

SUMMARY
Recent estimates of the human proteome suggest there are ~20,000 protein-coding genes, the protein products of which contain >145,000 phosphosites. Unfortunately, in-depth examination of the human phosphoproteome has outpaced the ability to annotate the kinases that mediate these post-translational modifications. To obtain actionable information about phosphorylation-driven signaling cascades, it is essential to identify the kinases responsible for phosphorylating sites that differ across disease states. To fill in these gaps, we have developed an unbiased, chemoproteomic approach for identifying high confidence kinase-substrate interactions with phosphosite specificity. Using this assay, we uncovered the role of cyclin-dependent kinase 4 (CDK4), a clinically validated kinase important for cell cycle progression, in regulating cap-dependent translation via phosphorylation of the tumor suppressor 4E-BP1. The discovery of this signaling axis sheds light on the mechanisms by which CDK4/6 inhibitors control cell proliferation and constitutes a successful example of kinase discovery using an activity-based, kinase-directed probe.
Graphical Abstract
eTOC Blurb
Mitchell et. al describe PhAXA, an improved chemoproteomic pipeline for mapping kinase-substrate interactions with phosphorylation-site specificity. Using this assay, the role of CDK4 in phosphorylating 4E-BP1 was identified, thereby influencing mTORC1-inhibitor resistant cap-dependent translation and specifically promoting c-Myc expression.
INTRODUCTION
mTOR is a highly-conserved protein kinase present in two distinct complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2) (Saxton and Sabatini, 2017). mTORC1 is essential for integrating stimuli from several signaling cascades to promote cap-dependent translation (CDT), a process by which proteins responsible for carrying out many anabolic processes are translated (Saxton and Sabatini, 2017). mTORC1 positively regulates CDT through phosphorylation-mediated inactivation of the translational gatekeeper protein 4E-BP1, which is achieved by hyperphosphorylation at the canonical mTORC1-dependent sites Thr37, Thr46, Ser65, and Thr70. However, phosphorylation of mTOR-independent sites may also play a role in 4E-BP1 activity (Martineau et al., 2013). Transcripts that are translated via CDT include cyclins, VEGF, c-Myc, and HIF-1α among other proteins involved in oncogenesis, highlighting the potential ramifications of dysregulated CDT in cancer (Bhat et al., 2015). As such, phosphorylated 4EBP1 has been implicated as a biomarker for interpreting the severity and aggressiveness of many human cancers (Armengol et al., 2007). Relatedly, analysis of patient samples has uncovered a positive correlation between hyperphosphorylated 4E-BP1 and poor prognosis (Castellvi et al., 2006; Graff et al., 2009; Lee et al., 2015; O’Reilly et al., 2009; Rojo et al., 2007).
As 4E-BP1 has been shown to be the principal effector of the mTORC1-controlled translational program (Dowling et al., 2010; Hsieh et al., 2012; Thoreen et al., 2012), which is dysregulated in nearly all cancers, significant effort has been put forth to develop mTORC1 inhibitors as therapeutics for targeting aberrant CDT. Unfortunately, the clinical impact of these drugs, including the allosteric mTORC1 inhibitors rapamycin and the rapalogs, has not lived up to expectations. Although many explanations for the limited efficacy of these drugs have been postulated (Choo and Blenis, 2009; Wendel et al., 2004), nearly all reports of rapalog insensitivity refer to the inability of these drugs to prevent the phosphorylation of 4E-BP1 (Choo et al., 2008; Feldman et al., 2009; Thoreen et al., 2009). This has fueled the development of ATP-competitive mTOR inhibitors capable of completely inhibiting 4E-BP1 phosphorylation (Feldman et al., 2009; Thoreen et al., 2009); however, drug resistance is still observed in part due to incomplete inhibition of 4E-BP1 phosphorylation and the downregulation of 4E-BP1 levels (Ducker et al., 2014; Wang et al., 2017; Wei et al., 2016; Zhang and Zheng, 2012). Thus, these reports hint at the presence of unidentified kinases that promote mTOR inhibitor resistance via phosphorylation of 4E-BP1 at canonical and non-canonical phosphosites.
RESULTS
A phosphosite-accurate kinase-substrate crosslinking assay
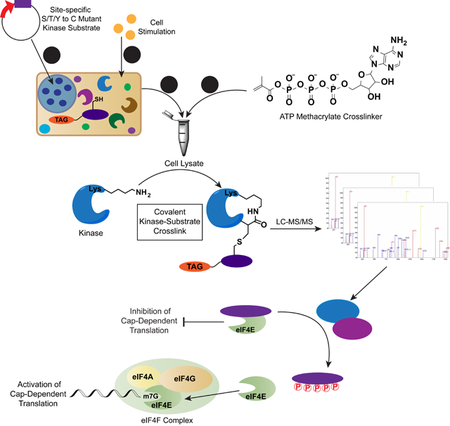
Because of the transient nature of kinase-substrate interactions, the mapping of kinases to their substrates remains a challenge (Chuh et al., 2016). Previous attempts at designing activity-based, phosphosite-specific probes for kinase identification have used two approaches: (1) assays that utilize peptide substrates that poorly recapitulate the activity of the bait protein (Maly et al., 2004; Riel-Mehan and Shokat, 2014; Statsuk and Shokat, 2012); or (2) photoaffinity probes, which are inherently promiscuous and low yielding, preventing the identification of low-abundance kinases (Dedigama-Arachchige and Pflum, 2016; Parang et al., 2002). To identify the kinase(s) that contribute to mTOR inhibitor resistance, we were inspired by a previously reported ATP crosslinker probe (1) in which the γ-phosphate of ATP has been modified with a methacrylate moiety (Figure 1A), allowing for the conversion of the highly conserved Lys residue within a kinase active site to an acrylamide (Riel-Mehan and Shokat, 2014). The position of this electrophilic handle enables a phosphosite-specific crosslinking reaction to occur via Michael addition with a Cys thiol which has been inserted in place of the Ser/Thr/Tyr of a substrate protein. The result is a hydrolytically stable bond formed between the acrylamide-kinase and Cys mutant probe.
Figure 1 |. Workflow for identifying site-specific kinase-substrate interactions.

A, PhAXA and structure of ATP crosslinker probe 1. B, HEK293T cells transfected with WT or 3XFLAG phosphosite-to-Cys mutant 4E-BP1 constructs were lysed, treated with 1 or ATP and analyzed via Western blot. A minimal mass shift is observed due to the negligible size of FLAG-4E-BP1 relative to mTOR. C, PP242 (10,000–1 nM) and D, rapamycin (100 nM) inhibit the crosslinking of mTOR to the T46C 4E-BP1 in the presence of 1.
To improve upon this activity-based method, which relied on the use of biotinylated peptide pseudosubstrates, we have developed a phosphosite-accurate kinase-substrate crosslinking assay, or PhAXA (Figure 1A), which allows a full-length kinase-substrate complex to be isolated via immunoprecipitation and the kinase(s) identified via mass spectrometry (MS)-based proteomics. The benefits of this approach are two-fold: (1) Kinases have been shown to achieve substrate specificity through complex docking interactions (Remenyi et al., 2006). As such, these interactions are impossible to achieve by using a peptide or partial substrate as bait for activity-based pulldown. In this method, the full-length protein is used, and thus, the method is more biologically accurate. (2) By transiently expressing the substrate protein, as opposed to adding exogenous protein or peptide, the substrate is present in situ and can engage in all physiologically relevant interactions prior to cell lysis, aiding in specificity. This is particularly relevant in the context of 4E-BP1, as it is thought to be exclusively phosphorylated while bound to eIF4E (Gingras et al., 1999).
To evaluate the potential of this approach, the assay was applied to 4E-BP1 Cys mutant probes at sites known to be phosphorylated by mTOR. Selective pulldown was achieved and mTOR was highly enriched from HEK293T lysate expressing the T37C and T46C 4E-BP1 mutants treated with 1 relative to the wild type (WT) and ATP controls (Figure 1B). Unsurprisingly, mTOR pulldown was not observed with S65C, T70C or S101C probes, as these sites are considered to be poor substrates of mTOR (Kang et al., 2013). We subsequently determined that kinase pulldown was dependent on the catalytic lysine by inhibiting mTOR pulldown with the ATP competitive inhibitor PP242 (Figure 1C). Importantly, enrichment of mTOR was only abrogated at concentrations exceeding the reported IC50 value (Apsel et al., 2008), providing evidence that the level of enrichment was representative of kinase activity towards the Cys mutant substrate. Specificity was further demonstrated by disrupting pulldown of mTOR using rapamycin (Figure 1D), 4E-BP1 probes containing mutations that inhibit raptor-mediated substrate recruitment, and with detergents that inhibit mTORC1 activity (Figures S1A and S1B) (Beugnet et al., 2003). Collectively, these results demonstrated successful proof-of-concept for PhAXA.
To validate PhAXA as a viable pipeline for the identification of kinases by MS, HEK293T cells were transiently transfected with T46C and WT 4E-BP1 probes, followed by treatment of lysate with 1 or ATP, affinity enrichment and LC-MS/MS analysis. Of all the proteins identified across biological replicates, only three were kinases. Of these, two were considered hits as they exhibited >2-fold enrichment from the T46C sample containing 1 relative to the WT and ATP controls using two methods for label-free quantification. We identified mTOR, which was the top hit across all proteins in the sample, and CDK4, which has never been described as a 4E-BP1 kinase (Figures 2A and 2B). To validate these findings, we used PhAXA to profile mTOR and CDK4 activity towards 4E-BP1 at each of its well-documented phosphosites by Western blot. CDK4 was enriched from lysate expressing the T37C, T46C, T70C, and S101C mutant probes, but showed no activity at S65C (Figure 2C).
Figure 2 |. MS-analysis of PhAXA pulldown identifies mTOR and CDK4 as 4E-BP1 kinases.

A, Table of kinases identified by at least 2 peptides in each biological replicate following filtering of common contaminants (Mellacheruvu et al., 2013). Average peptide-spectrum matches (PSMs) are from 2 biological replicates. ‘+’ refers to samples treated with 1; ‘-’ refers to ATP only controls. Dashes were added for ratios that could not be calculated due to absence of any high scoring PSMs. The ratio of MS1 intensities was determined using Skyline (MacLean et al., 2010a). Fold change is represented as +/−standard deviation (n = 3–13). B, Log2 fold change of spectral counts for non-filtered proteins plotted for the T46C probe relative to the appropriate controls. 1 PSM was added to each sample before averaging to enable calculation of the fold change for samples with no PSMs. C, CDK4 is enriched from lysate expressing 3x-FLAG-4E-BP1 phosphosite-to-Cys mutants. A representative input is shown to demonstrate the mass-shift of CDK4 upon crosslinking to 4E-BP1.
Separately, we established the broad applicability and high specificity of PhAXA by applying this assay to two other bait proteins of interest, c-Jun and Erk2 (Figures S1D–S1G). As expected, when subjected to MS analysis, MEK2 and MAPK8/9 were identified as top hits for the Erk2 and c-Jun pulldowns, respectively (Tables S1 and S2). Collectively, these results demonstrate that PhAXA is an activity-based assay, dependent upon kinase catalytic activity towards the Cys mutant substrate, and is amenable to the phosphosite-specific identification of kinase-substrate interactions.
4E-BP1 is a CDK4 substrate
The CDKs are conserved Ser/Thr kinases that control cell cycle progression via phosphorylation of cell cycle-regulating proteins, most notably the tumor suppressor protein RB1. Each CDK associates with a specific subset of binding proteins, the cyclins, that are required for kinase activity by providing substrate specificity via recognition of a required RXL motif (Anders et al., 2011; Choi and Anders, 2014). 4E-BP1 contains one RXL motif (R73DL) in a region of the protein that is free in solution while bound to eIF4E (Peter et al., 2015). CDK4 also demonstrates substrate specificity via recognition of the consensus sequence Ser/Thr-Pro with optimal substrates containing one or more basic amino acids downstream of this motif. 4E-BP1 has several phosphosites that fit the optimal CDK4 motif, including Thr10, Ser65, Thr70 and S101; while Thr37, Thr46 and S83 contain the minimal S/T-P motif. Moreover, 4E-BP1 can be found in the nucleus as well as the cytoplasm (Figure S2) (Rong et al., 2008), providing evidence that a difference in subcellular localization would not prevent CDK4 from phosphorylating 4E-BP1 (Gingras et al., 1999).
CDK4 associates with cyclins D1, D2 and D3; thus, we performed in vitro kinase assays with each of these complexes. The presence of a D-cyclin proved necessary for CDK4 activity, and large differences in the in vitro activities of each CDK4-cyclin D complex were observed with CDK4-cyclin D3 demonstrating the greatest kinase activity towards 4E-BP1 (Figure 3A). CDK4 appears to associate with D3 more readily than D1 or D2, which may account for the perceived increase in activity. However, the in vitro activity of CDK4-Cyclin D3 was only slightly higher than CDK4-Cyclin D2 complexes towards recombinant, full-length RB1 (Figures 3A and S3A). Nevertheless, these complexes efficiently phosphorylated 4E-BP1 at each of the canonical mTOR phosphorylation sites, while no in vitro activity was observed with any of the three CDK6-cyclin D complexes (not shown). To verify that the activity observed was due to CDK4 and not a co-purifying kinase, we utilized the clinically approved and highly selective active site CDK4/6 inhibitor, palbociclib (Fry et al., 2004). Phosphorylation of 4E-BP1 was inhibited at concentrations between 5–50 nM, mirroring the reported in vitro IC50 value (Figure 3B) (Fry et al., 2004), providing additional confidence in our finding that 4E-BP1 is a CDK4 substrate.
Figure 3 |. CDK4-Cyclin D complexes phosphorylate 4E-BP1 at canonical and non-canonical phosphorylation sites, including S101.

A, Immunopurified Flag-CDK4/myc-Cyclin D complexes phosphorylate recombinant 4E-BP1 and Rb. ***In vitro kinase assay using recombinant Rb as a substrate was run separately. Full Western blots for Rb shown in Figure S3A. B, Palbociclib (50 μM–500 pM) inhibits in vitro phosphorylation of 4E-BP1 by CDK4-cyclin D3. C, In vitro kinase assay using WT, S65A and S101A 4E-BP1 phosphorylated by CDK4-cyclin D3 complexes. D, MS analysis of in vitro phosphorylated 4E-BP1 by cyclin D3/CDK4. Phosphosites identified by manual curation of MS/MS assignments from ATP-treated sample are shown in red. No suitable phospho-peptides were identified in the no ATP control sample. Gold asterisk = canonical mTORC1 sites, Blue asterisk = non-canonical phosphorylation sites. E, HEK293T cells were transfected with indicated 3xFLAG-4EBP1 constructs. F, S101 (red) and the CDK4 recognition motif (blue) are highly conserved across mammals.
S101 is a poorly studied 4E-BP1 phosphosite (Wang et al., 2003); however, a large phosphoproteomics study observed hyperphosphorylation of this site in response to rapamycin treatment, suggesting a potential mechanism of resistance to mTORC1 inhibition (Yu et al., 2011). Therefore, we were interested in exploring the enrichment of CDK4 with the S101C mutant and its lack of activity with S65C (Figure 2C). Due to high sequence similarity, our phospho-specific antibody recognizes both phosphosites; thus, the relative contribution of each phosphorylation event was determined using Ala mutants (Figure 3C). Interestingly, the signal observed for phosphorylation at S65/101 was unaffected by mutation of S65; however, it was diminished with the S101A mutant. CDK4 was further validated as a S101 kinase using PhAXA, which, when analyzed by MS, identified only CDK4 and ERK2 as enriched in the S101C mutant over the controls (Table S3). While ERK2 is known to phosphorylate free 4E-BP1 in vitro, this relationship has never been validated in vivo or in cells (Gingras et al., 1999). We also analyzed CDK4-cyclin D3-mediated 4E-BP1 phosphorylation via phosphoproteomics, which confirmed S101 as a CDK4 substrate in addition to uncovering several potential non-canonical phosphorylation sites (Figure 3D). As some of the sites do not fit the CDK4 recognition motif, this may be due to STK38, a common contaminating kinase in FLAG pulldowns (Mellacheruvu et al., 2013).
4E-BP1 is known to be phosphorylated in an ordered fashion (Gingras et al., 2001); however, the role of S101 in the hierarchy has not been established. To probe this, we expressed nonphosphorylatable Ala mutants in HEK293T cells. We observed a near-complete loss of signal at S65/101 in S101A transfected cells, whereas S65A-transfected cells showed only a modest decrease compared to the WT control (Figure 3E). Intriguingly, phosphorylation of S101, which is highly conserved across mammals (Figure 3F), partially affected global 4E-BP1 inactivation, with S101A showing a moderate decrease in phosphorylation at each site that we investigated. This indicates a previously unknown contribution of S101 in initiating or maintaining the inactivation of 4E-BP1.
CDK4 promotes rapamycin-resistant cap-dependent translation via phosphorylation of 4EBP1
To further characterize CDK4-cyclin D-mediated inactivation of 4E-BP1, HEK293T cells were transfected with FLAG-CDK4 and/or myc-cyclin D2/D3 before stimulating with serum and insulin. Cells expressing cyclin D2 or D3 showed increased phosphorylation of 4E-BP1, an effect that became more evident in cells also treated with rapamycin (Figure 4A). Based on our in vitro kinase assay and PhAXA data, we assume this extends primarily to the non-canonical phosphosites such as S101, as other sites were largely unaffected (Figure S3B).
Figure 4 |. mTORC1 and CDK4/6 cooperate to regulate 4E-BP1 phosphorylation and initiation of cap-dependent translation.

A, Western blots of 3xFLAG-CDK4 and/or myc-D-cyclin transfected HEK293T cells stimulated and treated with rapamycin or DMSO following serum deprivation. B, Cells were deprived of serum, followed by stimulation with media containing serum and insulin with or without rapamycin and/or palbociclib. C, Colony formation of MCF-7, MDA-MB-468 and MDA-MB-231 cell lines treated with rapamycin and/or palbociclib. D, Quantification of cell density from (c). Signal intensity is normalized to the no treatment control. Error bars indicate standard deviation for 3 biological replicates. E, Western blot of MCF-7, MDA-MB-468 and MDA-MB-231 cell lines treated with rapamycin and/or palbociclib. Vertical bars separate samples run on separate blots or different exposures. F, Cap-dependent dual luciferase assay of cells treated with rapamycin, palbociclib and/or INK128. Normalized Renilla luciferase is shown relative to the no-treatment control for each cell line. Data represented as mean +/− standard deviation (n=4), and are representative of 3 independent replicates. *P<0.05, **P<0.01 ***P<0.001.
We next investigated if CDK4 contributes to the time-sensitive, rapamycin-resistant phosphorylation of 4E-BP1 (Choo et al., 2008). HEK293T cells were treated with rapamycin and palbociclib following serum starvation, and 4E-BP1 phosphorylation was monitored over time. At 2 h post stimulation, reduced phosphorylation at S65/101 was observed, indicated by a decrease in the highly phosphorylated, slow migrating phosphoform (Figure 4B). 6 h post-treatment, S65/101 phosphorylation had partially recovered in cells treated with rapamycin alone; those treated with the combination, however, showed a marked decrease in phosphorylation. Interestingly, most other sites for which phosphorylation-specific antibodies exist were only slightly affected by this co-treatment, strengthening our link between CDK4 and S101 phosphorylation (Figure S3C). This decrease in 4E-BP1 phosphorylation was accompanied by a decrease in cyclin D2, the translation of which has been demonstrated to be cap-dependent (Descamps et al., 2012). We verified palbociclib does not affect mTORC1 activity by assessing phosphorylation of ribosomal protein S6 (Figure 4B) and profiling the in vitro activity of mTORC1 isolated from palbociclib-treated cells (Figure S3D). These experiments clearly demonstrate that CDK4 phosphorylates 4E-BP1 in HEK293T cells under the same conditions in which PhAXA identified CDK4. Thus, our cumulative results serve as validation and strengthen the applicability of this assay to identify physiologically relevant kinases.
As palbociclib has recently been approved for the treatment of HR+, HER2- breast cancers (O’Leary et al., 2016), we sought to investigate whether a correlation exists between CDK4 inhibitor sensitivity and CDK4-mediated phosphorylation of 4E-BP1. In-line with previous findings, we found the ER+ MCF-7 and triple negative MDA-MB-231 cell lines to be palbociclib-sensitive and the triple-negative MDA-MB-468 cell line to be resistant (Figure 4C and 4D) (Finn et al., 2009). Palbociclib treatment induced a decrease in 4E-BP1 phosphorylation in the MCF-7 and MDA-MB-231 cell lines; this effect was magnified in cells co-treated with rapamycin (Figure 4E). However, in the MDA-MB-468 cell line, palbociclib had no effect on 4E-BP1 phosphorylation. Although this cell line expresses CDK4 at levels commensurate with other cell lines, D-type cyclin expression is nearly undetectable at the protein level (Figure S4A), likely contributing to palbociclib insensitivity.
Next, we investigated the impact of combined inhibition of CDK4 and mTORC1 using cells stably expressing a bicistronic reporter construct containing Renilla and Firefly luciferase separated by the poliovirus internal ribosomal entry site (IRES) (Poulin et al., 1998). Using this assay, we verified that the combination of palbociclib and rapamycin causes a functional impact by decreasing CDT in MCF-7 cells, whereas palbociclib provided no added benefit over rapamycin treatment alone in MDA-MB-468 cells (Figure 4F). The current understanding of rapamycin resistance suggests mTORC1 can carry-out rapamycin-insensitive activities, thus active-site mTOR inhibitors (mTORKIs) such as INK128 are used to promote robust 4E-BP1 reactivation (Thoreen et al., 2009). However, the combination of palbociclib and rapamycin was as effective as INK128 at reducing relative rates of CDT in this assay (Figure 4F), likely due to the significant reduction in total 4E-BP1 levels following chronic treatment with mTORKIs (Figure S4B) (Wang et al., 2017).
Inhibition of CDK4 downregulates c-Myc via reactivation of 4E-BP1
We next investigated the effect of this combination treatment on c-Myc expression, as MYC mRNA is translated via CDT (Gera et al., 2004). Intriguingly, we noted a decrease in c-Myc protein expression in palbociclib-sensitive cells treated with the combination, while no change was observed in the resistant MDA-MB-468 cells (Figure 5A). We determined that downregulation of c-Myc expression was due to decreased rates of translation, rather than an E2F-dependent decrease in MYC transcription (Lin et al., 2008), as mTORC1 inhibition resulted in an increase in MYC transcript levels (Figure 5B). We then profiled mRNA expression of several c-Myc target genes (Schmidt, 2004), including NPM1, EIF4E, ODC1 and CDK4, to investigate the functional consequences of diminished c-Myc. In MCF-7 cells, inhibition of mTORC1 and CDK4 decreased c-Myc transcriptional activity with the most robust decrease in activity observed in the combination treatment (Figure 5B and S5A). MDA-MB-468 cells showed little change in activity, in-line with the observed response at the protein level (Figure 5B and S5A).
Figure 5 |. CDK4 drives translation of c-Myc via phosphorylation of 4E-BP1.

A, Western blot of c-Myc expression in MCF-7, MDA-MB-468, and MDA-MB-231 cell lines treated with rapamycin and/or palbociclib. B, Relative RNA expression of transcripts from cells treated as in (a). Expression is normalized to the no treatment control sample for each cell line. UBB was used as an internal control for each gene. Error bars standard deviation (n=3). Data is representative of 2 independent replicates. C, Expression of c-Myc in MDA-MB-468 and MCF-7 cell lines stably expressing doxycycline inducible FLAG-4E-BP1 mutants. D, Western blot of c-Myc expression in 4E-BP1 knockout or control cells treated as in A. Vertical bars separate images obtained from separate exposures. E, Western blot of cyclin D3 expression in 4E-BP1 knockout or control cells treated +/− palbociclib. F, Cap-dependent dual luciferase assay of cells treated with rapamycin and/or palbociclib. Data represented as mean +/− SEM (n=3) after normalization to no treatment control for the appropriate cell line. *P<0.05, **P<0.01.
We next verified that this phenotype was the direct result of inhibition of 4E-BP1 phosphorylation by generating doxycycline-inducible cell lines that express nonphosphorylatable forms of 4E-BP1 at near-physiological levels. c-Myc downregulation was equivalent in MCF-7 cell lines expressing the S101A mutant and the fully nonphosphorylatable, 6A (T37A, T46A, S65A, T70A, S83A, and S101A) mutant (Figure 5C). Other mutations had minimal effect on c-Myc levels, whereas only the 4E-BP1 (T37/46A) expressing MDA-MB-468 cell lines showed any change (Figure 5C). These findings correlate with the effect these 4E-BP1 mutations have on the in vitro proliferation of these two cell lines (Figure S5B). Finally, to further explore the involvement of 4E-BP1 in the palbociclib-induced downregulation of c-Myc, we generated 4E-BP1 knockout (KO) cells. In MCF-7 cells lacking 4E-BP1, palbociclib treatment had no effect on c-Myc expression (Figure 5D). Alternatively, rapamycin induced in an increase in c-Myc (Figure 5D), which correlated with MYC transcript levels. As expected, palbociclib had no effect on c-Myc expression in MDA-MB-468 cells following 4E-BP1 knockout.
We next investigated the effects of CDK4 inhibition on cyclin D3, the translation of which is cap-dependent (Hsieh et al., 2012; Tsukumo et al., 2016). Treatment of MCF-7 cells with palbociclib resulted in a slight reduction in the expression of cyclin D3, an effect which was reversed by 4E-BP1 KO (Figure 5E). To further probe the effect of CDK4-mediated phosphorylation of 4E-BP1 on global regulation of CDT, we created 4E-BP1 KO and control cells stably expressing the dual luciferase CDT reporter construct. 4E-BP1 KO had very little effect on the rate of CDT in response to palbociclib treatment alone; however, treatment of 4EBP1 KO cells with both rapamycin and palbociclib resulted in a 2.2-fold increase in CDT relative to control cells subjected to the same conditions (Figure 5F). These experiments provide further evidence that CDK4 and mTOR converge on 4E-BP1 to regulate CDT.
4E-BP1 as a biomarker for palbociclib sensitivity.
CDK4/6 inhibitors are thought to function primarily by reducing phosphorylation-mediated inactivation of the RB1 tumor suppressor, thereby inducing a cell cycle arrest at the G1-S transition. While RB1 is a useful biomarker for predicting response to this class of inhibitors in cell lines and preclinical models (Finn et al., 2009; Fry et al., 2004), the utility of RB1 as a biomarker in the clinic is controversial (DeMichele et al., 2015; Knudsen and Witkiewicz, 2017; Rubio et al.). As more biomarkers are needed to allow for better predictors of drug sensitivity (Fang et al., 2018), we investigated the antiproliferative effects of palbociclib on 4E-BP1 KO cells to understand the value of 4E-BP1 in regulating response to CDK4/6 inhibition. Intriguingly, we found that MCF-7 cells lacking 4E-BP1 were less sensitive than control cells to both palbociclib and rapamycin (Figure 6A), indicating that inactivation of 4E-BP1 contributes to the global role of CDK4 as an oncogene. 4E-BP1 knockout had no effect on the sensitivity of MDA-MB-468 cells to rapamycin and palbociclib (Figure S6B).
Figure 6 |. CDK4-mediated phosphorylation of 4E-BP1 serves as a biomarker for predicting breast cancer survival.

A, Proliferation of MCF-7 cells +/− 4E-BP1 knockout treated with rapamycin and/or palbociclib assessed by CellTiter Glow assay. Treated samples are shown relative to no treatment control for that cell line. Bars represent mean +/− standard deviation (n=3) ***P<0.001, ****P<0.0001. B, List of proteins/antibodies with the highest correlation to survival in the BRCA dataset from TCPA. Shown are all antibodies with a Log-Rank P value of <0.01. P-values were used as reported by TCPA. C, Kaplain-Meyer curves showing the total survival time for BRCA patients from the TCPA dataset with high (red) and low (blue) p4E-BP1(S65/101). D, Histogram showing the Pearson Coefficient for pairwise regression between p4E-BP1 (S65/101) and every other antibody in the dataset.
To further evaluate the impact of CDK4-medated phosphorylation of 4E-BP1 in breast cancer patients, we mined Reverse-Phase Protein Array (RPPA) Breast Invasive Carcinoma (BRCA) datasets available through The Cancer Proteome Atlas (TCPA) (Li et al., 2013). We noted that phosphorylation of 4E-BP1 at S65/101 was the third most significant marker associated with overall survival across all 224 antibodies used in the array, while phosphorylation of T37/46 had no predictive value in this dataset (Figures 6B and 6C). We also found a strong positive correlation between phosphorylation of 4E-BP1 at S65/101 and Rb at S807/811 (Figure 6D), indicating that these phosphorylation events are upregulated in similar patient populations, which strengthens the link between, and the importance of, CDK4-mediated phosphorylation of S101 in BRCA.
DISCUSSION
We have demonstrated successful kinase discovery using an activity-based, kinase-directed probe. We have provided examples of the broad applicability of PhAXA by application to three distinct classes of substrate proteins, using Ser, Thr, and Tyr to Cys mutants in our proof-of-concept studies. In each case, only bona fide kinases were identified as top hits, highlighting the highly specific nature of this method. Despite the success of our approach, it is important to emphasize that the work-flow for this assay can be further optimized for each bait protein of interest. All three bait systems described here utilized the same assay conditions with respect to buffers, additives, detergents, lysis methods and cell lines; only the method of cellular stimulation was changed. It is possible that, with further optimization of these assay parameters, PhAXA can provide more robust enrichment of the appropriate kinase or kinases. However, the conditions outlined provide a starting point for pursuing other kinase-substrate systems of interest, and serve as a testament to the robust nature of this chemoproteomic approach.
One limitation is the need to perform crosslinking in lysate, as subcellular localization is known to contribute to kinase activity (Menon et al., 2014; Wang et al., 2015). It is also possible that a positive identification may arise from a kinase that is normally localized to a compartment that would preclude an interaction with the bait of interest. This hypothetical scenario highlights the importance of thoroughly validating any identified relationships in a more physiological setting. However, in light of potential drawbacks to the PhAXA workflow, we feel that the use of a full-length bait protein, expressed transiently in situ, marks a large improvement over previously reported methods that relied on exogenously added, biotinylated peptide pseudosubstrates. This is exemplified by the fact that, using PhAXA, we identified and validated CDK4 activity towards S101, but not S65, despite the very similar sequence shared by the two phosphorylation sites. Also, given that mTOR pulldown is not possible when the 4E-BP1 Cys mutant probes contain a very distant F114A mutation, a peptide-based method using only short phosphorylation site motifs would likely never work for this system. Thus, we believe that widespread adoption of this assay will help to fill in existing gaps in kinase signaling cascades, while facilitating the discovery of new druggable targets.
Using this pipeline, we have identified CDK4 as a 4E-BP1 kinase, demonstrating that CDK4 phosphorylates 4E-BP1 to maintain rapamycin-resistant cap-dependent translation. We also report the identification of S101, a poorly understood 4E-BP1 phosphosite, as a bona fide CDK4 substrate, and that phosphorylation of this residue promotes expression of c-Myc. However, it is currently unclear if this is strictly due to translational control, as the entirety of its function has yet to be elucidated. Finally, we provide preliminary evidence that 4E-BP1, namely 4E-BP1 hyperphosphorylated at S101, should be investigated as a biomarker for predicting sensitivity to palbociclib and other CDK4/6 inhibitors. Unfortunately, it is impossible to differentiate between the phosphorylation of S65 and S101 by Western blot, RPPA or IHC given the promiscuity of the antibody, thus targeted MS-based methods, such as Selected Reaction Monitoring (SRM), will need to be developed for accurate analysis.
Given this role of CDK4, it is likely that inhibition of this process is a previously unknown function of CDK4/6 inhibitors. Recently, several studies have reported potent in vivo antitumor effects by combined inhibition of mTOR and CDK4/6 (Cortes et al., 2017; Michaloglou et al., 2018; Olmez et al., 2017; Pikman et al., 2016), while a large phenotypic screen found the strongest cooperation between PI3K/mTORC1 and CDK4/6 inhibitors (Vora et al., 2014). In addition, many ongoing clinical trials are investigating the efficacy of this combination in treating a range of cancers; however, the rationale for this combination is lacking, as an in-depth understanding of the molecular mechanisms for this observed cooperativity is not understood. Our data clearly demonstrate that these drugs provide an additive benefit over standalone therapies, in part, by inhibiting cap-dependent translation, thereby decreasing the expression of anabolic proteins, and by altering the c-Myc transcriptional program that drives a wide range of cancers (Dang, 2012). These findings support the investigation of this combination for treating cancers displaying clear hallmarks of an addiction to cap-dependent translation (Pelletier et al., 2015).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amanda Garner (algarner@umich.edu)
EXPERIMENTAL MODELS AND SUBJECT DETAILS
HEK293T and HeLa cells (Female, 31 years old) were grown in DMEM (Corning) supplemented with 10% FBS, glutamine, penicillin, and streptomycin (Gibco). U2 OS cells (Female, 15 years old) and MCF-7 cells (Female, 59 years old) were cultured according to ATCC guidelines. MDA-MB-231 cells (Female, 51 years old) were grown in RPMI-1640 media supplemented with 10% FBS and glutamine. MDA-MB-468 cells (Female, 51 years old) were grown in DMEM (Corning) supplemented with 10% FBS and glutamine. Cells were grown at 37 °C with 5% CO2 in a humidified incubator, passaged at least twice before use for experiments and no more than 10 times before returning to low passage stocks. All cell lines were authenticated by STR profiling, and regularly tested for mycoplasma contamination.
METHOD DETAILS
Small molecule reagents
Palbociclib Isethionate (Selleckchem) was dissolved in water to 10 mM. Trametinib (Selleckchem), Rapamycin (Alfa Aesar), and SP600125 (ApexBio) were dissolved in DMSO. Human recombinant insulin was purchased from Sigma, PMA (Phorbol-12-Myristate-13-Acetate) was purchased from Acros. Anisomycin was purchased from Cayman Chemical. 3XFLAG peptide was purchased from ApexBio. All reagents were used as received.
Plasmids
Annealed oligos corresponding to the N-terminal 3XFLAG tag were ligated into pcDNA3 at BamHI and EcoRI. Those corresponding to the C-terminal myc-tag were cloned into pcDNA3 at XbaI and ApaI. 4E-BP1 cDNA was purchased from Promega in the pFN29K expression vector. HA-Erk2 (Rattus norvegicus), pcDNA3/Au1-mTOR, pRK5/HA-Raptor plasmids were generously shared by Dr. Diane Fingar. CDK4, CDK6, CCND1, CCND2 and CCND3 cDNA were cloned directly from A549 cDNA prepared using the Superscript III first-strand synthesis kit (Invitrogen). CDK4, CDK6, 4E-BP1, Erk2, c-Jun and Raptor were cloned into the pcDNA3/3XFLAG vectors. CCND1, CCND2, CCND3 were cloned into the pcDNA3/myc tag vectors. MBP-4E-BP1 (wild type and alanine mutants) was prepared by cloning into pMCSG9 (Univ. of Michigan Center for Structural Biology) using LIC cloning. HaloTag-4E-BP1 and -eIF4E have been described elsewhere.(Song et al., 2017) All mutations were generated by PCR mutagenesis. The sequences for all primers used have been listed in Supplementary Excel File. All constructs were fully sequenced by Sanger sequencing.
Lentivirus and stable cell lines
rLuc-PolIRES-fLuc was digested out of pcDNA3 (a kind gift from Dr. Peter Bitterman) using NheI and XhoI then ligated into pLentiLoxEV (UM vector core). The multiple cloning site of PLVX-Tre3G-mCherry (Clontech) was modified to include a XbaI restriction site by ligating annealed oligos into MluI and EcoRI. 3X-FLAG-4E-BP1 constructs were cloned from pcDNA3 into MluI and XbaI. PLVX-Tet3G (Clontech) was used for making rtTA-packaging lentivirus. Lentiviruses were packaged in HEK293T cells by cotransfection of the transfer plasmid, pMD2.G (a gift from D. Trono, Addgene plasmid 12259) and psPAX2 (a gift from D. Trono, Addgene plasmid 12260) using linear PEI (3:1 ratio of PEI to DNA). Media was changed 16 h after transfection, and viral supernatant was collected after an additional 24 and 48 h, before filtering through 0.45-μm filters and storing in aliquots at −80 °C. Titer was determined by colony formation assay where possible. Cells were infected at a MOI of <0.5 in the presence of polybrene (Santa Cruz Biotechnology) (8 μg/mL), then after 48 h, selected with geneticin (1 mg/mL) (Gibco) for 3 weeks or puromycin (2 μg/mL) (Sigma) for 6 d; no selection was used for the dual luciferase cell lines. Polyclonal cell lines were maintained in geneticin (300 μg/mL) and puromycin, 1 μg/mL for MDA-MB-231 and MDA-MB-468, and 0.25 μg/mL for MCF-7. Expression of 3X-FLAG-4E-BP1 was induced with doxycycline (Alfa Aesar), 1 μg/mL for MDA-MB-231 and MDA-MB-468 and 0.1 μg/mL for MCF-7.
CRISPR Knockouts
pSpCAS9(BB)-2A-GFP (a gift from Feng Zheng, Addgene plasmid #48138) was digested with BbsI and oligos targeting nothing (for non-target control) or exon 1 of EIF4EBP1 (designed using http://tools.genome-engineering.org) were added. 1e6 MCF-7 or MDA-MB-468 cells were transfected using Lipofectamine LTX with PLUS reagent according to the manufacturer’s instructions, and media was changed after 18 hours. After a 48-hour recovery, GFP-positive cells were sorted and saved using a MoFlo Astrios flow cytometer. Single cell clones were then isolated in 96-well plates by limiting dilution, and knockout was confirmed by western blot and sanger sequencing. For the final cell lines used in the experiments, five clones were pooled together (knockout or non-targeting control) and all experiments were then performed within 3 passages.
Kinase Assays
For CDK4/CDK6 kinase assays, HEK293T cells were grown in 10-cm plates to 60–70% confluence. Cells were subsequently transfected with 3XFLAG-CDK4/6 (4 μg) and myc-CCND1/2/3 or empty pcDNA3 (3 μg) using linear PEI (3:1 ratio of PEI to DNA). After 18 h, the media was changed to plain growth media. ~20 h later, cells were stimulated with insulin (150 nM) for 60 min, and then harvested by scraping into TBST (50 mM Tris pH 7.4, 150 mM NaCl, 0.1% Tween-20) with aprotinin (10 μg/mL), leupeptin (5 μg/mL), pepstatin (7 μg/mL), NaF (10 mM), sodium orthovanadate (2 mM), β-glycerophosphate (10 mM), and sodium pyrophosphate (2 mM). The cells were disrupted by pipetting up-and-down (20×) and rotated end-over-end for 15 min at 4 °C. The resulting lysate was cleared by centrifugation at 18,000×g for 10 min at 4 °C. CDK4/6 complexes were isolated from 1 mg of lysate (1 mL) by immunoprecipation with packed, prewashed FLAG-M2 magnetic resin (15 μL) for 90 min at 4 °C. The resin was then washed with lysis buffer (1 mL, 3×), TBS with β-glycerophosphate (10 mM) (1 mL, 1×), and TBS containing MgCl2 (10 mM) and β-glycerophosphate (10 mM) with or without ATP (1 mM) (1 mL, 1×). After washing, the resin was suspended in 1x TBS (30 μL) with MgCl2 (10 mM), DTT (1 mM) and β-glycerophosphate (10 mM), with or without ATP (1 mM) and palbociclib as described. Recombinant 4E-BP1 (1 μM) and HaloTageIF4E (2 μM), or p110RB (500 nM) were also added as described. The kinase reaction was incubated at 30 °C for 30 min, and subsequently quenched with 5× Laemmli buffer (7 μL).
mTOR kinase assays were performed similar to those with CDK4/6 complexes with a few notable exceptions. HEK293T cells were transfected with 3XFLAG-Raptor (4 μg) and Au1-mTOR (3 μg). Lysis was performed in HEPES buffer (pH 7.4, 20 mM) containing NaCl (100 mM), CHAPS (0.3% v/v) and protease/phosphatase inhibitors as described above. For assessing the effect of CDK4/6 inhibition on mTORC1 activity in vitro, palbociclib (5 μM) was included in all lysis buffers, wash buffers and assay buffers, in addition to using cells treated with palbociclib (5 μM) for 2 h before lysis.
All assays were performed in biological triplicate.
Immunoblotting
MCF-7, MDA-MB-231, MDA-MB-468 cells were lysed directly in-well using RIPA buffer (10 mM Tris-HCl, 150 mM NaCl, 1% Triton, 1% sodium deoxycholate, 0.1% SDS, pH 7.2) supplemented with 10 μg/mL aprotinin, 5 μg/mL leupeptin, 7 μg/mL pepstatin, 10 mM NaF, 2 mM sodium orthovanadate, 10 mM β-glycerophosphate, and 2 mM sodium pyrophosphate). Lysates were then sonicated thoroughly on ice. Protein concentrations were normalized by the BCA assay (Pierce), resolved on 4–20% Tris-glycine gels (Invitrogen), transferred to 0.45-μm PVDF (Thermo) using Towbin’s buffer (low amperage for ~4 h at 4 °C), blocked with 5% non-fat milk in TBST, then probed with primary antibodies overnight at 4 °C. Antibodies used in this study were the following: Actin-HRP (sc-47778) and Myc-9E10 (sc-40) from Santa Cruz Biotechnology; CDK4 (12790), CDK6 (13331), Cyclin D2, (3741), DYKDDDK tag (14793), eIF4E (9742), eIF4G (2498), MEK1/2 (9122), mTOR (2972), S6 (2217), pS6 (240/244) (2215), p4E-BP1 (T37/46) (2855), p4E-BP1 (S65/101) (9451), p4E-BP1 (T70) (9455), SAPK/Jnk (9252), 4E-BP1 (9644), c-Myc (13987), Rb (9313) and pRb (S780) (3590) from Cell Signaling Technology; and FLAG-M2 (F1804) from Sigma. All experiments were performed in biological triplicate.
Protein expression and purification
MBP-tagged 4E-BP1 proteins were expressed and purified from BL21 Rosetta 2 (DE3) E. coli. LB media supplemented with ampicillin and chloramphenicol (500 mL) was inoculated with an overnight culture (5 mL) and grown to an OD600 of 0.8 before expression was induced with IPTG (1 mM) for 2 h at 37 °C. After centrifugation (15 min at 7,500×g), cell pellets were re-suspended in NiA buffer (30 mL; 50 mM sodium phosphate, 100 mM NaCl, 20 mM imidazole, 2 mM DTT) containing guanidine-HCl (6M), and then sonicated on ice for 4 min (pulse: 1sec on/1sec off at 35% amplitude). Cleared lysate (18,000×g for 20 min at 4 °C) was added to packed Ni-NTA resin (4 mL; Qiagen) by gravity filtration. The resin was washed with NiA buffer with guanidine-HCl (6M) (25 mL, 1×), NiA buffer (25 mL, 1×), and then eluted with NiB buffer (NiA buffer with 500 mM imidazole). Eluted protein was cleaved with TEV protease overnight at 4 °C and dialyzed into Tris-buffered saline (TBS) with 2mM DTT (6 L). Dialyzed protein was passed through Ni-NTA resin and pure, cleaved 4E-BP1 was collected as flow through. Purity was verified as >95% by Coomassie stain. Protein concentration was determined by absorbance at 280 nM. Single-use aliquots were stored at −80 °C. HaloTag-eIF4E was purified as described previously (Song et al., 2017).
qRT-PCR
Total RNA was isolated using TRIzol reagent (InvitrogenO), and cDNA was subsequently prepared using the Superscript III first-strand synthesis kit (Invitrogen) according to manufacturer’s instructions. For gene expression analysis, qPCR was performed using the PowerUP sybr green master mix (Applied Biosystems) on a Viia 7 thermocycler using the fast-qPCR protocol. The relative fold change was calculated using the comparative threshold cycle (CT) method.(Schmittgen and Livak, 2008) PCR amplification efficiency controls were performed for each primer set and dissociation curves verified single product amplification. All experiments were performed in biological triplicate.
Dual Luciferase reporter assay
The cap-dependent translation luciferase reporter assay was performed using the dual glo luciferase assay system according to manufacturer’s instructions (Promega). Briefly, polyclonal rLuc-polIRES-fLuc expressing cell lines were plated at 20,000 cells per well in 96-well, white, tissue culture-treated plates, and treated as described in the figure legends. After 72 h, media was aspirated and OptiMEM (75 μL) (Gibco) was added to each well. Cells were lysed in firefly luciferase buffer, and total (firefly luciferase) luminescence was measured after 15 min. Total luminescence (renilla luciferase) was measured within one hour after addition of Stop & Glo reagent. All experiments were performed in biological triplicate.
PhAXA
For Western blot analysis, HEK293T cells were grown in 10-cm plates to 50% confluence and transfected with DNA (6 μg) by calcium phosphate precipitation. 18 h later, the media was changed to serum-free DMEM (4E-BP1 and rnErk2 pulldowns), or growth media (c-Jun pulldown). After 24 h (20 h for nocodazole experiments), cells were stimulated as follows: 10 min with growth media containing insulin (150 nM) for 4E-BP1 pulldown, 15 min with growth media containing PMA (100 nM) for rnERK2 pulldown, or 30 min with growth media containing anisomycin (10 μg/mL) for c-Jun pulldown. Cells were then harvested in NLB buffer (1 mL; 50 mM Tris pH 8.0, 150 mM NaCl, 10 mM MgCl2, 10 μg/mL aprotinin, 5 μg/mL leupeptin, 7 μg/mL pepstatin) per plate by scraping. Cells were lysed by forcefully passing through a 28.5G insulin syringe 5× consecutively on ice. Debris was pelleted at 18,000×g for 10 min at 4 °C. Cleared lysate was split into 500 μL aliquots (×2) in 1.5-mL tubes, and 1 or ATP was added to a final concentration of 250 μM. Lysate was incubated at 30 °C for 60 min under constant agitation. FLAG-BAIT complexes were then isolated by immunoprecipitation for 12–15 h at 4 °C with end-over-end rotation. The resin was subsequently washed 3× for 15 min under constant agitation with 1× TBS containing Triton X-100 (2% v/v), then thrice with TBS for 30 s each; 1 mL was used for each wash. Proteins were eluted with 2× Laemmli buffer and resolved by SDS-PAGE. All experiments were performed in biological triplicate.
For analysis by LC-MS/MS, the protocol is similar to that above with the following modifications: HEK293T cells were grown in 15-cm plates (×4) to 50% confluence and transfected with WT or Cys-mutant plasmid DNA (12 μg per plate) by calcium phosphate precipitation. Each plate was harvested in lysis buffer (2.5 mL), and samples were processed as above through the immunoprecipitation step. We found it critical to keep volumes at 500 μL per tube with 5 tubes per reaction condition. After overnight immunoprecipitation and following the final wash with TBS, complexes were eluted with elution buffer (250 μL; TBS, 0.1% sodium deoxycholate, 1 mg/mL 3XFLAG peptide) per tube. Elutions were carried out for 90 min at 25 °C with constant agitation on a plate shaker (120 rpm). Common eluents were pooled, tricholoracetic acid was added to a final concentration of 10% (w/v), and samples were incubated on ice for 60 min. Tubes were then centrifuged at 21,000×g for 15 min at 4 °C before discarding supernatants. Protein pellets were re-suspended in ice-cold acetone (1.5× eluate volume) for each wash using a water bath sonicator. After another 30 min on ice, the protein was again precipitated and the supernatant discarded. The acetone wash was repeated once more, and the protein pellets were re-suspended in 10 mM HEPES buffer (pH 8.0) containing 8 M urea (50 μL) and immediately frozen at −80°C until processing by in-solution digestion.
In-solution digestion
Protein samples were treated with ammonium bicarbonate buffer (pH ~8), which was added to a final concentration of 100 mM. Cysteine residues were reduced by adding 10 mM DTT (50 μl) and incubation at 45 °C for 30 min. Samples were cooled to room temperature, and alkylation of cysteines was achieved by incubating with 2-chloroacetamide (65 mM) under darkness for 30 min at room temperature. Upon diluting the urea to a final concentration of <1 M, overnight digestion with sequencing grade, modified trypsin (1 ug) was carried out at 37 °C. Digestion was stopped by acidification and peptides were desalted using SepPak C18 cartridges using manufacturer’s protocol (Waters). Samples were completely dried using a Vacufuge concentrator (Eppendorf).
In-gel digestion
TEV-cleaved 4E-BP1 (30 pmol) was used as a substrate for in vitro phosphorylation by cyclin D3-CDK4 complex with or without ATP (1 mM). The samples were separated by SDS-PAGE and stained with Coomassie R-250 (Bio-Rad). The protein samples were processed and analyzed at the Proteomics Resource Facility at the University of Michigan. The gel slice corresponding to TEV-cleaved 4E-BP1 was destained with 30% methanol for 4 h. Following reduction and cysteine alkylation as described above, proteins were digested overnight with sequencing grade, modified trypsin (Promega) (500 ng) at 37 °C. Peptides were extracted by incubating the gel with of 50% acetonitrile/0.1% TFA (150 mL) for 30 min at room temperature. A second extraction with 100% acetonitrile/0.1% TFA (150 mL) was also performed. Both extracts were combined and dried in a Vacufuge concentrator (Eppendorf).
Mass spectrometry
Peptides resulting from trypsin digestion were dissolved in 0.1% formic acid/2% acetonitrile solution (9 mL). 2 mL of the resulting peptide solution were resolved on a nano-capillary reverse phase column (Acclaim PepMap C18, 2 micron, 50 cm, ThermoScientific) using a 0.1% formic acid/acetonitrile gradient at 300 nl/min over a period of 90 min (in-gel digests) or 180 min (in-solution digests). Eluent was directly introduced into a Q Exactive HF mass spectrometer (Thermo Scientific, San Jose CA) using an EasySpray source. MS1 scans were acquired at 60K resolution (AGC target = 3 × 106; max IT = 50 ms). Data-dependent collision-induced dissociation MS/MS spectra were acquired on the 20 most abundant ions following each MS1 scan (NCE ~28%; AGC target = 1 × 105; max IT = 45 ms).
Data analysis
The resulting raw files were converted into mzXML files and centroided using MSConvert.(Holman et al., 2014) Spectra were searched against the Swiss-Prot Human protein database (2.15.17 Download) appended with all isoforms and cRAP contaminants using the COMET(Eng et al., 2013) search engine as part of the Trans-Proteomic Pipeline (TPP) (version 5.0).(Deutsch et al., 2010) Peptide mass tolerance was set to 10 ppm, fragment bin tolerance to 0.02 Da, and two missed cleavages were allowed. Met oxidation (+15.9949), Ser/Thr/Tyr phosphorylation (+79.966331) and Gln/Asn deamidation (+0.98402) were included as variable modifications; carbamidomethylated Cys (+57.021464) was set as a fixed modification. The resulting pep.xml files were analyzed for peptide probability using PeptideProphet,(Keller et al., 2002) where a minimum peptide probability of 0.95 was required, with only the expect score used as a discriminant. Protein level validation was performed using ProteinProphet;(Nesvizhskii et al., 2003) only proteins with a probability of >0.97 were considered.
Protein-level quantification was performed in two ways to account for the relatively low peptide-spectrum match (PSM) count of observed peptides. First, spectral counts were compiled using Abacus,(Fermin et al., 2011) and the “adjusted spectral count” was used to compare relative protein concentrations between samples. Each experiment was performed in biological duplicate, and PSMs from each duplicate sample were averaged. Second, relative quantification of proteins using MS1 intensity was accomplished using Skyline.(MacLean et al., 2010b) Briefly, spectral libraries were built from amino acid sequences for all kinases meeting initial refinement criteria (see below). MS1 intensities were extracted for proteotypic peptides within a mass error of 10 ppm that eluted within a 10 min window between runs. MS1 intensities were compared between samples where the corresponding peptide was positively identified by PeptideProphet with a probability >0.95. Comparisons were only made between samples that were simultaneously prepared, i.e. only within the first or second replicate, not between replicates. Between 1–15 peptides were compared for each protein depending on the number of positive identifications.
Data refinement
Initial data refinement for kinase identification was performed as follows. Only kinases found with at least 2 PSMs in both biological replicates (where applicable) were considered. These identified proteins were compared to a list of common contaminating proteins using CRAPome.(Mellacheruvu et al., 2013) Briefly, a repository was built from the top 20 experiments (in terms of number of positive identified proteins) using FLAG magnetic resin and total cell lysate from HEK293 cells. Any kinase found in more than 75% of these experiments was not considered for further characterization; this removed STK38, STK38L and PRKDC.
Phosphosite identification by MS/MS
To identify phosphorylation sites on 4E-BP1 following in vitro phosphorylation by CDK4/cyclin D3, MS/MS spectra were manually inspected after COMET search (as described above); however, a database of only cleaved 4E-BP1 was used. Only those phosphopeptides for which the phosphate could be clearly localized to one residue were considered putative CDK4 substrates. No other peptide or protein level validation was performed. This experiment was performed only once.
Cell Proliferation Assay
Cells were plated at a density of 2,000 cells per well in white-bottom, tissue culture treated, 96-well plates. The following day, cells were treated as stated in the figure legends; media (+/− drug) was changed every 48-hours. After 6-days, proliferation was assessed using Cell-Titer Glo (Promega) as according to the manufacturer’s instructions. All samples were normalized to the no-treatment control. All experiments were performed in biological triplicate.
Immunofluorescence
MCF-7 cells were grown in 24-well plates on poly-L-lysine-coated coverslips (Fisher) until ~50% confluent. Cells were washed twice with ice-cold 1× PBS, and then fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were washed twice with 1× PBS, permeabilized with 0.2% Triton X-100 in PBS for 30 min, blocked with 1% BSA in PBS and 0.1% Triton X-100 for 1 h at room temperature, and then probed with primary antibody (1:500 for 4E-BP1 and CCND3) in blocking buffer overnight at 4 °C. Coverslips were washed 3× with PBS for 10 min each before probing with AlexaFluor488-conjugated donkey anti-rabbit (Invitrogen) or AlexaFluor647-conjugated goat anti-mouse secondary antibodies (1:1000 dilution for each). Cover slips were subsequently washed 3× with 1× PBS for 10 min before mounting onto glass slides with Prolong diamond antifade mountant with DAPI. Images were taken using a Nikon A1 Spectral confocal microscope with a 60x objective. All images were compared to no-primary or cross-primary controls to ensure a specific signal. Images were compiled using Adobe Photoshop (CS6).
Colony formation assay
Cells were seeded in 6-well plates at a density of 20,000 cells per well for MCF-7 and MDA-MB-468 cells, and 1,000 cells per well for MDA-MB-231 cells. The next day, cells were treated with rapamycin (100 nM), palbociclib (5 μM) or DMSO (0.001% v/v). Fresh media containing the appropriate inhibitors was added after 5 days. On day 10, cells were fixed with glutaraldehyde (6% v/v) and stained with crystal violet (0.25% w/v) for 3 hours at room temperature. Excess stain was removed with water, then the plates were dried overnight before imaging and quantifying cell density using an Odyssey CLx (Licor). All experiments were performed in biological triplicate.
TCPA dataset mining
The TCGA-L4 breast invasive carcinoma (BRCA) Reverse-Phase Protein Array (RPPA) dataset (901 samples) was downloaded from The Cancer Proteome Atlas (TCPA) on 04/08/2018. Pairwise linear regressions were run for each antibody against p4EBP1_S65, and Pearson coefficients were extracted and plotted as a histogram using R version 3.4.4.
Synthesis of Crosslinker 1
The synthesis of 1 was adapted from that reported.(Riel-Mehan and Shokat, 2014) ATP-triethylammonium salt (0.1 g, 0.11 mmol) was dissolved in anhydrous DMSO (3 mL) under nitrogen. A solution of methacrylic anyhydride (0.44 mmol), anhydrous DMSO (1 mL), dioxane (1 mL), anhydrous DMF (1 mL) was added, and the mixture was stirred at room temperature under nitrogen. After 4 d, the reaction was quenched with water (5 mL) and extracted with ethyl acetate (3 × 5 mL). The aqueous layer was collected, flash frozen and lyophilized. Crude 1 was then dissolved in water and purified by preparative reverse-phase HPLC using an Agilent 1260 Infinity HPLC equipped with a PrepHT XDB-C18 column (21.2 × 150 mm; 5 μm) at a flow rate of 20 mL/min using 100 % water as a mobile phase and detection at 254 nm. Fractions were analyzed off-line using an Agilent Q-TOF HPLC-MS. 1-containing fractions were pooled and lyophilized to dryness. Purified compound 1 (0.028 g) was subsequently dissolved in D2O, and the stock concentration was determined by quantitative NMR using a 4,4-dimethyl-4-silapentane-1-sulfonic acid capillary normalized using calcium formate. Single-use aliquots (13.3 mM) were stored at −80 °C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two-sided t-tests were performed using Prism (v7); equal variance between samples being compared was established. Graphs show mean +/− S.E.M or +/− standard deviation as described in the figure legends.
DATA AND SOFTWARE AVAILABILITY
The software used in this study is listed in the Key Resources Table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013097. Protein identifications are included in Supplemental Table 4.
Supplementary Material
Supplemental Table 4 (Related to Figures 2 and S1) | Raw PSM counts for all replicates of PhAXA. Each bait is included as a separate tab
Supplemental Table 5 (Related to STAR Methods) | All primers used in this study for qPCR, cloning, mutagenesis and CRISPR plasmids.
SIGNIFICANCE.
The transient nature of kinase-substrate interactions has precluded the use of traditional approaches for mapping protein-protein interactions. Other methods currently in use provide little information about phosphosite specificity, which is critical for studying proteins with phosphosites that elicit differential effects on activity. Given these limitations, new tools were needed to assign kinases to specific phosphorylation events, allowing for a better understanding of the interplay between signaling networks and the identification of therapeutically relevant protein targets. Here we show that PhAXA allows for high confidence identification of kinases with phosphosite specificity. Using this assay, we identified CDK4-dependent phosphorylation of 4E-BP1, which is significant as mTORC1 is the only kinase known to phosphorylate 4E-BP1, and activation of unknown kinases that can act on 4E-BP1 has been attributed as a mechanism of drug resistance to mTORC1 inhibitors. These findings shed light on the mechanism behind the synergy between mTORC1 and CDK4/6 inhibitors, and provide rationale for the use of this combination in the clinic. To our knowledge, this work represents one of the first successful applications of such a chemical biology strategy for the unbiased discovery of a kinase-substrate interaction in a phosphosite-specific manner and provides a framework by which to deconvolute kinase signaling networks.
Highlights.
Development of a kinase-substrate crosslinking assay
A mechanism of CDK4/6 and mTORC1 inhibitor cooperativity in breast cancer cell lines
Mechanistic evaluation of an orphan 4E-BP1 phosphorylation site
Dissection of 4E-BP1 phosphorylation-dependent c-Myc translation
Acknowledgements
This work was supported by the University of Michigan College of Pharmacy (A.L.G.), the University of Michigan Rogel Cancer Center John S. and Suzanne C. Munn Cancer Fund (A.L.G.), the University of Michigan Rackham Predoctoral Fellowship (D.C.M) and the NIH (R01 CA202018 to A.L.G. and T32 CA140044 to D.C.M.). We thank Dr. Venkatesha Basrur, Professor Alexey Nesvizhskii and the University of Michigan Proteomics Resource Facility for assistance with MS experiments. We also thank Dr. Kenneth Takeuchi for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, et al. (2011). A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20, 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apsel B, Blair J,A, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, and Knight ZA (2008). Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengol G, Rojo F, Castellvi J, Iglesias C, Cuatrecasas M, Pons B, Baselga J, and Ramon y Cajal S (2007). 4E-Binding protein 1: a key molecular “funnel factor” in human cancer with clinical implications. Cancer Res 67, 7551–7555. [DOI] [PubMed] [Google Scholar]
- Beugnet A, Wang X, and Proud CG (2003). Target of rapamycin (TOR)-signaling and RAIP motifs play distinct roles in the mammalian TOR-dependent phosphorylation of initiation factor 4E-binding protein 1. J Biol Chem 278, 40717–40722. [DOI] [PubMed] [Google Scholar]
- Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, and Topisirovic I (2015). Targeting the translation machinery in cancer. Nat Rev Drug Discov 14, 261–278. [DOI] [PubMed] [Google Scholar]
- Castellvi J, Garcia A, Rojo F, Ruiz-Marcellan C, Gil A, Baselga J, and Ramon y Cajal S (2006). Phosphorylated 4E binding protein 1: a hallmark of cell signaling that correlates with survival in ovarian cancer. Cancer 107, 1801–1811. [DOI] [PubMed] [Google Scholar]
- Choi YJ, and Anders L (2014). Signaling through cyclin D-dependent kinases. Oncogene 33, 1890–1903. [DOI] [PubMed] [Google Scholar]
- Choo AY, and Blenis J (2009). Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 8, 567–572. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, and Blenis J (2008). Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci, U S A 105, 17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, and Pratt MR (2016). Chemical methods for encoding and decoding of posttranslational modifications. Cell Chem Biol 23, 86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J, Im S-A, Holgado E, Perez-Garcia JM, Schmid P, and Chavez-MacGregor M (2017). The next era of treatment for hormone receptor-positive, HER2-negative advanced breast cancer: triplet combination-based endocrine therapies. Cancer Treat Rev 61, 53–60. [DOI] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedigama-Arachchige PM, and Pflum MK (2016). K-CLASP: A Tool to Identify Phosphosite Specific Kinases and Interacting Proteins. ACS chemical biology 11, 3251–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMichele A, Clark AS, Tan KS, Heitjan DF, Gramlich K, Gallagher M, Lal P, Feldman M, Zhang P, Colameco C, et al. (2015). CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 995–1001. [DOI] [PubMed] [Google Scholar]
- Descamps G, Gomez-Bougie P, Tamburini J, Green A, Bouscary D, Maiga S, Moreau P, Le Gouill S, Pellat-Deceunynck C, and Amiot M (2012). The cap-translation inhibitor 4EGI-1 induces apoptosis in multiple myeloma through Noxa induction. Br J Cancer 106, 1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, et al. (2010). A guided tour of the Trans-Proteomic Pipeline. Proteomics 10, 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJO, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, et al. (2010). mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker GS, Atreya CE, Simko JP, Hom YK, Matli MR, Benes CH, Hann B, Nakakura EK, Bergsland EK, Donner DB, et al. (2014). Incomplete inhibition of phosphorylation of 4E-BP1 as a mechanism of primary resistance to ATP-competitive mTOR inhibitors. Oncogene 33, 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, and Hoopmann MR (2013). Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24. [DOI] [PubMed] [Google Scholar]
- Fang H, Huang D, Yang F, and Guan X (2018). Potential biomarkers of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer. Breast Cancer Res Treat 168, 287–297. [DOI] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, and Shokat KM (2009). Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PloS Biol 7, e1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermin D, Basrur V, Yocum AK, and Nesvizhskii AI (2011). Abacus: a computational tool for extracting and pre-processing spectral count data for label-free quantitative proteomic analysis. Proteomics 11, 1340–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. (2009). PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11, R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, et al. (2004). Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3, 1427–1438. [PubMed] [Google Scholar]
- Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL, and Lichtenstein AK (2004). AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression. The Journal of biological chemistry 279, 2737–2746. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, and Sonenberg N (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15, 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, Lynch RL, Dumstorf CA, Dowless MS, McNulty AM, Parsons SH, Brail LH, Colligan BM, Koop JW, et al. (2009). eIF4E activation is commonly elevated in advanced human prostate cancers and significantly related to reduced patient survival. Cancer Res 69, 3866–3873. [DOI] [PubMed] [Google Scholar]
- Holman JD, Tabb DL, and Mallick P (2014). Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr Protoc Bioinformatics 46, 13 24 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. (2012). The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, and Sabatini DM (2013). mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 341, 1236566–1236561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Knudsen ES, and Witkiewicz AK (2017). The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 3, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Lee EH, Lee JH, Kim J-E, Kim S-H, Kim TG, Hwang SW, and Kang KW (2015). Prognostic significance of phosphorylated 4E-binding protein 1 in non-small cell lung cancer. Int J Clin Exp Pathol 8, 3955–3962. [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, et al. (2013). TCPA: a resource for cancer functional proteomics data. Nature methods 10, 1046–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CJ, Cencic R, Mills JR, Robert F, and Pelletier J (2008). c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer research 68, 5326–5334. [DOI] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010a). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010b). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maly DJ, Allen JA, and Shokat KM (2004). A mechanism-based cross-linker for the identification of kinase-substrate pairs. Journal of the American Chemical Society 126, 9160–9161. [DOI] [PubMed] [Google Scholar]
- Martineau Y, Azar R, Bousquet C, and Pyronnet S (2013). Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene 32, 671–677. [DOI] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, et al. (2013). The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature methods 10, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, and Manning BD (2014). Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 156, 771–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curven J, Carnevalli L, Staniszweska A, Polanska U, Cheraghchi-Bashi A, Lawson M, et al. (2018). Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long term growth inhibition in estrogen receptor positive breast cancer. Cancer research 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- O’Leary B, Finn RS, and Turner NC (2016). Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13, 417–430. [DOI] [PubMed] [Google Scholar]
- O’Reilly KE, Warycha M, Davies MA, Rodrik V, Zhou XK, Yee H, Polsky D, Pavlick AC, Rosen N, Bhardwaj NB, et al. (2009). Phosphorylated 4E-BP1 is associated with poor survival in melanoma. Clin Cancer Res 15, 2872–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, Manigat L, Abbas T, Lee J, Nakano I, et al. (2017). Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res 23, 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parang K, Kohn JA, Saldanha SA, and Cole PA (2002). Development of photo-crosslinking reagents for protein kinase-substrate interactions. FEBS Lett 520, 156–160. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Graff J, Ruggero D, and Sonenberg N (2015). Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res 75, 250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Weichenrieder O, and Izaurralde E (2015). Molecular architecture of 4E-BP translational inhibitors bound to eIF4E. Mol Cell 57, 1074–1087. [DOI] [PubMed] [Google Scholar]
- Pikman Y, Alexe G, Roti G, Conway AS, Furman A, Lee ES, Place AE, Kim S, Saran C, Modiste R, et al. (2016). Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin Cancer Res 23, 1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin F, Gingras AC, Olsen H, Chevalier S, and Sonenberg N (1998). 4E-BP3, a new member of the eukaryotic initiation factor 4E-kinding protein family. Journal of Biological Chemistry 273, 14002–14007. [DOI] [PubMed] [Google Scholar]
- Remenyi A, Good MC, and Lim WA (2006). Docking interactions in protein kinase and phosphatase networks. Curr Opin Struct Biol 16, 676–685. [DOI] [PubMed] [Google Scholar]
- Riel-Mehan MM, and Shokat KM (2014). A crosslinker based on a tethered electrophile for mapping kinase-substrate networks. Chemistry & biology 21, 585–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo F, Najera L, Lirola J, Jimenez J, Guzman M, Sabadell MD, Baselga J, and Ramon y Cajal S (2007). 4E-binding protein 1, a cell signaling hallmark in breast cancer that correlates with pathologic grade and prognosis. Clin Cancer Res 13, 81–89. [DOI] [PubMed] [Google Scholar]
- Rong L, Livingstone M, Sukarieh R, Petroulakis E, Gingras AC, Crosby K, Smith B, Polakiewicz RD, Pelletier J, Ferraiuolo MA, et al. (2008). Control of eIF4E cellular localization by eIF4E-binding proteins, 4E-BPs. RNA 14, 1318–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio C, Martinez-Fernandez M, Segovia C, Lodewijk I, Suarez-Cabrera C, Segrelles C, Lopez-Calderon F, Munera-Maravilla E.A.-O.h.o.o., Santos M, Bernardini A.A.-O.h.o.o., et al. Cdk4/6-inhibitor as a novel therapeutic approach for advanced Bladder Cancer independently of RB1 status. LID - clincanres.0685.2018 [pii] LID - 10.1158/1078-0432.CCR-18-0685 [doi]. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EV (2004). The role of c-myc in regulation of translation initiation. Oncogene 23, 3217–3221. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C-T method. Nature protocols 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Song JM, Menon A, Mitchell DC, Johnson OT, and Garner AL (2017). High-Throughput Chemical Probing of Full-Length Protein-Protein Interactions. ACS Comb Sci 19, 763–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statsuk AV, and Shokat KM (2012). Covalent cross-linking of kinases with their corresponding peptide substrates. Methods in molecular biology 795, 179–190. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, and Sabatini DM (2012). A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, and Gray NS (2009). An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. The Journal of biological chemistry 284, 8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukumo Y, Alain T, Fonseca BD, Nadon R, and Sonenberg N (2016). Translation control during prolonged mTORC1 inhibition mediated by 4E-BP3. Nat Commun 7, 11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, et al. (2014). CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ye Q, Cao Y, Guo Y, Huang X, Mi W, Liu S, Wang C, Yang HS, Zhou BP, et al. (2017). Snail determines the therapeutic response to mTOR kinase inhibitors by transcriptional repression of 4E-BP1. Nat Commun 8, 2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W, et al. (2015). Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li W, Parra J-L, Beugnet A, and Proud CG (2003). The C terminus of initiation factor 4E-binding protein 1 contains multiple regulatory features that influence its function and phosphorylation. Mol Cell Biol 23, 1546–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Shin YS, Xue M, Matsutani T, Masui K, Yang H, Ikegami S, Gu Y, Herrmann K, Johnson D, et al. (2016). Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell 29, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel H-G, de Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, and Lowe SW (2004). Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428, 332–337. [DOI] [PubMed] [Google Scholar]
- Yu YH, Yoon SO, Poulogiannis G, Yang Q, Ma XJM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. (2011). Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate That Negatively Regulates Insulin Signaling. Science 332, 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Zheng XFS (2012). mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle 11, 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 4 (Related to Figures 2 and S1) | Raw PSM counts for all replicates of PhAXA. Each bait is included as a separate tab
Supplemental Table 5 (Related to STAR Methods) | All primers used in this study for qPCR, cloning, mutagenesis and CRISPR plasmids.
Data Availability Statement
The software used in this study is listed in the Key Resources Table. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013097. Protein identifications are included in Supplemental Table 4.