

Abstract
Activation of the heat shock response, and in particular up-regulation of stress-inducible Hsp70, herein referred to as Hsp70i, in newly transformed cells, appears to protect against protein damaging stimuli, induction of premature oncogene-induced terminal senescence (OIS) and apoptosis, thereby enabling tumor initiation and progression to an aggressive phenotype. Expressed at very low or undetectable levels in normal tissue, the cytoprotective effects of Hsp70i appear to be mediated through its activity as a molecular chaperone allowing proper folding of mutated proteins, and by blocking cell signaling pathways that regulate OIS and apoptosis. Identification of small-molecule inhibitors selective for Hsp70i could provide new therapeutic tools for cancer treatment. However, identification of selective inhibitors of Hsp70i has proven challenging largely because of the affinity of the protein for ATP. Additionally, its chaperone functions do not lend the protein amenable to traditional enzymatic high through put screens. Here we describe the use of fluorescence-linked enzyme chemoproteomic strategy (FLECS) to identify Hsp70i inhibitors. The FLECS assay is a simple binding assay that enables proteins tagged with fluorophors to be rapidly and quantitative screened against small molecule libraries. We show several case history examples of the methodology that led to discovery of the Fatty acid synthase inhibitor, FASNALL, the DAPK3 inhibitor HS38 and HS72, an allosteric inhibitor selective for Hsp70i.
Keywords: Heat shock protein 70, Hsp70 inhibitor, drug discovery, anticancer drug, antiviral drug
1. Introduction
The Heat shock protein 70 (Hsp70) family have broad chaperone functions in cells that include folding of nascent proteins, refolding of misfolded proteins, protein transport, regulation of breakdown of unstable proteins, removal of protein complexes, and control of regulatory proteins 1,2. These functions are driven by ATP hydrolysis in the N-terminal nucleotide-binding domain (NBD) of Hsp70. The Hsp70s are evolutionary conserved across species and there are 8 mammalian Hsp70 family members 3. The inducible form of Hsp70 (Hsp70i, also called Hsp72, Hsp70–1, HspA1A/HspA1B) is present in low or undetectable levels in most unstressed normal cells and tissues, however, expression levels rapidly increase in response to cellular stresses such as heat shock or transformation. Deletion of its immediate paralog, constitutively active heat shock protein cognate 70 (Hsc70) is developmentally lethal, whereas deletion of Hsp70i results in sterility of male mice, but no other overt phenotype in unstressed mice 4,5. Hsp70i and Hsc70 are highly related, sharing 90% over all sequence identity, however, most of the sequence variability is confined to the NBD (<80% identity). The close sequence similarity between Hsp70i and Hsc70 has certainly contributed to past difficulties in distinguishing the biological functions of the two proteins using both pharmacologic and RNA interference approaches. The broad structural organization of the Hsp70’s is similar; consisting of three functional domains, the NBD in the N-terminal region, a substrate-binding domain (SBD) in the C-terminal region, and a linker in the middle 6. The chaperone activity of Hsp70i is a function of the C-terminus in cooperation with other co-chaperones such as Hsp40, Hip, Hop, CHIP and Bag1 6. Crystallographic and NMR studies have shown that Hsp70i has distinct open and closed conformational states that change based on the presence of nucleotides and some of its co-chaperones 2,7,8. When complexed with ADP and substrate, there is little interaction between the NBD, SBD, and linker region. Upon substrate and ADP release and rebinding of ATP, the linker region and SBD make contact with the NBD 9. There is also allosteric regulation of ATP hydrolysis between the domains 10.
Hsp70 inhibitor Prior Art.
The involvement of Hsp70i in maintaining tumor stability and its potential to synergize with Hsp90 to selectively kill tumors, have been driving factors to develop inhibitors specifically targeting the protein in vivo 9,11. Confounding factors in the discovery of Hsp70i inhibitors include its affinity for ATP/ADP and their conformational influences, the close sequence similarity within the NBD with Hsc70. Indeed, conventional approaches have failed to consider off target binding to Hsc70, which could account for the poor performance in vivo of current Hsp70 inhibitors. Current inhibitors identified to target mammalian Hsp70 include NSC 630668-R/1, VER-155008, MAL3–101, MKT-077, Pifithrin, and Apoptozole (Fig.1). There is considerable structural diversity amongst the prior inhibitors, reflecting the various methodologies that were employed to define them, as well as whether they target the NBD or SBD on Hsp70 (REFS 30–33). The NDB domain is most favored for inhibitor development, albeit challenging. Inhibitors targeting the NBD to date have either employed rationale design approaches based on partial crystallographic structures with bound nucleotide or have relied on coupled enzymatic assays measuring ATP depletion. The polar interactions present in the NBD and its low μM affinity for ADP have certainly contributed to difficulties in selective inhibitor discovery 12. The crystal structure of the active form of Hsp70i shows the NBD to be completely enclosed around the nucleotide with almost no solvent accessibility to the surface 13. Aside from the nuances associated with targeting the NBD, none of the approaches adopted thus far by other groups have been able to separate Hsp70 family members, especially Hsp70i from Hsc70. NSC 630668-R/1, inhibits ATPase activity but does not discriminate Hsp70i from Hsc70 14. VER-155008 shows broad specificity with other Hsp family members, largely because it is a nucleotide derivative. It also contains two potentially labile (perhaps by design) benzyl groups 12. MAL3–101 has been shown to stimulate Hsp70 ATPase function, suggesting it is an allosteric regulator, although the exact binding site of this molecule remains unknown 15. Like NSC 630668-R/1, MAL3–101 is quite large and has a number of labile ester groups. MKT-077 targets the NBD and inhibits proliferation in tumor cell lines, however, severe renal dysfunction in patients was observed in phase I clinical trials 16,17. Pifithrin, binds to the SBD of both Hsc70 and Hsp70i disrupting client protein interaction in vitro. In tumor cells the molecule promotes caspase dependent cell death in tumor cells only suggesting it has some specificity to Hsp70i in vivo, although p53 binding has also been shown, which could explain its antitumor actions 18. MKT-O77 and Pifithrin have potential reactive groups that render them covalent modifiers, which may contribute to side effects in vivo. More promising inhibitors of the Hsp70 class are second and 3rd generation MKT-077 analogs JG18 and JG40. Their mechanism of action and specificity lies in interaction with Hsp70 co-chaperones such as NEF 19.
Figure1.
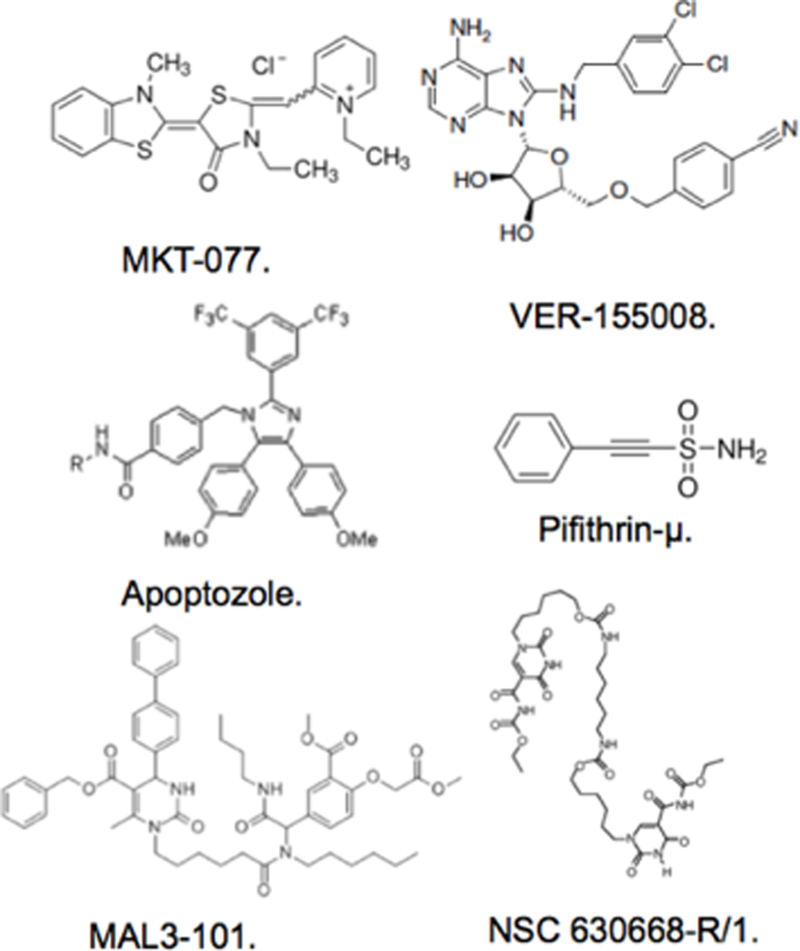
Prior Art Inhibitors Reported to Target Heat Shock Protein 70.
Discovery of HS-72 by FLECS.
In Howe et al. 20 we reported the results of screening Hsp70i by fluorescent-linked enzyme chemoproteomic strategy (FLECS) and identified several novel inhibitors that appeared to act by allosterically affecting the Hsp70i’s affinity for ATP. The most promising of these was HS-72 a ((S)-N-(1-propyl-1H-benzo[d]imidazol-2-yl)-1-(pyrazin-2-yl)piperidine-3-carboxamide (Fig.2). The FLECS screen is a variation of proteome mining technology utilized in the discovery of the Hsp90 inhibitor SNX5422 21. Proteome mining was designed to screen all purine binding proteins (the purinome) expressed in cells/tissues en masse against large directed chemical libraries, matching early chemical starting points with targets 22,23. The power of this approach enabled not only hundreds of diverse enzymes to be screened at a single step but also enabled the selectivity of a particular hit molecule to be determined simultaneously. The same assay could also then be used in subsequent iterative campaigns to monitor or improve selectivity as one strived to improve the molecules potency and bioavailability. The hypothesis being that this would avoid later stage failures due to unpredicted toxicity during in vivo studies in animals. This approach was put into practice in the discovery and development of SNX5422, and orally bioavailable inhibitor for Hsp90 that is currently advancing in clinical trials 21. A cell/tissue extract is applied to an ATP affinity resin in which the nucleotide is immobilized through its gamma-phosphate 21–23. This orientation favors reversible binding of proteins that bind purines through multiple contacts with the adenosine ribose moiety with the gamma phosphate showing solvent accessibility at the surface (the kinase orientation). This orientation of the purine is highly conserved across multiple gene families including protein kinases, non-protein kinases, dehydrogenases, hydrolyases formylases, most metabolic enzymes, helicases, DNA and RNA binding proteins, transcription factors and chaperones such as Hsp90. Over the years, by MS analysis we have documented several hundreds proteins that reversibly bind to ATP when tethered by its gamma phosphate from human, mammalian and pathogen sources. Typically we estimate that up to 10% of the expressed genome of most species express protein that bind the purine in the ‘kinase orientation’. In human tissues this represents up to 2000 distinct proteins. Importantly, the majority of the enzymes captured on this media are considered intractable to conventional methods of high through put (HT) screening. They often require radioisotopes to follow activity or have functions that do not lend themselves readily amenable HT screening e.g. protein folding. The method is a simple universal binding assay that enables ATP competitive inhibitors to be identified directly.
Figure2.

Structure of HS-72.
The FLECS assay is a variation on the proteome mining approach and is more targeted from the outset, yet still retains the selectivity profiling capabilities of proteome mining 20,24,25. Moreover, FLECS is more suited to academic based drug discovery than the original proteome mining approach which was really designed to identify chemical starting points that would likely derive novel composition of matter claims. For a FLECS screen one expresses the targeted purine binding protein of choice as a recombinant GFP-fusion protein (one could use YFP or RFP) either in a bacterial, yeast or mammalian cell line. The expressing cells are then lysed, homogenized, clarified and the entire extract applied to gamma phosphate ATP resin without further purification. The bound proteins are washed with high salt to remove any non-specifically associated proteins. To verify that the GFP-fusion protein has a functional ATP binding site the GFP-fusion protein charged resin is serially eluted with increasing concentrations of ATP [1–100mM]. The resin flow though is collected and aliquots measured for fluorescence in a plate reader. This method can also be used to determine the dissociation constant (Kd) of the bound fluorescent protein for ATP as described 25. Once competitive binding is established one is ready to screen the fusion protein against large libraries of small molecules. Figure 3 illustrates the diversity of proteins we have screened using the FLECS assay. In some instances native proteins can also be screened after labeling them with reactive fluorophors directly while bound to the ATP resin. This approach was used to identify fatty acid synthase (FASN) inhibitor Fasnall 24. Briefly, lactating pig mammary gland extract was applied to the ATP resin and the bound proteins labeled with cysteine reactive fluor probe 24. The reader should note that FASN is a highly induced in the lactating mammary gland and constitutes ~25% of the soluble cellular protein fraction.
Figure3. FLECS is a Universal assay that enables purine utilizing proteins of diverse function to be screened against small molecule libraries.
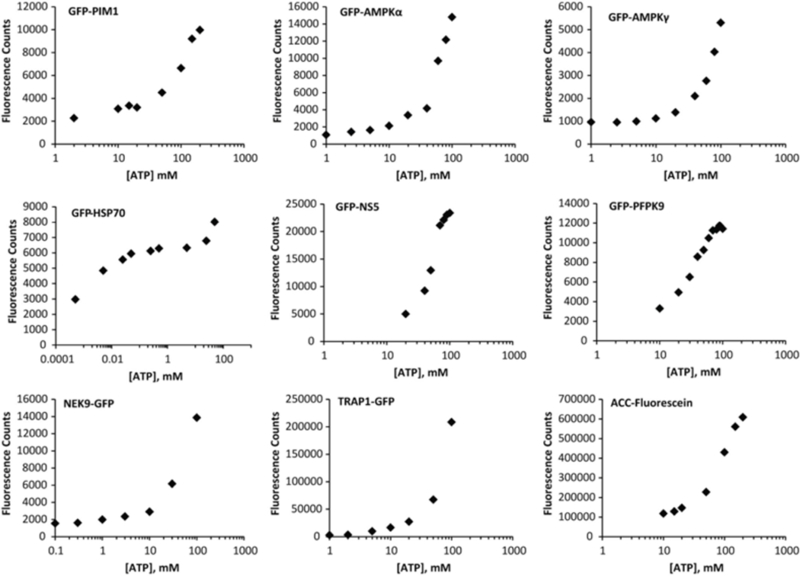
The indicated GFP-fusion proteins were expressed in various cell lines and captured from homogenates on ATP media. The proteins were then tested for their ability to be competitively released from the media with increasing concentrations of free ATP.GFP proteins tested-PIMK,Pim kinase 1;AMPKα,α subunit AMP activated protein kinase; AMPK γ,γ regulatory subunit AMPK; GFP-Hsp70, inducible form of heat shock protein 70; NS5 non-structural protein 5 of Dengue virus; PfPK9, plasmodium falciparum Protein kinase 9; NEK9 protein kinase; TRAP1;TNF Receptor Associated Protein 1; ACC Acetyl CoA Carboxylase.
Hsp70 and ATP binding.
The Hsp70 family have a very low affinity for ATP (<10 μM) compared with most ATP binding proteins we have examined and its ATPase activity turns over rapidly. Interestingly, based on the crystal structure of Hsc70 with bound nucleotide one would predict that the protein is sterically hindered from binding ATP tethered through its gamma phosphate. This is because the protein normally binds the nucleotide in the opposite direction to the majority of purine utilizing enzymes such as protein kinases or Hsp90. However, when one mixes cellular extract with gamma-phosphate linked ATP resin one can efficiently bind native forms of Hsp70. Interestingly however if one performs a titration experiment against ATP and ADP from μM to high [mM] one observes a strikingly different pattern of elution compared with the majority of proteins bound to gamma phosphate linked ATP resin (Fig.4). Figure 4 shows that very low [μM] of ATP are required to elute GFP-Hsp70i from gamma phosphate linked ATP compared to very high [mM] of ADP. This contrast with proteins like Hsp90 which generally require [mM] ATP to be released from the same resin. The underlying mechanisms for these different elution patterns are related to distinct mechanism of binding and release from the media. In the case of Hsp90 binding is due to first order binding to the immobilized ATP and the amount of free ATP required to release is therefore a function of the ligand density (10μmol/ml) and the proteins affinity for ATP (<10μM). In the case of Hsp70, since the nucleotide is bound in the opposite orientation. Therefore, in order for Hsp70 to bind ATP tethered through its gamma phosphate the protein would have to first be in its ‘apo’ open conformation state. Once bound the protein cannot be released by simple nucleotide competition as shown by the high [ADP] required to liberate the protein. However, as shown in figure 5, when exposed to even low [μM] ATP the protein turns over and is released. Since it is unlikely that the low ATP (<10μM) is able to compete with high ligand density the only possible mode of release is through an allosteric effect i.e. the protein is regulated allosterically via a nucleotide binding site distinct from its active site. For these reasons, when screening for inhibitors of Hsp70 using the FLECS assay one is unlikely to identify inhibitors that directly compete at its active site.
Figure.4. Nucleotide binding of the inducible form of Hsp70 is allosterically regulated.
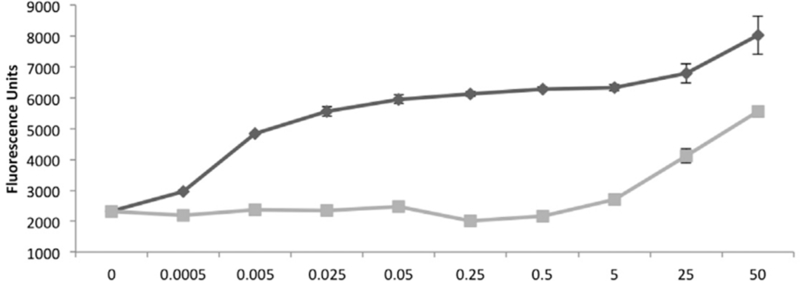
GFP-Hsp70 bound to γ–phosphate linked ATP is selectively released with low [μM]ATP compared with high mM[ADP],suggesting that protein is allosterically regulated by ATP.
Figure5. Steps involved in screening for inhibitors of Hsp70 by FLECS.
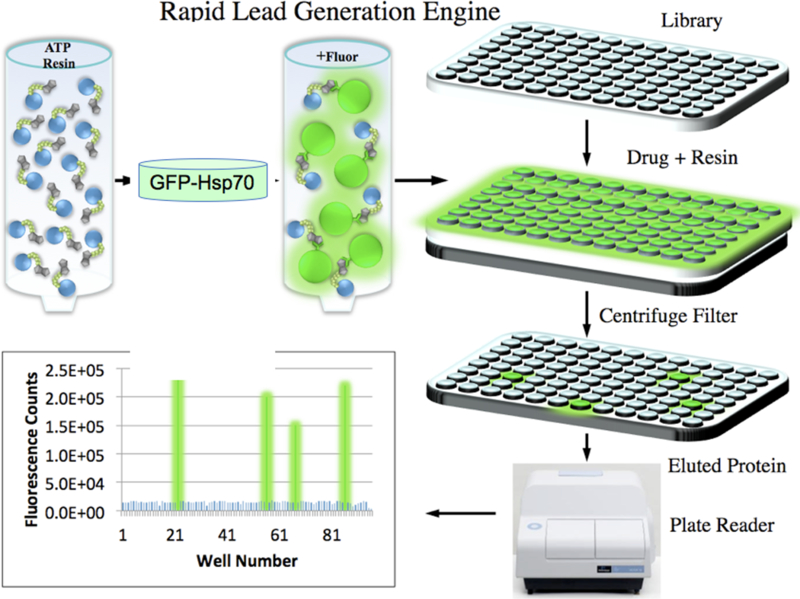
Extracts are prepared from cells expressing GFP-Hsp70 and mixed with a column of ATP resin.Following washing, the resin with bound protein is parsed into a 96 well filter plate and each well eluted by centrifugation with a drug like molecule. The eluted proteins are captured in a 96 well titer plate and measured for fluorescence in a standard fluorescence plate reader.
2. Materials.
γ-Linked ATP Sepharose Media (see Note 1).
Dry CNBr- Activated Sepharose 4B media (28.6 g, GE Healthcare) was equilibrated in HCl (1 mM, 333 mL) for 5−15 min, washed with HCl (1 mM, 600 mL) followed by deionized water (333 mL). The media was added to mixture A (NaHCO3, 0.97 g; NaCl, 3.4 g; H2O, 115 mL; 1,4-dioxane, 29 mL; 1,10-diaminodecane, 3.6 g; ethanolamine, 3.6 mL) and shaken for 2 h. Meanwhile, reaction mixture B (H2O, 143 mL; ATP, disodium salt, 7g; 1-methylimidazole, 5.2 mL; EDC, 12 g) was stirred for 1 h. Mixture A was removed and the media was washed with HCl (1 mM, 600 ml) then deionized water (333 mL). Mixture B and the media were combined rotated for 24 h. The resulting ATP Sepharose media was filtered and washed with HCl (1 mM, 600 mL), then deionized water (333 mL) and stored at 4 °C in phosphate buffer (0.1 M, pH 7.4) containing NaN3 (3 mM).
Cell Lysis Buffer 150 mM NaCl, 50 mM Tris pH 7.5, 1% Triton X-100, 1 mM EDTA, 1 mM dithiothreitol (DTT]).
Low Salt Buffer 150 mM NaCl, 25 mM Tris pH 7.5, and 60 mM MgCl2
High Stringency Wash Buffer 1 M NaCl, 25 mM Tris pH 7.5, 60 mM MgCl2, and 1 mM DTT
Low-Stringency Wash Buffer 150 mM NaCl, 25 mM Tris pH 7.5, 60 mM MgCl2, and 1 mM DTT.
3. Methods.
Screening compounds by FLECS.
Figure 5 illustrates the steps and equipment required to complete the FLECS assay. ATP media (25ml) is charged with clarified cellular extract expressing an N terminal GFP fused to Hsp70i. Typically for screening 1000 compounds we recommend charging the media with 1×107 cells/ml of ATP media. The media is washed with high and low ionic strength buffers to remove non-specifically bound proteins. Next one determines the signal to noise ratio for the assay by parsing out the charged resin without further purification into a 96 well filter plate (0.2μm cut off) and eluting the bound proteins with increasing [ATP, 0– 100mM] (See Note 2). As shown in figure 5, following addition of ATP (or drug) solution (50μl), the elution plate containing the resin is placed over a catch plate and centrifuged (3000xg) for 5 min. The eluted proteins are measure in a fluorescent plate reader. Signal to noise ratio is determined from the maximal signal one on obtains with ATP and should be 10–15 fold above background. Once one has established that the assay is working one is ready to screen small molecule libraries for compounds that will affect Hsp70i affinity for ATP. In the discovery of HS-72, we screened a ~4000 member library of purine-like analogs against GFP-Hsp70i en masse. Following parsing of the charged resin into filter plates, 50μl of 350μM solution (in 10% DMSO) of each compound from the library was added to each well. The plate was placed over the catch plate and centrifuged resulting in 96 compounds screened simultaneously for potential Hsp70i inhibitors. This operation can be carried out either manually or using standard liquid handling robot. All compounds yielding significant fluorescence should be verified by repeating the assay preferably in titration experiments. In our HS-72 study the primary screen identified 197 hits from the library, which were first sorted by their specificity toward GFP-Hsp70i over other purinome members that had also been screened against the same chemical library by FLECS. The compounds that were active in multiple assays were removed from consideration since these were considered non-selective. Next, the presence of GFP-Hsp70i in the eluates from the 197 primary hits was determined by Western blot. This reduced the collection to 60 compounds and also eliminated autofluorescent false-positive molecules. Then, we tested the ability of the 60 compounds for elution of native Hsp70 from the ATP resin using pig bladder extracts, a rich source of native Hsp70i, reducing the final collection to 22 diverse structures (0.65% of the library). This collection contained several diverse chemical structures which were further prioritized in a series of cell based assays of Hsp70i function that defined HS-72 as the most promising molecule.
FLECS Screen
A pEGFP-tagged Hsp70i was (plasmid 15215; Addgene) used in the FLECS assay and was originally cloned by Evan Eisenburg (Zeng et al., 2004). ATP 200 mM stock was prepared with low salt buffer. FuGENE 6 transfection reagent (Roche) was used for transfection of GFP-Hsp70i into HEK293T cells, following the manufacturer protocol. The transfection ensued for 48 hr, upon which time the cells were harvested and lysed in cell lysis buffer and one tablet Complete Mini protease inhibitor [Roche]). Cell lysates were stored at −80°C until further use. After binding, the resin lysates were washed three times with high stringency wash buffer and three times with low-stringency wash buffer. Next, the resin with bound proteins (50μl) were transferred to 0.2 μm polyvinylidene fluoride filter 96-well plate (Corning) sitting on top of a black flat-bottomed 96-well catch plate (Corning). Inhibitors or ATP were added to each well (50μl) and the plates were centrifuged using an Eppendorf Centrifuge5810 at 2,000 rpm for 2 min.
4. Notes.
Note 1.
As with any synthesis batches of gamma-phosphate linked ATP resin can vary considerably if protocols are not followed rigorously. The methods outlined give a specific density of ATP/ml of resin, typically 10μmols/ml. If the density is increased by 2 fold this increases the avidity of the media considerably reducing the sensitivity of the assay. This also leads to size exclusion due to ligand crowding i.e. high molecular proteins >50kDa start to be occluded. If one reduces the ligand density to 1μmol/ml, the resin begins to lose capacity. A lower density media can however be used with more purified proteins. We prefer the 10μmol/ml for 3 reasons; 1) this mimics intracellular ATP concentration (~10mM most cells); 2) this reduces the sensitivity of the assay to only define more potent inhibitors; 3) this enables both the GFP-fusion protein to be recovered as well as the entire cellular purinome. Later one can evaluate the intrinsic selectivity of a hit as well as its potency.
Note 2.
The major issue with the FLECs assay is the quality of the recombinant GFP-protein and ensuring adequate expression to complete the screen. One typically wants to achieve a signal to noise ratio of 10:1 over base line. This is determined by expressing the protein of interest, making a cell extract and applying the clarified mixture directly to the ATP resin. The resin is washed and the bound protein eluted with high Mg/ATP (60mM/100mM). The eluate is collected and amount of liberated GFP-fusion protein determined by fluorescence over low stringency buffer elution. Although it is possible to carry out the assay at lower ratio’s we would not recommend it. Once one has established an effective level of expression one should calculate how much expressed GFP fusion protein on requires to complete the entire screen i.e. how much extract needs to be applied to routinely give a signal to noise ratio of 10:1 or > for every compound in the library. One should then scale up expression to enable complete screening of the library in a single batch.
Acknowledgments
This work was supported by NIH grants R01-AI089526-04 to T.A.J.H. and a Department of Defense Transformative Vision Award to T.A.J H.
Footnotes
heat shock protein 70, fluorescence linked enzyme chemoproteomic strategy
References.
- 1.Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS letters 2007;581:3702–10. [DOI] [PubMed] [Google Scholar]
- 2.Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. Journal of medicinal chemistry 2010;53:4585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hunt C, Morimoto RI. Conserved features of eukaryotic hsp70 genes revealed by comparison with the nucleotide sequence of human hsp70. Proceedings of the National Academy of Sciences of the United States of America 1985;82:6455–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dix DJ, Allen JW, Collins BW, et al. Targeted gene disruption of Hsp70–2 results in failed meiosis, germ cell apoptosis, and male infertility. Proceedings of the National Academy of Sciences of the United States of America 1996;93:3264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wacker JL, Huang SY, Steele AD, et al. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. The Journal of neuroscience : the official journal of the Society for Neuroscience 2009;29:9104–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tavaria M, Gabriele T, Kola I, Anderson RL. A hitchhiker’s guide to the human Hsp70 family. Cell stress & chaperones 1996;1:23–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos C Molecular chaperones and protein quality control. Protein and peptide letters 2011;18:100. [DOI] [PubMed] [Google Scholar]
- 8.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences : CMLS 2005;62:670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell cycle 2010;9:1542–50. [DOI] [PubMed] [Google Scholar]
- 10.Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Molecular cell 2007;26:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle 2009;8:518–26. [DOI] [PubMed] [Google Scholar]
- 12.Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem 2010;53:7280–6. [DOI] [PubMed] [Google Scholar]
- 13.Qi R, Sarbeng EB, Liu Q, et al. Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol 2013;20:900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fewell SW, Day BW, Brodsky JL. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J Biol Chem 2001;276:910–4. [DOI] [PubMed] [Google Scholar]
- 15.Braunstein MJ, Scott SS, Scott CM, et al. Antimyeloma Effects of the Heat Shock Protein 70 Molecular Chaperone Inhibitor MAL3–101. J Oncol 2011;2011:232037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Propper DJ, Braybrooke JP, Taylor DJ, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol 1999;10:923–7. [DOI] [PubMed] [Google Scholar]
- 17.Britten CD, Rowinsky EK, Baker SD, et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin Cancer Res 2000;6:42–9. [PubMed] [Google Scholar]
- 18.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell 2009;36:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taguwa S, Frydman J. The significance of Hsp70 subnetwork for Dengue virus lifecycle. Uirusu 2015;65:179–86. [DOI] [PubMed] [Google Scholar]
- 20.Howe MK, Bodoor K, Carlson DA, et al. Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem Biol 2014;21:1648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fadden P, Huang KH, Veal JM, et al. Application of chemoproteomics to drug discovery: identification of a clinical candidate targeting hsp90. Chem Biol 2010;17:686–94. [DOI] [PubMed] [Google Scholar]
- 22.Haystead TA. The purinome, a complex mix of drug and toxicity targets. Curr Top Med Chem 2006;6:1117–27. [DOI] [PubMed] [Google Scholar]
- 23.Graves PR, Kwiek JJ, Fadden P, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol 2002;62:1364–72. [DOI] [PubMed] [Google Scholar]
- 24.Alwarawrah Y, Hughes P, Loiselle D, et al. Fasnall, a Selective FASN Inhibitor, Shows Potent Anti-tumor Activity in the MMTV-Neu Model of HER2(+) Breast Cancer. Cell Chem Biol 2016;23:678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carlson DA, Franke AS, Weitzel DH, et al. Fluorescence linked enzyme chemoproteomic strategy for discovery of a potent and selective DAPK1 and ZIPK inhibitor. ACS Chem Biol 2013;8:2715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]