

Abstract
The transmission fitness and pathogenesis of HIV-1 is disproportionately influenced by evolution in the five variable regions (V1–V5) of the surface envelope glycoprotein (gp120). Insertions and deletions (indels) are a significant source of evolutionary change in these regions. However, the rate and composition of indels has not yet been quantified through a large-scale comparative analysis of HIV-1 sequences. Here, we develop and report results from a phylogenetic method to estimate indel rates for the gp120 variable regions across five major subtypes and two circulating recombinant forms (CRFs) of HIV-1 group M. We processed over 26,000 published HIV-1 gp120 sequences, from which we extracted 6,605 sequences for phylogenetic analysis. We reconstructed time-scaled phylogenies by maximum likelihood and fit a binomial-Poisson model to the observed distribution of indels between closely related pairs of sequences in each tree (cherries). By focusing on cherries in each tree, we obtained phylogenetically independent indel reconstructions, and the shorter time scales in cherries reduced the bias due to purifying selection. Rate estimates ranged from to indels/nt/year and varied significantly among variable regions and subtypes. Indel rates were significantly lower in V3 relative to V1, and were also lower in HIV-1 subtype B relative to the 01_AE reference. We also found that V1, V2, and V4 tended to accumulate significantly longer indels. Furthermore, we observed that the nucleotide composition of indels was distinct from the flanking sequence, with higher frequencies of G and lower frequencies of T. Indels affected N-linked glycosylation sites more often in V1 and V2 than expected by chance, consistent with positive selection on glycosylation patterns within these regions. These results represent the first comprehensive measures of indel rates in HIV-1 gp120 across multiple subtypes and CRFs, and identifies novel and unexpected patterns for further research in the molecular evolution of HIV-1.
Keywords: HIV-1, gp120, phylogenetics, virus evolution, indel, subtype
1. Introduction
Human immunodeficiency virus type 1 (HIV-1) is a rapidly evolving retrovirus with enormous genetic diversity that is divided into four groups (M, N, O and P). The global HIV-1 pandemic that affects approximately 37 million people as of 2017 (World Health Organization 2018) is largely caused by group M, which is further partitioned into nine subtypes (A–D, F–H, J, K) that can differ by roughly 30 per cent of their genome sequence (Korber et al. 2001) and have distinct geographic distributions due to historical founder effects (Tebit and Arts 2011). In addition, there are a large number of circulating recombinant forms (CRFs) that are the result of recombination among two or more HIV-1 subtypes that have subsequently become established in particular regions at high prevalence. The HIV-1 subtypes and CRFs are clinically significant because of variation in pathogenesis, e.g. rates of disease progression, and evolution of drug resistance (Wainberg 2004; Vasan et al. 2006; Ariën, Vanham, and Arts 2007; Kiwanuka et al. 2008).
In the host cell-derived lipid membrane of every HIV-1 particle, there are numerous virus-encoded envelope glycoprotein complexes composed of three gp41 transmembrane units and three gp120 surface units (Tran et al. 2012). The HIV-1 gp120 glycoprotein is a potent surface-exposed antigen that plays a significant role in the recognition and binding of target cell receptors (Kwong et al. 1998). One reason for the difficulty in immunologically targeting this glycoprotein is the abundance of N-linked glycosylation sites: sequence motifs that encode the post-translational linkage of glycan groups to asparagine residues (Zhang et al. 2004). In addition, the HIV-1 gene encoding gp120 has a particularly high rate of evolution, especially within the five hypervariable regions that encode surface-exposed, disordered loop structures. These five variable regions (numbered V1–V5) can tolerate substantially higher amino acid substitution rates than the rest of the HIV-1 genome (Li, Tanimura, and Sharp 1988). Both the extensive glycosylation and rapid substitution rates in HIV-1 gp120 facilitate the escape of the virus from neutralizing antibodies (Wood et al. 2009).
There are multiple mechanisms by which mutations arise within the HIV-1 genome including nucleotide substitutions, insertions, and deletions (Abram et al. 2010). While substitution rates have been extensively characterized in HIV-1 and specifically in the env gene (Keulen, Boucher, and Berkhout 1996; Nielsen and Yang 1998), less attention has been given to sequence insertions and deletions (indels). The few studies that examine indels in the HIV-1 genome have focused on the location, behavior, and clinical significance of specifically recurring indels, such as indels in HIV-1 pol associated with drug resistance and indels in gag and vif associated with disease progression and infectivity (Rakik et al. 1999; Alexander et al. 2002; Aralaguppe et al. 2017). Only a small number of comparative studies have examined indel rates in the HIV-1 env gene encoding gp120 and gp41. Wood et al. (2009), for one, found that indels preferentially accumulate in the variable loops of gp120 compared to the remainder of this sequence, while other studies have suggested that variable loop indels correspond with HIV-1 transmission and modulate coreceptor switching (Derdeyn et al. 2004; Tsuchiya et al. 2013).
Despite the significant impact of indels within HIV-1 gp120 on virus transmission and adaptation, the overall rates of indel evolution in gp120 have not yet been measured through a comparative analysis. Furthermore, as previous studies on indels in HIV-1 have tended to focus on defined study populations, we have not found any study that has examined indel rates in a large database covering multiple HIV-1 subtypes and geographical regions. Here, we present results from a dated-tip phylogenetic analysis of HIV-1 env sequences from a public database. By comparing sequences from different hosts, our analysis focuses on fixed indel differences that are tolerated by the virus; for example, this analysis implicitly excludes indels that induce frameshifts in env. Our novel phylogenetic method specifically analyzes cherries (McKenzie and Steel 2000)—pairs of sequences directly descended from a common ancestor with no intervening ancestral nodes—in time-scaled phylogenies to estimate the rates of indel evolution in the gp120 variable loops of seven HIV-1 group M subtypes and CRFs (herein referred to collectively as clades). We focused on cherries to reduce the exposure of indels to purifying selection, and also the probability of multiple indel events occurring in the same variable region, as the divergence time in a cherry tends to be shorter on average than a random selection of two tips. Using this method, we evaluate the hypothesis that the mean rates of indels significantly vary among the gp120 variable loops and group M clades. Furthermore, we examine the nucleotide composition of indels to assess how this characteristic might be shaped by the virus genome, and quantify the impact of indels on N-linked glycosylation sites in HIV-1 gp120.
2. Results
We collected HIV-1 sequences covering gp120 from the Los Alamos National Laboratory (LANL) HIV Sequence Database (http://www.hiv.lanl.gov/) and filtered these data (as described in Section 3) to obtain a final data set of 6,605 sequences. To estimate the rates of indel evolution for different HIV-1 subtypes and CRFs in this data set, we reconstructed phylogenies for each of the seven group M clades using maximum likelihood, and then rooted and rescaled each tree based on the sample collection dates under a molecular clock model. Initially, we employed a strict clock model in root-to-tip regressions (Fig. 1) to assess whether the data sets contained sufficient signal to estimate rates of evolution (Table 1). Specifically, we confirmed that the lower bounds of the 95 per cent confidence intervals of rate estimates exceeded zero for all clades, which implied a gradual and measurable accumulation of mutations over the sampling time frame. Furthermore, we assessed the model fit with the coefficient of determination (R2), which was greatest for 01_AE and F1, and lowest for subtype C (Table 1). Next, we employed a more robust least-squares dating (LSD) method (To et al. 2016) to rescale the trees in time. Table 1 summarizes the substantial differences between the strict clock and least-squares estimates of the times to the most recent common ancestor (tMRCA) for each clade.
Figure 1.

The relationship between sequence root-to-tip branch lengths and sequence collection dates in seven clade-wise phylogenetic trees reconstructed from gp120 conserved region (C1–C5) alignments. Each panel is labeled by the clade that its tree represents. All plot axes have been adjusted to the same scales for comparison. Regions of greater color density indicate the clustering of multiple plotted points. The solid line on each plot describes the linear regression of branch lengths on collection dates.
Table 1.
Summary of the evolutionary rate estimates, tMRCAs, and R2 values generated by applying root-to-tip and LSD models to our seven clade-specific trees. The 95 per cent confidence intervals for the evolutionary rates of both models and for the tMRCAs estimates of the LSD model are enclosed in brackets. Both models are shown to illustrate the differences between fitting strict (root-to-tip) and relaxed clock models to our sequence data.
| Root-to-tip |
LSD |
||||
|---|---|---|---|---|---|
| Clades | Rate | tMRCA | R 2 | Rate | tMRCA |
| 01_AE | 1971.4 | 0.51 | 1.87 (1.85, 2.10) | 1968.6 (1965.5, 1974.6) | |
| 02_AG | 1957.4 | 0.35 | 2.27 (2.10, 2.68) | 1961.9 (1957.5, 1969.0) | |
| A1 | 1932.0 | 0.26 | 2.45 (2.32, 2.66) | 1966.3 (1964.0, 1969.5) | |
| B | 1941.2 | 0.34 | 1.47 (1.46, 1.57) | 1951.7 (1951.2, 1954.8) | |
| C | 1926.9 | 0.13 | 1.80 (1.78, 1.96) | 1939.8 (1937.5, 1946.7) | |
| D | 1944.5 | 0.33 | 1.88 (1.68, 2.11) | 1957.8 (1952.7, 1962.9) | |
| F1 | 1970.4 | 0.57 | 1.67 (1.34, 2.03) | 1956.2 (1943.9, 1965.2) | |
We extracted pairs of ‘cherries’ from these rescaled trees as phylogenetically independent observations on relatively short time frames. Next, we used these cherries to estimate the mean indel rates for each variable loop using a binomial-Poisson model, where the probability of detecting an indel event in a cherry increased exponentially with the divergence time. The indel rate estimates across the five variable loops and seven HIV-1 clades in this study ranged between and indels/nt/year (Fig. 2). We could not obtain an indel rate estimate for V3 in F1 due to low sample size for this sub-subtype, such that no cherries had discordant sequence lengths in V3. Similarly, we observed wide confidence intervals for the rate estimates for indels within V1 in 02_AG and F1, and for V5 in F1. The frequency of indels was significantly lower in subtype B than the reference clade, 01_AE (binomial generalized linear model [GLM], ; Supplementary Table S1). In addition, indels were significantly less frequent in V3 irrespective of clade relative to V1. Estimated interaction effects in the model also indicated that indels were significantly less frequent than expected in V2 within clades B and C.
Figure 2.

Indel rate estimates in the five gp120 variable loops of seven HIV-1 group M clades. Each group of five colored bars describes the indel rates of V1–V5 for one of the seven examined clades. Maximum likelihood estimation was applied to cherry indel outcomes using a binomial-Poisson model to determine the above indel rates. Error bars represent the 95 per cent confidence intervals within which indel rates were estimated. Arrows labeled with a * symbol indicate the presence and direction of significant differences among the mean indel rates of group M clades, relative to the CRF 01_AE reference. Arrows labeled by a † symbol denote significant differences among the variable loops irrespective of clade, relative to V1. Arrows labeled by a ‡ symbol denote individual interactions between variable loop and clade which are significantly different than their predicted value. No meaningful rate estimate was provided for V3 of clade F1 because no indels were detected in this data set.
Under the assumption that differences in sequence lengths of variable loops was caused by a single fixed indel (i.e. no multiple hits), we examined the distribution of indel lengths among variable loops and clades. Cherries with putative indels in the HIV-1 subtype C phylogeny tended to contain significantly longer indels than expected (Fig. 3). The variable loops V1, V2, and V4 tended to contain longer indels than expected irrespective of clade, whereas V3 and V5 tended to contain shorter indels.
Figure 3.

The distribution of indel lengths within (a) the seven group M clades and (b) the five gp120 variable loops. Indel lengths, measured in nucleotides, were classified into three categories: 3 , 6 , and 9 nt or longer. Box heights indicate the proportion of indels belonging to the given length category, while box widths indicate the proportion of indels belonging to (a) each clade or (b) each variable loop. Pearson residuals—quantified measures of the difference between observed and expected values—were calculated for every group on these plots to determine if, and in what direction, these proportions significantly deviated from the value. Pearson residuals are comparable to the number of standard deviations away from the value, meaning that values greater than 2, and especially those greater than 4, describe groups whose proportions significantly deviate from the predicted outcome. Blue shading indicates higher indel counts than expected, while red indicates lower counts.
Next, we examined the frequencies of nucleotides in indel- and non-indel regions of sequences in cherries with putative indels (Fig. 4). Because these frequencies measured for different clades tended to cluster by variable loop, we treated the clades as rudimentary replicates for this comparison (notwithstanding sample variation associated with variable loop V3 and subtype F1, e.g.). Overall, we observed that indels tended to contain higher proportions of G and lower proportions of T than the corresponding non-indel regions.
Figure 4.

Nucleotide proportions in indel sequences relative to flanking non-indel sequences for all examined variable loops and subtypes. Plots (a–d) illustrate these relations in adenine, cytosine, guanine, and thymine nucleotides, respectively. Each group denoted by a colored shape represents one of the five variable loops of gp120 and contains seven data points corresponding to each of the examined group M clades. The plotted line with a slope of 1 (y = x) represents the null result in which sequences inside and outside of indels show no difference in their nucleotide proportions. Plotted points that deviate from this line indicate differences between nucleotide proportions found in indels compared to those found outside indels. Larger data points indicate a significant test result testing for a difference between indel and non-indel counts in that particular data set.
Supplementary Fig. S1 summarizes the numbers of potential N-linked glycosylation sites (PNGSs) detected in each variable loop across the clades in our study. The mean counts in loops V1–V5 were 2.4, 2.1, 0.9, 4.1, and 1.3 PNGSs, respectively. We found significant differences in PNGS counts among variable loops (likelihood ratio test, ) and among clades (). For variable loop V1, 01_AE contained significantly more PNGS (mean 2.93 PNGS) than the other clades; the next highest count was obtained for subtype C (2.43 [95% CI 2.31, 2.57]). Subtypes B and C had significantly higher numbers of PNGS within V3 on average (0.96 [0.87, 1.05] and 0.95 [0.96, 1.05], respectively) than the reference clade 01_AE (mean 0.81). We observed substantial variation in the numbers of PNGS among clades in variable loop V4. For instance, clades 02_AG, A1, and B had significantly higher numbers, and subtype F1 significantly lower, than the reference clade 01_AE (mean 3.72). Finally, we mapped indels to PNGS in the variable loop sequences to determine how frequently indels were associated with the addition or removal of a PNGS (‘disruption’, Fig. 5). V1, V2, and V4 contained the highest proportions of indel-induced PNGS disruption among the five variable loops. Again, we observed that estimates for different clades visibly clustered by variable loop. When we adjusted for the relative proportions of the variable loops occupied by PNGS, only V1 and V2 markedly departed from this expectation.
Figure 5.
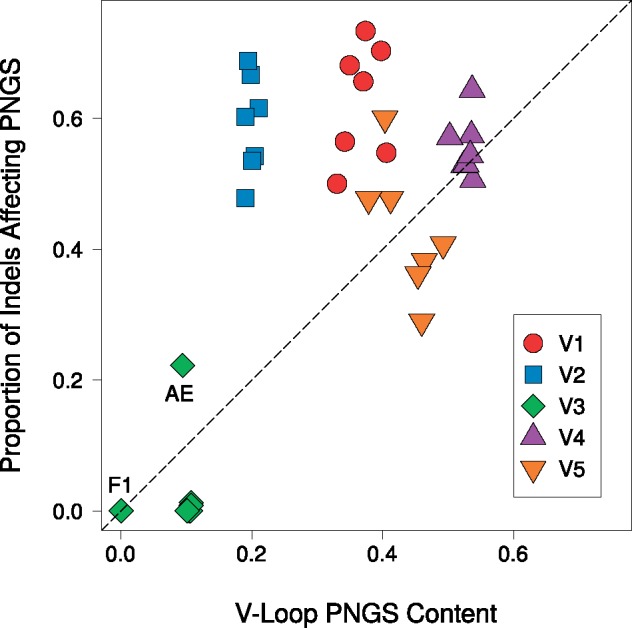
The proportion of indels in a variable region that knocked out at least one PNGS, relative to the PNGS content of the given variable loop. Data sets were represented by a colored shape to denote their variable loop and contained seven points derived from the group M clades. The dotted line of slope 1 provides a rough representation of the general trend expected if PNGSs were under no selection. Individual points that did not cluster with their variable loop were labeled with their clade. Specifically, V3 of subtype F1 did not have any records of indel events.
3. Discussion
To our knowledge, these results represent the first comprehensive measurement of indel rates in variable regions of HIV-1 gp120 across major virus subtypes and CRFs. Surprisingly, one of the only estimates of HIV-1 indel rates we have found dated back to 1995 (Mansky and Temin 1995), where Mansky and Temin used an in vitro assay of genetic mutations in HIV-1 reverse transcriptase (RT) and reported the observed counts of both nucleotide substitutions and indels. In contrast, our comparative study measures indel rates among different hosts, and as a result will inevitably underestimate these rates due to purifying selection on indels. We chose to focus on cherry sequences derived from between-host data as it provided phylogenetically independent observations of indel evolution while attempting to reduce the probability of multiple hits and the impacts of purifying selection. While the comparison of within-host HIV-1 sequences would provide even shorter time scales and thereby more accurate measures of indel rates before selection, some HIV-1 subtypes and CRFs remain underrepresented in publicly available, large and longitudinal same-patient sequence data sets. In addition, results from Wood et al. (2009) suggest that purifying selection against indels is relaxed in the variable regions of HIV-1 gp120.
To estimate indel rates, we needed to accurately rescale the HIV-1 phylogenies in chronological time. Estimates of the tMRCA can vary by genomic region, and estimates from regions within the HIV-1 gag and pol genes tend to be more recent than regions in env irrespective of subtype (Olabode et al. 2018). Overall, we determined that the diversity of HIV-1 sequences and sample collection dates were sufficient to fit a strict molecular clock model (Table 1). We note that for the purpose of rescaling the trees after this initial assessment with a strict clock, we employed an implementation of a relaxed clock model that allows for rate variation over time. However, we also observed that the goodness-of-fit used to assess support for the clock model was the lowest for subtype C. We attribute this poor model fit to both the relatively old age of subtype C (Wertheim, Fourment, and Kosakovsky Pond 2012) and the relative lack of HIV-1 C samples collected prior to 1995 (Fig. 1). Estimates of the tMRCA from the relaxed clock model implemented in the LSD program were generally comparable to previous estimates in the literature for the corresponding HIV-1 clades (Hemelaar 2012; Wertheim, Fourment, and Kosakovsky Pond 2012) except for subtype A1, for which we obtained a more recent range of estimates (1964–70). For instance, Tongo et al. (2018) recently estimated that (sub-)subtype A1 originated around 1946–57 from an analysis of full-length genome sequence data. We note that because our estimate relies on the ‘point estimate’ of the phylogeny reconstructed by maximum likelihood, the confidence intervals reported for our tMRCA estimates underestimate the true level of uncertainty and fixing the tree may skew the mean estimate. A Bayesian method would more accurately capture this substantial source of uncertainty, but would also be restricted to substantially reduced numbers of sequences due to the complexity of sampling from tree space. Furthermore, Wertheim, Fourment, and Kosakovsky Pond (2012) postulated that estimates of tMRCA among studies may be inconsistent due to the use of nucleotide substitution models that are an inadequate approximation of past molecular evolution.
Our estimates of region- and subtype-specific indel rates ranged from to indels/nt/year (Fig. 2). As expected, our average estimate ( indels/nt/year) was considerably lower than the rate inferred from Mansky and Temin’s in vitro experiments (about indels/nt/year) (Mansky and Temin 1995), where we used parameter estimates from Perelson and Nelson (1999) to convert the observed numbers of indel counts to a rate. Since our study compares HIV-1 sequences isolated from different hosts, the indels have been filtered by purifying selection so that only a subset become fixed within the respective hosts. We found that indel rates in subtype B gp120 were significantly lower than the reference clade 01_AE, and generally lower than the other clades in our study. The significantly lower indel rate estimates in V3 irrespective of HIV-1 clade (Fig. 2) were consistent with the functional importance of this variable loop. As V3 contributes to HIV entry by binding to the CCR5 or CXCR4 coreceptors, there is substantial purifying selection to conserve its overall structure (Liang et al. 1999; Jiang et al. 2010). This lower tolerance for mutational change, relative to other variable loops of gp120, is consistent with reduced numbers of fixed indels among hosts. The lower indel rates might also be attributed in part to compensatory mutations to preserve structural interactions in V3 (Poon et al. 2007). For example, an arginine insertion at position 11 of V3 confers CXCR4 tropism tends to be accompanied by a single amino acid deletion near the C-terminal of V3 (Tsuchiya et al. 2013).
The tendency of HIV-1 subtype C to accumulate longer indels in our analysis is consistent with results previously reported by Derdeyn et al. (2004). By examining HIV-1 heterosexual donor–recipient pairs, Derdeyn et al. (2004) first determined that subtype C viruses initially contained shorter V1–V4 sequences upon transmission, which then substantially lengthened by up to twenty-five amino acids after progressing to chronic late-stage infection. A follow-up study by Chohan et al. (2005) provided evidence that this trend was subtype-specific, as it was observed in infections by subtypes A and C, but not subtype B. Similarly, the observed preference for longer indels in V1 and V2 in our data is consistent with the role of these variable loops in facilitating immune evasion. For example, the insertion of five or more amino acids into V1/V2 is associated with reduced sensitivity of HIV-1 gp120 to neutralizing antibodies (Sagar et al. 2006; Curlin et al. 2010), which is more efficiently achieved by a single long insertion than a series of short insertions. Conversely, the tendency for shorter insertions to accumulate in V3 is consistent with the existence of functional and structural constraints as noted above.
The nucleotide composition of the HIV-1 genome is generally skewed to higher frequencies of A (adenine), in large part due to G-to-A hypermutation induced by host factors (Liddament et al. 2004). We have not found previous studies that have compared the nucleotide composition of indels to the flanking sequence in the HIV-1 genome. Overall, we observed that indel sequences tended to comprise higher frequencies of G and lower frequencies of T relative to the rest of the variable loop sequence. We note that some frequency estimates had greater sample variation due to limited numbers of indels and sequences in association with V3 and subtypes D and F, for instance. Because the env gene generally contains slightly higher proportions of A (40%) and lower proportions of G (18%) than the rest of the HIV-1 genome (35 and 24%, respectively), we propose that this outcome might reflect the derivation of insertions into env from outlying sequence. Since we have not individually resolved these numerous indels into insertions or deletions through ancestral reconstruction, however, we cannot determine whether this pattern reflects a tendency for sequence insertions to be G-rich, or whether G-rich sequences are specifically targeted for deletion.
The variable regions of HIV-1 gp120 contribute disproportionately to the mean number of PNGSs (11 out of 25), which make up the glycan shield of gp120 (Zhu et al. 2000). Overall, we found that the numbers of PNGS for each variable loop was fairly consistent across clades, with some significant but minor differences in means (Supplementary Fig. S1). Furthermore, we observed that PNGS were more frequently affected by indels in variable loops V1 and V2 irrespective of clade (Fig. 5). Put another way, the proportion of indels affecting PNGS in these variable regions was substantially greater than the proportion of the region encoding PNGS. This outcome supports the hypothesis of diversifying selection for the addition or removal of PNGS in V1/V2, where glycosylation plays a major role in mediating immune escape (Sagar et al. 2006) and transmission fitness (Derdeyn et al. 2004) at different stages in the natural history of HIV-1 infection.
Our estimates of indel rates in the variable regions of HIV-1 gp120 imply that fixed differences in variable loop lengths accumulate between infections on a time scale of about 10–20 years per variable loop with the exception of V3, which accumulates these differences an order of magnitude slower. This time frame is consistent with past observations that HIV-1 gradually ‘raises’ the glycan shield with insertions in V1/V2 several years post-infection (Sagar et al. 2006). The accumulation and composition of indels among infections is clearly heterogeneous among HIV-1 clades and variable loops. Some of the more exploratory results in this study, e.g. differences in nucleotide frequencies within indels, are particularly novel and suggest new areas for further research in the molecular evolution of HIV-1 to identify the biological or selective determinants of sequence insertions and deletions.
4. Methods
4.1 Data processing
We queried the LANL HIV Sequence Database (http://www.hiv.lanl.gov/) for all sequence records covering HIV-1 env gp120, limiting the records to one sequence per patient. The 26,359 matching sequences were downloaded with predicted subtype, collection year and GenBank accession number. We parsed the resulting FASTA file and removed sequences that lacked subtype or collection year fields, or were shorter than 1,400 nt (roughly 90% of full-length HIV-1 gp120), yielding a final data set of 6,605 sequences. To extract the interval encoding gp120 from each sequence and partition the result into the variable and conserved regions, we performed pairwise alignments using an implementation of the Altschul–Erickson (Altschul and Erickson 1986) modification of the Gotoh algorithm in Python (http://github.com/ArtPoon/gotoh2). Each nucleotide sequence was aligned against the HXB2 (GenBank accession number K03455) gp120 reference sequence with match/mismatch scores of +5/−4, gap open/extension penalties of thirty and ten, respectively, and no terminal gap penalty. The aligned query sequence was cut at the boundaries of the aligned HXB2 reference gene to extract the patient-derived subsequence homologous to gp120. Next, we removed any gaps in this result and then aligned the amino acid translation to the gp120 protein reference sequence using an empirical HIV amino acid scoring matrix (25% divergence [Nickle et al. 2007]) with the same gap penalties, except that terminal gaps were penalized at this stage. Finally, we used the aligned query to insert gap character triplets into the preceding nucleotide sequence as ‘in-frame’ codon deletions.
Using the HXB2 reference annotations, we extracted the five variable (V1–V5) and five conserved (C1–C5) regions of gp120. The conserved region sequences were concatenated and exported to separate files for phylogenetic reconstruction. We subsequently determined that our method was not reliably extracting the V5 region, based on the overabundance of multiple gap characters at the 5′ end of many outputs. To avoid further problems downstream, we implemented a modified extraction method specific to V5. We first extracted nine extra nucleotides beyond the 5′ boundary of the V5 reference to provide conserved sequence coverage outside this hypervariable region. The extended V5 sequence was then translated and aligned to a V5 amino acid reference sequence of matching length as above. Lastly, we used the first non-gap character (a matched amino acid) immediately following the first three conserved residues in the amino acid alignment as the adjusted V5 start position, thereby omitting any gap characters that preceded this first residue.
4.2 Phylogenetic analysis
We used the program MAFFT (version 7.271) with the default settings (Katoh and Standley 2013) to generate a multiple sequence alignment (MSA) from the concatenated sequences of conserved regions for each subtype. On manual inspection of the resulting MSAs, we found some alignment columns comprised mostly of gaps caused by rare insertions, so we removed all columns with gap characters in more than 95 per cent of sequences. Next, we reconstructed phylogenies for each subtype-specific MSA by approximate maximum likelihood using FastTree2 (version 2.1.8) compiled with double precision (Price, Dehal, and Arkin 2010). The resulting trees were manually screened for unusually long terminal branches indicative of problematic sequences, which we removed from the corresponding MSA before reconstructing a revised tree.
Effective estimation of indel rates required that all phylogenetic trees be scaled in time. To rescale the maximum likelihood trees, sequence accession numbers were used to query the GenBank database for more precise collection dates containing month and day fields; otherwise we retained the collection years from the LANL database. The R package ape was then used to change each tree into a strictly bifurcating structure and to root the tree using root-to-tip regression (Paradis, Claude, and Strimmer 2004) based on the associated tip dates. We evaluated the correlation between the time since the inferred root date (x-intercept) and the total branch length (in expected numbers of substitutions) to determine if the data were consistent with a molecular clock (Drummond, Pybus, and Rambaut 2003).
Using the same dates, we employed the LSD program (To et al. 2016) to adjust node heights and rescale the tree in time under a relaxed molecular clock model. Dates lacking either month or day fields were specified as bounded intervals. The time-scaled tree outputs from LSD were imported into R to extract the ‘cherries’: pairs of sequences directly descended from a common ancestor with no intervening ancestral nodes. Focusing on sequences in cherries provides phylogenetically independent observations and minimizes the divergence times, thereby reducing the chance of encountering multiple indel events as well as the effect of purifying selection on indels. Cherries with time-scaled branch lengths totaling zero years were removed from our analysis as they did not provide meaningful indel observations and caused problems for rate estimation.
4.3 Indel rate estimation
To estimate the rate of indels in the variable loops, homologous variable regions from each pair of sequences in a cherry were compared for length differences. The presence of a length difference was reported as a binomial outcome implying that an indel event had occurred along these branches; this approach does not account for the possibility of multiple indels causing reversion to the same sequence lengths. In addition, the total branch lengths comprising the cherry were employed as an estimate of divergence time in years (Supplementary Fig. S2). The indel rate was estimated from these data by fitting the following model using maximum likelihood, where the Bernoulli likelihood for the ith cherry is:
where Yi = 1 if the sequence lengths differ (implying one or more indels), and is 0 otherwise; ti is the total branch length, λ is the overall indel rate, and is the Poisson probability of no indels in the cherry. The total log-likelihood across cherries is thus:
We used the Brent minimization method implemented in the R package ‘bbmle’ to obtain a maximum likelihood estimate of λ for each clade and variable loop combination. A GLM with a logit link function was also applied to these data to evaluate statistical associations of the inferred distribution of indels on clades and variable loops; the model incorporated a divergence time term as a rudimentary adjustment for variation in ‘sampling effort’.
4.4 Analysis of indels
For every combination of five variable loops and seven clades, we categorized the inferred lengths of indels into three discrete classes: single-codon (3 nt), double-codon (6 nt), and long (9+ nt). Pearson residuals were calculated on these distributions to determine if, and in what direction, these observed proportions significantly deviated from their expected values. To further analyze the composition of indels, we generated pairwise alignments for each cherry with discordant sequence lengths to identify and extract indels. From these pairwise alignments, we calculated the proportions of adenine, thymine, guanine, and cytosine (A, C, G, and T) nucleotides in the indel and non-indel regions of the gp120 variable loops. In addition, we recorded the positions and numbers of PNGSs in the five gp120 variable loops by scanning the unaligned amino acid sequences with the regular expression ‘N[^P][ST][^P]’, where ‘^P’ maps to any symbol except P (proline). We then used these data to investigate how commonly indels tended to change PNGSs in the variable loops. By combining PNGS and indel location data, we searched for instances where an indel overlapped with a PNGS in one of the two sequences of a cherry, indicating either the deletion of a PNGS or the insertion of a sequence containing one. To avoid recording instances of partial indel overlap that leave the PNGS intact, we verified the PNGS was disrupted by scanning it again with a regular expression.
Data availability
Sequence data and scripts used for this study are available at https://github.com/PoonLab/indelrates.
Funding
This study was made possible by the HIV-1 sequence database curated by the Los Alamos National Laboratory. This work was supported in part by the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-131), and by grants from the Canadian Institutes of Health Research (CIHR PJT-153391, PJT-155990, and PJT-156178) and the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN-2018-05516).
Conflict of interest: None declared.
Supplementary Material
References
- Abram M. E. et al. (2010) ‘Nature, Position, and Frequency of Mutations Made in a Single Cycle of HIV-1 Replication’, Journal of Virology, 84: 9864–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander L. et al. (2002) ‘Inhibition of Human Immunodeficiency Virus Type 1 (HIV-1) Replication by a Two-Amino-Acid Insertion in HIV-1 Vif From a Nonprogressing Mother and Child’, Journal of Virology, 76: 10533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Erickson B. W. (1986) ‘Optimal Sequence Alignment Using Affine Gap Costs’, Bulletin of Mathematical Biology, 48: 603–16. [DOI] [PubMed] [Google Scholar]
- Aralaguppe S. G. et al. (2017) ‘Increased Replication Capacity Following Evolution of PYxE Insertion in Gag-p6 is Associated With Enhanced Virulence in HIV-1 Subtype C From East Africa’, Journal of Medical Virology, 89: 106–11. [DOI] [PubMed] [Google Scholar]
- Ariën K. K., Vanham G., Arts E. J. (2007) ‘Is HIV-1 Evolving to a Less Virulent Form in Humans?’ Nature Reviews Microbiology, 5: 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chohan B. et al. (2005) ‘Selection for Human Immunodeficiency Virus Type 1 Envelope Glycosylation Variants With Shorter V1–V2 Loop Sequences Occurs During Transmission of Certain Genetic Subtypes and May Impact Viral RNA Levels’, Journal of Virology, 79: 6528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curlin M. E. et al. (2010) ‘HIV-1 Envelope Subregion Length Variation During Disease Progression’, PLoS Pathogens, 6: e1001228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn C. A. et al. (2004) ‘Envelope-Constrained Neutralization-Sensitive HIV-1 After Heterosexual Transmission’, Science, 303: 2019–22. [DOI] [PubMed] [Google Scholar]
- Drummond A., Pybus O. G., Rambaut A. (2003) ‘Inference of Viral Evolutionary Rates From Molecular Sequences’, Advances in Parasitology, 54: 331–58. [DOI] [PubMed] [Google Scholar]
- Hemelaar J. (2012) ‘The Origin and Diversity of the HIV-1 Pandemic’, Trends in Molecular Medicine, 18: 182–92. [DOI] [PubMed] [Google Scholar]
- Jiang X. et al. (2010) ‘Conserved Structural Elements in the V3 Crown of HIV-1 gp120’, Nature Structural & Molecular Biology, 17: 955. [DOI] [PubMed] [Google Scholar]
- Katoh K., Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keulen W., Boucher C., Berkhout B. (1996) ‘Nucleotide Substitution Patterns Can Predict the Requirements for Drug-Resistance of HIV-1 Proteins’, Antiviral Research, 31: 45–57. [DOI] [PubMed] [Google Scholar]
- Kiwanuka N. et al. (2008) ‘Effect of Human Immunodeficiency Virus Type 1 (HIV-1) Subtype on Disease Progression in Persons From Rakai, Uganda, With Incident HIV-1 Infection’, The Journal of Infectious Diseases, 197: 707–13. [DOI] [PubMed] [Google Scholar]
- Korber B. et al. (2001) ‘Evolutionary and Immunological Implications of Contemporary HIV-1 Variation’, British Medical Bulletin, 58: 19–42. [DOI] [PubMed] [Google Scholar]
- Kwong P. D. et al. (1998) ‘Structure of an HIV gp120 Envelope Glycoprotein in Complex With the CD4 Receptor and a Neutralizing Human Antibody’, Nature, 393: 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. H., Tanimura M., Sharp P. M. (1988) ‘Rates and Dates of Divergence Between AIDS Virus Nucleotide Sequences’, Molecular Biology and Evolution, 5: 313–30. [DOI] [PubMed] [Google Scholar]
- Liang X. et al. (1999) ‘Epitope Insertion into Variable Loops of HIV-1 gp120 as a Potential Means to Improve Immunogenicity of Viral Envelope Protein’, Vaccine, 17: 2862–72. [DOI] [PubMed] [Google Scholar]
- Liddament M. T. et al. (2004) ‘APOBEC3F Properties and Hypermutation Preferences Indicate Activity Against HIV-1 In Vivo’, Current Biology, 14: 1385–91. [DOI] [PubMed] [Google Scholar]
- Mansky L. M., Temin H. M. (1995) ‘Lower In Vivo Mutation Rate of Human Immunodeficiency Virus Type 1 Than That Predicted From the Fidelity of Purified Reverse Transcriptase’, Journal of Virology, 69: 5087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie A., Steel M. (2000) ‘Distributions of Cherries for Two Models of Trees’, Mathematical Biosciences, 164: 81–92. [DOI] [PubMed] [Google Scholar]
- Nickle D. C. et al. (2007) ‘HIV-Specific Probabilistic Models of Protein Evolution’, PLoS One, 2: e503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R., Yang Z. (1998) ‘Likelihood Models for Detecting Positively Selected Amino Acid Sites and Applications to the HIV-1 Envelope Gene’, Genetics, 148: 929–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olabode A. S. et al. (2018) ‘Evidence for a Recombinant Origin of HIV-1 Group M From Genomic Variation’, Virus Evolution, 5: vey039. [DOI] [PMC free article] [PubMed]
- Paradis E., Claude J., Strimmer K. (2004) ‘APE: Analyses of Phylogenetics and Evolution in R Language’, Bioinformatics (Oxford, England), 20: 289–90. [DOI] [PubMed] [Google Scholar]
- Perelson A. S., Nelson P. W. (1999) ‘Mathematical Analysis of HIV-1 Dynamics In Vivo’, SIAM Review, 41: 3–44. [Google Scholar]
- Poon A. F. et al. (2007) ‘An Evolutionary-Network Model Reveals Stratified Interactions in the V3 Loop of the HIV-1 Envelope’, PLoS Computational Biology, 3: e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., Arkin A. P. (2010) ‘FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments’, PLoS One, 5: e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakik A. et al. (1999) ‘A Novel Genotype Encoding a Single Amino Acid Insertion and Five Other Substitutions Between Residues 64 and 74 of the HIV-1 Reverse Transcriptase Confers High-Level Cross-Resistance to Nucleoside Reverse Transcriptase Inhibitors’, JAIDS Journal of Acquired Immune Deficiency Syndromes, 22: 139–45. [DOI] [PubMed] [Google Scholar]
- Sagar M. et al. (2006) ‘Human Immunodeficiency Virus Type 1 V1–V2 Envelope Loop Sequences Expand and Add Glycosylation Sites Over the Course of Infection, and These Modifications Affect Antibody Neutralization Sensitivity’, Journal of Virology, 80: 9586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebit D. M., Arts E. J. (2011) ‘Tracking a Century of Global Expansion and Evolution of HIV to Drive Understanding and to Combat Disease’, The Lancet Infectious Diseases, 11: 45–56. [DOI] [PubMed] [Google Scholar]
- To T.-H. et al. (2016) ‘Fast Dating Using Least-Squares Criteria and Algorithms’, Systematic Biology, 65: 82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongo M. et al. (2018) ‘Unravelling the Complicated Evolutionary and Dissemination History of HIV-1M Subtype a Lineages’, Virus Evolution, 4: vey003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E. E. et al. (2012) ‘Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation’, PLoS Pathogens, 8: e1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K. et al. (2013) ‘Arginine Insertion and Loss of N-Linked Glycosylation Site in HIV-1 Envelope V3 Region Confer CXCR4-Tropism’, Scientific Reports, 3: 2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan A. et al. (2006) ‘Different Rates of Disease Progression of HIV Type 1 Infection in Tanzania Based on Infecting Subtype’, Clinical Infectious Diseases, 42: 843–52. [DOI] [PubMed] [Google Scholar]
- Wainberg M. A. (2004) ‘HIV-1 Subtype Distribution and the Problem of Drug Resistance’, AIDS, 18: S63–8. [DOI] [PubMed] [Google Scholar]
- Wertheim J. O., Fourment M., Kosakovsky Pond S. L. (2012) ‘Inconsistencies in Estimating the Age of HIV-1 Subtypes Due to Heterotachy’, Molecular Biology and Evolution, 29: 451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (2018) Global Health Sector Strategy on HIV, 2016–2021. <https://www.who.int/hiv/strategy2016-2021/ghss-hiv/en/> accessed 4 Oct 2018. [Google Scholar]
- Wood N. et al. (2009) ‘HIV Evolution in Early Infection: Selection Pressures, Patterns of Insertion and Deletion, and the Impact of APOBEC’, PLoS Pathogens, 5: e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M. et al. (2004) ‘Tracking Global Patterns of N-Linked Glycosylation Site Variation in Highly Variable Viral Glycoproteins: HIV, SIV, and HCV Envelopes and Influenza Hemagglutinin’, Glycobiology, 14: 1229–46. [DOI] [PubMed] [Google Scholar]
- Zhu X. et al. (2000) ‘Mass Spectrometric Characterization of the Glycosylation Pattern of HIV-gp120 Expressed in CHO Cells’, Biochemistry, 39: 11194–204. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data and scripts used for this study are available at https://github.com/PoonLab/indelrates.