

Abstract
Genetic variation in the NFκB inhibitors, ABIN1 and A20, increase risk for psoriasis. While critical for hematopoietic immune cell function, these genes are believed to additionally inhibit psoriasis by dampening inflammatory signaling in keratinocytes. We dissected ABIN1 and A20’s regulatory role in human keratinocyte inflammation using an RNA-seq based comparative genomic approach. Here we show subsets of the IL-17 and TNFα signaling pathways are robustly restricted by A20 overexpression. In contrast, ABIN1 overexpression inhibits these genes more modestly for IL-17, and weakly for TNFα. Our genome-scale analysis also indicates that inflammatory program suppression appears to be the major transcriptional influence of A20/ABIN1 overexpression, without obvious influence on keratinocyte viability genes. Our findings thus enable dissection of the differing anti-inflammatory mechanisms of two distinct psoriasis modifiers, which may be directly exploited for therapeutic purposes. Importantly, we report that IL-17 induced targets of A20 show similar aberrant epidermal layer-specific transcriptional upregulation in keratinocytes from diseases as diverse as psoriasis, atopic dermatitis, and erythrokeratodermia variabilis, suggesting a contributory role for epidermal inflammation in a broad spectrum of rashes.
INTRODUCTION
Specific blockade of immune cell signaling pathways has sharply expanded the efficacy and diversity of dermatologic therapies in the past two decades. Systemic biologic treatments that block tumor necrosis factor alpha (TNFα) and interleukin-17 (IL-17) function (e.g. infliximab and secukinumab, respectively) now represent some of the most effective psoriasis therapies (Langley et al. 2014; Reich et al. 2005). The cytokine TNFα, mainly produced by activated macrophages, is increased in psoriatic skin and believed to accelerate epidermal immune cell infiltration and induce dendritic cell activation (Uchi et al. 2000). IL-17, a cytokine produced by T helper 17 and innate lymphoid cells (Cua and Tato 2010), potentiates inflammatory states by stimulating keratinocyte secretion of proinflammatory mediators and recruiting neutrophils and other inflammatory cells (Ogawa et al. 2018). In inflammatory skin diseases such as psoriasis, TNFα and IL-17 synergistically augment dysregulated immune response (Krueger and Brunner 2018).
These infiltrating immune cells clearly play a fundamental role in cutaneous inflammation. However, recent attention has turned to the immunologic role of target tissues in inflammatory disease. In psoriasis, keratinocytes influence and establish the inflammatory milieu by producing an array of inflammatory cytokines, such as TNFα, interleukins, and interferons (Rupec et al. 2010), while also undergoing abnormal differentiation and hyperproliferation. Molecular abnormalities occurring primarily in the epidermis cause other rashes, including pityriasis rubra pilaris, ichthyoses, and erythroderma keratodermia variabilis. These conditions, which share the clinical phenotype of inflammatory scaly plaques, histopathologically all show scant immune cell infiltrates, underscoring the importance of keratinocyte-driven inflammation (Hanifin 2009).
Nuclear factor kappa B (NFκB) activation, a common pathway downstream of both TNFα and IL-17, has been recurrently implicated in keratinocyte-driven rashes. Gain-of-function mutations in CARD14, an NFκB activator, underlie familial forms of pityriasis rubra pilaris (Fuchs-Telem et al. 2012). The rare inherited ichthyoses, often caused by genes important for epidermal barrier establishment, typically manifest early in childhood with cutaneous inflammation, diffuse scaling, and barrier defects. It has recently been noted that ichthyotic skin displays a strong IL-17 dominant immune profile also suggestive of a link between epidermal-based dysfunction and NFκB-mediated inflammation (Paller et al. 2017). Erythrokeratodermia variabilis is a rare genetic disorder characterized by migratory erythematous scaly plaques. The majority of cases are due to loss-of-function connexin gap junction mutations (Scott et al. 2012), which appear to activate keratinocyte-autonomous inflammatory programs, although NFκB has not been established as the central pathway.
NFκB signaling is critical for both innate and adaptive immunity, as well as epidermal homeostasis. In its baseline inactive state, the NFκB transcription factor complex is bound to the inhibitory protein IκBα and sequestered in the cytoplasm (Mitchell et al. 2016). Canonical NFκB activation occurs through phosphorylation of IκBα by the IκB kinase (IKK) complex (Mercurio et al. 1997). IκBα is then ubiquinated and degraded by the proteasome, releasing NF-κB for nuclear translocation and subsequent transcriptional activation. Multiple human linkage studies and mouse models have demonstrated a role for NFκB signaling pathway alterations in autoimmune disease and inflammatory skin disease (Sun et al. 2013). For example, genome-wide association studies in psoriasis, rheumatoid arthritis, and systemic lupus erythematosus have identified the NFκB signaling pathway genes ABIN1 (G’Sell et al. 2015; Nair et al. 2009), A20 (Catrysse et al. 2014), and CARD14 (Jordan et al. 2012) as disease susceptibility loci.
The NFκB inhibitors A20 (encoded by TNFAIP3) and ABIN1 (encoded by TNIP1) are well established as regulators of hematopoietic immune cell activity (Zhou et al. 2011). The adapter protein ABIN1 physically links the A20 deubiquitinase to the IKK complex, where A20 deubiquinates and inactivates IKK to prevent phosphorylation and inactivation of the NFκB inhibitor IκBα. Immune cell-specific deletions of A20 or ABIN1 in mice produce a wide variety of systemic hyperinflammatory phenotypes, reminiscent of systemic lupus erythematosus and rheumatoid arthritis (Catrysse et al. 2014; G’Sell et al. 2015). Recently, however, experimental manipulation has also revealed a role for A20 and ABIN1 in keratinocyte-mediated inflammation. In response to inflammatory stimuli (imiquimod and IL-17), ABIN1 epidermal depletion in mice enhances chemokine expression (Ippagunta et al. 2016). Correspondingly, HaCaT human keratinocyte cultures exposed to the dsRNA mimic Poly (I:C) markedly increase inflammatory gene expression when ABIN1 is depleted (Rudraiah et al. 2018). While a link between A20 depletion and epidermal inflammation has not been shown yet, its overexpression in HaCaT cells represses a small set of profiled cytokines when exposed to Poly (I:C) (Sohn et al. 2016).
Inhibition of target tissue inflammation may improve a broad range of rashes and could potentially lead to localized treatments that avoid the serious side effects that can accompany systemic immunosuppression. While localized repression of NFκB signaling may represent one such strategy, a full accounting of the ability for A20 and ABIN1 to suppress keratinocyte inflammation has not been reported, nor has their potential non-inflammatory effects. Here we report a global transcriptional analysis of the inflammation repressing effects of the NFκB inhibitory binding partners, A20 and ABIN1, in human keratinocytes. We show that overexpressed A20 is a more potent inhibitor of keratinocyte inflammation than ABIN1, in the context of IL-17A or TNFα cytokine stimulation. Single cell RNA-sequencing of epidermis from diverse rashes revealed a common inflammatory gene expression signature, whose genes were repressed by A20 overexpression in primary keratinocyte culture.
RESULTS
We utilized lentivirus to overexpress A20 and ABIN1 in primary normal human epidermal keratinocyte (NHEK) culture, leading to a ~1300 or 500% increase in protein expression, respectively (Figure S1). RNA sequencing was performed on mock-infected (GFP), A20, or ABIN1 overexpressing NHEK cells to assess global expression patterns. The edgeR software package (Robinson et al. 2010), which uses an overdispersed Poisson model to address biological and technical variability, was used to identify genes differentially expressed between treatment conditions. We first examined downregulation of inflammation-related genes, given the known roles of A20 and ABIN1 in suppressing inflammation. Under standard (no cytokine stimulation) culture conditions, expression of relatively few inflammatory genes, or genes overall, were repressed by A20 or ABIN1 overexpression in NHEK cells (Figure 1). A20 overexpression downregulated 53 genes (log2FC<−0.5, FDR<0.05, Table S1A), amongst these were several inflammatory genes, such as CXCL1, C1R, IL24, IL32, MMP9, and SAA1. ABIN1 repressed fewer genes (31 overall genes with log2FC<−0.5, FDR<0.05, Table S1B), including a subset of the A20 repressed inflammatory genes (C1R, IL32, MMP9, and SAA1), with similar or greater effect than A20.
Figure 1. A20 and ABIN1 repress transcription of relatively few inflammatory genes under unstimulated conditions.
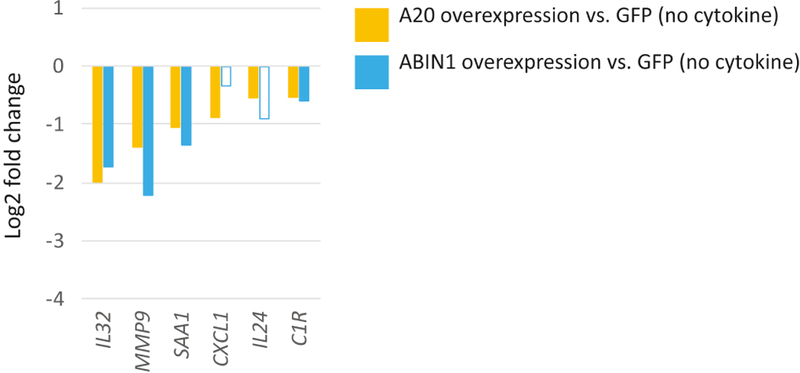
Relative expression (log2 fold change) of inflammatory genes in A20 or ABIN1 versus mock infected (GFP) overexpressing keratinocytes. Data shown represents edgeR RNA-seq differential expression results for 6 replicates for each permutation (FDR < 0.05 except for non-shaded bars).
Given the relatively modest gene regulatory effects exerted by overexpression of these two genes under standard culture conditions, we next sought to understand their regulatory power when challenged with inflammatory stimuli. We utilized the pro-inflammatory cytokines IL-17A and TNFα, given their known roles and specific blockade by biological therapeutics in cutaneous inflammatory disease. To better understand the specific roles for A20 and ABIN1 in each of these cytokines’ signaling pathways, we stimulated keratinocytes with single cytokine exposure. RNA-seq was performed on mock infected (GFP), A20, and ABIN1 overexpressing NHEK cells that were cytokine stimulated with either IL-17A or TNFα for 1 or 24 hours.
We used expression data from mock infected (GFP expressing) keratinocytes as a proxy for cytokine stimulation of phenotypically normal keratinocytes. Incubation of these keratinocytes with IL-17A for 24 hours led to greater transcriptional upregulation of genes than at 1 hour (131 to 36 genes, respectively (log2FC>0.5, FDR<0.05; Tables S1D and S1E). We thus focused our attention on the 24 hour IL-17A cytokine stimulation time point. Consistent with previous reports, 24 hour IL-17A exposure upregulated genes such as antimicrobial peptides (DEFB4A, LCN2, and S100A7), CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL8), and interleukins (IL1A, IL1B, IL23A, IL24, IL33, and IL36G) (Figure 2, Chiricozzi et al. 2011; Nograles et al. 2008).
Figure 2. A20 robustly and ABIN1 modestly represses IL-17A induced inflammatory transcriptional programs.
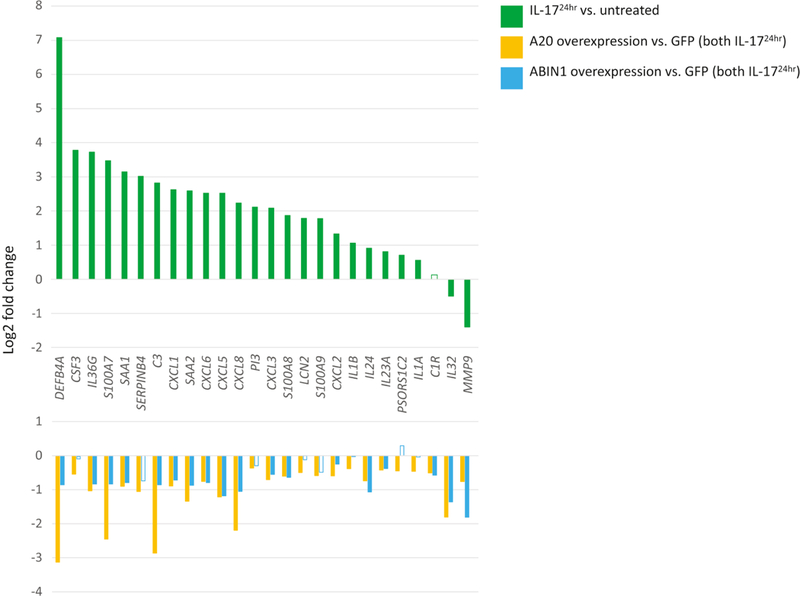
Top panel depicts relative expression (log2 fold change) of mock infected (GFP) keratinocytes incubated with IL-17A for 24 hours versus untreated mock infected keratinocytes. Bottom panel depicts relative expression (log2 fold change) for either A20 or ABIN1 overexpressing keratinocytes compared to mock infected (GFP) keratinocytes, all samples treated with IL-17A for 24 hrs. Data shown represents edgeR RNA-seq differential expression results for 6 replicates for each permutation (FDR < 0.05 except for non-shaded bars).
While overexpression of A20 in NHEK culture robustly inhibited IL-17A (24 hour) induced inflammatory genes, ABIN1 overexpression produced more modest effects (Figure 2, Tables S1E and S1F). With IL-17A exposure, most of the few inflammatory genes repressed by A20 overexpression under unstimulated conditions continued to be repressed by a similar magnitude; however, repression of an extended set of inflammatory genes became evident, e.g. DEFB4A, S100A7, S100A8, CCXL1, CXCL3, CXCL5, CXCL6, CXL8, IL24, and IL36G (Figure 2). Most of this set of genes were lowly expressed under standard culture conditions before IL17A stimulation. For ABIN1 overexpressing keratinocytes exposed to IL-17A (24 hour) stimulation, the small set of inflammatory genes downregulated under baseline conditions also continue to be repressed, along with repression of a similar extended set of inflammatory genes as with A20 overexpression, albeit with lesser effect (Figure 2). With 1 hour of IL-17A cytokine stimulation, a similar pattern emerged where A20 overexpression exhibited stronger inflammatory gene repressive activity (both in terms of number of genes and degree of repression) than ABIN1 (Figure S2, Tables S1D, S1G, and S1H).
We next tested the effects of A20 and ABIN1 overexpression on NHEK cells stimulated with either 1 or 24 hours of TNFα exposure. We noted that the greatest modulation by A20 or ABIN1 of inflammatory gene expression occurred with 1 hour of TNFα exposure, so we focused on this time point. In mock infected keratinocytes, 90 genes were upregulated with 1 hour of TNFα exposure (Figure 3, log2FC>0.5, FDR<0.05, Table S1I). Similar to previous findings, upregulated genes include neutrophil chemoattractants (e.g. CXCL1, CXCL2, CXCL3, CXCL5, and CXCL8), interleukins (IL16, IL1A IL1B, IL20, IL23A, IL36G, and IL6), CCL20, ICAM1, CSF1/2 and TNF itself (Banno et al. 2004).
Figure 3. A20 robustly and ABIN1 weakly represses TNFα induced inflammatory transcriptional programs.
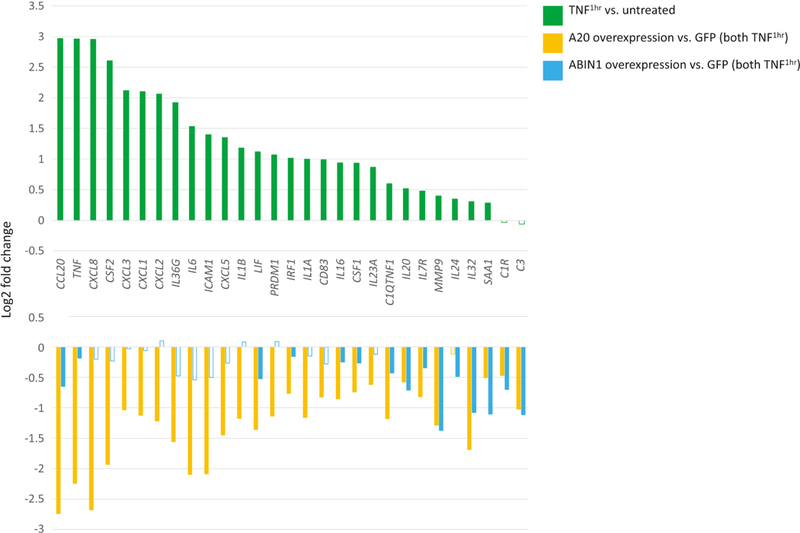
Top panel depicts relative expression (log2 fold change) of mock infected (GFP) keratinocytes incubated with TNFα for 1 hour versus untreated mock infected keratinocytes. Bottom panel depicts relative expression (log2 fold change) for either A20 or ABIN1 overexpressing keratinocytes compared to mock infected (GFP) keratinocytes, all samples treated with TNFα for 1 hour. Data shown represents edgeR RNA-seq differential expression results for 6 replicates for each permutation (FDR < 0.05 except for non-shaded bars).
As in the IL-17A stimulation experiments, A20 overexpression strongly inhibited expression of inflammatory genes induced by 1 hour of TNFα exposure, repressing ~79% of all induced genes (Figure 3, log2FC<−0.5, FDR<0.05; Table S1J). Similar to IL-17A exposure, genes downregulated under baseline conditions continue to be downregulated by A20 overexpression, despite an overall increase in their absolute expression levels with TNFα stimulation. A20 repressed inflammatory genes that were evident only upon TNFα stimulation including CSF1/2, ICAM1, TNF, chemokines (CCL20, CXCL1, CXCL2, CXCL3, CXCL5, and CXCL8), and interleukins/interleukin receptors (IL16, IL1A, IL1B, IL6, IL24, IL20, IL23A, IL36G, and IL7R). In contrast, ABIN1 overexpression produced only a small effect, repressing 4/89 TNFα stimulated genes, (e.g. IL20, CCL20, and C3) and continued to repress genes that were already repressed at baseline (e.g. C1R, IL32, MMP9, and SAA1, Figure 3, log2FC <−0.5, FDR < 0.05, Table S1K). 24 hour TNFα exposure for ABIN1 and A20 overexpressing keratinocytes showed similar trends with A20 clearly repressing inflammatory genes such as chemokines and interleukins although to a slightly lesser extent than with 1 hour of TNFα exposure. The ABIN1 overexpression effect was still weak, although slightly more appreciable, with extended TNFα stimulation (Figure S3, Tables S1L, S1M, and S1N).
Given a potential association between keratinocyte inflammation and differentiation (Schröder et al. 2006), we next focused on the effects of A20 and ABIN1 overexpression on keratinocyte differentiation. We noted that A20 repressed late differentiation genes, such as small proline rich proteins (SPRR; SPRR2A, SPRR2G, and SPRR3), late cornified envelope (LCE) genes (LCE1B, LCE3D, and LCE3E), as well as the early differentiation marker KRT1. ABIN1 did not consistently repress this set of differentiation genes (Figure 4, log2FC<−0.5, FDR<0.05; Supplemental Tables S1A and S1B).
Figure 4. A20 represses late differentiation gene transcription.
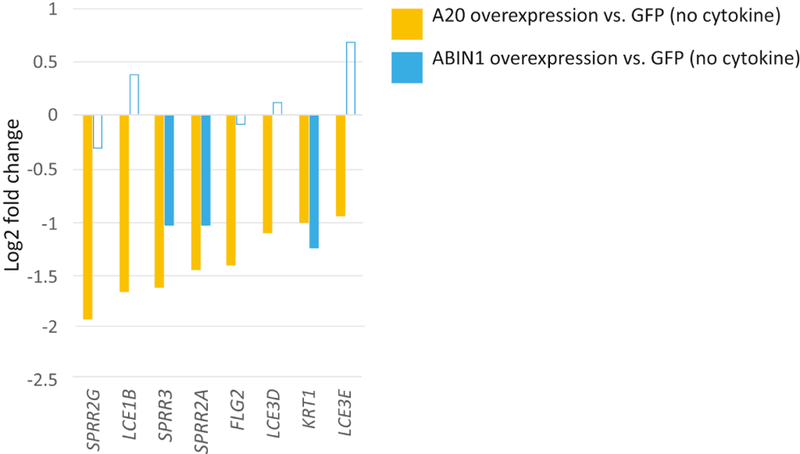
Relative expression (log2 fold change) of either A20 or ABIN1 overexpressing keratinocytes vs mock infected (GFP) keratinocytes. Data shown represents edgeR RNA-seq differential expression results for 6 replicates for each permutation (FDR < 0.05 except for non-shaded bars).
We were curious if our focus on inflammation had caused us to overlook other transcriptional programs controlled by A20 and ABIN1 that might complicate clinical treatment strategies. We performed unsupervised hierarchical clustering on 861 differentially expressed genes when comparing the A20, ABIN1, or GFP (control) keratinocyte overexpression, with and without 24 hours of IL-17A or TNFα stimulation permutations (FDR<1×10−5 for each condition, six replicates each). This data is depicted as a heatmap in Figure 5. As expected, we noted inflammatory gene clusters, with 3 groupings that were enriched for gene ontology terms such as immune response, immune system response, and immune system process. One cluster represents genes strongly upregulated by IL-17A and downregulated by A20 and ABIN1 overexpression, characterized by genes such as DEFB4A, CXCL6, SAA2, and CSF3. A second cluster generally encompasses genes upregulated by both TNFα and IL-17A, and repressed by A20 and ABIN1 (e.g. C3, CXCL5, CSF2, and CXCL3). The third cluster represents a third set of inflammatory genes, containing genes such as IL32, C1R, MMP9, and TNF, that are more strongly upregulated by TNFα and downregulated by A20 and/or ABIN1 overexpression.
Figure 5. Inflammation predominates the response to A20 and ABIN1 overexpression in the context of cytokine stimulation.
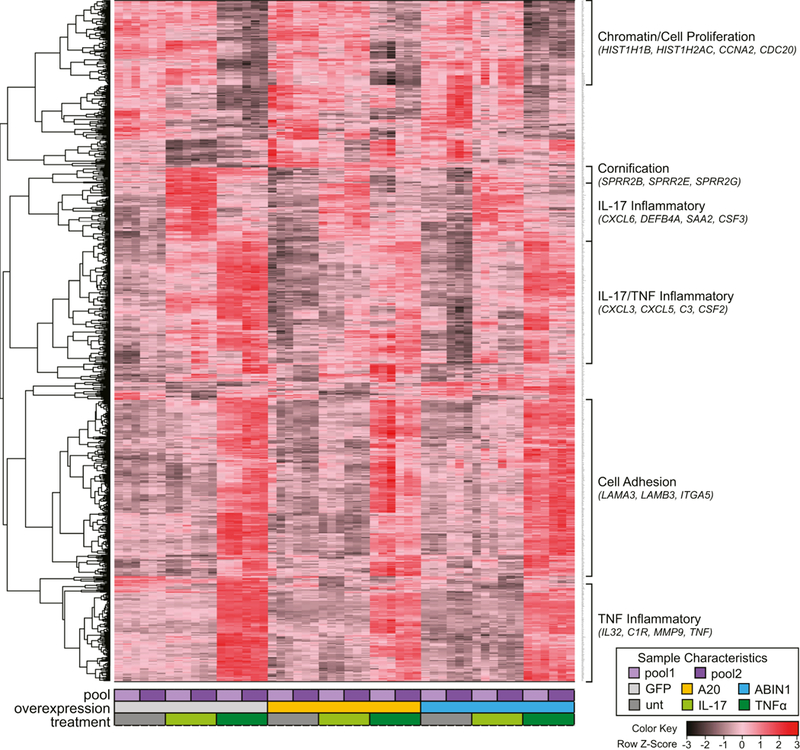
Unsupervised hierarchical clustering dendrogram and heatmap of gene RNA expression values. Each column represents RNA-seq CPM values for one of six replicates (3 replicates each for 2 pooled keratinocyte isolates) for 9 different treatment conditions (GFP expressing (negative control), A20 overexpressing, or ABIN1 overexpressing keratinocytes, with and without 24 hours of IL-17A or TNFα stimulation). Each row represents one of the 861 differentially expressed genes with FDR < 1 ×10-5. The color key represents for each row, deviation from the mean in standard deviations.
We also noted non-inflammatory gene clusters that showed regulation by TNFα and/or A20/ABIN1 overexpression. A small cluster is enriched for a set of cornification and keratinization genes, such as small proline rich proteins (SPRR2E, SPRR2B, and SPRR2G) that are modestly downregulated by A20 and weakly by ABIN1. Another cluster includes genes strongly downregulated by TNFα treatment and is enriched for GO terms such as chromatin (e.g. HIST1H1B and HIST1H2AC) and cell proliferation (e.g. CCNA2 and CDC20). This cluster of genes likely corresponds with genes affected by TNF mediated reduction in cell proliferation (Banno et al. 2004; Detmar and Orfanos 1990). Lastly, we noted a cluster enriched for cell adhesion genes (e.g. LAMA3, LAMB3, and ITGA5), which is strongly upregulated by TNFα without much effect from A20 or ABIN1 overexpression. There was no clearly assignable function or biological process associated with the remaining gene clusters.
Given the greater repression of inflammation by A20 compared to ABIN1, we sought to understand whether keratinocytes in psoriasis and other rashes display upregulation of the A20 repressed gene sets discovered in our NHEK experiments. Such a finding would suggest a potential inflammation-suppressive effect of A20 in certain human rashes that could be therapeutically targeted. We generated single cell RNA-seq (scRNA-seq) expression data from epidermis freshly isolated from three normal, three psoriasis, one atopic dermatitis, and one erythrokeratodermia variabilis skin samples, each of which was validated by a board-certified dermatopathologist. To assess for whole epidermis-level expression alteration, scRNA-seq data for keratinocytes from each sample type were aggregated in bulk and differential expression analysis between normal skin samples and each of the disease types was performed using limmatrend (Law et al. 2014a). Expression for 67 genes was strongly increased in psoriatic epidermis (log2FC >0.9, adj FDR<0.05; Table S2). In our NHEK experiments, 49 genes were upregulated by 24 hours of IL-17A exposure (log2FC >0.9, FDR<0.05, Table S1C). Ten genes overlapped between these sets (S100A7, S100A8, S100A9, SPRR2A, PDZK1I, PI3, SAA1, CRABP2, SERPINB3, and SERPINB4). Six of these ten genes also overlapped with upregulated transcripts in the EKV and atopic dermatitis samples (log2FC >0.9, FDR<0.05, Table S2). Of the ten genes that were upregulated in both psoriatic epidermis and IL-17A stimulated NHEK cells, all but two were repressed in our A20 overexpression 24 hour IL-17A exposure NHEK experiments, suggestive of in vivo relevance for A20-mediated gene repression in inflammatory skin disease (FDR<0.05, Table S1E).
If this set of A20 repressible inflammatory genes shared a regulatory mechanism, indicative of a potentially targetable common pathway, we might expect to see shared expression dynamics corresponding with keratinocyte differentiation (e.g. differing expression levels between basal and late differentiated keratinocytes). We examined our scRNA-seq data by displaying transcript abundance for these genes and KRT10 (which we used as a marker of keratinocyte differentiation state) on a single cell level (Figure 6). In normal skin, these ten genes all showed decreased expression in the most differentiated keratinocytes (as represented by highest KRT10 expression). However, 9 of these genes showed coordinated aberrant upregulation in the most differentiated keratinocytes in the psoriatic samples, suggesting an epidermal layer-specific expression signature and common therapeutically targetable mechanism. Many of these genes also showed similar differentiation-related transcript upregulation in the erythrokeratodermia variabilis and atopic dermatitis scRNA-seq data, suggesting that these A20 regulated inflammatory transcripts are more generally aberrantly expressed in diverse types of skin disease (Figure 6).
Figure 6. Shared single-cell RNA expression differentiation-related patterns in psoriatic, erythrokeratodermia variabilis, and atopic dermatitis keratinocytes, for selected IL-17A induced and A20 repressed genes.
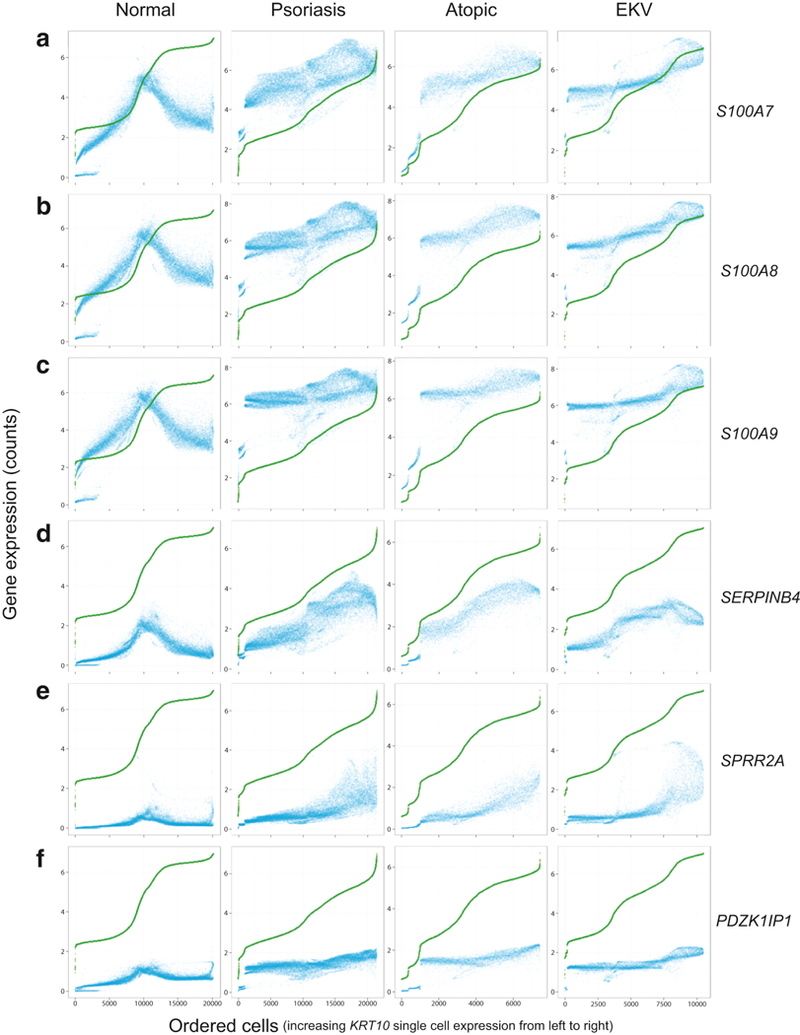
Each row of panels represents normalized/imputed single cell RNA expression data for a different gene (A) S100A7, B) S100A8, C) S100A9, D) SERPINB4, E) SPRR2A, or F) PDZK1IP1; see Methods). Each column of panels represents a distinct inflammatory state (normal, psoriasis, EKV, or atopic dermatitis-affected epidermis). In each panel, the dots represent single cells. For each panel, cells are ordered along the x-axis by KRT10 expression from low to high starting with the lowest expressing single cell on the far left and the highest expressing single cell on the far right (i.e. increasing differentiation status of single cells from left to right on the X-axis). The scale of the x-axis differs for each inflammatory state, reflective of the differing number of profiled single cells. On the y-axis of each panel, transcript abundance (in log2 counts per 10,000) for each single cell is displayed for a given gene (blue dot) or KRT10 (green dot). As there are small incremental differences in KRT10 expression between adjacent points and the large number of plotted cells, the green dots which represent KRT10 single cell expression merge into a contiguous line. In the normal skin samples, there is a decrease in gene expression in the highest KRT10 expressing single cells; however, in the inflamed disease samples, there is an aberrant coordinated increase in gene expression in the highest KRT10 expressing single cells.
DISCUSSION
Numerous lines of evidence, from human disease linkage analyses to mouse experimental models, attest to critical roles for the NFκB inhibiting partner proteins A20 and ABIN1 in systemic and cutaneous inflammatory disease. Given the increasing awareness of the role keratinocytes play in potentiating inflammatory cutaneous disease, we focused on the function of these two genes in keratinocyte inflammatory response. The combination of A20 and ABIN1 comparative analyses, RNA-sequencing, and cytokine stimulation treatments enabled us to greatly expand upon previous reports which focused on the effects of ABIN1 overexpression in unstimulated HaCaT keratinocytes (Ramirez et al. 2015) or on a small defined inflammatory gene set with A20 overexpression in Poly (I:C) stimulated NHEK cells (Sohn et al. 2016). Global transcriptional analysis of unstimulated ABIN1 overexpressing HaCaT cells had previously identified enrichment in repressed genes for the “immunological disease” biological process, without further exploration of specific inflammatory genes (Ramirez et al. 2015). We found that under baseline unstimulated conditions, A20 and ABIN1 both repress a small set of inflammatory genes (e.g. C1R, IL32, MMP9, and SAA1; Figure 1).
More importantly, we discovered a robust and broader role for A20 compared to ABIN1 in repressing inflammation induced by the critical pro-inflammatory cytokines, IL-17A or TNFα. This suggests that modulating A20 activity may be a more promising therapeutic target than ABIN1, given its greater inflammation repressing role. With IL-17A and TNFα exposure, both ABIN1 and A20 generally continued to downregulate the few baseline repressed genes to a similar extent despite a greater overall expression level for cytokine responsive genes. However, upon cytokine stimulation, differences in A20 and ABIN1’s repressive effect on inflammatory genes become more evident. With 24 hour IL-17A simulation, a large set of inflammatory genes were repressed by both A20 or ABIN1 overexpression: DEFB4A, C3, S100A7, CXCL8, SAA2, CXCL5, CSF2, IL36G, SAA1, CXCL1, CXCL6, IL24, IL74, CXCL3, S100A8, and CXCL22, although generally to a greater extent by A20. However, there were a few genes uniquely repressed by A20, including SERPINB4, S100A9, and LCN2. For TNFα exposure, common repressed genes by A20 or ABIN1 overexpression include CCL20, IL32, C3, SAA1, IL32, IL20, and C1R, but there were many genes uniquely repressed by A20 including CXCL8, IL6, IL36G, CXCL5, IL1B, IL1A, CXCL1, CXCL3, PI3, and IL23A. While A20 repressed a large shared set of genes for both IL-17A or TNFα stimulation, genes that were repressed only with IL-17A stimulation (e.g. DEFB4A, CSF3, SERPINB4, SAA2, CXCL6, and LCN2) or only with TNFα stimulation (e.g. TNF, CCL20, and ICAM1) generally had low baseline expression levels before strong upregulation with the respective cytokine. These results substantially expand upon the 5 repressed inflammatory genes seen with A20 overexpression in Poly (I:C) stimulated NHEK cells (Sohn et al. 2015). For ABIN1, given its greater effect in the context of IL-17A stimulation, genes such as CXCL5, CXCL8, DEFB4A, IL36G, CXCL1, and CXCL3, were repressed only with IL-17A stimulation and CCL20 was one of the few inflammatory genes uniquely repressed in the context of TNFα stimulation.
The differential repressive potency and gene targets of A20 and ABIN1 overexpression with IL-17A and TNFα exposure suggest unique roles for these two genes in modulating keratinocyte inflammatory response. In the context of TNFα signaling, ABIN1 is believed to primarily function as a facilitative adapter protein with A20 to inhibit NFκB signaling, by preventing phosphorylation and inactivation of the NFκB inhibitor IκBα (G’Sell et al. 2015). A20 also employs additional inhibitory mechanisms such as restricting TNF receptor initiation of NFκB and JNK signaling through TRAF2 and RIP1 (Catrysse et al. 2014). We speculate that overexpressed ABIN1 may readily saturate endogenous levels of A20 so that increased expression in keratinocytes only modestly correlates with functional activity when exposed to TNFα.
Both ABIN1 and A20 appear to independently inhibit IL-17 signaling. A20 inhibits TRAF6, a molecule required for IL-17 induced NFκB and MAPK signaling (Garg et al. 2013). A20 also directly binds to and inhibits the IL-17 receptor, an additional mechanism for inhibiting IL-17 induced NFκB and MAPK activation (Garg et al. 2013). The role of ABIN1 in IL-17 signaling (e.g. whether and how ABIN1 and A20 cooperatively interact to inhibit NFκB activation as in TNF signaling) is less well defined; however, ABIN1 can inhibit IL-17 signaling independent of A20 (Cruz et al. 2017). Whether ABIN1 also directly interacts with the IL-17 receptor like A20, also remains an open question. ABIN1’s greater inflammation repressing effects in the context of IL-17A stimulation (compared to TNFα), likely arises from its A20independent role in IL-17 repression. However, ABIN1’s lower overall activity compared to A20 may stem from its inability to bind the IL-17 receptor or inhibit other A20-specific functions. Alternatively, the greater levels of A20 protein overexpression compared to ABIN1 may account for its greater overall inflammatory repression. However, this appears unlikely given that expression levels for both proteins are increased by at least 5-fold.
Our results also punctuate the importance of keratinocyte-mediated inflammation and thus its potential as a target for clinical intervention. By utilizing single cell RNA-seq on rashaffected epidermis, we identified a set of epidermal layer-specific, IL-17A inducible inflammatory transcripts in diverse skin diseases, such as psoriasis, eczema, and EKV, that are repressed by A20 overexpression. While systemic blockade of cytokine signaling pathways, such as IL-17A and TNFα, have had immense therapeutic benefit in patients, the ability to specifically block their effect in a readily accessible target tissue, such as skin, would be highly desirable in minimizing the systemic side effects of these potent immunosuppressants. Based on our findings, we hypothesize that in vivo upregulation of cutaneous A20 activity may represent a therapeutic path to dampen target tissue inflammation in diverse inflammatory skin diseases.
MATERIALS AND METHODS
Keratinocyte isolation and primary culture
Primary human keratinocyte cultures were isolated from neonatal foreskins as previously described (Lowdon et al. 2014). Written informed consent for surgical tissue discards were obtained using protocols approved by the UCSF institutional review board. Briefly, skin was incubated overnight at 4 °C in 25 U/mL dispase solution (Corning Life Sciences, Corning, NY) followed by mechanical separation of epidermis from dermis. Epidermis was incubated in 0.05% trypsin for 15 minutes at 37 °C. Dissociated epidermal cells were filtered using a 100 µm nylon cell strainer (Corning Life Sciences) and cultured in keratinocyte growth media (KGM; Medium 154CF supplemented with 0.07 mM CaCl2 and Human Keratinocyte Growth Supplement; Life Technologies, Waltham, MA).
ABIN1 and A20 lentiviral overexpression and cytokine treatment
ABIN1, A20, and GFP open reading frame expression vectors were purchased from GeneCopoeia (Rockville, MD; Table S3). Lentivirus particles were prepared by the UCSF ViraCore. Briefly, HEK293T cells were seeded at 7×104 cells per cm2 in 15 cm tissue culture dishes in 20 mL media (DMEM, 10% FBS, Life Technologies). 24 hours after plating, 12 µg of lentiviral transfer vector was transfected alongside 7 µg psPAX2 (Addgene #12260) and 3 µg pMD2.G (Addgene #12259) with 50 µL jetPRIME transfection reagent (Polyplus, New York, NY) according to manufacturer’s protocol. 72 hours post-transfection, lentiviral supernatant was collected and passed through a 0.45 µm filter (EMD Millipore, Burlington, MA). Lentiviral titer was determined using the p24 ELISA kit from Takara (Mountain View, CA). Pooled primary cultured keratinocytes from three different individuals were transduced at a multiplicity of infection (MOI) of 10–100 as previously described (Kuhn et al. 2002). One day prior to transduction, keratinocytes were plated at 1×104 cells/cm2. Lentivirus supernatants were diluted using KGM and 4 ug/mL Polybrene (EMD Millipore). Keratinocytes were incubated with lentivirus for 16 hours after which they were allowed to recover for 24 hours in fresh KGM. Lentivirus infection was selected for using 2 µg/mL puromycin (Life Technologies) for 48 hours. At ~80% confluency, cells were incubated in fresh KGM or KGM containing cytokine (10 ng/ml TNFα or 200 ng/ml IL-17A, PeproTech, Rocky Hill, NJ) for 1 or 24 hours. Samples from triplicate experiments for 2 distinct pooled primary cultured keratinocytes from three different individuals (six total replicates) were harvested for protein and RNA extraction.
Western blot
Whole cell lysate was extracted using radioimmunoprecipitation assay (RIPA) buffer with freshly dissolved protease/phosphatase inhibitor as per manufacturer’s protocol (Life Technologies). Protein concentration was measured using DC™ Protein Assay (BioRad Laboratories, Hercules, CA). Equal amounts of protein were separated using a NuPAGE 4–12% Bis-Tris Protein Gel and transferred to PVDF membrane (Life Technologies). Membranes were blocked with Odyssey® PBS Blocking Buffer (Li-Cor, Lincoln, NE) for 30 minutes at room temperature and incubated with primary antibodies against ABIN1(TNIP1) (Proteintech, Rosemont, IL; 1:1000, rabbit) and A20 (TNFAIP3) (Cell Signaling, Danvers, MA; 1:1000, rabbit). GAPDH (Cell Signaling; 1:10000, mouse) was used as loading control. Anti-rabbit and anti-mouse secondary antibody conjugated to an infrared dye (IRDye800CW and IRDye 680RD respectively; Li-Cor) or anti-rabbit and anti-mouse secondary antibody conjugated to horse radish peroxidase were used (Cell Signaling) and the images were acquired on an Odyssey FC imaging instrument (Li-Cor) or peroxidase activity was detected using Pierce ECL Western Blotting Substrate (Life Technologies).
Statistical analysis for western blot band intensity analysis
When applicable, the results are presented as mean+SD. Statistical analysis was conducted using GraphPad Prism v5.0f (La Jolla, CA, USA). Student’s t-test was used to compare two separate sets of independent and identically distributed samples with a p-value < 0.05 considered as significant.
RNA isolation and RNA-sequencing
Total RNA was extracted using TRIzol reagent (Life Technologies) as per manufacturer’s protocol. RNA-seq libraries were prepared with 300–1000 ng of total RNA using KAPA Biosystems Stranded RNA-Seq Kits with RiboErase HMR (Roche, Pleasanton, CA). Technical duplicate sequencing libraries were generated for each RNA sample to minimize batch effect. Total RNA samples were depleted for rRNA through hybridization of complementary DNA oligonucleotides, followed by treatment with RNase H and DNase to remove rRNA duplexed to DNA and original DNA oligonucleotides, respectively. The resulting ribosomal depleted RNA then underwent RNA fragmentation using heat and magnesium. First strand complementary DNA (cDNA) synthesis was performed using random primers followed by second strand synthesis. To the 3’ ends of the dscDNA library fragments, dAMP was added (A-tailing). dsDNA adapters with 3’dTMP was ligated to the A-tailed library fragments. Library fragments with appropriate adapter sequences were amplified via ligation-mediated PCR. Post amplified cDNA libraries were quantitated with either Quant-iT™ dsDNA or Qubit™ dsDNA HS assay kits (Life Technologies). Quality assessment was performed using the LabChip GX Touch HT microfluidics platform (Perkin Elmer, Waltham, MA). 2 X 150 base pair sequencing on a NovaSeq 6000 instrument was performed on libraries with a PhiX Control v3 (Illumina, San Diego, CA).
RNA-seq analysis
The RNA-Seq by Expectation Maximization (RSEM) algorithm was used to quantify gene expected counts used for differential expression analysis and Counts Per Million (CPMs) used for heatmap clustering. Differential expression analysis was performed using edgeR (v3.22.3(Robinson et al. 2010); R v3.5.1). For each comparison, very low expressing genes with a CPM ≤ 1 in 6 (the number of replicate per treatment condition) or more samples were removed. An additive model formula was then used to adjust for batch effect differences between the two batches of keratinocyte pools within edgeR’s glmQLFit framework (i.e. genewise negative binomial generalized linear models with Quasi-Likelihood tests, which fits a quasi-likelihood negative binomial generalized log-linear model to count data to conduct genewise statistical tests for a given coefficient or contrast, Lund et al. 2012), and a quasi-likelihood F-test (glmQLFTest) was used to identify those genes significantly different (False Discovery Rate (FDR) p-value < 0.05 after correcting for multiple hypothesis testing using the Benjamini-Hochberg procedure) between each pairwise comparison. Gene ontology analysis was performed using Bioconductor’s goseq package (v1.32.0, Young et al. 2010).
To generate the heatmap, variation in gene expression across samples was visualized using R’s (v3.4.4) gplots package (v30.1). Pearson’s correlation distance was calculated between Z scores for each gene, and complete linkage clustering was performed to group genes by common patterns across samples. The resulting dendrogram was then cut to yield 14 clusters. Gene ontology was performed on genes in each cluster as compared to a background of all expressed genes using R’s goseq package (v1.28.0). The R scripts for these analyses as well as their resulting output are available on GitHub: https://github.com/SRHilz/ModOfKerInf_RNAseqAnalysis.
Single-cell RNA-sequencing
Human epidermal cells were isolated from normal surgical tissue discards or lesional skin from psoriasis, eczema, or erythrokeratodermia variabilis patients. Written informed consent for skin samples was obtained using protocols approved by the UCSF institutional review board. Skin was incubated for 2 hours at 37 °C in 25 U/mL dispase solution followed by mechanical separation of epidermis from dermis. Epidermis was incubated in 0.05% trypsin for 15 minutes at 37 °C. Dissociated epidermal cells were washed with KGM and filtered using a 40 µm nylon cell strainer (Corning Life Sciences). FACS was performed on dissociated cells to exclude debris, doublets and DAPI-positive cells. The sorted cells were resuspended in 0.04% BSA in PBS (Life Technologies) prior to Chromium Single cell 3’ Solution V2 (10x Genomics, Pleasanton, CA) library preparation, performed by the UCSF Institute for Human Genetics Core as per manufacturer’s protocol. Four scRNA-seq sample datasets were originally published in Cheng et al (“Transcriptional programming of normal and inflamed human epidermis at single-cell resolution”, in press) and were re-analyzed along with the new samples as described below.
ScRNA-seq data processing and QC filtering
Cellranger (10X genomics v2.0.2) was used to de-multiplex the raw Illumina sequencing data, from scRNA-seq libraries, quantify UMIs (using the GRCh38 v1.2.0 reference transcriptome), and aggregate data for the 8 samples. We managed and filtered the resulting data from 59,502 cells with Seurat (v2.2.0 (Macosko et al. 2015)). To control for damaged cells, we filtered out cells in the top 5th percentile of proportion of mitochondrial UMI, which corresponded to cells with greater than 14% of total UMI accounted for by mitochondrial transcripts. We used multiplet rate (MR) estimates provided by 10X genomics (https://assets.ctfassets.net/an68im79xiti/UhAMGmlaEMmYMaA4A4Uwa/274a813b81e42cba81345d49380432d7/CG00052_SingleCell3_ReagentKitv2UserGuide_RevD.pdf) to fit a linear model to estimate the percentage of multiplets in each sample based on the number of loaded cells. In order to avoid including partial cells and multiplets in our analysis, we filtered on the number of detected genes in each cell. On a per sample basis, we removed cells using a low threshold of 0.5% and high threshold of 100 – 2*MR on the percentiles of number of genes detected. These filters left 42,105 cells to include in our primary analyses.
ScRNA-seq dimensional reduction and data imputation
Using the pipeline described in Cheng et al (“Transcriptional programming of normal and inflamed human epidermis at single-cell resolution”, in press), we used ZINB-WaVE (v1.0.0)(Risso et al. 2018) to remove confounding signal due to the percent mitochondrial expression, the total number of UMIs detected (log scaled) and the sample membership from the raw count data of highly expressed (at least 500 counts per million in 0.1% of cells) genes in each cell. We used the 20 dimensional projection of the data produced by ZINB-WaVE to construct an affinity matrix (with adaptive distance parameters ka = 10 and k = 30) to input to the MAGIC (Dijk et al. 2017) imputation algorithm. For the imputation step we set the parameter t to 10.
ScRNA-seq differential expression analysis
We used limma-trend (Law et al. 2014b) to compare the single cell expression profiles from each disease to that in the normal skin samples. For this analysis, UMI count data was converted to library-size normalized counts per million (CPM) and log2 scaled with an offset of 1. We performed this analysis on 10,194 moderately expressed genes in our cohort of cells (at least 300 CPM in 0.1% of cells). We removed melanocytes by dropping cells with PMEL expression greater than 500 CPM. To remove immune cells, we used an HLA-DRA expression cutoff of greater than 2000 CPM. Limma-trend calculates the difference in mean log-CPM for each group of cells, and to evaluate statistical significance, moderated t-statistics are calculated based on an empirical Bayes approach. We use the false discovery rate (FDR) of the p-values associated with these statistics to correct for multiple hypothesis testing.
Supplementary Material
Supplemental Table 1A: edgeR differential expression results for A20 overexpressing keratinocytes vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1B: edgeR differential expression results for ABIN1 overexpressing keratinocytes vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1C: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1D: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1E: edgeR differential expression results for A20 overexpressing keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes with 24 hours IL17 (six replicates for each condition).
Supplemental Table 1F: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes with 24 hours IL17 (six replicates for each condition).
Supplemental Table 1G: edgeR differential expression results for A20 overexpressing keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes with 1 hour IL17 (six replicates for each condition).
Supplemental Table 1H: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes with 1 hour IL17 (six replicates for each condition).
Supplemental Table 1I: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1J: edgeR differential expression results for A20 overexpressing keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes with 1 hour TNFα (six replicates for each condition).
Supplemental Table 1K: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes with 1 hour TNFα (six replicates for each condition).
Supplemental Table 1L: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1M: edgeR differential expression results for A20 overexpressing keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes with 24 hour TNFα (six replicates for each condition).
Supplemental Table 1N: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes with 24 hour TNFα (six replicates for each condition).
Supplemental Table 2: Limma trend differential expression results for bulk aggregated epidermal single cell RNA-seq data for psoriasis, erythrokeratodermia variabilis, or atopic dermatitis compared to normal truncal skin.
ACKNOWLEDGEMENTS
We acknowledge Rachel Sevey for assistance with figure generation, using Adobe Illustrator CC. This work was supported in part by funds from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health K08AR067243 to JBC.
ABBREVIATIONS
- ABIN1
A20 binding inhibitor of NFκB
- EKV
erythrokeratodermia variabilis
- FDR
false discovery rate
- GFP
green fluorescent protein
- IL-17
interleukin-17
- KGM
keratinocyte growth media
- NFκB
nuclear factor κB
- NHEK
normal human epidermal keratinocyte
- TNFα
tumor necrosis factor α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Access
Sequence data has been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001002981. Further information about EGA can be found on https://ega-archive.org.
CONFLICTS OF INTEREST
AJS, CJV, and SCB are employees of Nantomics, LLC. CJV and SCB are equity holders of Nantomics, LLC.
REFERENCES
- Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem 2004;279(31):32633–42 [DOI] [PubMed] [Google Scholar]
- Catrysse L, Vereecke L, Beyaert R, van Loo G. A20 in inflammation and autoimmunity. Trends Immunol 2014;35(1):22–31 [DOI] [PubMed] [Google Scholar]
- Chen Y, Yan H, Song Z, Chen F, Wang H, Niu J, et al. Downregulation of TNIP1 Expression Leads to Increased Proliferation of Human Keratinocytes and Severer Psoriasis-Like Conditions in an Imiquimod-Induced Mouse Model of Dermatitis. PloS One 2015;10(6):e0127957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I, et al. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Invest. Dermatol 2011;131(3):677–87 [DOI] [PubMed] [Google Scholar]
- Cruz JA, Childs EE, Amatya N, Garg AV, Beyaert R, Kane LP, et al. IL-17 Signaling Triggers Degradation of the Constitutive NF-κB Inhibitor ABIN-1. ImmunoHorizons 2017;1(7):133–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol 2010;10(7):479–89 [DOI] [PubMed] [Google Scholar]
- Detmar M, Orfanos CE. Tumor necrosis factor-alpha inhibits cell proliferation and induces class II antigens and cell adhesion molecules in cultured normal human keratinocytes in vitro. Arch. Dermatol. Res 1990;282(4):238–45 [DOI] [PubMed] [Google Scholar]
- Dijk D van, Nainys J, Sharma R, Kathail P, Carr AJ, Moon KR, et al. MAGIC: A diffusionbased imputation method reveals gene-gene interactions in single-cell RNA-sequencing data. bioRxiv 2017;111591
- Fuchs-Telem D, Sarig O, van Steensel MAM, Isakov O, Israeli S, Nousbeck J, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet 2012;91(1):163–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AV, Ahmed M, Vallejo AN, Ma A, Gaffen SL. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci. Signal 2013;6(278):ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- G’Sell RT, Gaffney PM, Powell DW. A20-Binding Inhibitor of NF-κB Activation 1 is a Physiologic Inhibitor of NF-κB: A Molecular Switch for Inflammation and Autoimmunity. Arthritis Rheumatol. Hoboken NJ 2015;67(9):2292–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanifin JM. Evolving concepts of pathogenesis in atopic dermatitis and other eczemas. J. Invest. Dermatol 2009;129(2):320–2 [DOI] [PubMed] [Google Scholar]
- Ippagunta SK, Gangwar R, Finkelstein D, Vogel P, Pelletier S, Gingras S, et al. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc. Natl. Acad. Sci. U. S. A 2016;113(41):E6162–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT, Cao L, Roberson EDO, Duan S, Helms CA, Nair RP, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am. J. Hum. Genet 2012;90(5):796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JG, Brunner PM. Interleukin-17 alters the biology of many cell types involved in the genesis of psoriasis, systemic inflammation and associated comorbidities. Exp. Dermatol 2018;27(2):115–23 [DOI] [PubMed] [Google Scholar]
- Kuhn U, Terunuma A, Pfutzner W, Foster RA, Vogel JC. In Vivo Assessment of Gene Delivery to Keratinocytes by Lentiviral Vectors. J. Virol 2002;76(3):1496–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CEM, Papp K, et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N. Engl. J. Med 2014;371(4):326–38 [DOI] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014a;15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014b;15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowdon RF, Zhang B, Bilenky M, Mauro T, Li D, Gascard P, et al. Regulatory network decoded from epigenomes of surface ectoderm-derived cell types. Nat. Commun 2014;5:5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund PL, Nettleton D, McCarthy DJ, Smyth GK. Detecting Differential Expression in RNAsequence Data Using Quasi-likelihood with Shrunken Dispersion Estimates. Stat. Appl. Genet. Mol. Biol 2012;11:5. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015;161(5):1202–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997;278(5339):860–6 [DOI] [PubMed] [Google Scholar]
- Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med 2016;8(3):227–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet 2009;41(2):199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol 2008;159(5):1092–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of psoriasis and development of treatment. J. Dermatol 2018;45(3):264–72 [DOI] [PubMed] [Google Scholar]
- Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL-17–dominant immune profile is shared across the major orphan forms of ichthyosis. J. Allergy Clin. Immunol 2017;139(1):152–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez VP, Krueger W, Aneskievich BJ. TNIP1 reduction of HSPA6 gene expression occurs in promoter regions lacking binding sites for known TNIP1-repressed transcription factor. Gene 2015;555(2):430–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich K, Nestle FO, Papp K, Ortonne J-P, Evans R, Guzzo C, et al. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet Lond. Engl 2005;366(9494):1367–74 [DOI] [PubMed] [Google Scholar]
- Risso D, Perraudeau F, Gribkova S, Dudoit S, Vert J-P. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun 2018;9(1):284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma. Oxf. Engl 2010;26(1):139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudraiah S, Shamilov R, Aneskievich BJ. TNIP1 reduction sensitizes keratinocytes to postreceptor signalling following exposure to TLR agonists. Cell. Signal 2018;45:81–92 [DOI] [PubMed] [Google Scholar]
- Rupec RA, Boneberger S, Ruzicka T. What is really in control of skin immunity: lymphocytes, dendritic cells, or keratinocytes? facts and controversies. Clin. Dermatol 2010;28(1):62–6 [DOI] [PubMed] [Google Scholar]
- Schröder JM, Reich K, Kabashima K, Liu FT, Romani N, Metz M, et al. Who is really in control of skin immunity under physiological circumstances - lymphocytes, dendritic cells or keratinocytes? Exp. Dermatol 2006;15(11):913–29 [DOI] [PubMed] [Google Scholar]
- Scott CA, Tattersall D, O’Toole EA, Kelsell DP. Connexins in epidermal homeostasis and skin disease. Biochim. Biophys. Acta 2012;1818(8):1952–61 [DOI] [PubMed] [Google Scholar]
- Sohn K-C, Back SJ, Choi D-K, Shin J-M, Kim SJ, Im M, et al. The inhibitory effect of A20 on the inflammatory reaction of epidermal keratinocytes. Int. J. Mol. Med 2016;37(4):1099–104 [DOI] [PubMed] [Google Scholar]
- Sun S-C, Chang J-H, Jin J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol 2013;34(6):282–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchi H, Terao H, Koga T, Furue M. Cytokines and chemokines in the epidermis. J. Dermatol. Sci 2000;24 Suppl 1:S29–38 [DOI] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology 2010;11:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wu R, High AA, Slaughter CA, Finkelstein D, Rehg JE, et al. A20-binding inhibitor of NF-κB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein β activation and protects from inflammatory disease. Proc. Natl. Acad. Sci. U. S. A 2011;108(44):E998–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1A: edgeR differential expression results for A20 overexpressing keratinocytes vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1B: edgeR differential expression results for ABIN1 overexpressing keratinocytes vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1C: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1D: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1E: edgeR differential expression results for A20 overexpressing keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes with 24 hours IL17 (six replicates for each condition).
Supplemental Table 1F: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 24 hours IL17 vs mock infected (GFP expressing) keratinocytes with 24 hours IL17 (six replicates for each condition).
Supplemental Table 1G: edgeR differential expression results for A20 overexpressing keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes with 1 hour IL17 (six replicates for each condition).
Supplemental Table 1H: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 1 hour IL17 vs mock infected (GFP expressing) keratinocytes with 1 hour IL17 (six replicates for each condition).
Supplemental Table 1I: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1J: edgeR differential expression results for A20 overexpressing keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes with 1 hour TNFα (six replicates for each condition).
Supplemental Table 1K: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 1 hour TNFα vs mock infected (GFP expressing) keratinocytes with 1 hour TNFα (six replicates for each condition).
Supplemental Table 1L: edgeR differential expression results for mock infected (GFP expressing) keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes (six replicates for each condition).
Supplemental Table 1M: edgeR differential expression results for A20 overexpressing keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes with 24 hour TNFα (six replicates for each condition).
Supplemental Table 1N: edgeR differential expression results for ABIN1 overexpressing keratinocytes with 24 hours TNFα vs mock infected (GFP expressing) keratinocytes with 24 hour TNFα (six replicates for each condition).
Supplemental Table 2: Limma trend differential expression results for bulk aggregated epidermal single cell RNA-seq data for psoriasis, erythrokeratodermia variabilis, or atopic dermatitis compared to normal truncal skin.