

Abstract
Candida species are the second most frequent cause of fungal infections worldwide. Current knowledge of immunity to Candida has been gleaned almost exclusively from studies on Candida albicans, the most common disease-causing species. Knowledge of immunity to non-albicans Candida (NAC) species is still at an early stage due to the lack of tractable animal models with which to study these important pathogens. This is partly because many NAC species are not usually pathogenic in mouse models of candidiasis. In this study, we established an immunosuppressed mouse model of disseminated candidiasis by the two clinically important NAC species, C. glabrata and C. tropicalis. The inbred mouse strains, A/J and BALB/c, show distinct susceptibilities to disseminated Candida infection. A/J mice, deficient for complement C5, are more susceptible to disseminated infection with both C. glabrata and C. tropicalis compared to BALB/c mice, the latter having functional C5. Here we show that peptide-pulsed dendritic cell (DC) vaccination with a peptide derived from a C. tropicalis cell surface protein, significantly improved survival and reduced the fungal burdens of disseminated candidiasis in these immunocompromised mice. Importantly, this study is the first report of protective efficacy conferred by a peptide vaccine against medically important NAC species in immunosuppressed hosts. Establishing this experimental mouse model provides an important tool to further understand pathogenesis and host resistance in Candida infection. Significantly, our findings also demonstrate how this model can be used to evaluate new control strategies against candidiasis, such as vaccines.
Keywords: peptide vaccine, antibody protection, candidiasis, immunocompromised, neutropenia
Introduction
Disseminated candidiasis is the leading cause of nosocomial bloodstream infections in the United States. Despite the availability of antifungal therapy, crude mortality in the last decade has remained unacceptably high. Disseminated candidiasis is the cause of more fatalities than any other systemic mycosis,1,2 yet to date there are no vaccines against Candida or indeed any other fungal pathogen. Candida albicans is the most common disease-causing species (60–70%); however, the prevalence of disease caused by non-albicans Candida (NAC) species is on the rise, along with an increase in antifungal drug resistance.3Candida glabrata now accounts for approximately 15% to 20% of all Candida infections in the United States and is the most common NAC species isolated.1,4 A particular problem with C. glabrata is its resistance to the most common drug classes, azoles and echinocandins.5–7C. tropicalis, in particular, is associated with invasive infection, accounting for up to 45% of NAC infections.8 Strikingly, higher mortality caused by disseminated C. tropicalis rather than C. albicans infection was documented in patients with acute leukemia or undergoing bone marrow transplantation, for whom neutropenia appears to be a particularly important risk factor,8–11 although the basis for this is unclear.
Experimental animal models are a critical component of understanding pathogenesis and host resistance to infection. Such models allow the evaluation of potential antifungal vaccines and chemotherapies. To investigate systemic Candida infection, mouse models have been developed that mimic human disease, the most common being the intravenous infection model. The murine intravenous model of disseminated C. albicans infection is well characterized and reproducible, while tractable mouse models for NAC are lacking because many NAC species are usually nonpathogenic in mice. Therefore, developing faithful mouse models to understand host resistance to NAC species is key for future work. In this study, we first performed a simultaneous comparison of the relative pathogenicities of the three clinically most relevant Candida species in both immunocompetent and immunocompromised mouse models to understand species-related variations in virulence. We then successfully established immunocompromised mouse models of disseminated infection by C. tropicalis and C. glabrata. Furthermore, we showed that peptide-pulsed dendritic cell (DC) vaccination with peptide derived from the Candida cell wall protein 5-methyltetrahydropteroyltriglutamate homocysteine methyltransferase (Met6), significantly improved survival and reduced the fungal burdens of disseminated candidiasis in immunocompromised mice of both BALB/c and A/J strains. Vaccination of high-risk groups is a particularly promising strategy to prevent invasive Candida infection.12 There are extensive data confirming the immunogenicity and efficacy of vaccines even in patients with weakened immune systems. For example, those with neutropenia, active leukemia, human immunodeficiency virus (HIV) infections, or those receiving immunosuppressive corticosteroids.13–17 Neutropenia is one of the most common problems associated with disseminated candidiasis in humans, and here we are first to report that a synthetic cell wall-derived peptide vaccine can provide effective protection against disseminated C. tropicalis infection in immunocompromised hosts.
Methods
Candida strains and culture conditions
Three isolates of Candida were used to induce systemic infection in BALB/c and A/J mice. C. albicans SC5314 (ATCC MYA-2876), C. tropicalis (ATCC MYA-3404) and C. glabrata (ATCC 2001) were grown as stationary-phase yeast cells (24 h cells) in glucose-yeast extract-peptone (GYEP, 2% glucose, 1% peptone, 0.3% yeast extract) broth at 37°C, washed, and suspended to the appropriate cell concentration (5 × 106/ml, 1 × 108/ml, or 1 × 109/ml) in Dulbecco's phosphate-buffered saline (DPBS; Sigma) and used to infect mice intravenously (IV) as described.18,19
Mice
BALB/c and A/J female mice (National Cancer Institute Animal Production Program, Frederick, MD, USA) 5 to 7 weeks old were used throughout. Mice were maintained in our AAALAC–certified animal facility and all animal experiments were performed using a protocol approved by the Institutional Animal Care and Use committee (IACUC) at LSU Health Sciences Center (LSUHSC).
Peptide vaccines
14-mer peptide Met6, PRIGGQRELKKITE, was synthesized based on a sequence located within the N-terminus of C. albicans cell wall protein 5 methyltetrahydropteroyltriglutamate homocysteine methyltransferase. Met6 peptide (purity 98.5%) was produced commercially (GenScript).
Isolation and culture of dendritic cells (DCs) from mouse bone marrow
Dendritic cells (DCs) were generated from mouse bone marrow by a previously described method.20,21 Briefly, donor mice were euthanized by CO2 asphyxiation, their long bones, femurs, and tibias were aseptically removed. Bone marrow was flushed from the bones by forcibly injecting several ml of Roswell Park Memorial Institute medium (RPMI-1640) and clumps removed or dispersed by gentle pipetting through a sterile 70-mm cell strainer. Red blood cells were lysed (ACK lysing buffer, 0.15 M NH4Cl, 1.0 mM KHCO3, 0.1 mM EDTA) for 4 min, and the remaining bone marrow cells were suspended in complete medium (CM, RPMI-1640 supplemented with 10% FBS, 2 mM l-glutamine, 1% nonessential amino acids, and 100 units/ml penicillin and 100 μg/ml streptomycin). Cell density was adjusted to 2 × 105 cells per ml and plated in six-well plates at 5 ml per well. Cells were cultured for up to 9 days in the presence of 40 ng/ml of rmGM-CSF and rmIL-4 (R&D Systems) at 37°C, 5% CO2. On days 4 and 7 of culture, the same amount of fresh GM-CSF and interleukin (IL)-4 was added to the wells.
Active immunizations with peptide pulsed dendritic cells
A time course of the protocols for active immunization, the induction of immunosuppression, and survival studies after Candida infection is summarized in Figure 1. All active vaccinations were conducted as previously described.21–23 DCs were pulsed in vitro with Met6 peptide as described before.21 Briefly, DCs in culture were pulsed with the peptide antigen (1 μM) on day 6. On day 7, PGE2 (10−7 M) was added along with LPS (2 μg/ml, Sigma) for 24 h to induce DC maturation. On day 9, antigen-pulsed DCs were washed extensively and 5 × 105 cells in 200 μl DPBS were administered via the intraperitoneal (IP) route. The mice were boosted IP at day 14 and day 28 with fresh antigen-pulsed DCs without adjuvant.
Figure 1.

A time course of the protocols for active immunization, the induction of immunosuppression, and survival studies after Candida infection is summarized. (A) Before challenge, BALB/c and A/J mice were immunosuppressed by IP with CY or SC injection of cortisone acetate at day 3. The IP injection (150 mg/kg) of CY was repeated every 7 days after infection and SC injection of cortisone (125 mg/kg) was performed daily to maintain leukocytopenia. (B) All active vaccinations were conducted as previously described (21-23). On day 0, antigen-pulsed dendritic cells (DCs) were washed extensively and 5 × 105 cells in 200 μl DPBS were given by IP route as a priming dose to the mice. The mice were boosted IP twice 2 weeks apart with same number of fresh antigen-pulsed DCs without adjuvant. This Figure is reproduced in color in the online version of Medical Mycology.
To test the efficacy of the vaccine in immunocompromised mice, vaccinated mice were immunosuppressed by intraperitoneal injection of 200 mg/kg of cyclophosphamide (CY; Sigma-Aldrich) on day -3 followed by another 4 doses (150 mg/kg) every 7 days on days 7, 14, 21, and 28 post- infection (Fig. 1).
Immunosuppression
Before challenge, BALB/c and A/J mice were immunosuppressed by intraperitoneal injection of a 200 mg/kg dose of cyclophosphamide (CY; Sigma-Aldrich), or subcutaneous (SC) injections of cortisone acetate (Merck) at a dose of 125 mg/kg at day 3. The IP injection (150 mg/kg) was repeated every 7 days after infection (i.e., at days 7, 14, 21, and 28 post-infection) in order to maintain leukocytopenia and low neutrophil counts for the entire experimental procedure. The SC injection of cortisone (125 mg/kg) was performed daily to maintain leukocytopenia. Both regimens have been shown to render mice neutropenic (The absolute neutrophil count is <500 cells/mm3) within 3–4 day of the first cyclophosphamide/cortisone injection, and neutropenia lasted until the termination of the experiments (day 30). Leukocyte and differential counts from peripheral blood were monitored for up to 30 days to verify the induction of leukocytopenia. Total leukocyte and differential cell counts were determined on a hemacytometer and by Wright-Giemsa staining. Body weights of the mice were also measured and compared for 21 days after the first CY/cortisone or saline injection. With regard to the mouse model of C. glabrata disseminated candidiasis, on day -3 before C. glabrata (ATCC 2001) inoculation (day 0), some groups of BALB/c mice received a combination of cyclophosphamide plus cortisone acetate at the same dosage schedule as indicated above. To verify the induction of neutropenia, leukocytes and differential counts were monitored 48 h after the injection, at which time the total peripheral leukocyte count fell to approximately 500 cells per ml (from a baseline of 8000 cells per ml), with a polymorphonuclear content of less than 10% of the total leukocyte count. Peripheral leukocyte count was monitored daily thereafter.
Fungal challenge dose and assessment of protection
Due to defective fungal host defense and impaired survival in immunosuppressed mice, the C. albicans challenge dose was modified from 5 × 105 to 5 × 104 to obtain similar survival curves as shown in immunocompetent mice. Immunosuppressed mice and age- and sex-matched normal control mice (n = 5 in each group) were injected intravenously with different doses of live C. albicans yeast cells (5 × 105, and 5 × 106 in 0.1 ml DPBS), or live C. glabrata yeast cells (1 × 107, 1 × 108, and 1 × 109 in 0.1 ml DPBS), or live C. tropicalis yeast cells (1 × 107, 5 × 107, and 1 × 108 in 0.1 ml DPBS). After two independent experiments, 5 × 104 of C. albicans SC5314, 1 × 107 of C. tropicalis, and 1 × 108 of C. glabrata were determined as the lethal challenge dose for immunosuppressed BALB/c and A/J mice in all the following survival studies, producing an acute infection, with 60–100% of animals dying within 10–15 days. For active immunization experiments, 2 weeks after the second boost of Met6 peptide vaccine on day 0, immune and control immunosuppressed BALB/c and A/J mice were infected i.v. with a lethal dose of C. tropicalis yeast cells (1–107 in 100 ul DPBS). Protection was evaluated by monitoring animal survival for 30 days. The challenged mice were monitored for the development of a moribund state, defined as being listless, disinterested in food or water, and nonreactive to finger probing. At that time the mouse was euthanized, kidneys and brains were homogenized in DPBS, and plated onto nutrient agar to determine colony-forming units (cfu).
Analysis of organ clearance
Groups of 20 mice for each strain were studied: a normal group and three groups immunosuppressed by either cyclophosphamide (CY) or cortisone acetate or both as previously described. Mice were inoculated intravenously with spores of C. glabrata or C. tropicalis at day 0, on days 2, 4, 6, and 8 postinfection. Five mice per experimental group were selected at random and euthanized. The kidneys, livers, brains, and spleen of each of these animals were removed aseptically and homogenized in 5 ml of sterile DPBS. Samples were removed from each homogenate, serially diluted in DPBS, plated onto GYEP agar plates (with 50 μg/ml chloramphenicol), and incubated at 37°C for 48 h. The colonies were then enumerated, and cfu calculated per gram of each organ. All experiments were performed in duplicate.
Statistical analysis
Survival times were statistically evaluated by Kaplan–Meier (GraphPad Prism, version 6), and statistical significance was subsequently calculated for each preset time point of analysis. All fungal burden data were analyzed by a multivariate analysis of variance (MANOVA). This multivariate analysis allowed for the correlation among CFUs obtained from organs within the same animal. All statistical tests were performed at the 5% significance level.
Results
Comparison of virulence of C. albicans, C. glabrata and C. tropicalis in immunocompetent BALB/c mice
To compare the virulence of two NAC species with C. albicans in immunocompetent BALB/c mice, initial experiments were conducted to challenge naïve BALB/c mice with high doses of both C. tropicalis and C. glabrata intravenously. A lethal dose of C. albicans SC 5314 (5 × 105) was used in naïve BALB/c to induce acute invasive candidiasis as a positive control. BALB/c Mice were inoculated i.v. with C. glabrata (108 organisms/mouse) or with C. tropicalis (107 organisms/mouse) and observed once daily for 30 days for morbidity and mortality. As shown in Figure 2, all mice inoculated with C. glabrata (108 organisms/mouse) survived and appeared clinically normal. In contrast, systemic infection with C. albicans (5 × 105 organisms/mouse) resulted in 100% mortality by 12 days postinfection, which was associated with clinical signs of disease including weight loss, lethargy, and a ruffled appearance. With an infective dose 1 × 107 of C. tropicalis, only 20% mortality was observed in BALB/c mice within 30 days after challenge.
Figure 2.
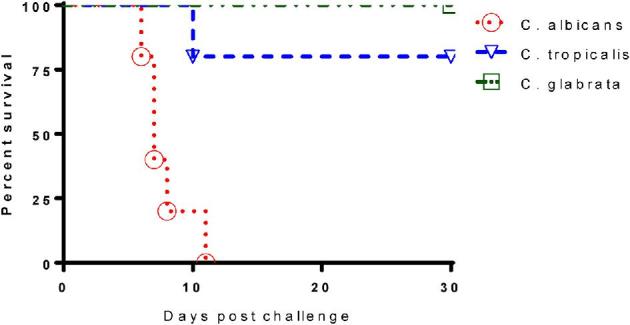
Comparison of virulence of C. albicans, C. tropicalis and C. glabrata in immunocompetent BALB/c mice. Naive immunocompetent BALB/c mice (5 weeks) were infected IV with either 5 × 105C. albicans SC5314 (ATCC MYA-2876) yeast cells, or 1 × 107 cfu C. tropicalis (ATCC MYA-3404) yeast cells, or 1 × 108 cfu C. glabrata ATCC 2001 yeast cells, respectively. Results are representative of five mice per inoculum. Experiments were repeated three times. This Figure is reproduced in color in the online version of Medical Mycology.
Induction of leukocytopenia in mice
BALB/c and A/J mice were rendered severely neutropenic (polymorphonuclear leukocyte count, <500/mm3) 3 days prior to the beginning of the challenge study (Day 0) with 200 mg/kg of body weight intraperitoneal (IP) cyclophosphamide (CY) or daily SC injection (125 mg/kg) of cortisone acetate. Significant decreases in total leucocytes and neutrophils were observed in the CY-treated or cortisone treated mice compared with those in the control mice. The effects of immunosuppression were monitored every 2–3 days during the entire experimental procedure. The total neutrophil counts were reduced (<500 cells/mm3) within 3 days of the first CY or cortisone injection, and severe neutropenia was maintained until the termination of the experiments at day 30 postinfection. Monocyte counts did not change significantly in cyclophosphamide-treated or cortisone-treated mice as compared to the normal control group (data not shown).
Comparison of virulence of C. glabrata and C. tropicalis in immunosuppressed BALB/c and A/J mice
Even though C. glabrata is not usually pathogenic in mouse models of candidiasis, C. glabrata infections often result in high morbidity and mortality in immunocompromised hospitalized patients.24,25 With the aim of mimicking the clinical situations in humans as closely as possible, experimental Candida infections were induced in mice rendered immunocompromised by pretreatment with cyclophosphamide (CY)26–28 or cortisone acetate treatments. In order to establish a reliable mouse model of disseminated candidiasis by C. tropicalis and C. glabrata, we treated BALB/c mice with cyclophosphamide and then infected immunosuppressed BALB/c mice with the same dose of C. tropicalis and C. glabrata as we used in naive untreated mice. Survival data show that immunosuppressed BABL/c mice are more susceptible than A/J to intravenous inoculation of 107C. tropicalis, where 100% mortality was achieved within 15 days postinfection (Fig. 3). Conversely, CY-treated BALB/c mice were still resistant against systemic infection by C. glabrata, where only 20% mortality was observed by 30 days post-infection (Fig. 3A). Due to the inability of CY to decrease the resistance of BALB/c mice to C. glabrata, we further immunosuppressed BALB/c mice with either cortisone or a combination of CY and cortisone. We then challenged these mice with the same inoculum of C. glabrata (1 × 108 cfu) and eventually achieved 60% mortality under these conditions (Fig. 3B). The need for combination immunosuppressive therapy to achieve greater than 50% mortality in BALB/c mice underlines the low pathogenicity of C. glabrata in this animal model.
Figure 3.

Comparison of virulence of C. glabrata and C. tropicalis in immunosuppressed BALB/c mice. Prolonged immunosuppression was maintained for 30 days by giving each animal a 150 mg/kg dose of CY by IP every 7 days after infection. (A) Survival data show immunosuppressed BABL/c mice are more susceptible to the intravenous inoculum of 107C. tropicalis, 100% mortality was achieved within 15 days postinfection. However, CY-treated BALB/c mice were still resistant against systemic infection by C. glabrata, only 20% mortality was observed within 30 days post-infection. (B) BALB/c mice were further immunosuppressed with either cortisone or combination of CY and cortisone, then challenged with the same inoculum of C. glabrata (1 × 108). Survival data show 60% mortality was achieved in immunocompromised BALB/c mice treated with CY/cortisone. This Figure is reproduced in color in the online version of Medical Mycology.
Analyses of the pathophysiology and host responses to acute C. albicans infection has demonstrated that A/J mice are extremely sensitive to infection. The greater susceptibility of A/J mice to systemic challenge with C. albicans prompted us to investigate whether A/J mice are more susceptible to both C. tropicalis and C. glabrata disseminated infection. To do this, we first tested the virulence of both NAC species in naïve and CY-treated A/J mice. Compared to BALB/c, both naive and immunosuppressed A/J mice are much more susceptible to C. glabrata; with the same inoculum of organism (108), 20% mortality was observed in naïve A/J mice within 30 days and 100% mortality in CY-treated A/J by 12 days post-challenge. In our intravenous mouse model of C. tropicalis systemic infection, A/J mice are more susceptible compared to BALB/c mice (20% mortality in Fig. 1), since 40% mortality was observed in immunocompetent A/J mice and 100% mortality was achieved in CY-treated immunosuppressed A/J mice (Fig. 4).
Figure 4.

A/J mice are more susceptible to disseminated candidiasis by both C. glabrata and C. tropicalis as compared to BALB/c. A/J mice were immunosuppressed with CY by IP and prolonged immunosuppression was maintained by the same CY regimen described as before. On day 0, both immunocompetent and immunosuppressed mice were intravenously infected with 1 × 108C. glabrata ATCC 2001 cells or 1 × 107 of C. tropicalis ATCC MYA-3404. As controls, immunocompetent A/J mice were challenged with the same dose of Candida cells as for immunosuppressed A/J mice. This Figure is reproduced in color in the online version of Medical Mycology.
The A/J mouse model was further evaluated and showed outcomes of a typical systemic infection of C. glabrata. Survival results in A/J mice treated with either CY or cortisone before IV challenge with C. glabrata, indicated 100% mortality within 10 days or 60% mortality within 30 days, respectively (Fig. 5). Therefore, the immunosuppressed CY-treated A/J mouse model can be considered an appropriate model of human disseminated C. glabrata infection. Importantly, this study represents the first demonstration of this relationship with two NAC species, C. glabrata and C. tropicalis, in both BALB/c and A/J mice.
Figure 5.

Immunosuppressed A/J intravenous mouse model of C. glabrata disseminated infection. A/J mice were immunosuppressed with either cyclophosphamide (CY) by IP or SC injection of cortisone acetate. Prolonged neutropenia were maintained for 30 days. On day 0, mice were intravenously infected with viable 1 × 108C. glabrata ATCC 2001 cells. As controls, immunocompetent A/J mice were challenged with the same dose of C. glabrata yeast cells. The C albicans disseminated A/J mouse model was used as positive control for Candida acute systemic infection. This Figure is reproduced in color in the online version of Medical Mycology.
Influence of immunosuppression on fungal burden and pathological alterations during the course of infection
Unlike C. albicans, C. glabrata does not induce a lethal phenotype in immunocompetent mice, even with infections of high inocula.29 In this study, we compared the effect of immunosuppression on the outcome of C. glabrata systemic infection in both A/J and BALB/c mice. We infected mice treated with either cyclophosphamide or cortisone acetate, or both, or left untreated (immunocompetent). Five mice per group were analyzed on days 2, 4, 6, 8 postinfection. Two days after infection, high fungal burdens of 105 to 107 of cfu/g were detected in all organs analyzed. Subsequently, the fungal burden in the spleen, brain and liver decreased over time in all groups (Fig. 6). However, cfu counts in the organs of cyclophosphamide-treated mice of both BALB/c (Fig. 6A) and A/J (Fig. 6B) strains were significantly higher (10- to 100-fold) through days 2 to days 8 than those from immunocompetent animals. During this period, the fungal burden declined about 1000-fold in brains and 100-1000-fold in spleen and livers of immunocompetent mice as compared to 10- to 100-fold decrease in organs of cyclophosphamide treated animals. In contrast to fungal clearance in the liver and spleen, the fungal burden in the brain of immunosuppressed A/J mice did not decrease until day 4 postinfection (Fig. 6B). In all examined organs, higher fungal burdens were always observed in immunosuppressed mice than those in immunocompetent animals (P < .001).
Figure 6.

Influence of immunosuppression on fungal burden in brain, liver and spleen during the course of systemic C. glabrata infection. To compare the effect of immunosuppression on the outcome of C. glabrata systemic infection in both A/J and BALB/c mice, we treated 20 mice of each strain with CY regimen or left untreated (immunocompetent) on day 3. Both immunosuppressed and untreated mice were inoculated IV with C. glabrata ATCC 2001 (108) on day 0. At each specific time point postinfection (day 2, 4, 6, and 8), five mice were euthanized, and net growth of C. glabrata quantified and compared in spleen, liver, and brain in BALB/c mice (A) and A/J mice (B). Results represent the mean SEM of CFU /tissue of five mice per time point. The CY- treated mice had significantly higher mean cfu in the organs examined than the normal group, and CY-treatment delayed fungus clearance in all tested organs of both mouse strains. This Figure is reproduced in color in the online version of Medical Mycology.
The kidney is known to be the major target organ for systemic candidiasis in mice. In contrast to other organs analyzed (brain, liver, spleen), the fungal burden in the kidneys of immunosuppressed mice of both strains did not decrease. Instead, it increased from day 2 to day 8 postinfection (Fig. 7). BALB/c mice treated with a combination of CY and cortisone had the most cfu in the kidney from day 2 to day 8 (Fig. 7A); A/J mice treated with CY had the most cfu through day 2 to 8 (Fig. 7B), as compared to a slow clearance trend observed in immunocompetent mice. Consistent with fungal burdens in other organs, A/J mice always had higher cfu than BALB/c mice at each time point examined. This was independent of host immunosuppression. In addition, our cfu data demonstrated that C. glabrata could persist for at least 3–4 weeks in the kidneys of immunocompetent mice and much longer in immunosuppressed mice (data not shown).
Figure 7.
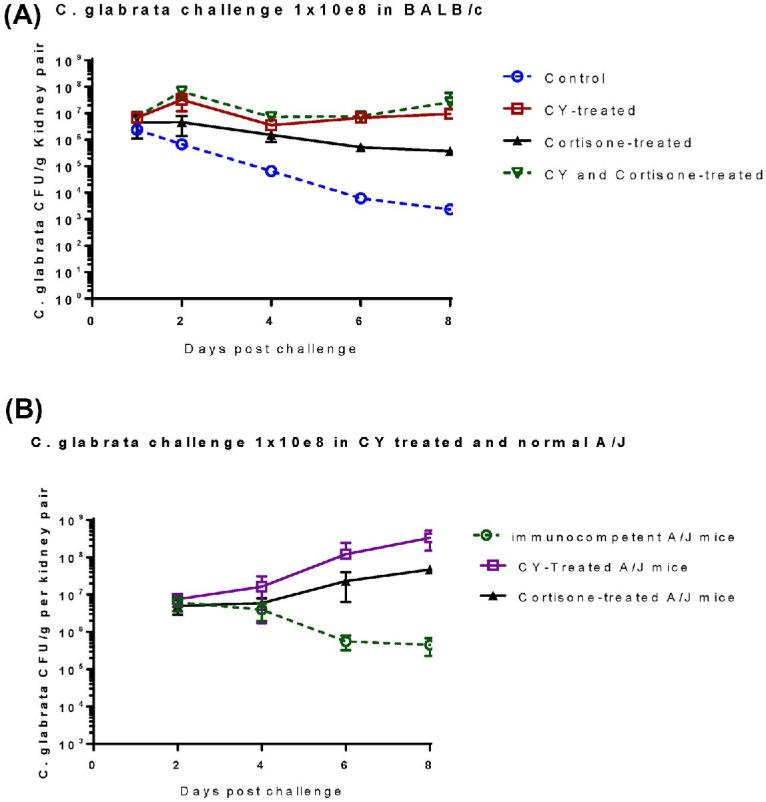
Dissemination and growth of C. glabrata in kidneys of immunocompetent and immunosuppressed BALB/c mice (A) and A/J mice (B). Immunosuppressed mice and untreated controls (n = 20 in each group) were inoculated IV with C. glabrata ATCC 2001 (108) on day 0. At each specific time point postinfection (day 2, 4, 6 and 8), five mice were euthanized, and net growth of C. glabrata was quantified and compared in kidney of BALB/c mice (A) and A/J mice (B). Results represent the mean SEM of cfu /tissue of five mice per time point. This Figure is reproduced in color in the online version of Medical Mycology.
The effect of immunosuppression on cfu for multiple organs of C. tropicalis systemic infection in BALB/c mice was also evaluated. Quantitative cultures taken from organs of normal and CY-treated BALB/c mice infected with 107 of live C. tropicalis showed that during the first 8 days of infection, the organisms had disseminated to various organs. The fungal burden in the spleen, liver and brain decreased over time in all groups. However, on day 8 postinfection, cfu in brain and liver of CY-treated BALB/c mice were 1000-fold higher than for immunocompetent controls, while only 10- to 100-fold higher cfu were observed in the spleens of CY-treated mice compared to controls (Fig. 8A). Compared to the rapidly decreasing cfu observed for spleen, brain, and liver of immunocompetent mice, the fungal burden in those organs of CY-treated mice decreased more slowly at all tested time points. Most cfu observed in kidneys of immunosuppressed BALB/c mice, were104-fold higher compared to immunocompetent controls on day 8 after challenge (Fig. 8B). Notably, fungal burden was persistently increased in kidneys of CY treated mice, in contrast to immunocompetent controls, where the fungus was gradually cleared from the kidney (Fig. 8B).
Figure 8.

Dissemination and growth of C. tropicalis in tissues of immunocompetent and CY-treated immunosuppressed BALB/c mice. In sum, 20 naive BALB/c mice were immunosuppressed by CY regimen as described before. Immunosuppressed BALB/c mice and untreated control mice (n = 20 in each group) were inoculated IV with C. tropicalis (107) on day 0. At each specific time point post-infection (day 2, 4, 6, and 8), five mice were euthanized, and net growth of C. tropicalis was quantified in spleen, livers and brains (A) and kidneys (B) by culture of tissue homogenates. Results represent the mean SEM of CFU /tissue of five mice per time point. The results of CFU determinations of infected kidneys were consistent with those of mortality, and number of cfu/g kidney tissue was significantly higher in CY treated mice as compared with those immunocompetent mice with same inoculum. This Figure is reproduced in color in the online version of Medical Mycology.
Vaccination with Met6 peptide vaccine significantly improved survival and decreased fungal burden in immunocompromised mice challenged intravenously with C. tropicalis
It is known that a significant fraction of immunocompromised patients respond to a variety of vaccines.12,13,30 In previous studies, we have demonstrated that active immunization with Met6 peptide-DC vaccine significantly improved the survival and reduced the fungal burden of disseminated candidiasis in immunocompromised mice. These mice were immunocompromised experimentally by multiple intraperitoneal injections of a 200 mg/kg dose of cyclophosphamide.22,23,31 In this study, we evaluated the potential for the Met6 peptide vaccine, which has 100% homology in C. tropicalis, to increase resistance in immunosuppressed mice against disseminated candidiasis by C. tropicalis. To establish severe acute infection of C. tropicalis in immunosuppressed animals, 1 × 107 of C. tropicalis (ATCC MYA-3404) yeast cells is required for both BALB/c and A/J strains. We used the same antigen-pulsed DC-based immunization strategy and schedules as described before20,21; age- and sex-matched control mice of both strains received untreated DCs only. Survival data show that the Met6 peptide vaccine was able to induce substantial protection in immunosuppressed mice of both strains as evidenced by significantly improved survival. Vaccinated mice of both strains showed 80% survival, as compared to 0% survival in A/J controls and 20% survival in BALB/c controls (Fig. 9A). Vaccination prior to rendering the mice immunocompromised provided significant protection, evidenced by prolonged survival and greatly reduced CFUs in the kidneys (Fig. 9B) and brains (Fig. 9C) of vaccinated immunosuppressed mice, compared to normal controls. We demonstrated that immunity induced by active immunization with the Met6 peptide vaccine was protective against C. tropicalis disseminated infection in immunocompromised mice.
Figure 9.

Vaccination with Met6 peptide vaccine significantly improved survival in immunosuppressed mice challenged intravenously with lethal dose of C. tropicalis. We used the same antigen-pulsed DC-based immunization strategy favoring production of antibodies as described before. Vaccinated BALB/c and A/J Mice were rendered severely neutropenic by CY 3 days prior to challenge. Control mice received DC only. Immunized and control mice were infected IV with a lethal dose of live C. tropicalis yeast cells (1 × 107). (A) Met6 peptide vaccine was able to induced good protection in immunosuppressed mice as evidenced by significantly improved survival (P < .01). Kaplan-Meier survival curves were compared for statistical significance with the log rank test between the control group (DC) and each of the Met6-DC groups. (B) In the vaccinated groups, fungal burdens were significantly reduced in kidney (P < 0.001), a major target organ in mouse model of disseminated candidiasis. (C) Fungal burdens were also significantly reduced in brains of the vaccinated groups (P < .01), another target organ in mouse intravenous models of disseminated candidiasis. The Mann-Whitney rank sum test was used to compare the vaccinated mice to the control mice. Data are reported as means ± standard errors of the means. This Figure is reproduced in color in the online version of Medical Mycology.
Discussion
The virulence of two major NAC species, C. tropicalis and C. glabrata, were compared with C. albicans in both naive wild type and immunosuppressed BALB/c mice. A combination of a large inoculum and immunosuppression was needed to establish severe acute infection of C. tropicalis in BALB/c mice; however, this strategy was not a prerequisite for C. glabrata disseminated candidiasis in the BALB/c intravenous mouse model. Eventually, 60% mortality was achieved in BALB/c mice treated with both CY and cortisone regimens. The need for combination immunosuppressive therapy to achieve greater than 50% mortality in BALB/c mice underlines the low pathogenicity of C. glabrata in this strain. To establish a simple and more reliable intravenous mouse model for acute infection of NAC, the C5-deficient A/J strain was used to evaluate C. glabrata and C. tropicalis virulence. Both immunocompetent and CY-treated A/J mice are more susceptible to NAC systemic infection compared to corresponding BALB/c controls. These findings indicate the importance of immunosuppression and C5 deficiency in the pathogenesis of C. glabrata infection. It is much more likely that C5 deficiency has its effect at the level of C5a, a chemotactic agent and anaphylatoxin. The most well-defined role for C5a during invasive candidiasis is as a chemotactic agent for neutrophils and other effector cells. The G-protein coupled C5a receptor (C5aR/CD88) is expressed on neutrophils, monocytes, macrophages, dendritic cells, basophils, eosinophils, and mast cells, among other cell types. In the absence of C5a, mice show reduced recruitment of effector cells to the site of infection. Similarly, an immunocompromised situation is one of the major risk factors for developing systemic candidiasis in humans, and the lack of functional C5 may mirror this process in mice. Therefore, we consider the CY-treated immunosuppressed A/J intravenous model to be an appropriate model for human disseminated C. glabrata infection, mimicking patients who develop deficiencies, including neutropenia. C. albicans has been shown to activate the complement system through the alternative pathway, with chemotactic and inflammatory activities of complement apparently crucial components for effective host defense. In particular, a relationship between a naturally occurring deficiency in complement component 5 (C5) and enhanced susceptibility to systemic C. albicans candidiasis is also evident.32,33 Even C. glabrata has been increasingly associated with opportunistic infections in immunocompromised individuals, yet relatively little is known about the host factors that mediate immunity to this organism. A/J mice, deficient in C5, have been reported by others to be highly susceptible to C. albicans disseminated infections. To our knowledge, this is the first study to report that C5-deficient A/J mice are more susceptible to C. glabrata systemic infection, suggesting the essential role of complement in immunity to this organism. Although this organism can induce the alternative pathway of complement activation,34 the role of complement in the control of C. glabrata has not yet been examined.
In this study, we show that the onset and outcome of systemic infection with two NAC species varies greatly between two inbred mouse strains, both in immunocompetent and immunosuppressed settings. The pathogenesis insusceptible strains resemble human candidiasis, in terms of the organs targeted for colonization and active replication, as well as the ensuing pathology. As compared to cfu obtained from spleens and livers of immunocompetent mice that decreased rapidly, the fungal burden in these organs under immunosuppressed conditions did not decrease efficiently at all tested time points.
Although both these inbred strains are considered to be resistant to C. albicans infection, immunocompetent A/J mice are more susceptible to disseminated candidiasis by two most common NAC species examined as compared to immunocompetent BALB/c mice. Interestingly, trends of fungal replication in the liver, spleen, and brain of A/J and BALB/c mice were similar. In particular, immunosuppressed A/J mice cleared infection less efficiently than immunosuppressed BALB/c mice. Furthermore, there was a significant difference in fungal burdens between the two strains. Fungal burdens in all the tested organs of CY-treated A/J mice were always 10- to 100-fold higher than that of CY-treated BALB/c at any time point tested. Particularly, in contrast to fungal clearance in the liver and spleen, the fungal burden in the brain of immunosuppressed A/J mice did not decrease until day 4 postinfection. The kidney is known to be the main target organ for systemic candidiasis in mice and humans. In contrast to a declining trend in other organs, kidney fungal burden in immunosuppressed mice of both strains remained comparatively high and even increased from day 4 postinfection throughout the experiment until death or euthanization. Common to NAC invasive infection in both strains was that the highest fungal loads were detected in the kidneys, with those of A/J mice containing an approximately 100-fold greater fungal burden at all-time points than BALB/c mice. The levels of several cytokines (IL-12, IFN-γ, and IL-6) that had previously been reported to be elevated during systemic C. albicans infection35,36 were measured in A/J and BALB/c mice. Cytokine levels in mouse serum isolated 48 h after infection were measured by ELISA. No detectable differences in these cytokines were observed post-infection between the two strains. However, A/J was found to have an elevated, although not significant (P = .25) level of IL-6 at the 48 h time point compared to BALB/c mice (data not shown). A/J is extremely sensitive to infection, with overwhelming fungal replication occurring primarily in the kidney, and to a lesser extent, the brain. Elevated levels of the pro-inflammatory cytokines TNF-α and IL-6 early in infection has been reported by other groups, suggesting that these mice undergo a heightened, and possibly unregulated, inflammatory response after challenge with Candida.35 Nevertheless, to improve prevention, management, and treatment of invasive NAC infection in immunocompromised patients, it is essential to study host pathogen interactions in murine models of candidiasis that mimic immunocompromised patients.
We have shown that active immunization with the Met6 peptide vaccine protects immunocompromised mice against C. tropicalis disseminated candidiasis. These findings indicate that neutrophils may not play a critical role in protection and this peptide vaccine-induced immunity may not modulate or be modulated by neutrophils. Neutropenic patients, who have a high risk of developing disseminated candidiasis, would be an obvious target population for instituting preventive measures through active immunization to induce protective immunity. This paradigm of vaccine-based immunity may potentially serve as a framework for new preventive strategies against infectious diseases in the immunocompromised host. Development of an effective Candida vaccine is dependent on defining immune effector mechanisms that display natural protective abilities in vivo. Antibody and CD4+ T-cell responses have evolved as independent mechanisms of fungal control, yet their precise mechanisms of protective immunity have not been thoroughly defined.37,38 We have previously shown that the Met6 peptide specific mAbs are protective in a mouse model of C. albicans systemic infection.23 It is important, however, to better understand protective mechanisms of vaccine-induced antibodies against invasive candidiasis by NAC in immunosuppressed mouse models. The identification of any protective mechanism is complex because antibodies against Candida may exert their effects through a variety of different effector mechanisms. Such mechanisms may include enhancement of phagocytosis by opsonization/complement fixation, antibody-dependent cellular cytotoxicity,39 and inhibition of adherence,40–42 growth, and germination.40,43,44 It is also important to investigate the Th1/Th17- mediated adaptive immunity induced by the Met6 peptide vaccine.
Although experimental infections of mice are considered valuable models for human disease, important differences between mice and humans must be taken into account when interpreting experimental data. For example, mice and humans differ with respect to their indigenous fungal gut flora. Indeed, mice are not naturally colonized by human pathogenic Candida species. Further, differences in biochemical pathways in laboratory mice and in humans must be considered when extrapolating experimental murine data into human treatment strategies. Therefore, conclusions drawn from animal studies depend on a critical understanding of the inherent limitations and parallels of mouse models to human disease states.
The association of C5 with high susceptibility in A/J mice provides strong evidence that a deficiency in C5 may be responsible for enhanced susceptibility to both C. tropicalis and C. glabrata systemic infection. In the future, to establish a reliable and simple mouse model of disseminated candidiasis by other NAC spices, the C5 status of the strains may be a useful predictor of their response to Candida. With a C5 deficiency associated with increased susceptibility, this A/J model emulates the most common host risk factor leading to susceptibility to disseminated candidiasis in humans.
A better understanding of the host–pathogen interaction is invaluable for the development of new prophylactics or therapies. Animal models provide a useful tool to study the response to systemic candidiasis and studies in mice have indicated the importance of the innate immune system, particularly the complement pathway, in controlling invasive fungal infection. As resistance to antifungal agents becomes a growing concern, especially for NAC, the animal model established in this study provides a useful tool to study the response to systemic candidiasis by the two most common forms of NAC.
Acknowledgments
This research was supported by the National Institutes of Health grant R21 AI113515. The author thanks the Research Institute for Children (RIC) at Children's Hospital in New Orleans and Louisiana State University Health Sciences Center (LSUHSC) for support of our research.
Declaration of interest
The author reports no conflicts of interest. The author alone is responsible for the content and the writing of the paper.
References
- 1. Pappas PG, Kauffman CA, Andes D et al.. Clinical practice guidelines for the management of candidiasis: 2009 update by the Infectious Diseases Society of America. Clin Infect Dis. 2009; 48: 503–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pappas PG, Rex JH, Lee J et al.. A prospective observational study of candidiemia: epidemiology, therapy, and influences on mortality in hosptilaized adult and pediatric patients. Clin Infect Dis. 2003; 37: 634–643. [DOI] [PubMed] [Google Scholar]
- 3. Whaley SG, Berknow EL, Rybak JM, Nishimoto AT, Barker KS, Rogers PD. Azole antifungal resistance in Candida albicans and emerging non-albicans Candida species. Front Microbiol. 2016; 17: 2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colombo AL, Perfect J, Dinubile M et al.. Global distribution and outcomes for Candida species causing invasive candidiasis: results from an international randomized double-blind study of caspofungin versus amphotericin B for the treatment of invasive candidiasis. Eur J Clin Microbiol Infect Dis. 2003; 22: 470–474. [DOI] [PubMed] [Google Scholar]
- 5. Alexander BD, Johnson MD, Pfeiffer CD, Jimenez-Ortigosa C, Catania J, Booker R. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated MIC. Clin Infect Dis. 2013; 56: 1724–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodrigues CF, Silva S, Henriques M. Candida glabrata: a review of its features and resistance. Eur J Clin Microbiol Infect Dis. 2014; 33: 673–688. [DOI] [PubMed] [Google Scholar]
- 7. Farmakiotis D, Tarrand JJ, Kontoyiannis DP. Drug-resistant Candida glabrata infection in cancer patients. Emerg Infect Dis. 2014; 20: 1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krcmery V, Barnes AJ. Non-albicans Candida spp. causing fungaemia: pathogenicity and antifungal resistance. J Hosp Infect. 2002; 50: 243–260. [DOI] [PubMed] [Google Scholar]
- 9. Wingard JR. Importance of Candida species other than C. albicans as pathogens in oncology patients. Clin Infect Dis. 1995; 20: 115–125. [DOI] [PubMed] [Google Scholar]
- 10. Wingard JR, Vaughan WP, Braine HG, Merz WG, Saral R. Prevention of fungal sepsis in patients with prolonged neutropenia: a randomized, double-blind, placebo-controlled trial of intravenous miconazole. Am J Med. 1987; 83: 1103–1110. [DOI] [PubMed] [Google Scholar]
- 11. Kontoyiannis DP, Vaziri I, Hanna HA et al.. Risk factors for Candida tropicalis fungemia in patients with cancer. Clin Infect Dis. 2001; 33: 1676–1681. [DOI] [PubMed] [Google Scholar]
- 12. Spellberg B. Vaccines for invasive fungal infections. F1000 Med Rep. 2011; 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dockrell DH, Poland GA, Steckelberg JM, Pomeroy C. Immunogenicity of three Haemophilus influenzae type b protein conjugate vaccines in HIV seropositive adults and analysis of predictors of vaccine response. Vaccine. 1999; 17: 2779–2785. [DOI] [PubMed] [Google Scholar]
- 14. Levin MJ, Gershon AA, Weinberg A et al.. Immunization of HIV-infected children with varicella vaccine. J Pediatr. 2001; 139: 305–310. [DOI] [PubMed] [Google Scholar]
- 15. Tedaldi EM, Baker RK, Moorman AC et al.. Hepatitis A and B vaccination practices for ambulatory patients infected with HIV. Clin Infect Dis. 2004; 38: 1478–1484. [DOI] [PubMed] [Google Scholar]
- 16. Leung TF, Hung E, Chan PK, Mo C, Wong RP, Chik KW. Immunogenicity of a two-dose regime of varicella vaccine in children with cancers. Eur J Haematol. 2004; 72: 353–357. [DOI] [PubMed] [Google Scholar]
- 17. Klugman KP, Madhi SA, Huebner RE, Kohberger R, Mbelle N, Pierce N. A trial of a 9-valent pneumococal conjugate vaccine in children with and those without HIV infection. N Engl J Med. 2003; 340: 1341–1348. [DOI] [PubMed] [Google Scholar]
- 18. Han Y, Cutler JE. Antibody response that protects against disseminated candidiasis. Infect Immun. 1995; 63: 2714–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han Y, Riesselman MH, Cutler JE. Protection against candidiasis by an immunoglobulin G3 (IgG3) monoclonal antibody specific for the same mannotriose as an IgM protective antibody. Infect Immun. 2000; 68: 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Son Y-I, Egawa S, Tatsumi T, Redlinger RE, Kalinski P, Kanto T. A novel bulk-culture method for generating mature dendritic cells from mouse bone marrow cells. J Immunol Meth. 2002; 262: 145–157. [DOI] [PubMed] [Google Scholar]
- 21. Xin H, Dziadek S, Bundle DR, Cutler JE. Synthetic glycopeptide vaccines combining β-mannan and peptide epitopes induce protection against candidiasis. Proc Natl Acad Sci U S A. 2008; 105: 13526–13531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xin H, Cutler JE. Vaccine and monoclonal antibody that enhance mouse resistance to candidiasis. Clin Vaccine Immunol. 2011; 18: 1656–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xin H. Double chimeric peptide vaccine and monoclonal antibodies that protect against disseminated candidiasis. J Vaccines Vaccin. 2014; 5: 241. [Google Scholar]
- 24. Sardi JCO, Scorzoni T, Bernardi T, Fusco-Almeida AM, Mendes-Gianini MJS. Candida species: current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J Med Microbiol 2013; 62: 10–24. [DOI] [PubMed] [Google Scholar]
- 25. Fidel PL, Vazquez JA, Sobel JD. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin Microbiol Rev. 1999; 12: 80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segal E, Frenkel M. Experimental in vivo models of candidiasis. Journal of Fungi 2018; 4: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Semis R, Mendlovic S, Polacheck I, Segal E. Activity of an Intralipid formulation of nystatin in murine systemic candidiasis. Int J Antimicrob Agents. 2011; 38: 336–340. [DOI] [PubMed] [Google Scholar]
- 28. Frenkel M, Mandelltat M, Alastruey-Izquierdo A, Mendlovic S, Semis R, Segal E. Pathogenicity of Candida albicans isolates from bloodstream and mucosal candidiasis assessed in mice and Galleria mellonella. J Mycol Med. 2016; 26: 1–8. [DOI] [PubMed] [Google Scholar]
- 29. Jacobsen ID, Brunke S, Seider K et al.. Candida glabrata persistence in mice does not depend on host immunosuppression and is unaffected by fungal amino acid auxotrophy. Infect Immun. 2010; 78: 1066–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Luo G, Ibrahim AS, French SW, Edwards JE Jr., Fu Y. Active and passive immunization with rHyr1p-N protects mice against hematogenously disseminated candidiasis. PLoS One. 2011; 6: e25909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xin H. Active immunizations with peptide-DC vaccines and passive transfer with antibodies protect neutropenic mice against disseminated candidiasis. Vaccine. 2016; 34: 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tuite A, Elias M, Picard S, Mullick A, Gros P. Genetic control of susceptibility to Candida albicans in susceptible A/J and resistant C57BL/6 J mice. Genes Immun. 2005; 6: 672–682. [DOI] [PubMed] [Google Scholar]
- 33. Tuite A, Mullick A, Gros P. Genetic analysis of innate immunity in resistance to Candida albicans. Genes Immun. 2004; 5: 576–587. [DOI] [PubMed] [Google Scholar]
- 34. Tsoni SV, Kerrigan AM, Marakalala MJ et al.. Complement C3 plays an essential role in the control of opportunistic fungal infections. Infect Immun. 2009; 77: 3679–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mullick A, Elias M, Picard S et al.. Dysregulated inflammatory resopnse to Candida albicans in a C5-deficient mouse strain. Infect Immun. 2004; 72: 5868–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mullick A, Leon Z, Min-Oo G et al.. Cardiac failure in C5-deficient A/J mice after Candida albicans infection. Infect Immun. 2006; 74: 4439–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanco JL, Garcia ME. Immune response to fungal infections. Vet Immunol Immunopathol. 2008; 125: 47–70. [DOI] [PubMed] [Google Scholar]
- 38. Richardson JP, Moyes DL. Adaptive immune responses to Candida albicans infection. Virulence. 2015; 6: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Casadevall A, Pirofski L-A. Antibody-mediated protection through cross-reactivity introduces a fungal heresy into immunological dogma. Infect Immun. 2007; 75: 5074–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Torosantucci A, Chiani P, Bromuro C et al.. Protection by anti-beta-glucan antibodies is associated with restricted beta-1,3 glucan binding specificity and inhibition of fungal growth and adherence. PLoS One. 2009; 4: e5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Bernardis F, Boccanera M, Adriani D, Spreghini E, Santoni G, Cassone A. Protective role of antimannan and anti-aspartyl proteinase antibodies in an experimental model of Candida albicans vaginitis in rats. Infect Immun. 1997; 65: 3399–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bromuro C, Romano M, Chiani P et al.. Beta-glucan-CRM 197 conjugates as candidates antifungal vaccines. Vaccine. 2010; 28: 2615–2623. [DOI] [PubMed] [Google Scholar]
- 43. Torosantucci A, Bromuro C, Chiani P et al.. A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med. 2005; 202: 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brena S, Omaetxebarria MJ, Elguezabal N, Cabezas J, Moragues MD, Pontón J. Fungicidal monoclonal antibody C7 binds to Candida albicans. Infect Immun. 2007; 75: 3680–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]