

Summary
Allosteric regulation of methylmalonyl-CoA mutase (MCM) by the G-protein chaperone CblA is transduced via three ‘switch’ elements that gate the movement of the B12 cofactor to and from MCM. Mutations in CblA and MCM cause hereditary methylmalonic aciduria. Unlike the bacterial orthologs used previously to model disease-causing mutations, human MCM and CblA exhibit a complex pattern of regulation that involves interconverting oligomers, which are differentially sensitive to the presence of GTP versus GDP. Patient mutations in the switch III region of CblA perturb the nucleotide-sensitive distribution of the oligomeric complexes with MCM, leading to loss of regulated movement of B12 to and/or from MCM and explain the molecular mechanism of the resulting disease.
Keywords: cobalamin, GTPase, cofactor, trafficking, vitamin B12, cblA, MMAA, G-protein
Graphical Abstract
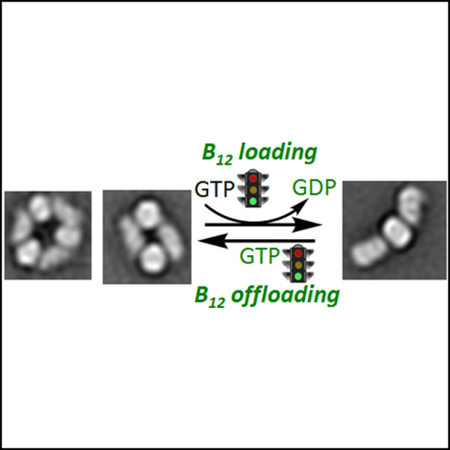
eTOC Blurb
Nucleotide-sensitive changes in oligomeric complexes between B12-dependent methylmalonyl-CoA mutase and the trafficking GTPase CblA, distinguish them from their bacterial orthologs. Mutations in a signaling element in CblA impair the regulated distribution of these complexes, impact B12 loading/off-loading function and lead to disease.
Introduction
An elaborate intracellular pathway sequesters, assimilates and escorts vitamin B12 from its point of entry into the cell to its two client proteins: cytoplasmic methionine synthase (MS) and mitochondrial methylmalonyl-CoA mutase (MCM) (Banerjee et al., 2009; Gherasim et al., 2013; Watkins and Rosenblatt, 2011). Insights into the mitochondrial B12 trafficking pathway, which supports MCM function, have emerged primarily from studies on bacterial orthologs of the human chaperones, adenosyltransferase (ATR, also known as MMAB) and the G- protein CblA (also known as MMAA). Mutations in ATR, CblA or MCM lead to methylmalonic aciduria, an autosomal recessive disorder (Dobson et al., 2002a; Dobson et al., 2002b; Ledley et al., 1988). In contrast, insights into B12 delivery to methionine synthase are relatively limited (Gulati et al., 1997; Yamada et al., 2006).
MCM utilizes 5’-deoxyadenosylcobalamin (AdoCbl) to initiate radical chemistry during the 1,2 rearrangement of methylmalonyl-CoA to succinyl-CoA (Banerjee, 2003). AdoCbl is synthesized by ATR in a reaction in which cob(I)alamin is adenosylated by ATP forming AdoCbl and PPPi (Leal et al., 2004; Yamanishi et al., 2005) (Fig. 1A). The newly formed AdoCbl product is then directly transferred from ATR to MCM in a process that is gated by the GTPase activity of CblA (Padovani et al., 2008). Detailed structural enzymology insights into the mitochondrial B12 pathway have emerged primarily from our studies on the homologous proteins in Methylobacterium extorquens (Campanello et al., 2017; Lofgren et al., 2013a; Lofgren et al., 2013b; Padovani and Banerjee, 2006b, 2009a, b; Padovani et al., 2006; Padovani et al., 2008) and on IcmF, a fusion between a sister isobuyryl-CoA mutase and its G-protein chaperone (Cracan and Banerjee, 2012a, b; Cracan et al., 2010; Jost et al., 2015a; Jost et al., 2015b; Li et al., 2017).
Figure 1. Chaperone functions and structure of human CblA.

(A) AdoCbl is loaded onto MCM from ATR in a process gated by the GTPase activity of CblA. MCM has GAP function and enhances the GTPase activity of CblA when the two proteins form a complex. Holo- MCM catalyzes the isomerization of methylmalonyl-CoA (M-CoA) to succinyl-CoA (S-CoA). When MCM is inactivated, the GTPase activity of CblA is used to off-load cob(II)alamin to the ATR active site. ATR completes the repairs process, reforming AdoCbl. (B) Cartoon showing differences between human and M. extorquens MCM and CblA and the M extorquens M2C1 complex that was previously characterized. (C) Structure of human CblA (PDB: 2WWW) with GDP bound. Switch I (green), II (yellow) and III (orange) loops are highlighted. Dashed lines represent disordered regions in switch I and III loops. (D) Since switch III residues 268–274 in human CblA are disordered, a close-up of the homologous region in MeMeaB (PDB: 4YJB) is shown with the location of residues (human numbering) mutated in this study.
The M extorquens ortholog of CblA (known as MeaB but hereafter referred to as MeCblA) is a multifunctional chaperone. It gates AdoCbl transfer from ATR to MCM, protects MCM from inactivation, and off-loads inactive cob(II)alamin from MCM to ATR for repair (Padovani and Banerjee, 2006b, 2009a). MeCblA exhibits low intrinsic GTPase activity that is enhanced ~100-fold by MCM, which binds to it with high affinity (Padovani et al., 2006). As a member of the P-loop GTPases (Leipe et al., 2002), CblA communicates with MCM via conformationally mobile switch I and II loops that classically function as loaded springs for signaling via nucleotide binding- and hydrolysis-responsive conformational changes (Wittinghofer and Vetter, 2011). While the conserved residues in the switch I and II loops in MeCblA are not essential for GTP hydrolysis, they are important for allosteric signaling to and from MCM and for suppressing its intrinsic GTPase activity (Campanello et al., 2017; Lofgren et al., 2013a).
Multiple crystal structures of MeCblA with and without various ligands captured a third conformationally plastic switch III loop, in various nucleotide-sensitive poses (Hubbard et al., 2007; Lofgren et al., 2013a; Lofgren et al., 2013b). Mutations in switch III disrupt bidirectional communication, muting the GTPase-activating protein (GAP) activity of MCM and disrupting the varied chaperone functions of MeCblA (Padovani and Banerjee, 2009a). While the switch I, II and III elements of MeCblA are conserved in human CblA, biochemical studies on the human CblA-MCM complex and on the range of CblA chaperone functions have been very limited to dat (Takahashi-Iniguez et al., 2011). Unfortunately, the switch I and III regions are disordered in the crystal structure of human CblA (Froese et al., 2010).
Despite their ~50% sequence identity, the human and bacterial MCMs and G proteins, exhibit significant topological differences (Fig. 1B), limiting the utility of the bacterial orthologs as models of the human proteins. While both bacterial and human CblAs are α2 homodimers (Froese et al., 2010; Hubbard et al., 2007), their interfaces are distinct and result in the nucleotide binding sites residing on the same (human Fig. 1C) or opposite (bacterial) faces at distances of 20 Å and 50 Å, respectively. Furthermore, while human MCM is an α2 homodimer (Froese et al., 2010) and each of the two B12-binding subunits can potentially interact with one CblA, MeMCM is an α/β-heterodimer and binds one AdoCbl. The proteins form a 2MeMCM-1MeCblA complex (hereafter referred to as the M2C1 complex, Fig. 1B) as visualized by negative stain EM analysis (Campanello et al., 2017). IcmF exhibits yet another architecture in which the G-domains reside at opposite ends of the α2 IcmF dimer while the mutase domains build the interface (Jost et al., 2015a; Jost et al., 2015b).
Herein, we report that human MCM-CblA exhibits complex nucleotide-sensitive interconversion between oligomeric states, which were not observed with the homologous M extorquens proteins as visualized by EM. The pathogenic (G274S, K276E and G278D) (Dempsey-Nunez et al., 2012; Girisha et al., 2012) and nonpathogenic (Q273A and K276A) mutations in the switch III region of CblA (Fig. 1D) perturb the nucleotide-regulated distribution of oligomers and cause pleiotropic chaperoning deficits. CblA mutations are correlated with a significantly lower AdoCbl pool size in patient fibroblasts compared to controls. These functional perturbations explain the molecular basis of the methylmalonic aciduria associated with the pathogenic mutations.
Results
Switch III mutations disrupt nucleotide-sensitive equilibration of MCM-CblA complexes
In the M2C1 complex formed between the M. extorquens MCM and CblA (Campanello et al., 2017), each GTP binding site in α2 MeCblA is predicted to interact with the single B12 binding site in αβ MCM (Fig. 1B). In contrast, the human protein complex exists in multiple oligomeric states (Fig. 2, Table 1). In the presence of the nonhydrolyzable GTP analog, GMPPNP, wild-type, Q273A and G274S CblA, exist predominantly in higher order oligomeric complexes while the M2C1 complex is a minor species (Fig. 2A, Fig. 3A,B). In contrast, in the presence of K276A/E and G278D CblA, the M2C1 population is larger and the higher order oligomers are proportionately smaller (Fig 3C–E).
Figure 2. Oligomeric states of MCM-wild-type CblA complexes.

(A) Distribution of complexes formed upon mixing MCM (25 μM) and CblA A (50 μM) in the presence of GMPPNP or GDP (500 μM each). Positions of the calibration standards are indicated in the light gray trace. (B) SEC-MALS analysis of a mixture of MCM (25 μM) and wild-type CblA (25 μM) in the presence of GMPPNP (250 μM). The black trace represents the normalized refractive index (left axis). The red line indicates the molecular weight as detected by MALS (right axis). (C) 2D class averages of the 395 kDa peak shows heterogeneity of oligomeric forms. The orange and blue arrows denote CblA and MCM, respectively.
Table 1.
Summary of comparative data on wild-type versus mutant CblAs.1
| Wild type CblA |
Q273A CblA |
G274S CblA |
K276A CblA |
K276E CblA |
G278D CblA | |
|---|---|---|---|---|---|---|
| Complex distribution + GMPPNP | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 |
| Complex distribution + GDP2 | M2C1, free MCM | M2C1, free MCM | Higher order oligomers, M2C1, free MCM | M2C1, M1C1, free MCM | M2C1, free MCM | free MCM |
| Complex distribution + GTP, after 30 min2 | M2C1, M1C1, free MCM | Higher order oligomers, M2C1 | Higher order oligomers, M2C1 | M2C1, M2C1 free MCM | Higher order oligomers, M2C1 free MCM | M2C1, freeMCM |
| 2GTPaseactivity (min−1) | 0.06 ± 0.04 | 0.03 ± 0.02 | 0.03 ± 0.02 | 0.04 ± 0.02 | 0.03 ± 0.02 | 0.06 ± 0.04 |
| 2GTPase activity + MCM (min−1) | 2.78 ± 0.41 | 0.86 ± 0.22 | 1.04 ± 0.16 | 2.98 ± 0.17 | 0.92 ± 0.09 | 3.07 ± 0.11 |
| 2Kact [μM] | 0.3 ± 0.1 | 10.0 ± 0.4 | 1.7± 0.3 | 5.0 ± 0.3 | 3.8 ± 0.2 | 3.9 ± 0.3 |
| AdoCbl transfer from ATR to MCM + GDP | complete | complete | partial | complete | complete | complete |
| AdoCbl transfer from ATR to MCM + GMPPCP | blocked | blocked | blocked | partial | partial | partial |
| Cob(II)alamin transfer from MCM to ATR + GTP | complete | impaired | almost complete | impaired | impaired | impaired |
Differences between wild-type and mutant CblA are highlighted in italics; the dominant oligomeric states are highlighted in bold lettering. 2Kact denotes the concentration of MCM needed for half maximal GTPase activity of CblA. The data represent the mean ± SD of three independent experiments.
Figure 3.

Oligomeric distribution of MCM-CblA complexes in the presence of GMPPNP. MCM (25 μM) and wild-type or mutant CblA (50 μM each) were incubated with GMPPNP (500 μM) for 4 min at 4°C. The black trace in each panel corresponds to the elution profile with wild-type CblA and the red trace with the following CblA mutants: (A) Q273A (B) G274S (C) K276A, (D) K276E, and (E) G278D. The M2C1 complex is simply denoted as 2:1 in the panels. The data are representative of two independent experiments.
GTP hydrolysis mimicked by the presence of GDP, leads to resolution of the higher order oligomers to a mixture of M2C1 and the individual proteins as seen with wild-type, Q273A, and K276E CblA (Figs. 2A, Fig. 4A,D). The K276A mutation exhibits a similar profile, albeit the concentration of the M2C1 complex is lower while the shoulder on the free MCM peak suggests the presence of an M1C1 complex. (i.e. a 1:1 complex of MCM and CblA). With G274S CblA and GDP, the M2C1 complex is also seen, although a sizeable fraction of the higher order oligomers remains and only a small proportion of MCM is free (Fig 4B). The profile of MCM-G274S CblA is, in fact, similar to those with K276A/E and G278D CblA in the presence of GMPPNP (Fig 3 C–E), and indicates that the G274S mutation impairs GTPase- driven resolution of the higher order oligomers. With G278D CblA, only the free proteins were seen (Fig 4E), indicating that this mutation destabilizes the M2C1 complex following GTP hydrolysis.
Figure 4.

Oligomeric distribution of MCM-CblA complexes in the presence of GDP. MCM (25 μM) and wild-type or mutant CblA (50 μM each) were incubated with GDP (500 μM) for 4 min at 4°C. In each panel, the black trace corresponds to the elution profile with wild-type CblA and the red trace in the presence of the following CblA mutant: (A) Q273A, (B) G274S, (C) K276A, (D) K276E, and (E) G278D. The data are representative of two independent experiments.
GAP activation by MCM enhances the GTPase activity of CblA ~50-fold with wild-type, K276A and G278D CblA (Table 1). As expected in the presence of GTP, the distribution of the MCM-CblA complexes changed with time. With wild-type and K276A CblA, peaks corresponding to M2C1, M1C1 complexes and the free proteins were observed as GTP is hydrolyzed and the profiles progress to the distribution seen in the presence of GDP (Fig. S1A,D). With G278D CblA, the M1C1 complex and the free proteins were seen, consistent with destabilization of MCM-CblA complexes in the presence of GDP (Fig S1F). The lower (~30-fold) GAP activation of the remaining mutants led to a higher proportion of the large oligomeric complexes (Fig S1 B,C,E) that were most enriched with G274S CblA, which exhibits impaired GTPase-regulated switching. These profiles did not change substantially upon prolonged incubation (30 min). The largest number of oligomeric species was resolved with K276E CblA.
Structural heterogeneity of MCM-CblA complexes
Size exclusion chromatography with multi-angle light scattering detection (SEC-MALS) of a mixture of MCM and CblA in the presence of GMPPNP showed a peak with an average molecular mass of 395 kDa, in good agreement with the predicted mass of 414 kDa for the M2C1 complex, and was initially selected for EM studies (Fig. 2B). 2D class averages of negative stain EM images of this peak revealed heterogeneity of oligomeric forms (Fig. 2C). The predominant species was the linear M2C1 complex, which displays considerable conformational flexibility. In addition, annular M2C2 and M3C3 complexes were observed (Fig. 2C).
Next, three fractions collected across the higher order oligomeric peak seen in the presence of GMPPCP (Fig. 5A), were analyzed by negative stain EM. In fractions 2 and 3, the dominant complexes were M4C4 and M3C3 (Fig. 5B, Fig S2 A,B). In fraction 1, linear fibrils and aggregates were observed in addition to circular complexes (Fig. S2C). In comparison to wild-type CblA, the K276E and G274S mutants formed a higher proportion of fibrils and protein aggregates, respectively (Fig. S2C). Consequently, the number of circular complexes in fraction 3 was reduced ~2-fold with K276E and G274S CblA compared to the wild-type protein (Fig. 5C).
Figure 5. Switch III mutations affect MCM-CblA oligomeric forms.

(A) MCM (25 μM) was mixed with wild-type (50 μM, black line), G274S (red line) or K276E CblA (blue line) CblA and GMPPCP (500 μM) and separated by analytical gel filtration. The fractions indicated by the numbers 1–3 (at 8.2, 9.0 and 9.8 ml) were diluted to a 10 μg/ml protein concentration and transferred onto the EM grid. (B) Selected reference-free 2D class averages showing the circular organization of MCM-wild-type CblA•GMPPCP complexes in fractions 2 and 3. Box length, 50.4 nm. The number of particles in each average shown in right corner. The lower resolution of the class averages in fraction 2 is due to the exhibit higher flexibility of the particles. (C) The number of circular particles averaged for 100 negative stained images for each MCM-CblA complex (fraction 3).
AdoCbl loading from ATR to MCM
Large spectroscopic differences between 5-coordinate base-off (ATR, 456 nm) and 6- coordinate base-on (MCM, 528 nm) AdoCbl allow ready distinction between the cofactor bound in their respective active sites. In the presence of GDP, AdoCbl transferred completely from ATR to MCM (Fig. 6A) while GMPPCP blocked the transfer (Fig. 6A, inset). GMPPCP also blocked cofactor transfer in the presence of the Q273A and G274S mutants, while GDP supported either full (Q273A) or partial (G274S) AdoCbl transfer (Fig. S3, Table 1). These data are consistent with full resolution of the higher order oligomeric complexes in the presence of GDP with wild-type and Q273A CblA but only partial resolution with the G274S mutant (Fig. 4).
Figure 6. Model for AdoCbl loading and offloading in human.

(A) Transfer of AdoCbl (15 μM) from ATR (15 μM trimer) to MCM (15 μM) in the presence of wild-type CblA (45 μM) and 1 mM GDP or GMPPCP (inset). The initial and final spectra (30 min) are in black and red, respectively. (B) Transfer of cob(II)alamin (15 μM) bound to MCM (9 μM, black trace) in the presence of CblA (30 μM) following addition of ATR (15 μM), ATP (5 mM) and 1 mM GTP or GMPPCP (inset). The final spectrum after 15 min is in red. (C) Model for cofactor loading/off-loading highlighting a central role for the GTPase activity of CblA. Resolution of the higher order oligomers (left) to a “loading ready” M2C1 complex is driven by GTP hydrolysis, which following AdoCbl transfer separates into the individual proteins. During catalysis, occasional inactivation of MCM leads to the loss of deoxyadenosine and accumulation of cob(II)alamin, which is off-loaded in the presence of CblA in a GTPase- dependent step. For simplicity, the offloading complex is shown as M2C1 although it is currently unclear whether it can also occur from the M1C1 or the higher order complexes.
In the remaining switch III mutants, a loss of the GTPase-gated AdoCbl transfer function was observed (Fig. S4, Table 1). The behavior of these mutants is correlated with the presence of a sizeable population of the M2C1 complex. Collectively, these data indicate that the annular complexes are not “loading-ready” and that their GTPase-driven resolution to the M2C1 complex is needed to load MCM with AdoCbl.
Switch III CblA mutations affect GAP activity of MCM
GDP binding to CblA (KD = 1.1 ± 0.3 μM) is enthalpically driven and is largely unaffected by switch III mutations (Table S1). The kcat for GTPase in wild-type CblA is unaffected by the presence of the cofactor (0.06 ±0.04 min−1 ± AdoCbl); however, the Kact for MCM increases 17-fold from 0.3 ± 1 μM in the absence to 5.0 ± 0.8 μM in the presence of AdoCbl, which could indicate weakening of the MCM-CblA complex once MCM is loaded with AdoCbl. The intrinsic GTPase activity of CblA, which is low, was unaffected by the switch III mutations (Table 1). GAP activation was reduced 1.5–2-fold with Q273A, G274S and K276E CblA but was unaffected with K276A and G278D CblA (Table 1). The Kact for MCM increased 6- to 33-fold in the CblA mutants.
Switch III CblA mutations affect MCM repair
Loss of the 5’-deoxyadenosine moiety results in the inability to form AdoCbl at the end of the MCM catalytic cycle and triggers the repair cycle (Fig. 1A) (Padovani and Banerjee, 2009a). Repair of inactive cob(II)alamin-containing MCM is achieved by cofactor offloading to ATR in a process gated by CblA. The M extorquens system uses the binding energy of GTP to facilitate transfer of cob(II)alamin from MeMCM to MeATR. Based on the relative affinities for cob(II)alamin binding to human MCM (KD ≥5 μM) versus ATR (0.08±0.01 μM) (Campanello et al., 2018), the reverse transfer of cob(II)alamin from MCM to ATR is expected to be thermodynamically favored. Binding of cob(II)alamin to MCM induces small changes in the absorption spectrum (Fig. S5A). The EPR spectrum of MCM-bound cob(II)alamin is characteristic of the base-on species showing hyperfine coupling to an I=7/2 nucleus. The superhyperfine triplet splittings in the high field region of the spectrum are indicative of coordination to an axial nitrogen (I=1) ligand (Fig. S5B). While the g values (2.24, 2.21 and 2.00) for MCM-bound and free cob(II)alamin are similar, they are distinct from base-off cob(II)alamin bound to ATR (Fig. S5B, lower panel). These differences in the EPR spectra confirm the presence of 5- versus 4-coordinate cob(II)alamin bound to MCM versus ATR, respectively.
Translocation of cob(II)alamin from MCM (472 nm) to ATR (464 nm) is accompanied by an increase in intensity and a blue shift in the absorption spectrum (Fig. 6B). In the presence of CblA, GTP and ATP, complete transfer of cob(II)alamin from MCM to ATR was seen, but blocked in the presence of GDP or GMPPCP. The switch III mutants showed various degrees of impairment in cofactor offloading with the G274S mutant being very similar to wild-type CblA (Fig. S5 C–G, Table 1).
Patient mutations in CblA decrease AdoCbl pool size
To assess the effect of CblA mutations on the mitochondrial B12 trafficking pathway, the size of the AdoCbl pool relative to the total B12 pool was determined. For this, B12 levels were determined in fibroblasts from control versus cblA patients. AdoCbl represents 15.1 ± 3.4% of the total B12 pool in control human fibroblasts (n=192), but is reduced to 3.3±1.6 % of the total pool in fibroblasts from cblA patients (n=72, p<0.001).
Discussion
Allostery, described by the concerted (Monod et al., 1965) and sequential (Koshland et al., 1966) models in the 1960s, is a fundamentally important mechanism of biological regulation. The current view of allostery has however, shifted from the classical two-state models to accommodate biomolecular dynamics and the existence of interconverting conformational ensembles in which ligand binding shifts the equilibrium between conformational sub-states (Hilser et al., 2012). In this study, we report a ligand-induced shift in heteromeric populations of MCM and its G-protein chaperone CblA, and the corruption of this process by pathogenic mutations housed within a signaling element within CblA.
Human CblA and MCM associate with variable stoichiometry ranging from 1:1 (e.g. M2C2 to M3C3) in the presence of the GTP analogs, GMPPN(C)P to a mixture of 2:1 and free proteins in the presence of GDP (Fig 6C). In comparison, the M. extorquens orthologs exist only in an M2C1 complex (Fig. 1B) with affinities between the two proteins ranging between 34–524 nM depending on the absence or presence of ligands in the two active sites (i.e. GMPPNP and AdoCbl) (Padovani et al., 2006). Characterization of the energetics of interaction between human MCM and CblA with different ligands was precluded by the heterogeneity of the resulting complexes and the instability of CblA upon prolonged exposure to GMPPN(C)P. However, unlike MeCblA, which exhibits similar affinities for GDP and GTP (1.3 and 6.6 μM respectively (Padovani et al., 2006)), human CblA exhibits a higher affinity for GDP (Kd = 1.1±0.3 μM) than for GTP (Km =150±4 μM in the presence of MCM assuming Kd ≈ Km). AdoCbl-loading onto MCM following GTP hydrolysis promotes dissociation of MCM from CblA as indicated by the gel filtration profile (Fig. 2A) and suggested by the 15-fold increase in the Kact for GAP activation of CblA by AdoCbl-loaded MCM. These data lead to the prediction that the catalytically active form of MCM is free (Fig 6C) while apo-MCM is associated with CblA in “holding” complexes of varied oligomeric composition. Free CblA on the other hand, should be predominantly GDP-bound given intracellular GTP (~470 μM) versus GDP (160 μM) concentrations in mammalian tissues (Traut, 1994) and the Kd values of free CblA for GDP (1.1±0.3 μM) versus GTP (Km =740± 90 μM in the absence of MCM, assuming Kd ≈ Km).
In our model, GTP hydrolysis energy is needed to form the “loading-ready” M2C1 complex for AdoCbl transfer from ATR (Fig. 6C). In the bacterial orthologs, the M2C1 complex exists independently of the presence or type of nucleotide bound to MeCblA and the complex remains intact even after GTP hydrolysis triggers AdoCbl loading onto MCM (Padovani and Banerjee, 2009a). In contrast, when human MCM is loaded with AdoCbl, it dissociates from CblA and catalyzes multiple rounds of radical-dependent isomerization reactions. With the bacterial orthologs we have shown that the repair cycle is triggered when deoxyadenosine is accidently lost from MCM during catalytic turnover and leads to the transfer of cob(II)alamin from MCM to ATR (Padovani and Banerjee, 2009a). In contrast to the bacterial system where GTP binding energy is utilized for cofactor offloading, GTP hydrolysis is needed to offload cob(II)alamin from human MCM to ATR. Following AdoCbl synthesis, MCM is reloaded from ATR, completing the repair cycle. While the nature of the active off-loading complex in human is unclear, the GTP requirement indicates that reformation of the heteromeric complexes and perhaps minimally, M2C1, is needed. Intriguingly, the higher repair efficiency of the G274S followed by Q273A CblA relative to the other mutants (Fig. S5 C–G), parallels their ability to stabilize higher order oligomers in the presence of GTP (Table 1), and suggests that the annular forms are needed for cofactor offloading from MCM.
The model raises obvious questions as to why the human proteins form higher order oligomers and what role they serve. In addition to a potential role in cofactor offloading, we speculate that sequestering MCM in the loading-blocked M3C3/M2C2 complexes, might serve a regulatory role for prioritizing B12, which is present at low concentrations in mammalian cells (Hsu et al., 1966), for the cytoplasmic pathway. Cytoplasmic methionine synthase is an essential enzyme that is found in a predominantly B12-loaded state while MCM serves an anaplerotic role and exhibits fractional B12 saturation (~25%) in liver (Chen et al., 1995; Kennedy et al., 1990). In contrast, in M extorquens, which can produce vitamin B12 (Ivanova et al., 2006), such regulation might not be needed. In our model, the GTPase activity of CblA would however, result in constant cycling between the oligomeric states and wasteful GTP consumption, requiring an additional mechanism for controlling MCM-CblA complex dynamics. The identity of such a control mechanism if it exists, is not known. The pathogenic G274S mutation stabilizes the higher order oligomers in the presence of GDP and impairs AdoCbl loading. The same mutation however, has the mildest impact on cob(II)alamin off-loading. The GAP-dependent GTPase activity of G274S CblA while lower than wild-type CblA, is comparable to that of other mutants (e.g. Q273A, K276E), which do not stabilize higher order oligomers. Hence, the defect in the G274S mutation is not reduced GTPase activity per se but rather, impaired nucleotide-sensitive signaling, which in turn regulates oligomer dynamics.
Interestingly, the pathogenic K276E and G278D mutations exhibit uncoupling between nucleotide-sensitive signaling and chaperone function albeit with opposite effects compared to the G274S mutation. Both mutants partially bypass the block on AdoCbl loading in the presence of GMPPCP, presumably due to the higher abundance of the M2C1 complex seen in the presence of the nonhydrolyzable GTP analog (Table 1). MCM-dependent GTPase activation in the G278D mutant is similar to wild-type CblA and 3-fold lower in the K276E mutant. However, while the G278D mutation leads predominantly to the resolution of oligomers into individual proteins in the presence of GDP, the K276E mutant exhibits a distribution that is similar to wild-type CblA (i.e. a mix of free proteins and the M2C1 complex). Neither mutation supports MCM repair. Hence, the switch III mutations have pleiotropic effects on the stability of MCM-CblA complexes and on the regulation of bidirectional cofactor transfer.
We have recently described a strategy for B12 conservation in the mitochondrial trafficking pathway in which ATR catalyzes a sacrificial cobalt-carbon bond homolysis if it is unable to transfer AdoCbl to MCM (Campanello et al., 2018). This strategy potentially averts the release of AdoCbl into solution as the resulting cob(II)alamin remains tightly bound to ATR. CblA mutations are predicted to lead to accumulation of inactive MCM by impairing cofactor off-loading. ATR, which is mitochondrial, presumably receives cob(II)alamin via transport from the cytoplasm or from inactivated MCM. We speculate that impaired AdoCbl loading from ATR to MCM or cob(II)alamin offloading from MCM to ATR in cblA cell lines would increase the probability of sacrificial cobalt-carbon bond homolysis on ATR, thereby decreasing the cellular AdoCbl pool size (Fig. S6). Indeed, the AdoCbl pool is significantly lower in fibroblasts carrying cblA mutations compared to controls. Surprisingly, the magnitude of diminution in cblA fibroblasts is comparable to that in cblB fibroblasts carrying mutations in ATR (3.0±2.1, n=45) (Campanello et al., 2018), which can directly impact AdoCbl synthesis.
Dynamic changes in quaternary states are known to regulate function in other proteins like ribonucleotide reductase (Ando et al., 2011) and porphobilinogen synthase (Breinig et al., 2003). However, unlike the MCM-CblA complex, the oligomer state changes in these cases involve single proteins with either one or two types of subunits. In porphobilinogen synthase, allosteric Mg2+ causes switching between the less active α6 and the more active α8 form. In class Ia ribonucleotide reductase, the allosteric inhibitor dATP induces reorganization of the active α2β2 to an inactive α4β4 complex that is ring shaped (Ando et al., 2011). The increased distance between the tyrosine and cysteine residues that represent the storage and working radical sites, respectively, preclude radical transfer between them in the inhibited α4β4 structure.
While our early studies relied on the more readily available bacterial homologs of B12 trafficking proteins, the current study makes clear that significant differences in their form, inform differences in their function (Padovani and Banerjee, 2006a, b, 2009a, b; Padovani et al., 2006; Padovani et al., 2008). Thus, complex oligomer dynamics of the human MCM-CblA complex regulate the availability of M2C1, which we assign as the “loading ready” complex. Repair of cob(II)alamin bound to MCM in turn, appears to rely on reformation of oligomeric complexes. While additional insights into the regulated movement of the rare and reactive B12 cofactor await further studies, characterization of pathogenic mutations in the key switch III signaling element in CblA demonstrates how this process can be corrupted leading to disease.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ruma Banerjee (rbanerje@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Control and cblA fibroblasts were obtained from the Mutant Human Cell Strains Repository (McGill University Health Centre). Information about the sex of the cell line donors was not provided.
METHOD DETAILS
Materials
GDP, GTP, GMPPCP, GMPPNP, ATP, AdoCbl were from Sigma Aldrich, HEPES and HPLC grade methanol and acetonitrile were purchased from Fisher Scientific.
Construction and purification of human CblA mutants
Mutations were introduced using the Quickchange kit and their presence was confirmed by nucleotide sequencing (University of Michigan DNA Sequencing Core). Wild-type and mutant CblAs were expressed in E. coli BL21(DE3). Starting cultures were grown overnight in lysogeny broth (LB) medium containing 50 μg/ml kanamycin at 37°C. The initial culture (1 ml) was transferred to 1 liter of terrific broth containing 50 μg/ml kanamycin and grown at 37°C until the OD 600 reached 1.0–1.2. Protein expression was induced with 500 μM isopropyl-β-D-1-thiogalacto- pyranoside and the culture was grown for an additional 18–20 h at 37°C. The cells were harvested by centrifugation and stored at −80°C until further use. The cell pellet was suspended (5 ml/g wet weight) in lysis buffer (50 mM Tris, 500 mM NaCl, 10 mM imidazole and 5 % glycerol pH 8.0) containing 100 μM PMSF and stirred for 30 min at 4°C. Following sonication, the cell debris was removed by centrifugation at 38,500 × g for 45 min at 4°C. The supernatant was loaded onto a Ni-NTA column (5 × 2.5 cm) and washed with 500 ml lysis buffer. CblA was eluted from the column with linear (0–200 mM) imidazole gradient. Fractions containing CblA were pooled and concentrated to ~20 ml with Amicon filters (30 kDa MWCO). TEV protease (0.02 mg TEV/mg protein) was added and the solution was dialyzed against buffer containing 50 M Tris, 300 mM NaCl, 1 mM DTT 0.5 mM EDTA and 5% glycerol pH 8.0. The His tag-cleaved CblA was purified on a second Ni-NTA column (5 × 2.5 cm). CblA was collected in the flow through and concentrated using an Amicon filter (30 kDa MWCO) to ~ 20 ml and dialyzed against Buffer A (50 mM HEPES pH 7.5, 150 mM KCl, 2 mM MgCl2, 2 mM TCEP, 5% glycerol). CblA was further purified by size exclusion chromatography on a 1.6 × 60 cm Hiload Superdex 200 column (GE Healthcare) pre-equilibrated with Buffer A) Fractions containing CblA were concentrated and flash frozen in liquid nitrogen.
Purification of human MCM
Human MCM was expressed in E. coli BL21(DE3). A starting culture was grown overnight in LB medium containing 50 μg/ml kanamycin at 37°C. Then, 1 liter of terrific broth was inoculated with 1 ml of the starting culture and grown at 37°C until OD 600 reached 1.0–1.2 at which point the temperature was reduced to 16°C and 30 ml DMSO was added (3% final concentration). After 1 h, 500 μM IPTG was added to induce protein expression and the culture was grown for an additional 20–24 h. The cells were harvested by centrifugation and stored at −80°C.
The cell pellet was suspended (5 ml/g wet cell pellet) in lysis buffer (described above) containing 100 μM phenylmethane sulfonyl fluoride and stirred for 30 min at 4°C. The suspension was sonicated, and the supernatant purified using a Ni-NTA column (6 × 2.5 cm) as described for CblA. Fractions containing MCM were pooled and concentrated to ~20 ml with Amicon filters (50 kDa MWCO). TEV protease (0.03 mg TEV/mg protein) was added to the solution and dialyzed against 50 mM Tris, 300 mM NaCl, 1 mM DTT 0.5 mM EDTA and 5% glycerol pH 8.0). His-tag cleaved MCM was purified by loading onto a second Ni-NTA column (6 × 2.5 cm). MCM, present in the flow through, was concentrated using an Amicon filter (50 kDa MWCO) to ~20 mL and dialyzed against 50 mM HEPES pH 7.3, 25 mM NaCl, 5% glycerol). MCM was purified by ion exchange chromatography on a 2.5 × 10 cm Source Q column (Omnifit) and eluted with 50 mM HEPES, pH 7.3 and 5% glycerol containing either 25 mM (Buffer 1) or 500 mM NaCl (Buffer 2). MCM was eluted with a linear gradient ranging from 0 – 50 % Buffer 2 in 20 min at a flow rate of 4 ml/min. Fractions containing MCM were concentrated and dialyzed overnight against Buffer A. Aliquots were flash frozen in liquid nitrogen.
AdoCbl transfer from ATR to MCM
The transfer of AdoCbl from ATR to MCM in the presence of wild-type or mutant CblA was monitored by UV/visible spectroscopy at 25 °C in Buffer A. The MCM-CblA complex was prepared by mixing 100 μM MCM (dimer) with 300 μM CblA (dimer). GMPPCP or GDP was added to a final concentration of 6.67 mM. ATR (15 μM trimer)) was loaded with AdoCbl (15 μM) and mixed with the MCM-CblA complex. The final concentration of reactants was: 15 μM MCM, 45 μM CblA and 1 mM GMPPCP or GDP in 200 μl reaction mixture. Absorption spectra were recorded every minute between 250–700 nm for 30 min. The extent of AdoCbl transfer to MCM was estimated by using Δε525 nm of 7,500 M−1cm−1. The change in absorbance at 525 nm was fitted to a single exponential equation below where k represents the rate constant,
A represents the amplitude change and y0 is the final absorbance. After 30 min, the reaction mixture was filtered through a Nanosep filtration device (10 kDa MWCO). The concentration of free AdoCbl in the filtrate was estimated using an ε 527nm = 8,000 M−1 cm−1.
Cob(II)alamin transfer from MCM to ATR
A stock solution of cob(II)alamin was prepared by photolysis of AdoCbl under anaerobic conditions. The transfer of cob(II)alamin bound to MCM to ATR in the presence of CblA was carried out in an anaerobic chamber (O2 <0.3 ppm). Cofactor transfer was followed by UV/visible spectroscopy in Buffer A. For this, MCM (9 μM) was mixed with 15 μM cob(II)alamin and 30 μM CblA (wild-type or mutant) was added. Transfer was initiated by addition of a premixed mixture of 150 μM ATR, 50 mM ATP and 10 mM GTP. The final concentration was 15 μM ATR, 5 mM ATP and 1 mM GTP. Spectra were recorded every 30 sec for 15 min. The amount of cob(II)alamin transferred was estimated using Δε465nm = 11,000 M−1cm−1. At the end of the reaction, titanium (III) citrate was added to reduce cob(II)alamin to cob(I)alamin, to trigger ATR-catalyzed synthesis of AdoCbl. After 5 min incubation, the proteins were precipitated with trifluoroacetic acid (1% final concentration) and removed by centrifugation at 15870 × g for 5 min. AdoCbl present in the supernatant was analyzed by HPLC using a Phenomenex Luna C-18-2 column. AdoCbl was eluted using Buffer 1 containing 0.1% TFA in H2O and Buffer 2 containing 0.1% TFA in acetonitrile. Elution was achieved using the following gradient: 0–2 min: 100% Buffer 1 isocratic; 2 – 20 min: 0 – 40% Buffer 2; 20 – 25 min: 40 – 60% Buffer 2; 25 – 30 min: 60% Buffer 2 isocratic; 30 – 32 min: 60 – 0% Buffer 2; 32–35 min: isocratic at 100% Buffer 1.
GTPase activity of CblA
The GTP hydrolysis rate for wild-type and mutant CblA (2.5 μM dimer) in the presence of MCM was determined by an HPLC assay at a fixed concentration of GTP (3 mM) at 30°C in 50 mM HEPES pH 7.5, 150 mM KCl, 2 mM MgCl2, 2 mM DTT and 5% glycerol. Aliquots (50 μl) were removed after 10 and 20 min and the reaction was terminated with 1 M trichloroacetic acid (2.5 μl). The precipitate was removed by centrifugation at 15,870 × g for 10 min. The supernatant was analyzed by HPLC using an Ultrasphere C-18 column and the following buffers: Buffer 1 (50 mM KH2PO4, pH 6.0 and 4 mM tetrabutyl ammonium bromide) and Buffer 2 (50% MeOH and 50% buffer 1, adjusted to pH 7.2) with 10 M KOH. Nucleotides were eluted using the following gradient: 0–4.2 min: 100% Buffer 1; 4.2–8.3 min: 0 – 20% Buffer 2; 8.3–16.7 min: 20 – 40% Buffer 2; 16.7–21.7 min 100 % Buffer 2; 21.7–28.3 min: isocratic at 100% Buffer 2; 28.3–30.0 min 100–0% Buffer 2; 30–35 min: isocratic at 100% Buffer 1; Under these conditions GDP eluted at 15.9 min and GTP at 19.8 min. The Kact value for MCM was obtained from the dependence of kobs for GTP hydrolysis on the concentration of MCM. The data were fitted to a single site model using Dynafit.
The Km for GTP for CblA alone and in the presence of a 3-fold excess of MCM was determined using the HPLC assay described above. Briefly, CblA (5 μM) with or without MCM (15 μM) was incubated with GTP (0–3 mM) at 30 °C and aliquots were removed after 10 and 20 min. The KM values were obtained by plotting the dependence of kcat on the concentration of GTP and fitting the data to the Michaelis Menten equation:
GDP binding to CblA
Isothermal titration calorimetry experiments were performed at 20°C in Buffer A using a 300 μl injection syringe and a 1.43-ml injection cell. Samples were prepared by filtration through a 0.2 μm filter and then degassed under vacuum at 15 °C using a ThermoVac sample degasser. Each titration was performed at least in duplicate. GDP (5 μl injections of 300–500 μM stock) was added to 15 – 20 μM CblA. The data were analyzed with a one-site binding model using the MicroCal Origin program.
Analytical gel filtration chromatography
The MCM-CblA complex was analyzed on a 1.0 × 30 cm Superdex 200 Increase GL 10/300 column (GE Healthcare) pre-equilibrated with Buffer A. The column was calibrated with gel filtration standards (Biorad). MCM (25 μM) was mixed with CblA (50 μM) and GDP or GMPPCP (500 μM) and incubated at 4°C for 1 min followed by centrifugation at 21,130 × g for 4 min. An aliquot of the supernatant (100 μl) was injected on the column and monitored at 280 nm. SEC MALS analysis of the MCM-CblA complex was performed as described previously (Campanello et al., 2017).
Electron microscopy
The MCM-GMPPCP-CblA complexes containing wild-type, K276E or G274S CblA were separated on a Superdex 200 column as described above except that glycerol was omitted from Buffer A. Fractions eluting at 8.2, 9.0 and 9.8 ml were collected and diluted to 10 μg/ml protein with the elution buffer. The samples were then added to carbon-coated grids, and stained with uranyl formate (0.75%,w/v) as described (Ohi et al., 2004). The grids were imaged either on a Thermo Fisher (Hillsboro, OR) 100 kV Morgagni transmission electron microscope equipped with a 1K × 1K CCD camera (Gatan) or a Thermo Fisher 120 kV Technai transmission electron microscope equipped with a Gatan Ultrascan 4k × 4k CCD camera (Pleasanton, CA). To quantify circular MCM-CblA•GMPPCP complexes, 100 micrographs each for wild-type and mutant CblA, were analyzed on the Thermo Fisher 100 kV Morgagni instrument from negatively stained grids made from fraction 3 (9.8 ml). Nikon Elements software was used to count the circular particles per micrograph. The data represent the average number of circular particles per micrograph ± SD.
2D class averages of the MCM-CblA•GMPPCP complex were generated from negative-stain micrographs of fractions 2 and 3 (9.0 and 9.8 ml). The micrographs were imaged on the Thermo Fisher 120 kV Technai instrument at a nominal magnification of 67,000X with a pixel size of 1.68 Å. 3,810 and 3,650 particles, were manually picked from fractions 2 and 3, respectively using E2boxer (EMAN2 (Tang et al., 2007)). Box size was 300 pixels (50.4 nm). The total particle set from each fraction was classified into reference-free 2D class averages in 10 classes using EMAN2.
EPR spectroscopy
EPR spectra were recorded on a Bruker EMX 300 equipped with a Bruker 4201 cavity and a ColdEdge cryostat. An Oxford Instruments MercuryiTC temperature controller was used. EPR spectra were recorded at 80 K using the following parameters: 9.33 GHz microwave frequency, power 2 mW, modulation amplitude 10 G, modulation frequency 100 kHz, 3000 G sweep width centered at 3500 G, conversion time 164 ms, time constant, 82 msec. Five scans were collected per sample. MCM (100 μM) in anaerobic 50 mM HEPES pH 7.5, 150 mM KCl, 2 mM MgCl2, 2 mM TCEP and 10 % glycerol buffer was mixed with cob(II)alamin (100 μM). After 15 min incubation at room temperature, the sample was either frozen in liquid nitrogen or 5 mM ATP (final concentration) was added and incubation was continued for 10 min at room temperature before freezing in liquid nitrogen. Spin concentration was estimated by comparison to 125 μM Cu EDTA standard.
AdoCbl estimation in fibroblasts
Cell lines for control and cblA fibroblasts were obtained from the Mutant Human Cell Strains Repository (McGill University Health Centre) and cultured for 96 h in the presence of [57Co]- cyanocobalamin (final concentration 25 pg/l) as described previously (Campanello et al., (2018). The percentage of radioactivity in the AdoCbl pool relative to the total cobalamin B12 pool, was determined by HPLC analysis as described (Jacobsen et al., 1982).
QUANTIFICATION AND STATISTICAL ANALYSIS
Kinetic data are presented as mean ± SD with the number of technical repeats noted in the corresponding figure legends. A Student’s T test was used to estimate the significance of the difference in AdoCbl pool size as a fraction of the total B12 pool in control versus cblA fibroblasts (p<0.05 was considered to be statistically significant).
Supplementary Material
Highlights.
Unlike the bacterial orthologs, human MCM-CblA form multiple oligomeric complexes
Annular and linear MCM-CblA complexes are seen with GMPPCP and GDP, respectively
Switch III mutations in CblA perturb the distribution of oligomeric complexes
These CblA mutations also impair B12 loading and off-loading leading to disease
Significance.
While a majority of enzymes rely on cofactors, our understanding of the strategies deployed for their assimilation and for achieving target specificity remains limited. Vitamin B12 is a complex organometallic cofactor that is targeted to two enzymes in mammals: methionine synthase and MCM. We demonstrate that cofactor loading and repair needed for the proper functioning of MCM, require the services of the CblA chaperone, which helps load AdoCbl onto MCM from ATR and to offload inactive cob(II)alamin from MCM to ATR. Mutations in MCM, CblA or ATR lead to methylmalonic aciduria, an inborn error of metabolism. CblA, a P-loop GTPase, transduces signals via three “switch” motifs including the canonical switch I and II loops found in other G-proteins. Unlike the bacterial orthologs that exist in a high affinity 2MCM-1CblA complex, our EM analysis revealed that the human MCM-CblA complex exists in a variety of forms ranging from linear 2MCM-1CblA to annular 1:1 complexes with 2–4 repeating units of MCM-CblA per oligomer. The distribution of the oligomeric species was sensitive to the presence and identity of the nucleotide bound to CblA and influenced its cofactor loading/offloading chaperone functions. We found that patient mutations in the switch III region of CblA, impacted the distribution of the MCM-CblA complexes shifting them either towards the higher order oligomers or the free proteins, and perturbed the GTPase-gated cofactor loading and repair functions. CblA mutations were correlated with a significantly decreased coenzyme B12 pool size in patient fibroblasts compared to controls. Our study predicts that the phenotypic alterations in CblA observed in vitro would lead to accumulation of inactive MCM and to a buildup of toxic methylmalonic acid levels in vivo, causing disease. The intricate mechanism for gating cofactor loading/offloading and its corruption by pathogenic mutations, could be broadly relevant to other cofactor trafficking pathways.
Acknowledgements
This work was supported in part by the National Institutes of Health (DK45776 to RB) and NIGMS Training Grant support (5 F32 GM113405 to GCC). We thank Drs. Chung and Porta for help with EM data analysis, Drs. Min and Bondy for maintenance of the electron microscopes, and acknowledge financial support for the cryo-EM facility from the Life Sciences Institute (University of Michigan).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare that no competing financial interests exist.
References
- Ando N, Brignole EJ, Zimanyi CM, Funk MA, Yokoyama K, Asturias FJ, Stubbe J, and Drennan CL (2011). Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc Natl Acad Sci U S A 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R (2003). Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem Rev 103, 2083–2094. [DOI] [PubMed] [Google Scholar]
- Banerjee R, Gherasim C, and Padovani D (2009). The Tinker, Tailor, Soldier in intracellular B12 Trafficking. Cur Op Chem Biol 13, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinig S, Kervinen J, Stith L, Wasson AS, Fairman R, Wlodawer A, Zdanov A, and Jaffe EK (2003). Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat Struct Biol 10, 757–763. [DOI] [PubMed] [Google Scholar]
- Campanello GC, Lofgren M, Yokom AL, Southworth DR, and Banerjee R (2017). Switch I-dependent allosteric signaling in a G-protein chaperone-B12 enzyme complex. J Biol Chem 292, 17617–17625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanello GC, Ruetz M, Dodge GJ, Gouda H, Gupta A, Twahir UT, Killian MM, Watkins D, Rosenblatt DS, Brunold TC, et al. (2018). Sacrificial Cobalt-Carbon Bond Homolysis in Coenzyme B12 as a Cofactor Conservation Strategy. J Am Chem Soc 140, 13205–13208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chakraborty S, and Banerjee R (1995). Demonstration that the mammalian methionine synthases are predominantly cobalamin-loaded. J. Biol. Chem 270, 19246–19249. [DOI] [PubMed] [Google Scholar]
- Cracan V, and Banerjee R (2012a). Novel B12-Dependent Acyl-CoA Mutases and Their Biotechnological Potential. Biochemistry 51, 6039–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracan V, and Banerjee R (2012b). Novel coenzyme B12-dependent interconversion of isovaleryl-CoA and pivalyl-CoA. J Biol Chem 287, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracan V, Padovani D, and Banerjee R (2010). IcmF is a fusion between the radical B12 enzyme isobutyryl-CoA mutase and its G-protein chaperone. J Biol Chem 285, 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey-Nunez L, Illson ML, Kent J, Huang Q, Brebner A, Watkins D, Gilfix BM, Wittwer CT, and Rosenblatt DS (2012). High resolution melting analysis of the MMAA gene in patients with cblA and in those with undiagnosed methylmalonic aciduria. Mol Genet Metab 107, 363–367. [DOI] [PubMed] [Google Scholar]
- Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, and Gravel RA (2002a). Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Genet 11, 3361–3369. [DOI] [PubMed] [Google Scholar]
- Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, and Gravel RA (2002b). Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci U S A 99, 15554–15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froese DS, Kochan G, Muniz JR, Wu X, Gileadi C, Ugochukwu E, Krysztofinska E, Gravel RA, Oppermann U, and Yue WW (2010). Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation. J Biol Chem 285, 38204–38213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherasim C, Lofgren M, and Banerjee R (2013). Navigating the B12 road: assimilation, delivery and disorders of cobalamin. J Biol Chem 288, 13186–13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girisha KM, Shrikiran A, Bidchol AM, Sakamoto O, Gopinath PM, and Satyamoorthy K (2012). Novel mutation in an Indian patient with Methylmalonic Acidemia, cblA type. Indian journal of human genetics 18, 346–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati S, Chen Z, Brody LC, Rosenblatt DS, and Banerjee R (1997). Defects in auxiliary redox proteins lead to functional methionine synthase deficiency. J Biol Chem 272, 19171–19175. [DOI] [PubMed] [Google Scholar]
- Hilser VJ, Wrabl JO, and Motlagh HN (2012). Structural and energetic basis of allostery. Annu Rev Biophys 41, 585–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JM, Kawin B, Minor P, and Mitchell JA (1966). Vitamin B12 concentrations in human tissue. Nature 210, 1264–1265. [Google Scholar]
- Hubbard PA, Padovani D, Labunska T, Mahlstedt SA, Banerjee R, and Drennan CL (2007). Crystal structure and mutagenesis of the metallochaperone MeaB: insight into the causes of methylmalonic aciduria. J Biol Chem 282, 31308–31316. [DOI] [PubMed] [Google Scholar]
- Ivanova EG, Federov DN, Doronina NV, and Trotensko YA (2006). Production of Vitamin B12 in Aerobic Methylotrophic Bacteria. Microbiology 75, 494–496. [PubMed] [Google Scholar]
- Jacobsen DW, Green R, Quadros EV, and Montejano YD (1982). Rapid analysis of cobalamin coenzymes and related corrinoid analogs by high-performance liquid chromatography. Anal Biochem 120, 394–403. [DOI] [PubMed] [Google Scholar]
- Jost M, Born DA, Cracan V, Banerjee R, and Drennan CL (2015a). Structural Basis for Substrate Specificity in Adenosylcobalamin-dependent Isobutyryl-CoA Mutase and Related Acyl-CoA Mutases. J Biol Chem 290, 26882–26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost M, Cracan V, Hubbard PA, Banerjee R, and Drennan CL (2015b). Visualization of a radical B12 enzyme with its G-protein chaperone. Proc Natl Acad Sci U S A 112, 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DG, Cannavan A, Molloy A, O’Harte F, Taylor SM, Kennedy S, and Blanchflower WJ (1990). Methylmalonyl-CoA mutase (EC 5.4.99.2) and methionine synthetase (EC 2.1.1.13) in the tissues of cobalt-vitamin B12 deficient sheep. Br J Nutr 64, 721–732. [DOI] [PubMed] [Google Scholar]
- Koshland DE Jr., Nemethy G, and Filmer D (1966). Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5 365–385. [DOI] [PubMed] [Google Scholar]
- Leal NA, Olteanu H, Banerjee R, and Bobik TA (2004). Human ATP: Cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J Biol Chem 279, 47536–47542. [DOI] [PubMed] [Google Scholar]
- Ledley FD, Lumetta M, Nguyen PN, Kolhouse JF, and Allen RH (1988). Molecular Cloning of L-Methylmalonyl CoA Mutase: Gene Transfer and Analysis of mut Cell Lines. Proc. Natl. Acad. Sci. U.S.A 85, 3518–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, and Aravind L (2002). Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 317, 41–72. [DOI] [PubMed] [Google Scholar]
- Li Z, Kitanishi K, Twahir UT, Cracan V, Chapman D, Warncke K, and Banerjee R (2017). Cofactor editing by the G-protein metallochaperone domain regulates the radical B12 enzyme IcmF. J Biol Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren M, Koutmos M, and Banerjee R (2013a). Autoinhibition and signaling by the switch II motif in the G-protein chaperone of a radical B12 enzyme. J Biol Chem 288, 30980–30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofgren M, Padovani D, Koutmos M, and Banerjee R (2013b). A switch III motif relays signaling between a B12 enzyme and its G-protein chaperone. Nat Chem Biol 9, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, and Changeux JP (1965). On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol 12, 88–118. [DOI] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, and Walz T (2004). Negative staining and image classification — powerful tools in modern electron microscopy. Biological Procedures Online 6 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovani D, and Banerjee R (2006a). Alternative pathways for radical dissipation in an active site mutant of B12-dependent methylmalonyl-CoA mutase. Biochemistry 45, 2951–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovani D, and Banerjee R (2006b). Assembly and protection of the radical enzyme, methylmalonyl-CoA mutase, by its chaperone. Biochemistry 45, 9300–9306. [DOI] [PubMed] [Google Scholar]
- Padovani D, and Banerjee R (2009a). A G-protein editor gates coenzyme B12 loading and is corrupted in methylmalonic aciduria. Proc Natl Acad Sci U S A 106, 21567–21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovani D, and Banerjee R (2009b). A Rotary Mechanism for Coenzyme B12 Synthesis by Adenosyltransferase. Biochemistry 48, 5350–5357. [DOI] [PubMed] [Google Scholar]
- Padovani D, Labunska T, and Banerjee R (2006). Energetics of interaction between the G-protein chaperone, MeaB and B12-dependent methylmalonyl-CoA mutase. J. Biol. Chem 281, 17838–17844. [DOI] [PubMed] [Google Scholar]
- Padovani D, Labunska T, Palfey BA, Ballou DP, and Banerjee R (2008). Adenosyltransferase tailors and delivers coenzyme B12. Nat Chem Biol 4 194–196. [DOI] [PubMed] [Google Scholar]
- Takahashi-Iniguez T, Garcia-Arellano H, Trujillo-Roldan MA, and Flores ME (2011). Protection and reactivation of human methylmalonyl-CoA mutase by MMAA protein. Biochem Biophys Res Commun 404, 443–447. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Watkins D, and Rosenblatt DS (2011). Inborn errors of cobalamin absorption and metabolism. Am J Med Genet C Semin Med Genet 157C, 33–44. [DOI] [PubMed] [Google Scholar]
- Wittinghofer A, and Vetter IR (2011). Structure-function relationships of the G domain, a canonical switch motif. Annu Rev Biochem 80, 943–971. [DOI] [PubMed] [Google Scholar]
- Yamada K, Gravel RA, Toraya T, and Matthews RG (2006). Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc Natl Acad Sci U S A 103, 9476–9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi M, Labunska T, and Banerjee R (2005). Mirror “base-off” conformation of coenzyme B12 in human adenosyltransferase and its downstream target, methylmalonyl- CoA mutase. J Am Chem Soc 127, 526–527. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.