year 1. No behavioral symptoms reported by family at subsequent visits. |
| Patient 3 | 65 | M | 1 | Mild executive dysfunction. Improved at subsequent testing. | No definitive Atrophy | Bipolar disorder and substance abuse | Depression (onset in 30s), diagnosed with bipolar disorder in 50s | Prior episodes of impulsivity. Developed constant impulsivity, hypersexuality, apathy, poor judgment for over two years. Behaviors improved by time of year 2 visit. |
| Patient 4 | 54 | F | 3 | Low average executive function initially. No decline over time. | Initially called mild biparietal atrophy. Later interpreted as normal. | AD vs FTD in one family member, psychiatric illness in others | Depression since teens, signs of functional movement disorder developed after behavior change. | Self-endorsed apathy, social withdrawal, sweet preference. Mild loss of empathy and compulsions reported by spouse. |
| Patient 5 | 47 | M | 1 | Low average executive function initially. No decline over time. | No definitive atrophy. | Parent with bipolar illness and dementia diagnoses, sibling with early onset dementia. | Mood disorder since college, potential history of hypomanic episodes, longstanding history of risk taking | Spouse reported loss of empathy, mild disinhibition, intensified and narrow interests. He endorsed marital discord. |
| Patient 6 | 63 | M | 1 | Impaired executive function, mild verbal memory impairment. Stable to mildly improved over time. | Mild generalized atrophy for age. | Late-life dementia, mental illness | Lifelong personality differences (little empathy, excessive joking, collecting) | Decompensation of longstanding traits – more distant, worse hoarding, overeating, more frequently crosses social boundaries |
| Patient 7 | 75 | M | 1 | Within normal limits at both time points. | Frontal, parietal, and temporal atrophy identified at first visit. Thought normal at second visit. | Parent with late-life impulsivity, alcohol abuse in the other parent | Day to day fluctuations in mood, energy level, and cognitive function from elated to lethargic, suggestive of, but excessively fast for bipolar illness | Mild loss of empathy, disinhibition, repetitive behavior. Non-progressive, consistent with some longstanding traits.. |
The frequency of change significantly differed between patients with bvFTD who met FTDC probable criteria (15.3%) and those who met only FTDC possible criteria (70.6%, X2 (1) = 19.2, p < .001). Diagnostic changes were also observed in 5 of 10 patients who were given a best-fit diagnosis of bvFTD, but did not meet the threshold for FTDC possible criteria. These patients were not included in subsequent comparisons of clinical features. While patients with stable diagnoses presented at the same age as those with diagnosis changes, patients with stable diagnoses showed greater overall impairment (MMSE, CDR, CDR-SB), greater frequency or severity of behaviors associated with bvFTD (more frequent apathy, more severe disinhibition, repetitive behaviors, and eating behaviors), and worse lexical fluency, with a trend towards worse design fluency and more design fluency repetitions (both p = .05). The patients with changes in diagnosis had more severe NPI depression scores (Table 4), and more often had potentially contributory secondary factors (e.g., vascular disease or depression).
Table 4 –
First time point features of patients meeting possible or probable bvFTD criteria who had stable diagnoses or change in diagnosis
| Total n | Stable diagnosis | Changed diagnosis | p value | |
|---|---|---|---|---|
| Age at presentation | 89 | 62.1 (7.6) | 61.9 (8.8) | 0.92 |
| Gender (M/F) | 89 | 40/26 | 17/6 | 0.37 |
| MMSE | 87 | 24.3 (5.1) | 26.8 (3.1) | 0.04* |
| CDR total | 87 | 1.3 (0.6) | 0.8 (0.4) | < .001* |
| CDR sum of boxes | 87 | 6.9 (2.7) | 4.4 (2.1) | < .001* |
| FTDC criteria core features | ||||
| Disinhibition | 89 | 92.40% | 91.30% | 1 |
| Apathy | 89 | 90.90% | 65.20% | 0.01* |
| Loss of sympathy/empathy | 89 | 78.80% | 56.50% | 0.07 |
| Compulsive behavior | 89 | 80.30% | 73.90% | 0.73 |
| Hyperorality | 88 | 80.00% | 73.90% | 0.75 |
| Neuropsychological profile | 87 | 53.10% | 47.80% | 0.85 |
| Additional diagnoses listed | ||||
| Secondary (contributing) factors | 24.2% | 56.5% | 0.01* | |
| Secondary (alternative) diagnoses | 21.2% | 34.8% | 0.31 | |
| NPI (frequency x severity scores) | ||||
| Delusions | 85 | 1.5 (2.7) | 1.8 (3.4) | 0.67 |
| Hallucinations | 85 | 0.5 (1.8) | 0 (0) | 0.21 |
| Agitation/Aggression | 83 | 2.5 (3.4) | 3.2 (3.4) | 0.4 |
| Depression/Dysphoria | 85 | 1.1 (2.2) | 2.4 (3.1) | 0.04* |
| Anxiety | 84 | 2.1 (3.1) | 3.5 (3.8) | 0.11 |
| Elation/Euphoria | 84 | 3.9 (3.9) | 3.3 (3.6) | 0.51 |
| Apathy/Indifference | 83 | 8.2 (2.9) | 6.6 (4.3) | 0.06 |
| Disinhibition | 83 | 7.3 (3.7) | 4.7 (3.6) | 0.006* |
| Irritability/Lability | 83 | 2.9 (3.7) | 3.8 (4.6) | 0.36 |
| Motor Disturbance | 83 | 7.2 (4.2) | 4.2 (4) | 0.007* |
| Nighttime Behaviors | 84 | 2.8 (3.4) | 2.9 (4.2) | 0.9 |
| Appetite/Eating | 83 | 7.4 (3.1) | 4.7 (4.8) | 0.004* |
| NPI Total | 79 | 47.8 (19.2) | 42.8 (24.3) | 0.36 |
| Neuropsychological testing | ||||
| CVLT-SF long delay free recall | 81 | 3.5 (2.7) | 3 (3.1) | 0.49 |
| Modified Trails Time | 80 | 77.1 (42) | 65.5 (36.7) | 0.26 |
| Design Fluency Correct Responses | 79 | 5.2 (3.9) | 7.2 (3.7) | 0.05 |
| Design Fluency Repetitions | 79 | 5 (5.8) | 2.3 (3.7) | 0.05 |
| Benson figure copy | 85 | 14.6 (1.9) | 15 (2.1) | 0.4 |
| D-Word Fluency | 84 | 8 (4.8) | 10.9 (5.3) | 0.02* |
Neuroimaging features of patients with diagnostic change
Patients with stable diagnoses had significantly greater atrophy (p < .05 FWE) in core right hemisphere bvFTD regions, including medial prefrontal/anterior cingulate cortex (peak T value = 6.82), insula (T = 6.59), lateral prefrontal cortex (T = 6.23), striatum (T = 5.85), and ventromedial prefrontal cortex (T = 5.40) (Figure 3). There were no regions of significantly greater atrophy among patients with diagnostic change compared to those without.
Figure 3.
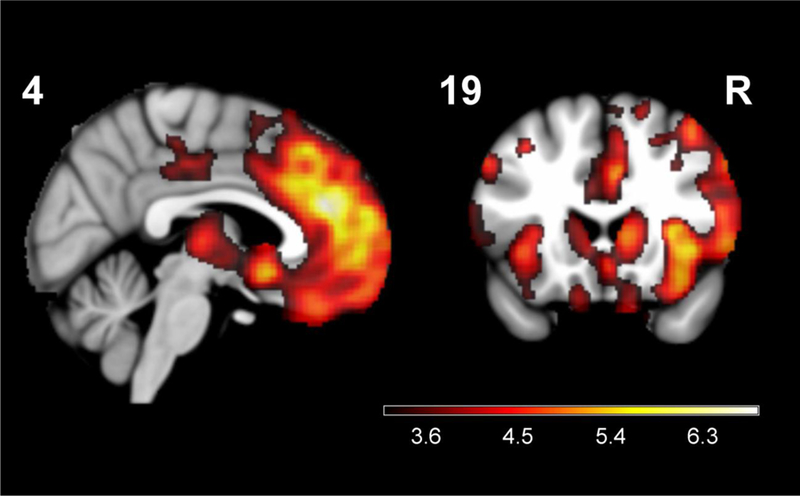
Regions of greater gray matter atrophy in patients with bvFTD and stable diagnoses compared to those with change in diagnosis at subsequent visits. Maps displayed at p < .001 uncorrected for multiple comparisons. N = 89
Pathological diagnoses among those with clinical diagnostic change
Of 99 patients with bvFTD, 38 had pathological diagnoses. Twenty seven of 38 had stable bvFTD diagnoses, and 26 of these had a congruent FTLD pathological diagnosis. The remaining patient had argyrophilic grain disease thought to be insufficient to have caused the behavioral syndrome. Eleven of 38 with pathological diagnoses had a change in clinical diagnosis. For two of these patients the clinical diagnosis changed to one that better predicted the pathological diagnosis (both involved changes to AD-type dementia with AD at autopsy). For seven of eleven the change in clinical diagnosis had a neutral effect on predicting the pathological diagnosis since both clinical diagnoses could be associated with the pathological diagnosis (e.g., change from bvFTD to CBS with underlying corticobasal degeneration (CBD)). In one patient the etiology remained unclear after autopsy, and in the remaining patient, neither clinical diagnosis (bvFTD and frontal variant AD) accurately predicted the patient’s Huntington’s disease.
Predictors of diagnostic change in bvFTD
Using stepwise logistic regression the combination of variables that predicted diagnostic stability included greater functional impairment (CDR-SB) and presence or greater severity of bvFTD-specific features (presence of loss of empathy, presence of apathy, and higher NPI disinhibition scores). Higher NPI anxiety or delusion scores predicted unstable diagnoses. The leave-one-out, cross-validated accuracy of predicting diagnostic stability was 80.0%.
DISCUSSION
The desire to give neurodegenerative diagnoses as early as possible, to guide treatment, inform research participation, and provide prognostic information, must be balanced with the need for confidence that diagnoses will be accurate and stable. In a large, prospectively followed cohort with FTD-spectrum diagnoses and AD-type dementia we found that while certain diagnoses were quite stable over follow-up others were more likely to change in the second or third visits and beyond. For bvFTD there is a particularly wide range of possible diagnostic changes. Certain factors help predict the stability of a bvFTD diagnosis, such as meeting bvFTD probable criteria, having a more severe and classic bvFTD presentation, and displaying greater atrophy in known regions of gray matter vulnerability.
Diagnostic stability
Three clinical diagnoses, svPPA, nfvPPA, and PSP-RS, were highly stable over follow-up. Their clinical and/or neuroimaging features have good specificity with a narrow differential diagnosis. If a confident first-visit diagnosis is made there is high likelihood that subsequent visits would only reinforce the diagnostic impression. The rare diagnostic changes reflect known diagnostic issues and clinicopathological overlap. Patients with focal right temporal atrophy can display overlapping bvFTD and svPPA features, potentially meeting criteria for either, both, or possibly neither syndrome. Patients with nfvPPA often develop motor features that reflect their underlying molecular etiology, most often CBD or PSP [26]. In these cases, rather than focusing on a “top” diagnosis, patients may simultaneously meet criteria for more than one syndrome, with both pointing towards a common prediction of the pathological diagnosis.
Patients with AD-type dementia showed changes that reflect development or greater specificity. Co-pathology of AD with vascular or Lewy body disease is common [27, 28]. Following these patients and predicting co-pathology does not alter the prediction of AD, but identifying these entities could be relevant for clinical prognostication and understanding participants’ responses in clinical trials. Patients with AD were often enrolled in the FTD Program Project Grant because of their young age or atypical presentations, leading this cohort to have a high frequency of focal cortical AD syndromes (e.g., frontal variant).
Diagnostic change was most common in patients with CBS; however, the changes were often within a narrow range reflecting syndromic overlap (e.g., nfvPPA) or other pathological entities that can cause CBS besides CBD (e.g., PSP and AD) [29].
While the majority of diagnostic changes occurred at the second time point, receiving the same diagnosis or even a different one at the second visit did not guarantee future stability. Determining the optimal number of visits would help observational study design, but this target cannot be derived from these results, because patients were not universally brought back for 3rd or 4th visits. Those with longer follow-up may have differed in diagnostic certainty from those who did not.
Features associated with diagnostic change in bvFTD
Diagnostic changes in bvFTD included a wide range of other neurodegenerative and non-neurodegenerative syndromes. Specific themes characterized these diagnostic changes. The development of new symptoms that pointed to a different neurodegenerative syndrome was rare. This is unsurprising since the first symptoms are typically the most valuable in diagnosing dementia. Amyloid PET results drove some diagnostic changes, suggesting that early biomarker availability would lead to fewer misdiagnoses. Of note, ¾ patients who changed to a final AD-type dementia diagnosis due to amyloid PET positivity continued to meet FTDC possible criteria (discordant biomarkers excluding probable bvFTD per the criteria), allowing for potential co-pathology. A lack of insidious, neurodegenerative progression influenced more changes in diagnosis than did new symptom development. While two non-progressive patients had features of another neurological disease, the most common cause for diagnostic change in bvFTD involved features that suggested a psychiatric explanation for the behavioral symptoms. While only 2/7 of these patients met probable bvFTD criteria at first visit, none met bvFTD criteria at final diagnosis. In some cases the change in level of criteria related to a perceptual shift regarding whether behavioral symptoms were severe or pervasive enough to satisfy criteria. In other cases it related to the criteria’s stipulations, which exclude any bvFTD diagnosis if behavioral changes are better accounted for by psychiatric illness. While some of these patients had mild impairment on cognitive tests, particularly in the area of executive function, they did not progress over time. In some cases the clinicians felt there was mild atrophy on imaging, though sometimes interpretations of the images differed in follow-up. Positive family history may have influenced the likelihood for some patients to receive a bvFTD diagnosis. Our recent study of 117 autopsied patients with a bvFTD diagnosis at any time point found none who lacked underlying neurodegenerative disease [18]. In contrast, another recent study reported that after two years of follow-up a shift to a psychiatric diagnosis occurred in 49% of patients who initially met possible or probable bvFTD criteria [4]. This discrepancy in the described frequency of bvFTD due to a psychiatric cause is in part related to the fact that patients with psychiatric disease do not come to autopsy as quickly as patients with bvFTD of the same age. The distinction between bvFTD (particularly patients without striking atrophy) and psychiatric disease can be challenging. Adding objective measures that distinguish neurodegenerative from psychiatric behavioral features at the first visit could help to refine existing bvFTD diagnostic criteria.
The FTDC diagnostic criteria provided valuable information to predict future diagnostic change. Patients who only met possible criteria changed diagnoses 70.6% of the time, even more frequently than those who met no criteria at all. This calls into question whether patients who only meet bvFTD possible criteria should be included in future bvFTD clinical trials or observational research publications, barring the discovery of a disease-causing mutation, which would satisfy bvFTD definite criteria.
We have previously shown that in spite of heterogeneity in both symptoms and underlying pathological diagnosis, patients with bvFTD show substantial clinical and neuroimaging overlap [18]. Our findings support the importance of this clinicoanatomic overlap in predicting diagnostic certainty. Patients with stable diagnoses displayed greater severity of bvFTD behaviors and greater atrophy in core bvFTD regions [18]. Core bvFTD behaviors also helped drive a logistic regression model that predicted stable bvFTD diagnoses with 80% accuracy. These results indicate to clinicians that while patients with mild behavioral symptoms and minimal atrophy may have underlying FTLD, diagnosing bvFTD in such patients carries a risk of reversing the diagnosis to one with a much less grave prognosis.
A limitation of the study is that patients were seen for an unequal number of visits. Differences may exist between patients seen once compared to those seen multiple times; these differences could be relevant to diagnostic stability. The pathological diagnoses found in autopsied patients suggest that a stable bvFTD diagnosis accurately predicts underlying FTLD. Future studies can prospectively follow patients with bvFTD systematically and can link diagnostic certainty with pathological diagnoses to determine the true ramifications of diagnostic changes on predictive accuracy. The observed rate of diagnostic change also occurred at a tertiary referral center with significant experience in evaluating FTD and atypical Alzheimer’s disease and access to investigations that are not part of common practice, such as amyloid PET. The rate of change in diagnosis may differ in other practice settings.
In spite of these limitations, this study provides guidance to clinicians who evaluate patients with neurodegenerative syndromes. Knowing who has a more unstable diagnosis can influence follow-up frequency, family counseling, and research enrollment. Future assessment of diagnostic criteria for sensitivity and specificity should incorporate information from each visit separately to best simulate the clinical scenario.
Acknowledgements:
This study was supported by grants K23AG045289 (DCP) and P01AG019724 (BLM) from the NIH. Dr. Perry, Mr. Datta, Dr. ZA Miller, Dr. Rankin, Dr. Gorno-Tempini, Dr. Kramer, Dr. Rosen, and Dr. BL Miller report no relevant disclosures. Dr. Seeley reports receiving consulting fees from Biogen Idec, Third Rock Ventures, Merck, Inc., and Bristol-Myers Squibb.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
COMPLIANCE WITH ETHICAL STANDARDS
Ethical approval
This study was approved by the UCSF Committee on Human Research.
Consent
Written informed consent was obtained from patients or surrogates, in accordance with the Declaration of Helsinki.
Conflicts of interest
The authors report no conflicts of interest.
REFERENCES
- 1.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R and Brashear HR (2014) Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N Engl J Med 370: 322–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sevigny J, Suhy J, Chiao P, Chen T, Klein G, Purcell D, Oh J, Verma A, Sampat M and Barakos J (2016) Amyloid PET Screening for Enrichment of Early-Stage Alzheimer Disease Clinical Trials: Experience in a Phase 1b Clinical Trial. Alzheimer Dis Assoc Disord 30: 1–7 [DOI] [PubMed] [Google Scholar]
- 3.Kipps CM, Nestor PJ, Fryer TD and Hodges JR (2007) Behavioural variant frontotemporal dementia: not all it seems? Neurocase 13: 237–247 [DOI] [PubMed] [Google Scholar]
- 4.Krudop WA, Dols A, Kerssens CJ, Eikelenboom P, Prins ND, Moller C, Schouws S, Rhebergen D, van Exel E, van der Flier WM, Sikkes S, Scheltens P, Stek ML and Pijnenburg YAL (2017) The Pitfall of Behavioral Variant Frontotemporal Dementia Mimics Despite Multidisciplinary Application of the FTDC Criteria. J Alzheimers Dis 60: 959–975 [DOI] [PubMed] [Google Scholar]
- 5.Morris JC (1993) The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43: 2412–2414 [DOI] [PubMed] [Google Scholar]
- 6.Knopman DS, Kramer JH, Boeve BF, Caselli RJ, Graff-Radford NR, Mendez MF, Miller BL and Mercaldo N (2008) Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 131: 2957–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA and Gornbein J (1994) The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 44: 2308–2314 [DOI] [PubMed] [Google Scholar]
- 8.Kramer JH, Jurik J, Sha SJ, Rankin KP, Rosen HJ, Johnson JK and Miller BL (2003) Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease. Cogn Behav Neurol 16: 211–218 [DOI] [PubMed] [Google Scholar]
- 9.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J and Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51: 1546–1554 [DOI] [PubMed] [Google Scholar]
- 10.Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M and Miller BL (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain [DOI] [PMC free article] [PubMed]
- 11.Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM and Grossman M (2011) Classification of primary progressive aphasia and its variants. Neurology 76: 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKhann G, Drachman D, Folstein M, Katzman R, Price D and Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: 939–944 [DOI] [PubMed] [Google Scholar]
- 13.Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, Goetz CG, Golbe LI, Grafman J, Growdon JH, Hallett M, Jankovic J, Quinn NP, Tolosa E and Zee DS (1996) Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 47: 1–9 [DOI] [PubMed] [Google Scholar]
- 14.Boxer AL, Geschwind MD, Belfor N, Gorno-Tempini ML, Schauer GF, Miller BL, Weiner MW and Rosen HJ (2006) Patterns of brain atrophy that differentiate corticobasal degeneration syndrome from progressive supranuclear palsy. Arch Neurol 63: 81–86 [DOI] [PubMed] [Google Scholar]
- 15.Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, Feiwell R, Kramer JH and Miller BL (2002) Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 58: 198–208 [DOI] [PubMed] [Google Scholar]
- 16.Mueller SG, Laxer KD, Barakos J, Cheong I, Garcia P and Weiner MW (2009) Subfield atrophy pattern in temporal lobe epilepsy with and without mesial sclerosis detected by high-resolution MRI at 4 Tesla: preliminary results. Epilepsia 50: 1474–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bettcher BM, Wilheim R, Rigby T, Green R, Miller JW, Racine CA, Yaffe K, Miller BL and Kramer JH (2012) C-reactive protein is related to memory and medial temporal brain volume in older adults. Brain Behav Immun 26: 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry DC, Brown JA, Possin KL, Datta S, Trujillo A, Radke A, Karydas A, Kornak J, Sias AC, Rabinovici GD, Gorno-Tempini ML, Boxer AL, De May M, Rankin KP, Sturm VE, Lee SE, Matthews BR, Kao AW, Vossel KA, Tartaglia MC, Miller ZA, Seo SW, Sidhu M, Gaus SE, Nana AL, Vargas JNS, Hwang JL, Ossenkoppele R, Brown AB, Huang EJ, Coppola G, Rosen HJ, Geschwind D, Trojanowski JQ, Grinberg LT, Kramer JH, Miller BL and Seeley WW (2017) Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 140: 3329–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee VM-, Miller BL, Trojanowski JQ and Grossman M (2006) Frontotemporal dementia: Clinicopathological correlations. Ann Neurol 59: 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tartaglia MC, Sidhu M, Laluz V, Racine C, Rabinovici GD, Creighton K, Karydas A, Rademakers R, Huang EJ, Miller BL, DeArmond SJ and Seeley WW (2010) Sporadic corticobasal syndrome due to FTLD-TDP. Acta Neuropathol 119: 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ and Mann DM (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM and Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122: 111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyman BT, Trojanowski JQ (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56: 1095–1097 [DOI] [PubMed] [Google Scholar]
- 24.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV and Montine TJ (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.R Core team (2015) R: A Language and Environment for Statistical Computing
- 26.Spinelli EG, Mandelli ML, Miller ZA, Santos-Santos MA, Wilson SM, Agosta F, Grinberg LT, Huang EJ, Trojanowski JQ, Meyer M, Henry ML, Comi G, Rabinovici G, Rosen HJ, Filippi M, Miller BL, Seeley WW and Gorno-Tempini ML (2017) Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol 81: 430–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalaria RN, Ballard C (1999) Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 13 Suppl 3: S115–23 [DOI] [PubMed] [Google Scholar]
- 28.Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM, Yan N, Yousef A, Hurtig HI, Siderowf A, Grossman M, McMillan CT, Miller B, Duda JE, Irwin DJ, Wolk D, Elman L, McCluskey L, Chen-Plotkin A, Weintraub D, Arnold SE, Brettschneider J, Lee VM and Trojanowski JQ (2018) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain [DOI] [PMC free article] [PubMed]
- 29.Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, Huang EJ, Trojanowski JQ, Growdon ME, Jang JY, Sidhu M, See TM, Karydas AM, Gorno-Tempini M, Boxer AL, Weiner MW, Geschwind MD, Rankin KP and Miller BL (2011) Clinicopathological correlations in corticobasal degeneration. Ann Neurol 70: 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]