

Abstract
Objective
A well-behaved model chemistry previously validated for the study of the chemical reactivity of peptides was considered for the calculation of the molecular properties and structure of the Taltobulin anticancer peptide. A methodology based on Conceptual Density Functional Theory (CDFT) was chosen for the determination of the reactivity descriptors.
Results
The molecular active sites were associated with the active regions of the molecule were associated with the nucleophilic and electrophilic Fukui functions. Finally, the bioactivity scores for the Taltobulin peptide are predicted through a homology methodology relating them with the calculated reactivity descriptors.
Keywords: Taltobulin, Conceptual DFT, Bioactivity scores
Introduction
The biodiversity of the marine environment and the associated chemical diversity constitute a practically unlimited resource of new antitumor agents in the field of the development of marine bioactive substances [1].
Hemiasterlins comprise a small family of naturally occurring N-methylated tripeptides with highly alkylated unnatural amino acids, was originally isolated from the sponge Hemiasterella minor (class Demospongiae, order Hadromedidia, family Hemiasterllidae). Hemiasterlins act as potent tumor growth inhibitors [2, 3].
A synthetic analogue of hemiasterlin, taltobulin (HTI-286) has been synthesized in which the 3-substituted indole ring was replaced by phenyl group. Taltobulin inhibits the polymerization of purified tubulin and disrupts microtubule organization in cells. Then, it is considered as a potent inhibitor of proliferation and has substantially less interaction with multidrug resistance protein than currently used antimicrotubule agents [4].
Assuming that an understanding of the chemical interactions is essential for the development of new pharmaceutical drugs, in this work we will be studying the chemical reactivity properties of Talbodulin by resorting to the Conceptual DFT methodology, which will allow the determination of the global properties as well as the local properties for the prediction of the active reaction sites, both electrophilic and nucleophilic. In a similar way, the descriptors of bioactivity (bioactivity scores) will be established through a procedure described in the literature [5, 6] trying to relate them with the global and local CDFT reactivity descriptors that result from a calculation protocol based on DFT already validated by our group in previous research [7–14].
Main text
Computational methodology
The ChemAxon Calculator Plugins for Conformers Searching available in Marvin View 17.15.0 were used to generate 3D structures from SMILES strings, and to propose low energy conformers for structure property prediction and calculation. For the molecule considered in the current study, the lowest energy conformers were used as a starting point for the geometry optimization. The geometries of all the selected conformers were optimized with the DFTBA program. The molecular structures of the five lowest energy conformers were reoptimized by resorting to the MN12SX/Def2TZVP/H2O model chemistry. The optimized structures were confirmed to be real minima by vibrational frequency analysis (no imaginary frequency).
Results and discussion
The molecular structures of the optimized conformers of Taltobulin obtained as mentioned in the previous section, and whose graphical sketch is shown in Fig. 1, has been submitted to optimization in absence of solvent by resorting the DFTBA model available in Gaussian 09 [15] and then reoptimized using the MN12SX/Def2TZVP/H2O model chemistry mentioned in that section. Having verified that each of the structures corresponded to the minimum energy conformations by running a frequency calculation analysis, the electronic properties were determined by using the same model chemistry.
Fig. 1.
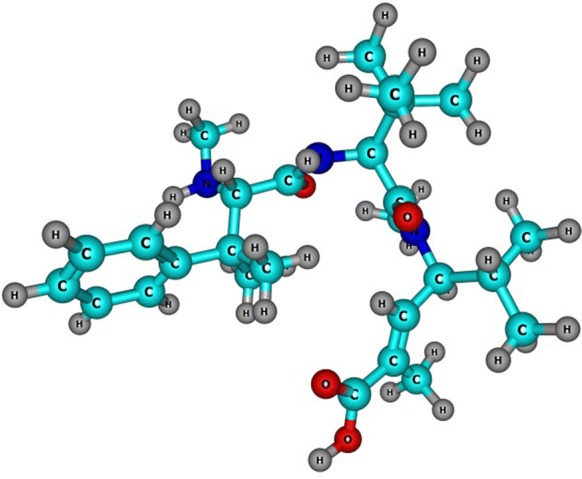
Graphical sketch of the molecular structure of the Taltobulin peptide
Becke has recently mentioned that the adiabatic connection and the ideas of Hohenberg, Kohn, and Sham applying only to electronic ground states is a common misconception [16]. In this regard, the HOMO–LUMO gap within the KS model represents a nice estimation of the lowest excitation energy [17]. Under this assumption, the determination of the maximum wavelength absorption of the Taltobulin peptide was performed by conducting a ground-state calculation with the aforementioned density functional at the same level of model chemistry and theory to obtain the HOMO–LUMO gap and subsequently, the . The electronic energy of the neutral molecular system of Taltobulin, the HOMO and LUMO orbital energies, and the maximum absorption wavelengths calculated with the MN12SX/Def2TZVP/H2O model chemistry are − 1517.422 au, − 6.240 eV, − 1.733 eV, and 275 nm, respectively.
According to the results obtained when studying melanoidins [7–13] as well as peptides from marine sources [14], it can be said that the calculations performed with the MN12SX density functional render HOMO and LUMO energies that satisfy the approximate Koopmans’ theorem. Thus, the application of the KID procedure will be justified. The global reactivity descriptors Electronegativity [18, 19], Global Hardness [18, 19], Electrophilicity [20], Electrodonating Power [21], Electroaccepting Power [21] and Net Electrophilicity [22] were calculated by resorting to the HOMO and LUMO energies determined with the MN12SX density functional with results being = 3.986 eV, = 4.507 eV, = 1.763 eV, = 5.800 eV, = 1.814, and = 7.614 eV. The interested reader in the mathematical formulations of these reactivity descriptors is referred to the original works and to our previous research on the field [7–14]. As expected from the molecular structure of this species, its electrodonating ability is more important that its electroaccepting character.
We now turn our attention to the local descriptors of chemical reactivity, namely the Electrophilic Fukui function [18, 19, 23], the Nucleophilic Fukui function [18, 19, 23] and the Dual Descriptor (DD) [24–28]. As for the case of the global reactivity descriptors, the interested reader in the mathematical formulations of these reactivity descriptors is referred to the original works and to our previous research on the field [7–14]. The Electrophilic Fukui functions and Nucleophilic Fukui functions for the Taltobulin peptide are shown in Fig. 2.
Fig. 2.

Graphical representation of the Electrophilic Fukui function (left) and Nucleophilic Fukui functions (right) of the Taltobulin peptide
Within the field of Chemoinformatics applied to the discovery of new pharmaceutical drugs, it is usual to verify the drug-likeness of the involved molecules resorting to some empirical rules, as for example, the Lipinski Rule of Five (Ro5) [29]. This can be achieved by using the readily available Molinspiration software and the results for the case of Taltobulin are presented next as miLogP (the octanol/water partition coefficient) = 4.43, TPSA (the molecular polar surface area) = 98.73, nAtoms (the number of atoms of the molecule) = 34, nON (the number of hydrogen bond acceptors) = 7, nOHNH (the number of hydrogen bond donors) = 3, nviol (the number of violations of the Ro5) = 0, nrotb (the number of rotatable bonds) = 11, volume (the molecular volume) = 479.94 and MW (the molecular weight) = 473.66. Although this criteria cannot always be applied in general to peptides, it can be seen from the previous results that for Taltobulin the number of violations of the Ro5 is 0, which means that Taltobulin can be considered as a druggable molecule.
As the next step, a new task was accomplished by resorting to the same software for the determination of the bioactivity scores for different drug targets whose values for the Taltobulin peptide are GPCR Ligand = 0.43, Ion Channel Modulator = 0.15, Kinase Inhibitor = - 0.12, Nuclear Receptor Ligand = 0.19, Protease Inhibitor = 0.68 and Enzyme Inhibitor = 0.42.
These bioactivity scores for organic molecules can be interpreted as active (when the bioactivity score is > 0), moderately active (when the bioactivity score lies between − 5.0 and 0.0) and inactive (when the bioactivity score 5.0). That means that the Taltobulin peptide can be considered a potentially bioactive as a Protease inhibitor, besides being able to act a ligand for GPCR and as an Enzyme inhibitor.
Limitations
In this research note, we have presented the results of a study of the chemical reactivity of a the Taltobulin anticancer peptide on the basis of the Conceptual DFT as a tool to explain the molecular interactions, the molecular properties related to bioavailability and the bioactivity scores.
However, this information is insufficient to be considered for peptidomimetics studies and additional results from other peptides will be necessary.
Authors' contributions
DGM conceived and designed the research and headed, wrote and revised the manuscript, while JF and NFH contributed to the writing and the revision of the article. All authors read and approved the final manuscript.
Acknowledgements
This work has been partially supported by CIMAV, SC and Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico). Daniel Glossman-Mitnik conducted this work while a Visiting Lecturer at the University of the Balearic Islands from which support is gratefully acknowledged. Norma Flores-Holguín and Daniel Glossman-Mitnik are researchers of CIMAV and CONACYT.
Funding
Consejo Nacional de Ciencia y Tecnología (CONACYT, Mexico) through Grant 219566-2014 for Basic Science Research financed the building of a supercomputing cluster and the purchase of the software used for the calculations. This work was also cofunded by the Ministerio de Economía y Competitividad (MINECO) and the European Fund for Regional Development (FEDER) (CTQ2014-55835-R) which supported DGM as a Visiting Lecturer at the University of the Balearic Islands.
Availability of data and materials
The data set from the current study is available upon request from the corresponding author.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- DFT
Density Functional Theory
- CDFT
Conceptual Density Functional Theory
- SMILES
Simplified Molecular Input Line Entry Specification
- DFTBA
Density Functional Tight Binding Model A
- HOMO
Higher Occupied Molecular Orbital
- LUMO
Lower Unoccupied Molecular Orbital
- KS
Kohn–Sham
- KID
Koopmans in DFT
- DD
Dual Descriptor
- Ro5
Lipinski Rule of Five
- GPCR
G Protein-coupled Receptor
Contributor Information
Norma Flores-Holguín, Email: norma.flores@cimav.edu.mx.
Juan Frau, Email: juan.frau@uib.es.
Daniel Glossman-Mitnik, Email: daniel.glossman@cimav.edu.mx.
References
- 1.Muller WEG, Schroder HC, Wang X (eds.): Blue biotechnology: from gene to bioactive product. Springer, Cham (2017)
- 2.La Barre S, Kornprobst J-M. Outstanding marine molecules. Weinheim: Wiley-Blackwell; 2014. [Google Scholar]
- 3.Kim S-K. Marine proteins and peptides—biological activities and applications. Chichester: Wiley-Blackwell; 2013. [Google Scholar]
- 4.Zheng L-H, Wang Y-J, Sheng J, Wang F, Zheng Y, Lin X-K, Sun M. Antitumor peptides from marine organisms. Mar Drugs. 2011;9(10):1840–1859. doi: 10.3390/md9101840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta GK, Kumar V. Chemical drug design. Berlin: Walter de Gruyter GmbH; 2016. [Google Scholar]
- 6.Gore M, Jagtap UB. Computational drug discovery and design. New York: Springer; 2018. [Google Scholar]
- 7.Frau J, Glossman-Mitnik D. Molecular reactivity and absorption properties of melanoidin Blue-G1 through conceptual DFT. Molecules. 2018;23(3):559. doi: 10.3390/molecules23030559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frau J, Glossman-Mitnik D. Conceptual DFT study of the local chemical reactivity of the dilysyldipyrrolones A and B intermediate melanoidins. Theor Chem Acc. 2018;137(5):1210. doi: 10.1007/s00214-018-2244-x. [DOI] [Google Scholar]
- 9.Frau J, Glossman-Mitnik D. Conceptual DFT study of the local chemical reactivity of the colored BISARG melanoidin and its protonated derivative. Front Chem. 2018;6(136):1–9. doi: 10.3389/fchem.2018.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frau J, Glossman-Mitnik D. Molecular reactivity of some Maillard reaction products studied through conceptual DFT. Contemp Chem. 2018;1(1):1–14. [Google Scholar]
- 11.Frau J, Glossman-Mitnik D. Computational study of the chemical reactivity of the Blue-M1 intermediate melanoidin. Comput Theor Chem. 2018;1134:22–29. doi: 10.1016/j.comptc.2018.04.018. [DOI] [Google Scholar]
- 12.Frau J, Glossman-Mitnik D. Chemicalreactivity theory applied to the calculation of the local reactivity descriptors of a colored Maillard reaction product. Chem Sci Int J. 2018;22(4):1–14. doi: 10.9734/CSJI/2018/41452. [DOI] [Google Scholar]
- 13.Frau J, Glossman-Mitnik D. Blue M2: an intermediate melanoidin studied via conceptual DFT. J Mol Model. 2018;24(138):1–13. doi: 10.1007/s00894-018-3673-0. [DOI] [PubMed] [Google Scholar]
- 14.Frau J, Flores-Holguín N, Glossman-Mitnik D. Chemical reactivity properties, pKa Values, AGEs inhibitor abilities and bioactivity scores of the mirabamides A-H peptides of marine origin studied by means of conceptual DFT. Mar Drugs. 2018;16(9):302–19. doi: 10.3390/md16090302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann: Gaussian 09 Revision E.01. Gaussian Inc, Wallingford CT, 2016 (2016)
- 16.Becke AD. Vertical excitation energies from the adiabatic connection. J Chem Phys. 2016;145(19):194107. doi: 10.1063/1.4967813. [DOI] [PubMed] [Google Scholar]
- 17.van Meer R, Gritsenko OV, Baerends EJ. Physical meaning of virtual Kohn–Sham orbitals and orbital energies: an ideal basis for the description of molecular excitations. J Chem Theory Comput. 2014;10(10):4432–4441. doi: 10.1021/ct500727c. [DOI] [PubMed] [Google Scholar]
- 18.Parr RG, Yang W. Density-functional theory of atoms and molecules. New York: Oxford University Press; 1989. [Google Scholar]
- 19.Geerlings P, De Proft F, Langenaeker W. Conceptual density functional theory. Chem Rev. 2003;103:1793–1873. doi: 10.1021/cr990029p. [DOI] [PubMed] [Google Scholar]
- 20.Parr RG, Szentpaly LV, Liu SB. Electrophilicity index. J Am Chem Soc. 1999;121:1922–1924. doi: 10.1021/ja983494x. [DOI] [Google Scholar]
- 21.Gázquez JL, Cedillo A, Vela A. Electrodonating and electroaccepting powers. J Phys Chem A. 2007;111(10):1966–1970. doi: 10.1021/jp065459f. [DOI] [PubMed] [Google Scholar]
- 22.Chattaraj PK, Chakraborty A, Giri S. Net electrophilicity. J Phys Chem A. 2009;113(37):10068–10074. doi: 10.1021/jp904674x. [DOI] [PubMed] [Google Scholar]
- 23.Chermette H. Chemical reactivity indexes in density functional theory. J Comput Chem. 1999;20:129–154. doi: 10.1002/(SICI)1096-987X(19990115)20:1<129::AID-JCC13>3.0.CO;2-A. [DOI] [Google Scholar]
- 24.Morell C, Grand A, Toro-Labbé A. New dual descriptor for chemical reactivity. J Phys Chem A. 2005;109:205–212. doi: 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- 25.Morell C, Grand A, Toro-Labbé A. Theoretical support for using the descriptor. Chem Phys Lett. 2006;425:342–346. doi: 10.1016/j.cplett.2006.05.003. [DOI] [Google Scholar]
- 26.Martínez-Araya JI. Revisiting Caffeate’s capabilities as a complexation agent to silver cation in mining processes by means of the dual descriptor—a conceptual DFT approach. J Mol Model. 2012;18:4299–4307. doi: 10.1007/s00894-012-1405-4. [DOI] [PubMed] [Google Scholar]
- 27.Martínez-Araya JI. Explaining reaction mechanisms using the dual descriptor: a complementary tool to the molecular electrostatic potential. J Mol Model. 2012;19(7):2715–2722. doi: 10.1007/s00894-012-1520-2. [DOI] [PubMed] [Google Scholar]
- 28.Martínez-Araya JI. Why is the dual descriptor a more accurate local reactivity descriptor than Fukui functions? J Math Chem. 2015;53(2):451–465. doi: 10.1007/s10910-014-0437-7. [DOI] [Google Scholar]
- 29.Leeson P. Drug discovery: chemical beauty contest. Nature. 2012;481(7382):455–456. doi: 10.1038/481455a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data set from the current study is available upon request from the corresponding author.