

SUMMARY
Activation of the unfolded protein response (UPR)-associated transcription factor ATF6 has emerged as a promising strategy to reduce the secretion and subsequent toxic aggregation of destabilized, amyloidogenic proteins implicated in systemic amyloid diseases. However, the molecular mechanism by which ATF6 activation reduces the secretion of amyloidogenic proteins remains poorly defined. We employ a quantitative interactomics platform to define how ATF6 activation reduces secretion of a destabilized, amyloidogenic immunoglobulin light chain (LC) associated with Light Chain Amyloidosis (AL). Using this platform, we show that ATF6 activation increases the targeting of this destabilized LC to a subset of pro-folding ER proteostasis factors that retains the amyloidogenic LC within the ER, preventing its secretion. Our results define a molecular basis for the ATF6-dependent reduction in destabilized LC secretion and highlight the advantage for targeting this UPR-associated transcription factor to reduce secretion of destabilized, amyloidogenic proteins implicated in AL and related systemic amyloid diseases.
Keywords: ER quality control, ER proteostasis, Unfolded Protein Response (UPR), ATF6, XBP1s, quantitative proteomics, ER chaperones
eTOC BLURB
Activating the ATF6 arm of the unfolded protein response selectively reduces the secretion and subsequent toxic aggregation of destabilized amyloidogenic light chains. Plate et al use a quantitative proteomics platform to identify specific ATF6-regulated ER proteostasis factors responsible for regulating the secretion of an amyloidogenic light chain from mammalian cells.
Graphical Abstract
INTRODUCTION
The toxic extracellular aggregation of destabilized, amyloidogenic proteins is implicated in the onset and pathogenesis of diverse systemic amyloid diseases including Light Chain Amyloidosis (AL) and the transthyretin (TTR)-related amyloid diseases (Blancas-Mejia and Ramirez-Alvarado, 2013; Powers et al., 2009). A critical determinant in dictating the pathologic protein aggregation central to these diseases is the aberrant secretion of destabilized, aggregation-prone proteins to the extracellular space (Plate and Wiseman, 2017). The efficient secretion of these proteins increases their extracellular populations available for concentration-dependent aggregation into toxic oligomers and amyloid fibrils that deposit in distal tissues such as the heart, inducing organ dysfunction. The importance of amyloidogenic protein secretion in disease pathogenesis suggests that targeting the biologic pathways responsible for regulating the secretion of destabilized, amyloidogenic proteins offers a unique opportunity to broadly ameliorate the pathologic extracellular protein aggregation implicated in the pathogenesis of diverse amyloid diseases (Plate and Wiseman, 2017).
Protein secretion through the secretory pathway is regulated by a process referred to as endoplasmic reticulum (ER) quality control (Balchin et al., 2016; Braakman and Bulleid, 2011; Feige and Buchner, 2014; Guerriero and Brodsky, 2012; Kim et al., 2013). In this process, newly synthesized proteins are co-translationally imported into the ER where they interact with ER chaperones and folding factors. These interactions facilitate the folding of proteins into their native conformations and prevent their misfolding and/or aggregation within the ER. Once folded, these proteins are packaged into vesicles for trafficking to downstream secretory environments including the extracellular space. Proteins unable to attain a native, folded conformation within the ER are instead recognized by ER degradation factors and directed toward degradation pathways such as ER-associated degradation (ERAD). Through this partitioning between ER protein folding, trafficking and degradation pathways (i.e., ER quality control), cells prevent the secretion of destabilized, aggregation-prone proteins to downstream secretory environments.
In the context of systemic amyloid diseases, destabilized, amyloidogenic proteins escape ER quality control, allowing their efficient secretion to the extracellular space (Blancas-Mejia and Ramirez-Alvarado, 2013; Plate and Wiseman, 2017). This suggests that enhancing ER quality control capacity could offer a unique opportunity to reduce the aberrant secretion and toxic extracellular aggregation associated with these disorders. One strategy to improve ER quality control for amyloidogenic proteins is by activating the unfolded protein response (UPR) (Hetz and Saxena, 2017; Plate and Wiseman, 2017). The UPR regulates ER quality control through activation of UPR-associated transcription factors such as XBP1s and ATF6. These transcription factors induce overlapping, but distinct, subsets of ER chaperones, folding factors, and degradation factors (collectively ER proteostasis factors) that dictate ER quality control (Adachi et al., 2008; Lee et al., 2003; Shoulders et al., 2013; Yamamoto et al., 2004). The differential remodeling of ER quality control pathways afforded by XBP1s or ATF6 activation indicates that the independent activation of these pathways offers unique opportunities to correct pathologic defects in ER quality control for destabilized, amyloid disease-associated proteins (Plate and Wiseman, 2017).
Previous results show that stress-independent activation of XBP1s or ATF6 differentially influence ER quality control for destabilized amyloidogenic proteins such as ALLC – a destabilized Vλ6 immunoglobulin light chain (LC) associated with AL pathogenesis (Arendt et al., 2008). Stress-independent XBP1s activation increases ALLC targeting to ER degradation pathways, while only modestly affecting its secretion (Cooley et al., 2014). In contrast, ATF6 activation does not increase ALLC degradation, but significantly reduces the secretion and extracellular aggregation of ALLC. It does so without affecting secretion of an energetically normal Vλ6 LC or the endogenous secretory proteome (Cooley et al., 2014; Plate et al., 2016). ATF6 activation also selectively reduces the secretion and toxic aggregation of destabilized variants of other aggregation-prone proteins, without significantly impacting secretion of the wild-type protein (Chen et al., 2014; Chiang et al., 2012; Shoulders et al., 2013; Smith et al., 2011b). These results identify ATF6 as a potential therapeutic target that can be pharmacologically accessed to improve ER quality control and selectively reduce the secretion and subsequent aggregation of destabilized, amyloidogenic proteins implicated in amyloid disease pathogenesis (Plate and Wiseman, 2017).
Despite this potential, the molecular mechanism responsible for ATF6-dependent reductions in destabilized, amyloidogenic protein secretion remains poorly defined. Quantitative affinity purification mass spectrometry (q-AP-MS) has proven a powerful tool to identify changes in protein-protein interactions induced by genetic or environmental perturbations (Hosp et al., 2015; Huttlin et al., 2015; Katrina Meyer, 2015). This indicates that q-AP-MS provides a unique opportunity to define proteins and pathways responsible for dictating the selective, ATF6-dependent reduction in amyloidogenic LC secretion. Here, we implement a tandem mass tag (TMT)-based q-AP-MS platform to define how ATF6 activation improves ER quality control to selectively reduce secretion of the destabilized, amyloidogenic LC, ALLC. We demonstrate that this platform allows efficient multiplexed quantification of ALLC interaction changes induced by activation of different UPR-associated transcriptional programs. Using this TMT-based q-AP-MS approach, we show that ATF6 activation reduces ALLC secretion by increasing its targeting to ATF6-regulated ER chaperones that selectively retain this destabilized LC within the ER lumen. Overexpression of these ATF6-regulated ER chaperones only partially mimics the reduction in ALLC observed following ATF6 activation. This indicates that ATF6 activation improves LC ER quality by global remodeling the ER proteostasis environment and not through upregulation of a specific chaperone. Our results define a mechanistic framework that explains the ATF6-dependent regulation of LC ER quality control and further motivates the development of therapeutic strategies that enhance ER quality control to ameliorate amyloid pathology in AL and related amyloid diseases.
RESULTS
A TMT-based AP-MS platform to define ER proteostasis factors that interact with LCs
ER quality control processes are governed by interactions between non-native protein conformations and ER proteostasis factors (Kim et al., 2013; Powers et al., 2009; Wiseman et al., 2007). Thus, defining the molecular interactions between destabilized, amyloidogenic proteins and ER proteostasis factors allows identification of the components of specific biologic pathways responsible for dictating ER quality control for a given protein under defined conditions, such as ATF6 activation. However, many challenges exist in defining interactions between ER proteostasis factors and destabilized protein substrates. These include the transient nature of substrate interactions with ER proteostasis factors and the difficulty in multiplexing interactome profiling to improve throughput without sacrificing sensitivity (Budayeva and Cristea, 2014; Kean et al., 2012; Miteva et al., 2013; Pankow et al., 2016; Pankow et al., 2015; Taipale et al., 2014).
To address these challenges in the context of amyloidogenic LCs such as ALLC, we implemented an affinity-purification mass spectrometry (AP-MS) platform that utilizes Tandem Mass Tags (TMTs). The TMT reagents enable multiplexed quantification of changes in protein abundances across multiple conditions or biological replicates within a single mass spectrometry experiment (Huttlin et al., 2015; McAlister et al., 2012; McAlister et al., 2014; Papachristou et al., 2018; Roumeliotis et al., 2017). We utilized the platform to define the specific ER proteostasis factors important for ATF6-dependent regulation of LC ER quality control (Fig. 1A). For these experiments, we employed HEK293DAX cells (Shoulders et al., 2013), which exhibit ATF6-dependent reductions in the secretion of destabilized ALLC (Arendt et al., 2008), but not the energetically-normal Vλ6 LC JTO (Cooley et al., 2014; Wall et al., 1999). ALLC exhibits both reduced thermodynamic stability and a faster rate of unfolding compared to JTO, which could explain the difference in amyloidogenicity (Cooley et al., 2014; Morgan and Kelly, 2016). We transiently transfected HEK293DAX cells with flag-tagged ALLC (FTALLC), flag-tagged JTO (FTJTO), or an untagged ALLC (Fig. 1B and Fig. S1A). We previously showed that the presence of the FLAG tag does not influence ER proteostasis of LCs (Cooley et al., 2014). We subjected cells expressing these different LCs to in situ crosslinking using the cell-permeable crosslinker dithiobis(succinimidyl propionate) (DSP) (Lomant and Fairbanks, 1976; Nittis et al., 2010; Smith et al., 2011a). We optimized DSP crosslinking to stabilize known interactions between ER proteostasis factors (e.g., BiP) and LCs in the ER (Fig. S1B,C). After crosslinking, we immunopurified (IP’d) FTALLC or FTJTO using anti-Flag beads. Following stringent washing in high-detergent RIPA buffer to remove non-specific interactors, the samples were reduced to cleave the disulfide bond comprising the crosslinks, alkylated, and digested with trypsin. The digested peptides arising from individual experiments were then labeled with distinct TMT reagents, combined, and analyzed by Multi-dimensional Protein Identification Technology (MuDPIT) proteomics (Washburn et al., 2001; Yates et al., 2009). Specific recovery of peptides under different conditions was then quantified by comparing the recovered signals from the TMT reporter ions in the MS2 spectra (Fig. 1A).
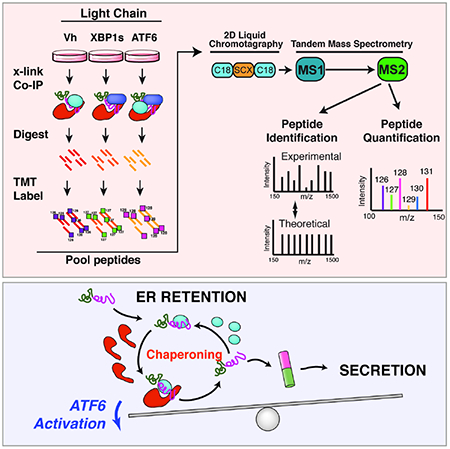
Figure 1. Establishing an AP-MS platform to identify ER proteostasis factors that interact with destabilized, amyloidogenic ALLC.
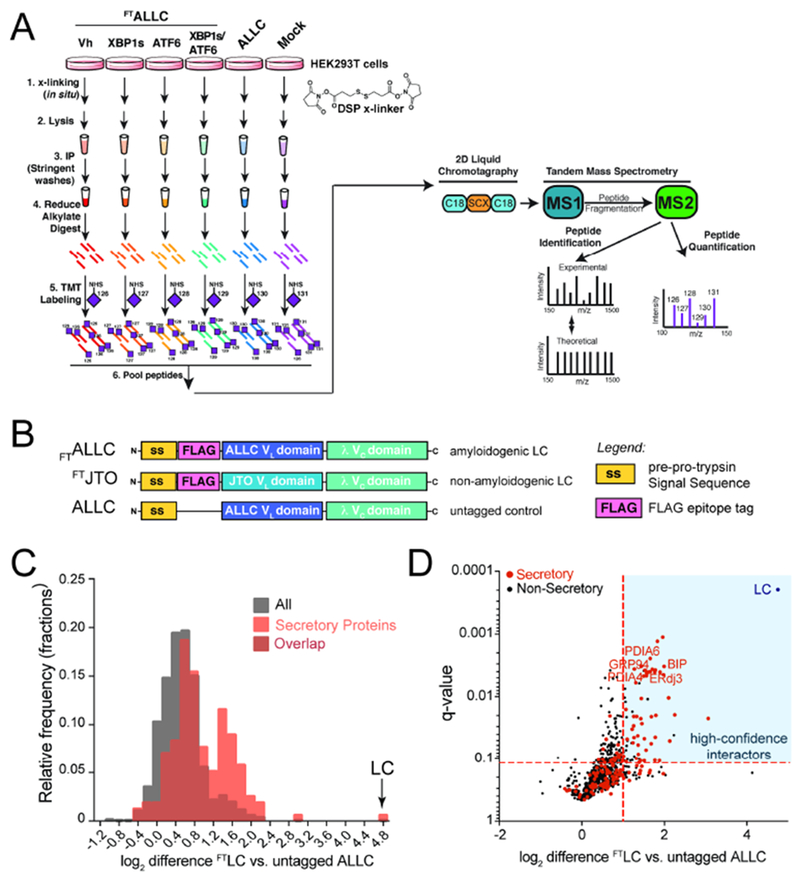
A. Schematic of the multiplexed quantitative interactomics methodology, which combines affinity purification mass-spectroscopy (AP-MS) with in situ DSP cross-linking to capture transient, low affinity interactions with proteostasis network components. Sixplex tandem mass tags (TMT) are used for relative quantification of proteins in individual AP samples, followed by MuDPIT (2D LC coupled to Tandem mass spectrometry).
B. Illustration showing the domain organization for the flag-tagged destabilized, amyloidogenic LC ALLC (FTALLC), the flag-tagged energetically normal LC JTO (FTJTO), and untagged ALLC. A sequence alignment of ALLC and JTO showing the differences in amino acid sequence is shown in Fig. S1A.
C. Histogram displaying TMT ratios of FTLC (combined n=4 FTALLC and n=6 FTJTO replicates) vs. untagged ALLC (mock) channels for all protein (grey) and filtered secretory protein (red).
D. Plot showing TMT ratio (log2 difference FTLC vs. untagged ALLC) vs. q-value (Storey) for proteins that co-purify with FTLC (either FTALLC or FTJTO) compared to untagged ALLC in anti-FLAG IPs (n=10 biological replicates; n = 4 for FTALLC and n = 6 for FTJTO). High confidence interactors are identified in the blue quadrant showing TMT ratio >2 and a q-value < 0.11. Secretory proteins are shown in red. Full data included in Supplemental Table 1.
Initially, we used this AP-MS platform to identify the ER quality control factors that bind LCs in situ by comparing TMT ratios for proteins that co-purify in anti-FLAG IPs from lysates prepared on HEK293DAX cells expressing untagged ALLC or either FTALLC or FTJTO (collectively FTLC). We defined the TMT ratio as: TMT signal FTLC IPs / TMT signal in untagged ALLC IPs. We observed two populations of proteins isolated in these samples separated by their TMT ratio (Fig. 1C and Supplemental Table 1). The first population exhibits a low TMT ratio of ~1.3, which represents proteins that non-specifically co-purify in both FTLC and untagged ALLC anti-FLAG IPs. However, a second population of 72 proteins displayed a ratio of >2, indicating selective interaction with FTALLC and FTJTO. This second population was highly enriched for secretory proteins (51 out 72, based on gene ontology terms describing localization in the secretory pathway) and included ER proteostasis factors known to interact with LCs in the ER such as BiP, GRP94, ERdj3, HYOU1, and PDIA1 (Behnke et al., 2015; Behnke et al., 2016; Cole et al., 2018; Davis et al., 1999; Hellman et al., 1999; Hsu et al., 1996; Melnick et al., 1992; Melnick et al., 1994; Shen and Hendershot, 2005; Skowronek et al., 1998). We defined these 72 interacting proteins as ‘high confidence interactors’ of FTLC (Fig. 1D), and we used these proteins as the basis for subsequent AP-MS experiments focused on defining the ER quality control pathways responsible for the selective, ATF6-dependent regulation of ALLC secretion.
TMT-based q-AP-MS allows multiplexed quantification of ALLC interaction changes induced by XBP1s and/or ATF6 activation
In order to define the specific ER proteostasis factors responsible for the differential impact of ATF6 or XBP1s activation on ALLC ER quality control (Fig. 2A), we used our TMT-based q-AP-MS proteomic platform to identify high confidence interactors that show altered interaction with FTALLC following stress-independent activation of these UPR-associated transcription factors in HEK293DAX cells. These cells express both doxycycline (dox)-inducible XBP1s and a trimethoprim (TMP)-regulated DHFR.ATF6, allowing stress-independent XBP1s or ATF6 activation through the administration of dox or TMP, respectively (Shoulders et al., 2013). We compared the recovery of TMT signals for high-confidence LC interactors that co-purify with FTALLC in lysates prepared from HEK293DAX cells following 24 h treatment with vehicle, dox (activates XPB1s), or TMP (activates ATF6) – a treatment paradigm we previously showed is sufficient to induce efficient remodeling of ER proteostasis pathways (Shoulders et al., 2013). A challenge in comparing the TMT signals across different IPs is the variability of bait protein (e.g., FTALLC). FTALLC can vary in concentration owing to factors including differences in transfection across biologic replicates, variability in sample preparation or alterations in protein secretion or degradation caused by XBP1s and/or ATF6 activation (Cooley et al., 2014). This results in a large variance in unnormalized interaction ratios for high confidence interactors (Fig. 2B, blue, Fig. S2A). To address this variability, we normalized the recovery of high confidence interactors to the amount of FTALLC identified in each channel. Normalization significantly improved the variance across samples (Fig. 2B, orange) and allowed quantification of interactions changes using this approach. Importantly, we showed high reproducibility of FTALLC interaction changes induced by XBP1s or ATF6 activation across 7 independent biological replicates (Fig. S2B,C).
Figure 2. Stress-independent XBP1s or ATF6 activation differentially influence interactions between FTALLC and ER proteostasis factors.
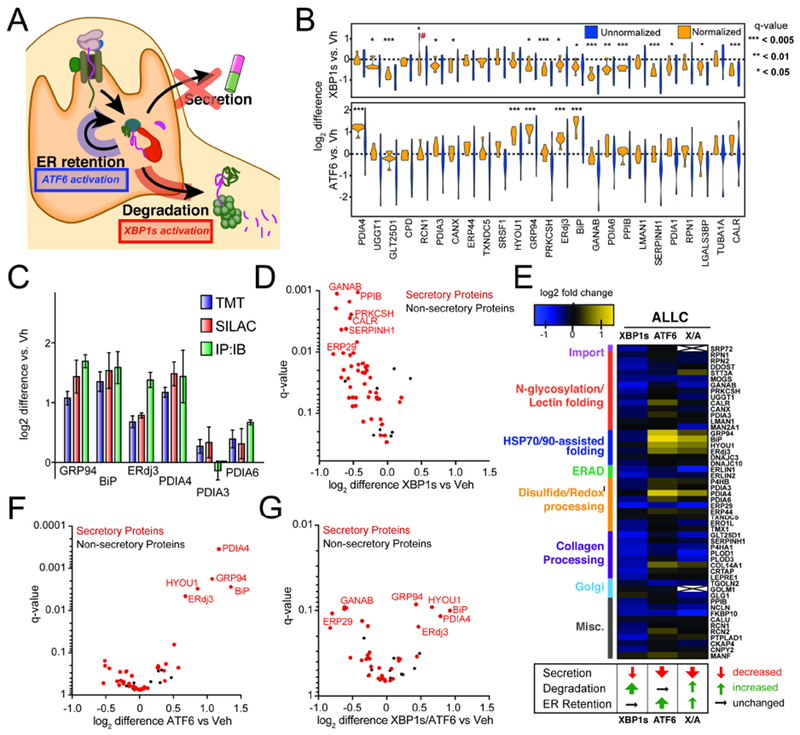
A. Illustration summarizing previous data showing how stress-independent activation of XBP1s (red) or ATF6 (blue) leads to reduced secretion of destabilized, amyloidogenic ALLC (Cooley et al., 2014). XBP1s activation modestly reduces ALLC secretion through increased targeting to degradation, while ATF6 activation significantly reduces ALLC secretion through its increased ER retention.
B. Plot showing the distribution of unnormalized (blue) and FTALLC-bait-normalized (orange) TMT interaction ratios for n = 7 biological replicates comparing the recovery of high confidence ALLC interacting proteins in anti-FLAG IPs from cells following XBP1s activation (top) or ATF6 activation (bottom). A simple normalization procedure of the protein TMT signal against the FTALLC bait protein signal across each TMT channel greatly diminishes the variance in interaction ratios. *q-value (Storey) < 0.15, **p < 0.05, ***p < 0.01; # denotes excluded outlier
C. Comparison of interaction fold changes between FTALLC and selected proteostasis factors in response to XBP1s or ATF6 activation as quantified by three independent methods: TMT-based q-AP-MS (n=7 biological replicates), SILAC-based q-AP-MS (n= 4 or 5 biological replicates), or Co-immunoprecipitation followed by quantitative immunoblotting (IP:IB; n=3–6 biological replicates).
D. Plot showing TMT interaction ratio vs. q-value (Storey) for high confidence FTALLC interacting proteins that co-purify with FTALLC in HEK293DAX cells following stress-independent XBP1s activation from n=7 biological replicates. Full data included in Supplemental Table 2.
E. Heatmap displaying the observed interactions changes between FTALLC and high confidence ER proteostasis network components following stress-independent XBP1s or ATF6 activation (data from n=7 biological replicates for ATF6 or XBP1s activation, and n = 4 for XBP1s/ATF6 co-activation). Interactors are organized by pathway or function. The previously defined impact of activating these pathways on ALLC secretion, degradation, and ER retention is shown below (Cooley et al., 2014).
F. Plot showing TMT interaction ratio vs. q-value (Storey) for high confidence FTALLC interacting proteins that co-purify with FTALLC in HEK293DAX cells following stress-independent ATF6 activation from n=7 biological replicates. Full data included in Supplemental Table 2.
G. Plot showing TMT interaction ratio vs. q-value (Storey) for high confidence FTALLC interacting proteins that co-purify with FTALLC in HEK293DAX cells following stress-independent XBP1s and ATF6 co-activation from n=4 biological replicates. Full data included in Supplemental Table 2.
TMT-based quantification of proteomics data is challenged in complex proteomes by reporter ion ratio compression. This is caused by co-isolation of interfering precursor ions and subsequent co-fragmentation, resulting in an underestimation of the abundance ratios (Savitski et al., 2013; Ting et al., 2011). Synchronous Precursor Selection (SPS) MS3 analysis is commonly employed to overcome this ratio compression challenge, however, this analysis is only possible using specialized Tribrid MS instruments, such as the Fusion Lumos (Huttlin et al., 2015; McAlister et al., 2014; Papachristou et al., 2018; Roumeliotis et al., 2017). Due to the simplicity of our proteomes afforded by the AP isolation of FTALLC, we predicted that ratio compression would be minimized and allow accurate quantification using MS2 reporter ion quantification. Furthermore, we performed our analysis using multi-dimensional protein identification technology (MuDPIT), which increases the chromatographic separation of peptides – another parameter that has been shown to reduce TMT ratio compression (Ow et al., 2011). To ensure that ratio compression is minimized, we compared FTALLC interaction ratios for high confidence interactors measured by TMT-based proteomics with ratios determined using Stable Isotope Labeling with Amino acids in Cell culture (SILAC), an independent quantitative proteomics approach that relies on quantification of light and heavy-isotope labeled precursor ions in the MS1 spectra (Fig. S2D) (Ong et al., 2002). As with TMT-based AP-MS, we performed SILAC-based AP-MS to monitor changes in FTALLC interactome induced by XBP1s and/or ATF6 activation in HEK293DAX cells (Fig. S2D–F). Alterations in the interactions between ER proteostasis factors and FTALLC observed using the alternative SILAC-based quantitation were nearly identical to those obtained using TMT-based quantification (Fig. S2G). To further confirm the accuracy of our TMT-quantification AP-MS assay we confirmed XBP1s or ATF6-dependent interaction changes between FTALLC and select high confidence interactors using AP followed by quantitative immunoblotting (IP:IB) (Fig. 2C and Fig. S2H). This proteomic-independent approach showed similar interaction ratio changes. Collectively, these results show that our TMT-based AP-MS platform can accurately monitor changes in the FTALLC interactome induced by XBP1s and/or ATF6 activation.
Comparing our multiplexed TMT-based interatomic approach to SILAC quantification also demonstrated enhanced throughput capacity. Since SILAC quantification only enables binary comparisons, a higher number of mass spectrometry runs and more instrument time was needed to generate the quantitative comparisons between different conditions (Fig. S2I–J). Furthermore, the number of proteins that could be reliably quantified in at least 3 biological replicates was at least 4-fold greater using our TMT-based platform than in any of the pairwise SILAC comparisons. (Fig. S2K–L). The improved analysis time and more reliable protein identification in our TMT-based AP-MS platform highlights additional advantage of this platform over more commonly used label free or SILAC-based approaches for defining interactome changes for destabilized proteins such as FTALLC in response to selective ATF6 or XBP1s activation.
XBP1s or ATF6 activation differentially influence interactions between ER quality control factors and FTALLC
Stress-independent activation of XBP1s or ATF6 differentially influence ALLC ER quality control (Fig. 2A) (Cooley et al., 2014). XBP1s activation increases targeting of ALLC to degradation, while only modestly reducing ALLC secretion. In contrast, ATF6 activation significantly reduces ALLC secretion and subsequent aggregation by >50%, but does not increase ALLC degradation, indicating that activating ATF6 increases the ER retention of this destabilized LC. In order to define the specific ER proteostasis factors responsible for the differential impact of ATF6 or XBP1s activation on ALLC ER quality control, we compared the changes in the FTALLC interactome induced by XBP1s and/or ATF6 activation.
Interestingly, XBP1s or ATF6 activation induce distinct changes in the interactions between FTALLC and ER proteostasis factors, reflecting the distinct impact of these UPR-associated transcription factors on ALLC ER quality control (Fig. 2D–F and Supplemental Table 2)(Cooley et al., 2014). XBP1s activation globally reduces interactions between FTALLC and ER proteostasis factors (Fig. 2D,E). This is consistent with the XBP1s-dependent increase in ALLC targeting to ER degradation pathways such as ERAD or autophagy – the latter being the predominant pathway responsible for degrading ALLC following ER stress (Cooley et al., 2014). Unfortunately, components of degradation pathways were poorly detected in our proteomics samples, which likely reflects poor crossinking between ERAD factors and FTALLC or that these mainly membrane-associated proteins require specific detergents for solubilization (Christianson et al., 2011). In contrast, ATF6 activation increases interactions between FTALLC and select ER proteostasis factors, including the ATP-dependent ER chaperones BiP and GRP94, the BiP co-chaperones ERdj3 and HYOU1, and the protein-disulfide isomerase PDIA4 (Fig. 2E,F). The increase in FTALLC interactions with these ER proteostasis factors is consistent with the ATF6-dependent increase in ALLC ER retention (Cooley et al., 2014) and suggests that ATF6 activation reduces secretion of ALLC through the increased targeting of this destabilized LC to specific ER proteostasis pathways.
Despite impacting ALLC ER quality control through distinct mechanisms, co-activation of XBP1s and ATF6 does not synergistically influence destabilized ALLC secretion (Cooley et al., 2014). Instead, XBP1s and ATF6 co-activation reduces ALLC secretion to the same extent observed with ATF6 activation alone and modestly increases ALLC degradation (Cooley et al., 2014). This indicates that co-activation of these transcription factors integrates distinct functional aspects of independent XBP1s or ATF6 activation to influence ALLC ER quality control. Consistent with this, AP-MS shows that XBP1s and ATF6 co-activation remodels the FTALLC interactome by promoting specific changes also observed following independent transcription factor activation (Fig. 2E,G and Supplemental Table 2). For example, XBP1s and ATF6 co-activation reduces interactions between FTALLC and numerous high confidence interactors, consistent with the moderate increase in ALLC degradation observed under these conditions (Cooley et al., 2014). Alternatively, co-activation of these transcription factors increases interactions between FTALLC and ER proteostasis factors including BiP, GRP94, ERdj3, HYOU1, and PDIA4 – all of which are also increased following ATF6 activation alone.
Comparing the functional impact of XBP1s and/or ATF6 activation on ALLC ER quality control to the changes in the interactions between FTALLC and ER proteostasis pathways provides an opportunity to identify the ER proteostasis factors likely responsible for the regulation of ALLC secretion. ATF6 activation, in both the presence or absence of XBP1s activation, reduces ALLC secretion by 50% (Cooley et al., 2014). Based on our AP-MS analysis, this reduced secretion correspond to increased interactions with a specific subset of ER proteostasis factors including BiP, GRP94, HYOU1, ERdj3, and PDIA4. We confirmed the ATF6-dependent increase in the interactions between these ER proteostasis factors and FTALLC by IP:IB (Fig. 2C). This suggests that these proteostasis factors are involved in dictating the selective, ATF6-dependent reduction in destabilized ALLC secretion.
ATF6 activation increases the interactions between ER proteostasis factors and an energetically normal LC
ATF6 activation selectively reduces secretion of destabilized ALLC relative to the energetically normal LC JTO (Cooley et al., 2014). Thus, we sought to define how ATF6 activation influences the interactions between JTO and ER proteostasis factors. Initially, we directly compared the interactomes of FTALLC and FTJTO in vehicle-treated HEK293DAX cells using our AP-MS proteomic platform (Fig. 1A). In order to normalize the recovery of ER proteostasis factors in these IPs, we used peptides from the λ Vc domain of these LCs, which is identical between ALLC and JTO (Fig. S1A). This allowed us to accurately monitor the differential interactions between ER proteostasis factors and specific LCs in this experiment (Fig. 3A). Using this approach, we identified numerous high confidence LC interacting proteins that showed increased association with the destabilized ALLC, relative to the stable JTO (Fig. 3A, Fig. S3A, and Supplemental Table 3). This includes many ER proteostasis factors identified to increase association upon ATF6 activation such as BiP and GRP94, indicating that these proteins are key determinants in dictating LC ER quality control. We confirmed the increased association of select ER proteostasis factors with ALLC by IP:IB (Fig. S3B).
Figure 3. ATF6 Activation Increases Interactions between Non-amyloidogenic FTJTO and ER Proteostasis factors.
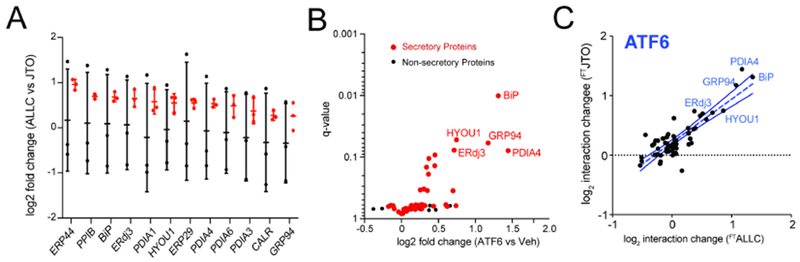
A. Distribution of unnormalized (black) and normalized (red) TMT ratios of FTALLC vs. FTJTO for proteins with significant interaction changes (n=3 biological replicates). Peptides of the λ Vc domain, which is identical for ALLC and JTO (Fig. S1A), were used to normalize the TMT signal of each individual protein against the λ Vc domain peptide signal across each TMT channel.
B. Plot showing TMT interaction ratio vs. q-value for high confidence ALLC interacting proteins that copurify with FTJTO from HEK293DAX cells following treatment with vehicle or TMP (to activate ATF6) for 16 h (n=6 biological replicates). Secretory proteins are shown in red. Full data available in Supplemental Table 3.
C. Plot comparing the interaction changes of high confidence ALLC interacting proteins with either FTALLC (n=7 biological replicates) or FTJTO (n=6 biological replicates) following stress-independent ATF6 activation. The dashed line represents least-squares linear regression. The solid lines show 95% confidence intervals
Next, we evaluated how ATF6 activation influences the interactions between FTJTO and ER proteostasis factors. ATF6 activation induced a similar remodeling of the FTJTO interactome to that observed for FTALLC (Fig. 3B,C, Fig. S3C and Supplemental Table 3). These results indicate that ATF6-dependent increases in the interactions with ER proteostasis factors occur independent of the energetic stability of the LC. However, interactions between ER proteostasis factors and FTJTO after ATF6 activation are nonetheless lower than observed for FTALLC because basal FTJTO interactions are less compared to FTALLC (Fig. 3A and Fig. S3A,B). This indicates that increased targeting of FTALLC to specific ER proteostasis factors represents a likely molecular mechanism responsible for selective, ATF6-dependent retention of this destabilized LC sequence.
ATF6 transcriptionally regulates ER proteostasis factors that show increased interactions with FTALLC
ATF6 activation transcriptionally regulates the expression of multiple ER proteostasis factors that show increased association with FTALLC following stress-independent ATF6 activation (e.g., BiP, GRP94) (Shoulders et al., 2013; Yamamoto et al., 2004). This suggests that the increased interaction between these ER proteostasis and FTALLC is regulated by ATF6-dependent increases in ER proteostasis factor expression. Consistent with this, ATF6-dependent changes in mRNA for high confidence interactors correlate to changes in interactions with FTALLC (Fig. 4A and Supplemental Table 4)(Plate et al., 2016). A similar relationship was observed when we compared ATF6-dependent increases in the protein levels for these ER proteostasis factors (measured by whole cell quantitative proteomics (Plate et al., 2016)) to increases in FTALLC interactions (Fig. 4B). These results indicate that the increased interactions between FTALLC and ER proteostasis factors is primarily dictated by ATF6-dependent increases in their expression.
Figure 4. ATF6-dependent increases in ALLC interactions correlate with ER proteostasis factor expression.
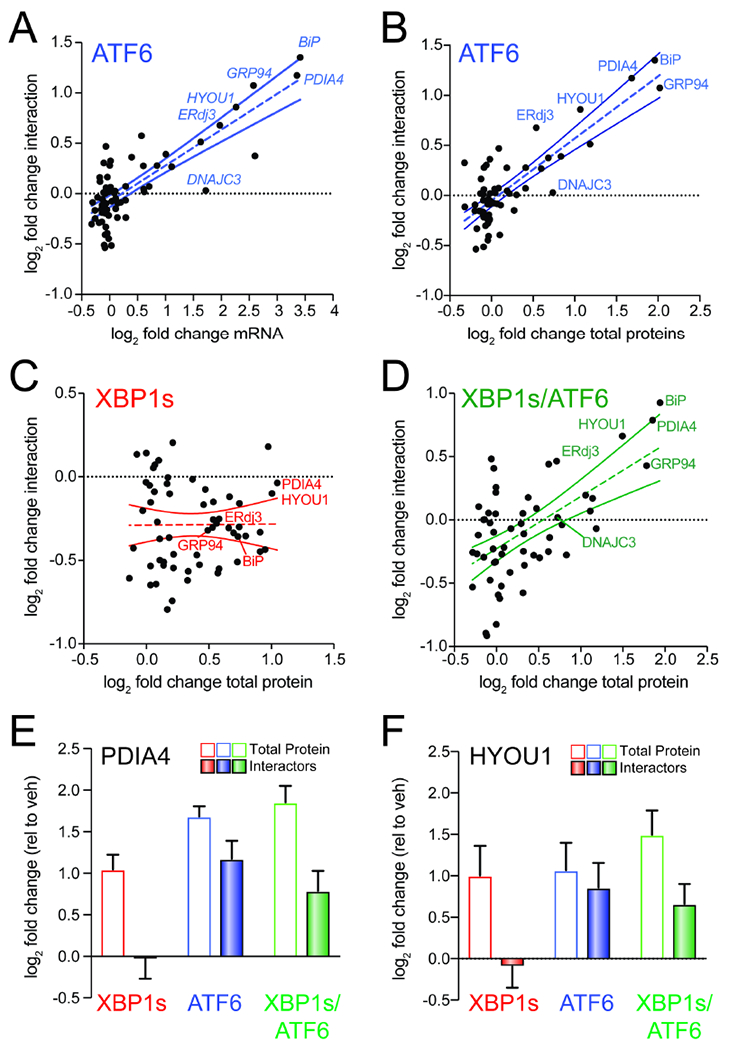
A. Plot comparing mRNA level for high confidence ALLC interacting proteins (measured by RNAseq in (Plate et al., 2016); n=3 biological replicates) vs. their increased interactions with FTALLC in HEK293DAX cells following stress-independent ATF6 activation (n=7 biological replicates). The dashed line shows least-squares linear regression. The solid lines show 95% confidence intervals.
B. Plot comparing cellular protein level for high confidence ALLC interacting proteins (measured by whole cell quantitative proteomics in (Plate et al., 2016); n=3 biological replicates) vs. their increased interactions with FTALLC in HEK293DAX cells following stress-independent ATF6 activation (n=7 biological replicates). The dashed line shows least-squares linear regression. The solid lines show 95% confidence intervals.
C. Plot comparing cellular protein level for high confidence ALLC interacting proteins (measured by whole cell quantitative proteomics in (Plate et al., 2016); n=3 biological replicates) vs. their increased interactions with FTALLC in HEK293DAX cells following stress-independent XBP1s activation (n=7 biological replicates). The dashed line shows least-squares linear regression. The solid lines show 95% confidence intervals.
D. Plot comparing cellular protein level for high confidence ALLC interacting proteins (measured by whole cell quantitative proteomics in (Plate et al., 2016); n=3 biological replicates) vs. their increased interactions with FTALLC in HEK293DAX cells following stress-independent ATF6 and XBP1s co-activation (n=4 biological replicates). The dashed line shows least-squares linear regression. The solid lines show 95% confidence intervals.
E. Graph showing changes in protein levels (open symbols; n=3) or FTALLC interactions (solid bars; n=4-7) for PDIA4 in HEK293DAX cells following stress-independent XBP1s (red), ATF6 (blue), or XBP1s and ATF6 (green) activation. Error bars show SEM for the individual replicates.
F. Graph showing changes in protein levels (open symbols; n=3) or FTALLC interactions (solid bars; n=4-7) for HYOU1 in HEK293DAX cells following stress-independent XBP1s (red), ATF6 (blue), or XBP1s and ATF6 (green) activation. Error bars show SEM for the individual replicates.
Despite this general correlation, increased expression of ER proteostasis factors does not appear sufficient to increase FTALLC interactions. This is evident by monitoring the recovery of the high confidence LC interactor DNAJC3 in FTALLC IPs (Fig. 4A,B). DNAJC3 is an ER HSP40 co-chaperone that binds to misfolded proteins within the ER and directs them to the ER HSP70 BiP for ATP-dependent chaperoning (Petrova et al., 2008; Rutkowski et al., 2007). ATF6 activation increases the expression of DNAJC3 >2-fold; however, we observe no significant increase in the association between DNAJC3 and FTALLC by AP-MS (Fig. 4A,B and Fig. S4A). This suggests that while ATF6-dependent increases in the expression of ER proteostasis factors such as BiP or GRP94 are important for dictating their increased interactions with FTALLC, increased expression does not appear sufficient to increase these interactions.
ATF6 and XBP1s induce overlapping, but distinct, subsets of ER proteostasis factors (Shoulders et al., 2013; Yamamoto et al., 2004). This provides a unique opportunity to identify key ER proteostasis factors specifically required for ATF6-dependent reductions in ALLC secretion. Towards that aim, we compared XBP1s-dependent changes in ER proteostasis factor expression to changes in their interaction with FTALLC. Unlike what we observed with ATF6 activation, XBP1s-dependent ER proteostasis factor expression does not correlate with FTALLC interactions (Fig. 4C). However, co-activation of XBP1s and ATF6 largely restored the correlation between ER proteostasis factor expression and FTALLC interactions (Fig. 4D). Interestingly, specific ER proteostasis factors such as HYOU1 and PDIA4 were transcriptionally induced by XBP1s or ATF6 activation alone, but only show increased interactions with FTALLC following ATF6 activation (Fig. 4E,F). This is in contrast to other ER proteostasis factors such as BiP and GRP94 that are primarily regulated by ATF6 and show increased association with FTALLC following ATF6 activation (Fig. S4B,C). The inability for XBP1s-dependent upregulation of PDIA4 and HYOU1 to increase interactions with FTALLC suggests that the increased expression of these ER proteostasis factors is not sufficient to influence LC ER quality control. Instead, these results suggest increased targeting to ATF6-regulated, ATP-dependent chaperones such as BiP and GRP94 is primarily responsible for the ATF6-dependent increase in LC ER quality control.
Overexpression of specific ER proteostasis factors recapitulates selective, ATF6-dependent reductions in destabilized LC secretion
Many of the ER proteostasis factors found to increase interactions with destabilized FTALLC following ATF6 activation (e.g., BiP, GRP94, and ERdj3) were previously reported to function as ‘pro-folding’ factors for LCs within the ER. BiP and GRP94 function sequentially in the folding of LCs in the ER (Melnick et al., 1994). Furthermore, BiP and ERdj3 can bind multiple hydrophobic sites localized throughout a non-secreted LC, preventing its aggregation and/or premature degradation (Behnke et al., 2016). In contrast, other BiP co-chaperones such as ERdj4 and ERdj5 – neither of which is regulated by ATF6 (Shoulders et al., 2013) – bind rarer, aggregation-prone sequences within the LC to increase its targeting to degradation. This indicates that ATF6 activation induces selective remodeling of ER chaperoning pathways that increase targeting of LCs to ATF6-regulated ‘pro-folding’ factors.
Our results indicate that ATF6 activation increases LC targeting to these ‘pro-folding’ factors by increasing their expression. Thus, we predicated that overexpression of specific pro-folding chaperones should mimic the capacity for ATF6 activation to selectively reduce secretion of destabilized, aggregation-prone LCs. To test this prediction, we co-overexpressed FTALLC and the ATF6-regulated chaperones BiP, GRP94, or ERdj3 in HEK293DAX cells and evaluated FTALLC secretion by ELISA. In this experiment, we collected lysates and conditioned media from cells following 0 or 4 h incubation with cycloheximide (CHX) in fresh media. We then calculated fraction FTALLC secreted using the equation: fraction secreted = FTALLC media at t = 4h / FTALLC lysate at t = 0 h. Overexpression of BiP or GRP94 decreased FTALLC fraction secreted by >20% (Fig. 5A). In contrast, ERdj3 overexpression reduced FTALLC secretion by a more modest 10%, likely reflecting the modest intracellular increase in this co-chaperone afforded by overexpression owing to its efficient secretion to the media (Genereux et al., 2015). Similar results were observed by [35S] metabolic labeling (Fig. S5A,B). Importantly, we do not observe significant loss of FTALLC over a 4 h time course in our [35S] metabolic labeling experiment, indicating that the reduction in FTALLC secretion observed upon overexpression of ER chaperones does not correspond to an increase in degradation (Fig. S5C). This result is identical to that observed upon ATF6 activation and indicates overexpression of ER chaperones attenuate ALLC secretion through the same ER retention mechanism afforded by ATF6 activation (Cooley et al., 2014).
Figure 5. Overexpression of ATF6-regulated pro-folding ER proteostasis factors preferentially reduces ALLC secretion.
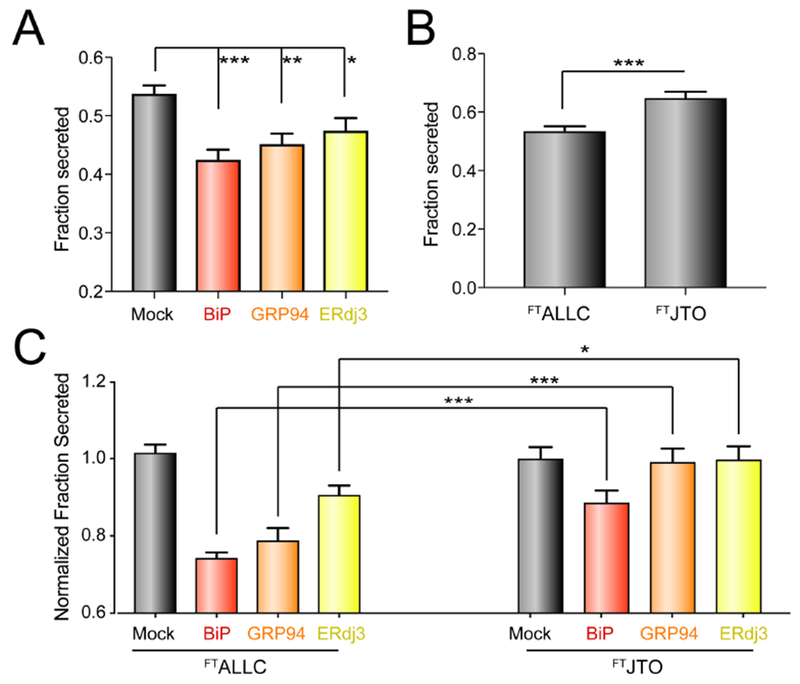
A. Graph showing the fraction secreted of FTALLC from HEK293DAX cells overexpressing the indicated ER proteostasis factor and treated with cycloheximide (CHX) for 0 or 4 h, as measured by ELISA. Fraction secreted was quantified using the following equation: fraction secreted = [FTALLC in media at t=4 h] / [FTALLC in lysate at t = 0 h]. Error bars show SEM for n > 14 replicates across > 4 independent experiments. *p<0.05, **p<0.01, ***p<0.005 for unpaired t-tests are shown.
B. Graph showing fraction secretion for FTALLC or FTJTO from HEK293DAX cells treated with CHX for 0 or 4 h, as measured by ELISA. Fraction secreted was calculated as described in Fig. 4A. Error bars show SEM for n>9 replicates across n>3 independent experiments. ***p<0.005 for unpaired t-test is shown
C. Graph showing the normalized fraction secreted of FTALLC or FTJTO from HEK293DAX cells overexpressing the indicated ER chaperoning factor. Normalized fraction secreted was calculated by the following equation: fraction secretion in cells overexpressing a given chaperone / fraction secretion in mock-transfected cells. Fraction secreted was calculated as in Fig. 4A. Error bars show SEM for n > 9 replicates collected across > 3 independent experiments. *p<0.05, ***p<0.005 for unpaired t-tests are shown.
ATF6 activation selectively reduces secretion of destabilized, amyloidogenic ALLC relative to the energetically-normal, non-amyloidogenic JTO. Thus, we sought to define whether overexpression of BiP, GRP94, or ERdj3 influenced secretion of FTJTO using our ELISA assay (Cooley et al., 2014; Plate et al., 2016). ALLC and JTO are secreted from cells with different secretion efficiencies, reflecting differences in the ER quality control for LCs with distinct stabilities (Cooley et al., 2014) – a difference further supported herein by the differential interactions between these LCs and ER proteostasis factors defined by our AP-MS analysis (Fig. 3A, Fig. S3A–B). Consistent with this, we found that the fraction ALLC secreted measured by CHX/ELISA is less than that observed for the more stable JTO (Fig. 5B). Thus, in order to compare the secretion of ALLC and JTO in cells co-overexpressing specific ER proteostasis factors, we normalized the secretion of these two LCs to control cells overexpressing each LC alone. Using this approach, we show that overexpression of BiP modestly reduces FTJTO secretion; however, this reduction is significantly less than that observed for FTALLC (Fig. 5C). Alternatively, despite significantly reducing FTALLC secretion, neither GRP94 nor ERdj3 overexpression impacted FTJTO secretion (Fig. 5C). These results show that overexpression of these ER proteostasis factors preferentially reduce secretion of the destabilized, amyloidogenic ALLC, mirroring the improved LC ER quality control observed upon ATF6 activation (Cooley et al., 2014).
BiP Overexpression only Partially Recapitulates the ATF6-Dependent Reduction in ALLC secretion
RNAi-depletion of core ER chaperones such as BiP or GRP94 activates the UPR, preventing us from defining the importance of these ER proteostasis factors for the ATF6-dependent reduction in destabilized ALLC secretion (Cooley et al., 2014). Instead, we evaluated how overexpression of the ATF6-regulated ER chaperone BiP influences ALLC secretion in the presence or absence of ATF6 activation. We selected BiP for this experiment because it is a core ER proteostasis factor whose overexpression reduces ALLC secretion to the greatest extent (Fig. 5C). Interestingly, the 20% reduction in ALLC secretion afforded by BiP overexpression is significantly less than the 40% reduction in secretion observed following ATF6 activation (Fig. 6A). The combination of BiP overexpression and ATF6 activation shows no further reduction in destabilized ALLC secretion, as compared to ATF6 activation alone (Fig. 6A). Similar results were observed by [35S] metabolic labeling where we show that the rate of ALLC secretion observed by ATF6 activation is less than that observed following BiP overexpression (Fig. 6B and Fig. S6A–C). These results indicate that overexpression of a core ER proteostasis factors only partially mimics the improved LC ER quality control observed following ATF6 activation and suggests that maximal reductions in ALLC secretion can only be achieved upon global, ATF6-dependent remodeling of ER quality control pathways.
Figure 6. Overexpression of BiP partially recapitulates ATF6-dependent reductions in ALLC secretion.
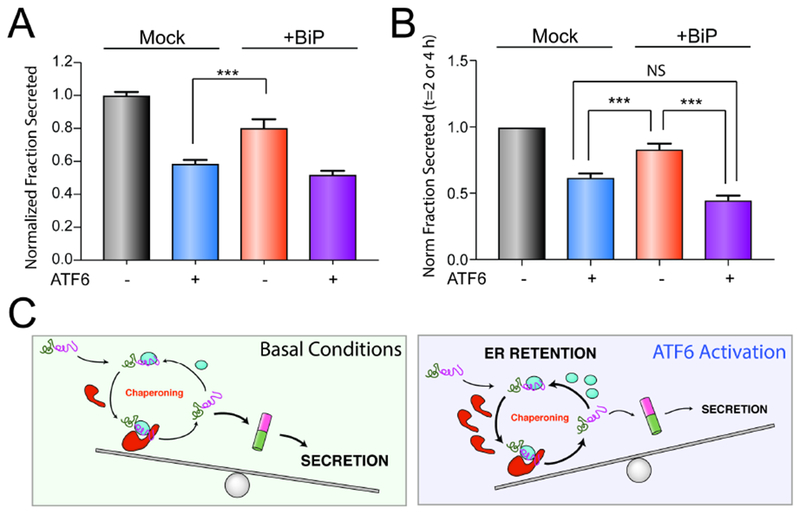
A. Graph showing the normalized fraction secretion of FTALLC in HEK293DAX cells mock-transfected or overexpressing BiP subjected to a 16 h pretreatment with vehicle or ATF6 activation, as measured by ELISA. ATF6 was activated in these cells using trimethoprim (TMP; 10 μM), as previously described (Shoulders et al., 2013). Error bars show SEM for n=6 replicates across two independent experiments. ***p<0.005 for unpaired t-test.
B. Graph showing the normalized fraction secretion of FTALLC at t = 2 or 4 h in HEK293DAX cells mock-transfected or overexpressing BiP subjected to 16 h pretreatment with vehicle or ATF6 activation, as measured by [35S] metabolic labeling. Representative autoradiograms are included in Fig. S6A. Error bars show SEM for n=4 separate measurements (2 measurements at t =2 h and 2 measurements at t=4 h) across two independent experiments. *p<0.05, ***p<0.005 for paired t-test.
C. Illustration showing a molecular model that explains the selective, ATF6-dependent reduction in destabilized ALLC secretion. The increased chaperoning environment afforded by ATF6 activation promotes iterative rounds of ALLC chaperoning to reduce ALLC folding into a trafficking competent conformation. This leads to increased ER retention of destabilized ALLC in chaperone-bound complexes that prevent its secretion to downstream secretory environments.
DISCUSSION
Here, we show that ATF6 activation improves ER quality control for destabilized LCs through increased targeting to select ‘pro-folding’ ER proteostasis factors. Interestingly, despite the fact that activating ATF6 selectively reduces secretion of destabilized LCs, ATF6 activation increases interactions between ‘pro-folding’ ER proteostasis factors and both destabilized (e.g., ALLC) and stable (e.g., JTO) LCs. This suggests the increased activity of these ‘pro-folding’ factors improves their capacity to ‘read-out the energetic stability of LCs and more efficiently regulate their ER quality. A potential explanation for this effect is that increased targeting to ‘pro-folding’ ER proteostasis factors increases iterative rounds of chaperone-assisted folding that selectively prevents destabilized, amyloidogenic LCs such as ALLC from adopting a secretion-competent conformation (Fig. 6C). In this model, destabilized ALLC is unable to complete its folding upon release from ER chaperoning pathways. Instead, the enhanced activity of these proteostasis factors afforded by ATF6 activation promotes reengagement of ALLC prior to folding, preventing trafficking to downstream secretory environments. This reengagement of destabilized ALLC with ‘pro-folding’ factors similarly prevents targeting to degradation pathways, resulting in the ER retention observed following ATF6 activation (Cooley et al., 2014). In contrast, energetically normal LCs such as JTO can efficiently fold following release from chaperoning pathways in the ATF6-remodeled ER environment due to its increased stability relative to ALLC (Cooley et al., 2014; Morgan and Kelly, 2016). This allows JTO to adopt a trafficking-competent conformation that can then be secreted to the extracellular space. Thus, while ATF6 activation increases interactions between JTO and select ER proteostasis factors, the capacity for this energetically-normal LC to fold following release from ER chaperones prevents ATF6 activation from significantly impairing its secretion. This indicates that the selective, ATF6-mediated remodeling of ‘pro-folding’ LC chaperoning pathways provides a unique opportunity to engage non-native LC conformations through interactions with multiple ER chaperones and co-chaperones to selectively reduce secretion of destabilized LCs implicated in AL disease pathogenesis.
Interestingly, overexpression of specific ER chaperones such as BiP only partially mimic the increases in LC ER quality afforded by ATF6 activation. This highlights a unique advantage for targeting endogenous transcriptional signaling pathways such as ATF6 to influence ER quality control for disease-associated proteins, as compared to targeting the activity of specific chaperones. The ATF6 transcriptional signaling pathway evolved to restore ER quality and function following diverse types of ER insults. As such, ATF6 regulates a distinct subset of ER proteostasis factors that can coordinate to impact ER quality control, providing an optimized environment to selectively influence the secretion of destabilized, amyloidogenic proteins such as amyloidogenic LCs. Consistent with this, our results show that ATF6 activation improves LC ER quality control to greater extents to that achieved by overexpression of specific ER proteostasis factors such as BiP or GRP94. This reflects the more global, ATF6-dependent remodeling of the ALLC interactome described herein, where ATF6 activation increases the interactions between FTALLC and multiple ‘pro-folding’ ER proteostasis factors.
The capacity for ATF6 activation to optimize ER proteostasis remodeling to improve LC ER quality control suggests that pharmacologic ATF6 activation provides an opportunity to reduce the secretion and subsequent aggregation of amyloidogenic LCs and other amyloidogenic proteins involved in diverse diseases (Plate and Wiseman, 2017). Interestingly, small molecule ER proteostasis regulators that activate ATF6 in AL patient-derived plasma cell models increase ALLC targeting to ER chaperones including BiP and GRP94 and reduce ALLC secretion (Plate et al., 2016), indicating that pharmacologic targeting of ER proteostasis can reduce ALLC secretion through a mechanism analogous to that described herein. Stress-independent ATF6 activation also reduces the secretion and toxic aggregation of destabilized variants of multiple other disease-associated proteins including TTR, rhodopsin, and α1-antitrypsin (Chen et al., 2014; Chiang et al., 2012; Shoulders et al., 2013; Smith et al., 2011b). Thus, our results defining the global remodeling of ALLC interactions afforded by ATF6 activation provides a molecular basis to deconvolute the impact of ATF6 activation on the ER quality control for these and other disease-relevant proteins. Our results also further motivate the discovery and development of pharmacologic ATF6 activating compounds that have the potential to ameliorate the aberrant secretion and toxic aggregation of destabilized, aggregation-prone proteins implicated in etiologically-diverse protein aggregation diseases(Plate and Wiseman, 2017).
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, R. Luke Wiseman (wiseman@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Transfections
The creation and maintenance of HEK293DAX cells has been described previously (Shoulders et al., 2013). Briefly, HEK293DAX cells were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM; Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (Gibco), 100 U*mL−1 penicillin, and 100 μg*mL”1 streptomycin (Gibco). All cells were cultured under typical tissue culture conditions (37°C, 5% CO2). Cells were routinely tested for mycoplasma every 6 months. No further authentication of cell lines was performed by the authors. Cells were transfected using calcium phosphate precipitation, as previously described (Shoulders et al., 2013). All plasmids for transfection were prepared using the Qiagen Midiprep kit according to the manufacturers protocol. For SILAC experiments, the SILAC Protein Quantification kit – DMEM (ThermoFisher) was purchased. In addition to the supplied 13C6-L-Lys, the heavy media was also supplemented with 13C6,15N4-L-Arg (Cambridge Isotopes Laboratories, Inc.). Light- and heavy-labeled HEK293DAX cells were generated for a previously study (Shoulders et al., 2013). Cells were cultured additionally for a minimum of 5 passages in heavy SILAC DMEM media prior to transfection to ensure full heavy-isotope incorporation.
METHOD DETAILS
Plasmids and Antibodies
Plasmids expressing FTALLC, FTJTO, or untagged ALLC in the pCMV1 or pDEST40 vector were described previously (Cooley et al., 2014). FTBiP and ERdj3 overexpression plasmids were used as described previously (Genereux et al., 2015). The GRP94 overexpression plasmid was prepared using GRP94.pDONR223 (Addgene; Cat #82130), which was recombined into pDEST40 using Gateway cloning according to the manufacturers protocol. Primary antibodies were acquired from commercial sources and used at the indicated dilutions in Antibody Buffer (50 mM Tris [pH 7.5], 150 mM NaCl supplemented with 5% BSA and 0.1% NaN3). Mouse monoclonal antibodies were used to detect KDEL (1:1000, Enzo Life Sciences), M2 anti-FLAG (1:500, Sigma Aldrich), Tubulin [B-5-1-2] (1:4000,Sigma), BiP/GRP-28 (1:500, Santa Cruz Biotechnology), β-actin (1:10000, Sigma Aldrich). Polyclonal rabbit antibodies were used to detect GRP94 (1:1000, GeneTex), HYOU1 (1:1000, GeneTex), ERdj3 (DNAJB11) (1:1000, ProteinTech), PDIA4 (1:1000, ProteinTech), PDIA6 (1:1000, GenTex), PDIA3 (ERp57) (1:1000, CellSignaling).
Affinity-purification of LC samples and MS Sample Preparation
In general, a 10 cm tissue culture plate of HEK293DAX cells was transfected with the appropriate LC expression plasmids and a fully confluent plate (approximately 107 cells) was used per condition. Cell harvest, cross-linking, lysis and co-immunoprecipitation were carried out as described in the Supplemental Materials and Methods. Proteins were eluted from anti-M1 FLAG agarose beads (Sigma) twice in 75μL elution buffer (10mM Tris [pH 7.5], 2% SDS, 1mM EDTA) by heating to 95°C for 5 min. Eluted fractions were combined and proteins were precipitated in methanol/chloroform, washed twice in methanol, and then air dried. For SILAC experiments, protein pellets were resuspended in 50μL 8M urea, 50mM Tris pH 8.0, reduced with 10mM TCEP (ThermoFisher) for 30 min at room temperature, and alkylated with 12 mM iodoacetamide (Sigma) for 30min in the dark. Samples were then diluted four-fold in 50mM Tris to lower the urea concentration. For TMT experiments, the protein pellets were resuspended in 3 – 5μL 1% RapiGest SF Surfactant (Waters) followed by addition of HEPES buffer (pH 8.0, 50 mM) to a volume of 50μL. Samples were reduced with 5mM TCEP for 30min at room temperature and alkylated with 10mM iodoacetamide for 30min in the dark. Trypsin (0.5μg, Sequencing grade, Promega) was then added to the SILAC or TMT samples and incubated for 16 hours at 37°C while shaking. After digestion, SILAC peptides samples were acidified with formic acid (5% final concentration) and directly proceeded to LC-MS analysis. TMT samples were first reacted with NHS-modified TMT sixplex reagents (ThermoFisher) in 40% v/v acetonitrile and incubated for 60 min at room temperature. Reactions were then quenched by addition of 0.4% (w/v) ammonium bicarbonate. The digested and labeled samples for a given sixplex experiment were pooled and acidified with formic acid (5% final concentration). Samples were concentrated on a SpeedVac and rediluted in buffer A (94.9% water, 5% acetonitrile, 0.1 formic acid, v/v/v). Cleaved Rapigest SF and debris was removed by centrifugation for 30min at 18,000× g.
Mass Spectrometry and Interactome Characterization
MuDPIT microcolumns were prepared as described (Fonslow et al., 2012), peptide samples were directly loaded onto the columns using a high-pressure chamber (Shotgun Proteomics Inc), and the columns were washed for 30min with buffer A. LC-MS/MS analysis was performed using a Q-Exactive mass spectrometer equipped with an EASY nLC 1000 (Thermo Fisher). MuDPIT experiments were performed by 10 pL sequential injections of 0, 20, 50, 80, 100% buffer C (500 mM ammonium acetate in buffer A) and a final step of 90% buffer C / 10% buffer B (19.9% water, 80% acetonitrile, 0.1% formic acid, v/v/v) and each step followed by a gradient from buffer A to buffer B on a 18 cm fused silica microcapillary column (ID 100μm) ending in a laser-pulled tip filled with Aqua C18, 3μm, 100Å resin (Phenomenex). Electrospray ionization (ESI) was performed directly from the analytical column by applying a voltage of 2.5 kV with an inlet capillary temperature of 275Ό. Data-dependent acquisition of MS/MS spectra was performed with the following settings: eluted peptides were scanned from 400 to 1800 m/z with a resolution of 70,000 and the mass spectrometer in a data dependent acquisition mode. The top ten peaks for each full scan were fragmented by HCD using normalized collision energy of 30%, 2.0 m/z isolation window, 120 ms max integration time, a resolution of 7500, scanned from 100 to 1800 m/z, and dynamic exclusion set to 60s. Peptide identification and SILAC- or TMT-based protein quantification was performed using the Integrated Proteomics Pipeline Suite IP2 (Integrated Proteomics Applications, Inc.) and modules ProLuCID, DTASelect and Census (Park et al., 2014; Tabb et al., 2002; Xu et al., 2015). MS2 spectra were extracted from Thermo XCalibur .raw file format using RawConverter (He et al., 2015). Spectra were searched using ProLuCID against a Uniprot human proteome database (release date 05/2014). The database was curated to remove redundant protein and splice-isoforms, and the sequences for the variable domains of FTALLC and FTJTO and the shared constant domain were added. Searches were carried out using a decoy database of reversed peptide sequences and the following search parameters: 50 ppm peptide precursor tolerance, 0.6 Da fragment mass tolerance, 6 amino acid minimum peptide length, trypsin cleavage (max. 2 missed cleavage events), static Cys modification of 57.0215 (carbamidomethylation), and static N-terminal and Lys modification of 229.1629 (TMT-sixplex). ProLuCID search results were filtered using DTASelect using combined XCorr and DeltaCN scores to minimize the peptide false discovery rate at 1% and minimum of 2 peptides per protein ID. TMT reporter ion intensities were extracted in Census using a mass tolerance of 0.05 Da and summed for individual peptides belonging to the same protein. SILAC data was processed similarly, except static modification from the TMT-sixplex were omitted, and heavy [15N, 13C]-Lys and Arg modifications were included in the ProLuCID search.
Light Chain ELISA
Transfected HEK293DAX were plated 150,000 cells/well in 2 identical 48-well plates (Genessee Scientific) containing 500 μL of media. Media was removed and wells were washed two times with 250 μL media containing 50 μg/mL cycloheximide (CHX). One plate was washed two times with 1× PBS and cell lysates prepared in RIPA buffer. This sample was used to monitor lysate levels of LC at t=0 h. The second plate was incubated 250 μL media with CHX for 4 h and conditioned media was collected. This sample was used to monitor secreted LC levels at 4 h. Free LC concentrations were determined by ELISA in 96-well plates (Immulon 4HBX, Thermo Fisher), as previously described (Cooley et al., 2014; Plate et al., 2016). Briefly, wells were coated overnight at 37 °C with sheep polyclonal free λ LC antibody (Bethyl Laboratories, A80-127A) at a 1:500 dilution in 50 mM sodium carbonate (pH 9.6). In between all incubation steps, the plates were rinsed extensively with Tris-buffered saline containing 0.05% Tween-20 (TBST). Plates were blocked with 5% non-fat dry milk in TBST for 1 hr at 37°C. Media analytes were diluted between 5 – 200-fold in 5% non-fat dry milk in TBST and 100 μL of each sample was added to individual wells. Light chain standards ranging from 3 – 300 ng/mL were prepared from purified human Bence Jones λ light chain (Bethyl Laboratories, P80-127). Plates were incubated at 37 °C for 1.5 h with shaking. Finally, HRP-conjugated goat anti-human λ light chain antibody (Bethyl Laboratories, A80-116P) was added at a 1: 5,000 dilution in 5% non-fat dry milk in TBST, followed by a 1.5 h incubation of the plates at 37 °C. The detection was carried out with 2,2’-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, 0.18 mg/mL) and 0.03% hydrogen peroxide in 100 mM sodium citrate pH 4.0. Detection solution (100 μL) was added to each well and the plates were incubated at room temperature. The absorbance was recorded at 405 nm and the values for the LC standards were fitted to a 4-parameter logistic function. LC concentrations were averaged from at least 3 independent replicates under each treatment and then normalized to vehicle conditions. Fraction secreted was then calculated using the equation: fraction secreted = [LC] in media at t=4 h / [LC] lysate at t = 0 h.
Immunoblotting, SDS-PAGE and Immunoprecipitation
Cell lysates were prepared as previously described in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1 % SDS, 1% Triton X-100, 0.5% deoxycholate and protease inhibitor cocktail (Roche). Total protein concentration in cellular lysates was normalized using the Bio-Rad protein assay. Lysates were then denatured with 1X Laemmli buffer + 100 mM DTT and boiled before being separated by SDS-PAGE. Samples were transferred onto nitrocellulose membranes (Bio-Rad) for immunoblotting and blocked with 5% milk in Tris-buffered saline, 0.5 % Tween-20 (TBST) following incubation overnight at 4°C with primary antibodies. Membranes were washed in TBST, incubated with IR-Dye conjugated secondary antibodies and analyzed using Odyssey Infrared Imaging System (LI-COR Biosciences). Quantification was carried out with LI-COR Image Studio software. For immunoprecipitations, cells were washed with PBS and then treated with the indicated concentration Dithiobis(succinimidiyl propionate) (DSP) for 30 min at room temperature. The crosslinking reaction was quenched by addition of 100 mM Tris pH 7.5 for 15 min, then lysates were prepared in RIPA buffer. Total protein concentration in cellular lysates was normalized using Bio-Rad protein assay. Cell lysates were then subjected to preclearing with Sepharose 4B beads (Sigma) at 4 °C for 1 h with agitation. The precleared lysates were then subjected to immunoprecipitation with a M1 anti-Flag agarose resin (Sigma) at 4 °C overnight. After four washes in RIPA buffer, proteins were eluted by boiling in 6× Laemmli buffer and 100 mM DTT. Blots from IPs and inputs were probed with the primary antibodies. Membranes were then treated as described above.
[35S] Metabolic Labeling
[35S] metabolic labeling experiments were performed as previously described (Cooley et al., 2014; Shoulders et al., 2013). Briefly, transfected HEK293DAX were plated on poly-D-lysine coated 6-well plates and metabolically labeled in DMEM-Cys/-Met (Corning CellGro, Mediatech Inc., Manassas, VA) supplemented with glutamine, penicillin/streptomycin, dialyzed fetal bovine serum, and EasyTag EXPRESS [35S] Protein Labeling Mix (Perkin Elmer) for 30 min. Cells were washed twice with complete media and incubated in pre-warmed DMEM for the indicated times. Media or lysates were harvested at the indicated times. Lysates were prepared in RIPA buffer (50mM Tris [pH 7.5], 150mM NaCl, 1% Triton X100, 0.5% sodium deoxycholate, 0.1% SDS) containing proteases inhibitors cocktail (Roche). FLAG-tagged LC variants were immunopurified using M1 anti-FLAG agarose beads (Sigma Aldrich) and washed four times with RIPA buffer. The immunoisolates were then eluted by boiling in 6X Laemmli buffer and separated on 12% SDS-PAGE. Gels were stained with Coomassie Blue, dried, exposed to phosphorimager plates (GE Healthcare, Pittsburgh, PA) and imaged by autoradiography using a Typhoon Trio Imager (GE Healthcare). Band intensities were quantified by densitometry in ImageQuant. Fraction secreted was calculated using the equation: fraction secreted = [extracellular [35S]-LC signal at t / (extracellular [35S]-LC signal at t=0 + intracellular [35S]-LC signal at t=0)]. Fraction remaining was calculated using the equation: [(extracellular [35S]-LC signal at t + intracellular [35S]-LC signal at t) / (extracellular [35S]-LC signal at t=0 + intracellular [35S]-LC signal at t=0)].
QUANTIFICATION AND STATISTICAL ANALYSIS
Mass Spectrometry Interactomics TMT and SILAC Quantification
Quantification of heavy and light peptide intensities was carried out in Census using MS1 spectral files extracted using RawConverter. Data normalization for TMT reporter intensities and SILAC ratios was carried out manually. For SILAC experiments, the SILAC heavy/light ratios for each quantified protein were normalized to the ratio observed for FTALLC or FTJTO. Each experiment represented a comparison of an experimental condition (light sample) against a common heavy reference sample (FTALLC, vehicle treated). Comparisons between experimental conditions were expressed as ratios of the LC-normalized SILAC ratios. For TMT experiments, the unnormalized TMT reporter ion intensities for each quantified protein were normalized against the intensities observed for FTALLC according to the following formula: (1) , where and are the normalized and unnormalized TMT intensities, respectively, for a given protein n in the TMT channels i-j. Channels that did not contain LC (e.g. control transfections with untagged ALLC) were omitted from the normalization. For interactome comparison between FTALLC and FTJTO, only shared peptides from the λ Vc constant domain were considered for the normalization. Interaction fold changes were expressed as log2 differences of the normalized TMT intensities for a given protein between respective TMT channels (experimental conditions), according to the following formula: (2) . The mean of the log2 interaction difference was calculated from multiple MuDPIT LC-MS runs, which each represented an individual biological replicate. Significance of interaction differences was assessed by a two-tailed unpaired student’s t-test of the normalized log2-transformed TMT intensities, followed by multiple-testing correction via FDR estimation using the method of Storey et al. (Storey and Tibshirani, 2003).
Statistical Analysis of Biochemical Experiment
Data are presented as mean ± SEM and were analyzed by two-tailed Student’s t test to determine significance, unless otherwise indicated.
DATA AND SOFTWARE AVAILABILITY
The datasets for the mass spectrometry interactomics experiments showing protein identifications and quantifications are included as supplemental tables:
Supplemental Table 1 (Supplement to Figure 1). Excel spreadsheets including the interactome data comparing the interactions between ER proteostasis factors and either FTLC (combined replicates of FTALLC and FTJTO) or untagged ALLC. Two sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance and 2) a sheet containing all of the raw data for the included analyses.
Supplemental Table 2 (Supplement to Figure 2). Excel spreadsheets describing the interactome data comparing interactions between ER proteostasis factors and FTALLC following stress-independent XBP1s and/or ATF6 activation in HEK293DAX cells. Two sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance and 2) a sheet containing all of the raw data for the included analyses.
Supplemental Table 3 (Supplement to Figure 3). Excel spreadsheet describing the interactome data comparing the interactions between ER proteostasis factors and FTALLC and FTJTO in HEK293DAX cells or FTJTO in HEK293DAX cells following stress-independent ATF6 activation. Four sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance comparing the interaction ratios between FTALLC and FTJTO and 2) a sheet containing all of the raw data used to compare the interactomes of FTALLC and FTJTO, 3) a summary sheet including only the final TMT ratios and significance comparing the interaction ratios for FTJTO in the absence or presence of ATF6 activation in HEK293DAX cells and 4) a sheet containing all of the raw data used to compare the interactome FTJTO in the presence or absence of ATF6 activation.
Supplemental Table 4 (Supplement to Figure 4). Excel spreadsheets comparing changes in the mRNA or protein levels and FTALLC interactions for high confidence LC interacting proteins in HEK293DAX cells following stress-independent activation of ATF6, XBP1s, or ATF6 and XBP1s co-activation. This table contains four sheets. Data for changes in mRNA or protein levels in HEK293DAX cells following these treatments is from (Plate et al., 2016; Shoulders et al., 2013).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KDEL (mouse monoclonal 10C3) | Enzo Life Sciences | Cat#ADI-SPA-827-D |
| M2 anti-FLAG (mouse monoclonal) | Sigma Aldrich | Cat#F3165 |
| Anti-α-Tubulin (mouse monoclonal B-5-1-2) | Sigma Aldrich | Cat#T5168 |
| BiP/GRP-78 (mouse monoclonal E-4) | Santa Cruz Biotechnology | Cat#sc-166490 |
| β-actin (mouse monoclonal AC-15) | Sigma Aldrich | Cat#A1978 |
| GRP94 (rabbit polyclonal N1N3) | GenTex | Cat#GTX103203 |
| HYOU1 (ORP150) (rabbit polyclonal C2C3) | GenTex | Cat#GTX102255 |
| ERdj3 (DNAJB11) (rabbit polyclonal) | ProteinTech | Cat#15484-1-AP |
| PDIA4 (ERp72) (rabbit polyclonal) | ProteinTech | Cat#14712-1-AP |
| PDIA6 (rabbit polyclonal N1N3) | GenTex | Cat#GTX121275 |
| PDIA3 (ERp57) (rabbit polyclonal G117) | CellSignaling | Cat#2881S |
| free λ LC antibody (sheep polyclonal) | Bethyl Laboratories | Cat#P80-127 |
| HRP-conjugated goat anti-human λ light chain antibody | Bethyl Laboratories | Cat#A80-116P |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 13C6,15N4-L-Arginine | Cambridge Isotopes Laboratories | Cat#CNLM-539-H |
| Anti-FLAG M1 agarose affinity gel | Sigma Aldrich | Cat#A4596-5ML |
| Tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) | ThermoFisher | Cat#20490 |
| Trypsin (Sequencing grade modified) | Promega | Cat#5111 |
| Iodoacetamide | Sigma Aldrich | Cat#I6125 |
| RapiGest SF Surfactant | Waters | Cat#186001861 |
| purified human Bence Jones λ light chain | Bethyl Laboratories | Cat#P80-127 |
| EasyTag EXPRESS [35S] Protein Labeling Mix | PerkinElmer | Cat#NEG772002MC |
| Critical Commercial Assays | ||
| SILAC Protein Quantification kit – DMEM | ThermoFisher | Cat#A33969 |
| TMTsixplex Isobaric Label Reagent Set | ThermoFisher | Cat#90066 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HEK293DAX cells | this group, Shoulders et al., 2013) | HEK293DAX |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Recombinant DNA | ||
| Mammalian expression plasmid (pCMVI) for FTALLC | this group, Cooley et al., 2014 | ss.FT.ALLC-pCMV1 |
| Mammalian expression plasmid (pCMVI) for FTJTO | this group, Cooley et al., 2014 | ss.FT.JTO-pCMV1 |
| Mammalian expression plasmid (pDEST40) for untagged ALLC | this group, Cooley et al., 2014 | ss.ALLC-pDEST40 |
| Entry plasmid (gateway cloning) for GRP94, GRP94.pDONR223 | Addgene | Cat #82130 |
| Mammalian expression plasmid (pDEST40) for GRP94 | this paper, Genereux et al., 2015 | GRP94.pDEST40 |
| Mammalian expression plasmid for FTBiP | this group, Genereux et al., 2015 | pFLAG.BiP.WT |
| Mammalian expression plasmid (pDEST40) for ERdj3 | this group, Genereux et al., 2015 | ss.ERdj3.pDEST40 |
| Software and Algorithms | ||
| Integrated Proteomics Pipeline Suite (IP2) | Integrated Proteomics Applications, Inc. | N/A |
| RawConverter | He et al., 2015 | N/A |
| ProLuCID (IP2 Suite) | Xu et al., 2015 | N/A |
| DTASelect (IP2 Suite) | Tabb et al., 2002 | N/A |
| Census (IP2 Suite) | Park et al., 2014 | N/A |
| Other | ||
SIGNIFICANCE.
Remodeling of endoplasmic reticulum (ER) proteostasis pathways has emerged as a promising strategy to correct protein quality control defects associated with diverse protein misfolding diseases such as Light Chain Amyloidosis. In particular, remodeling ER proteostasis pathways through stress-independent activation of the unfolded protein response-associated transcription factor ATF6 has been shown to reduce secretion and extracellular aggregation of destabilized, amyloidogenic proteins, while not affecting secretion of the stable wild-type proteome. However, the molecular mechanism by which ATF6 activation influences amyloidogenic protein secretion remains poorly defined. ATF6 transcriptionally regulates a subset of ER chaperones and quality control factors, yet the molecular details of how these factors enhance ER quality control decisions and prevent the secretion of destabilized, amyloidogenic proteins has been elusive. Here, we use a multiplexed quantitative proteomics platform to define the molecular mechanism by which ATF6 activation selectively influences secretion of the destabilized, amyloidogenic immunoglobulin light chain ALLC. We show that ATF6 activation increases interactions between ALLC and ER chaperones that increases retention of this destabilized protein within the ER. These increased interactions are mediated through ATF6-dependent transcriptional upregulation of ER chaperones and reflects remodeling of ER proteostasis pathways afforded by ATF6 activation. Interestingly, overexpression of specific ATF6-regulated chaperones only partially recapitulates the reduced secretion of ALLC afforded by ATF6 activation, highlighting the benefit of global remodeling of ER proteostasis for improving ER quality control of destabilized proteins through activation of endogenous stress-responsive signaling pathways. These results inform on the utility of therapeutic strategies targeting stress-independent activation of the ATF6 pathway to reduce secretion of destabilized protein variants associated with diverse protein aggregation diseases.
HIGHLIGHTS.
Quantitative proteomics defines the interactome for the amyloidogenic light chain ALLC
ATF6 or XBP1s activation distinctly impact interactions between ALLC and ER proteins
ATF6 activation reduces ALLC secretion through increased targeting to ER chaperones
Enhanced quality control is based on global interaction changes coordinated by ATF6
ACKNOWLEDGEMENTS
We thank Evan Powers for critical reading of the manuscript. We thank NIH (DK107604, RLW) for financial support. LP and BR were both supported by Leukemia and Lymphoma Society postdoctoral fellowships. JCG was supported by an American Heart Association postdoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
The authors declare that they have no competing interest.
REFERENCES
- Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, and Mori K (2008). ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct 33, 75–89. [DOI] [PubMed] [Google Scholar]
- Arendt BK, Ramirez-Alvarado M, Sikkink LA, Keats JJ, Ahmann GJ, Dispenzieri A, Fonseca R, Ketterling RP, Knudson RA, Mulvihill EM, et al. (2008). Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood 112, 1931–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Behnke J, Feige MJ, and Hendershot LM (2015). BiP and its nucleotide exchange factors Grp170 and Sil1: mechanisms of action and biological functions. J Mol Biol 427, 1589–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, and Hendershot LM (2016). Members of the Hsp70 Family Recognize Distinct Types of Sequences to Execute ER Quality Control. Mol Cell 63, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blancas-Mejia LM, and Ramirez-Alvarado M (2013). Systemic amyloidoses. Annu Rev Biochem 82, 745–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, and Bulleid NJ (2011). Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem 80, 71–99. [DOI] [PubMed] [Google Scholar]
- Budayeva HG, and Cristea IM (2014). A mass spectrometry view of stable and transient protein interactions. Adv Exp Med Biol 806, 263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Genereux JC, Qu S, Hulleman JD, Shoulders MD, and Wiseman RL (2014). ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem Biol 21, 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang WC, Hiramatsu N, Messah C, Kroeger H, and Lin JH (2012). Selective activation of ATF6 and PERK endoplasmic reticulum stress signaling pathways prevent mutant rhodopsin accumulation. Invest Ophthalmol Vis Sci 53, 7159–7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, and Kopito RR (2011). Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 14, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole KS, Grandjean JMD, Chen K, Witt CH, O’Day J, Shoulders MD, Wiseman RL, and Weerapana E (2018). Characterization of an A-Site Selective Protein Disulfide Isomerase A1 Inhibitor. Biochemistry 57, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley CB, Ryno LM, Plate L, Morgan GJ, Hulleman JD, Kelly JW, and Wiseman RL (2014). Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci U S A 111, 13046–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DP, Khurana R, Meredith S, Stevens FJ, and Argon Y (1999). Mapping the major interaction between binding protein and Ig light chains to sites within the variable domain. J Immunol 163, 3842–3850. [PubMed] [Google Scholar]
- Feige MJ, and Buchner J (2014). Principles and engineering of antibody folding and assembly. Biochim Biophys Acta 1844, 2024–2031. [DOI] [PubMed] [Google Scholar]
- Fonslow BR, Niessen SM, Singh M, Wong CC, Xu T, Carvalho PC, Choi J, Park SK, and Yates JR 3rd (2012). Single-step inline hydroxyapatite enrichment facilitates identification and quantitation of phosphopeptides from mass-limited proteomes with MudPIT. J Proteome Res 11, 2697–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, and Wiseman RL (2015). Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J 34, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, and Brodsky JL (2012). The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92, 537–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Diedrich J, Chu Y-Y, and Yates JR (2015). Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. In Anal Chem, pp. 11361–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman R, Vanhove M, Lejeune A, Stevens FJ, and Hendershot LM (1999). The in vivo association of BiP with newly synthesized proteins is dependent on the rate and stability of folding and not simply on the presence of sequences that can bind to BiP. J Cell Biol 144, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, and Saxena S (2017). ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13, 477–491. [DOI] [PubMed] [Google Scholar]
- Hosp F, Vossfeldt H, Heinig M, Vasiljevic D, Arumughan A, Wyler E, Landthaler M, Hubner N, Wanker EE, Lannfelt L, et al. (2015). Quantitative Interaction Proteomics of Neurodegenerative Disease Proteins In Cell Reports (Cell Press; ), pp. 1134–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TA, Watson S, Eiden JJ, and Betenbaugh MJ (1996). Rescue of immunoglobulins from insolubility is facilitated by PDI in the baculovirus expression system. Protein Expr Purif 7, 281–288. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex Network: A Systematic Exploration of the Human Interactome. In Cell (Elsevier), pp. 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrina Meyer MS (2015). Quantitative affinity purification mass spectrometry: a versatile technology to study protein–protein interactions. In Front Genet (Frontiers Media SA), pp. 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kean MJ, Couzens AL, and Gingras AC (2012). Mass spectrometry approaches to study mammalian kinase and phosphatase associated proteins. Methods 57, 400–408. [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, and Hartl FU (2013). Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82, 323–355. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, and Glimcher LH (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23, 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomant AJ, and Fairbanks G (1976). Chemical probes of extended biological structures: synthesis and properties of the cleavable protein cross-linking reagent [35S]dithiobis(succinimidyl propionate). J Mol Biol 104, 243–261. [DOI] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, et al. (2012). Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. In Anal Chem, pp. 7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch mS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. In Anal Chem, pp. 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick J, Aviel S, and Argon Y (1992). The endoplasmic reticulum stress protein GRP94, in addition to BiP, associates with unassembled immunoglobulin chains. J Biol Chem 267, 21303–21306. [PubMed] [Google Scholar]
- Melnick J, Dul JL, and Argon Y (1994). Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370, 373–375. [DOI] [PubMed] [Google Scholar]
- Miteva YV, Budayeva HG, and Cristea IM (2013). Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal Chem 85, 749–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan GJ, and Kelly JW (2016). The Kinetic Stability of a Full-Length Antibody Light Chain Dimer Determines whether Endoproteolysis Can Release Amyloidogenic Variable Domains. J Mol Biol 428, 4280–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nittis T, Guittat L, LeDuc RD, Dao B, Duxin JP, Rohrs H, Townsend RR, and Stewart SA (2010). Revealing novel telomere proteins using in vivo cross-linking, tandem affinity purification, and label-free quantitative LC-FTICR-MS. Mol Cell Proteomics 9, 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S-E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002). Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. In Molecular & Cellular Proteomics (American Society for Biochemistry and Molecular Biology), pp. 376–386. [DOI] [PubMed] [Google Scholar]
- Ow SY, Salim M, Noirel J, Evans C, and Wright PC (2011). Minimising iTRAQ ratio compression through understanding LC-MS elution dependence and high-resolution HILIC fractionation. In Proteomics, pp. 2341–2346. [DOI] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Bamberger A, and Yates JR 3rd (2016). Deep interactome profiling of membrane proteins by co-interacting protein identification technology. Nat Protoc 11, 2515–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Martinez-Bartolome S, Lavallee-Adam M, Balch WE, and Yates JR 3rd (2015). F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528, 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papachristou EK, Kishore K, Holding AN, Harvey K, Roumeliotis TI, Chilamakuri CSR, Omarjee S, Chia KM, Swarbrick A, Lim E, et al. (2018). A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes In Nat Commun (Nature Publishing Group; ), pp. 986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SKR, Aslanian A, McClatchy DB, Han X, Shah H, Singh M, Rauniyar N, Moresco JJ, Pinto AFM, Diedrich JK, et al. (2014). Census 2: isobaric labeling data analysis In Bioinformatics (Oxford University Press; ), pp. 2208–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova K, Oyadomari S, Hendershot LM, and Ron D (2008). Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EmBO J 27, 2862–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, Cooley CB, Chen JJ, Paxman RJ, Gallagher CM, Madoux F, Genereux JC, Dobbs W, Garza D, Spicer TP, et al. (2016). Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L, and Wiseman RL (2017). Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy. Trends Cell Biol 27, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, and Balch WE (2009). Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem 78, 959–991. [DOI] [PubMed] [Google Scholar]
- Roumeliotis TI, Williams SP, Gongalves E, Alsinet C, Del Castillo Velasco-Herrera M, Aben N, Ghavidel FZ, Michaut M, Schubert M, Price S, et al. (2017). Genomic Determinants of Protein Abundance Variation in Colorectal Cancer Cells. In Cell Reports, pp. 2201–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Kang SW, Goodman AG, Garrison JL, Taunton J, Katze MG, Kaufman RJ, and Hegde RS (2007). The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell 18, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Mathieson T, Zinn N, Sweetman G, Doce C, Becher I, Pachl F, Kuster B, and Bantscheff M (2013). Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. In J Proteome Res, pp. 3586–3598. [DOI] [PubMed] [Google Scholar]
- Shen Y, and Hendershot LM (2005). ERdj3, a stress-inducible endoplasmic reticulum DnaJ homologue, serves as a cofactor for BiP’s interactions with unfolded substrates. Mol Biol Cell 16, 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR 3rd, Su AI, Kelly JW, and Wiseman RL (2013). Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep 3, 1279–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronek MH, Hendershot LM, and Haas IG (1998). The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc Natl Acad Sci U S A 95, 1574–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AL, Friedman DB, Yu H, Carnahan RH, and Reynolds AB (2011a). ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PLoS One 6, e16206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Granell S, Salcedo-Sicilia L, Baldini G, Egea G, Teckman JH, and Baldini G (2011b). Activating transcription factor 6 limits intracellular accumulation of mutant alpha(1)-antitrypsin Z and mitochondrial damage in hepatoma cells. J Biol Chem 286, 41563–41577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR (2002). DTASelect and Contrast: Tools for Assembling and Comparing Protein Identifications from Shotgun Proteomics. In J Proteome Res (American Chemical Society), Npp. 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, and Lindquist S (2014). A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. In Nat Methods, pp. 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, and Solomon A (1999). Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity. Biochemistry 38, 14101–14108. [DOI] [PubMed] [Google Scholar]
- Washburn MP, Wolters D, and Yates JR 3rd (2001). Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19, 242–247. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, and Balch WE (2007). An adaptable standard for protein export from the endoplasmic reticulum. Cell 131, 809–821. [DOI] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. In J Proteomics (Elsevier), pp. 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, and Mori K (2004). Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem 136, 343–350. [DOI] [PubMed] [Google Scholar]
- Yates JR, Ruse CI, and Nakorchevsky A (2009). Proteomics by mass spectrometry: approaches, advances, and applications. Annu Rev Biomed Eng 11, 49–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets for the mass spectrometry interactomics experiments showing protein identifications and quantifications are included as supplemental tables:
Supplemental Table 1 (Supplement to Figure 1). Excel spreadsheets including the interactome data comparing the interactions between ER proteostasis factors and either FTLC (combined replicates of FTALLC and FTJTO) or untagged ALLC. Two sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance and 2) a sheet containing all of the raw data for the included analyses.
Supplemental Table 2 (Supplement to Figure 2). Excel spreadsheets describing the interactome data comparing interactions between ER proteostasis factors and FTALLC following stress-independent XBP1s and/or ATF6 activation in HEK293DAX cells. Two sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance and 2) a sheet containing all of the raw data for the included analyses.
Supplemental Table 3 (Supplement to Figure 3). Excel spreadsheet describing the interactome data comparing the interactions between ER proteostasis factors and FTALLC and FTJTO in HEK293DAX cells or FTJTO in HEK293DAX cells following stress-independent ATF6 activation. Four sheets are included within this file: 1) a summary sheet including only the final TMT ratios and significance comparing the interaction ratios between FTALLC and FTJTO and 2) a sheet containing all of the raw data used to compare the interactomes of FTALLC and FTJTO, 3) a summary sheet including only the final TMT ratios and significance comparing the interaction ratios for FTJTO in the absence or presence of ATF6 activation in HEK293DAX cells and 4) a sheet containing all of the raw data used to compare the interactome FTJTO in the presence or absence of ATF6 activation.
Supplemental Table 4 (Supplement to Figure 4). Excel spreadsheets comparing changes in the mRNA or protein levels and FTALLC interactions for high confidence LC interacting proteins in HEK293DAX cells following stress-independent activation of ATF6, XBP1s, or ATF6 and XBP1s co-activation. This table contains four sheets. Data for changes in mRNA or protein levels in HEK293DAX cells following these treatments is from (Plate et al., 2016; Shoulders et al., 2013).