

Abstract
Pompe disease is a rare inherited metabolic disorder of defective lysosomal glycogen catabolism due to a deficiency in acid alpha-glucosidase (GAA). Alglucosidase alfa enzyme replacement therapy (ERT) using recombinant human GAA (rhGAA ERT) is the only approved treatment for Pompe disease. Alglucosidase alfa has provided irrefutable clinical benefits, but has not been an optimal treatment primarily due to poor drug targeting of ERT to skeletal muscles. Several critical factors contribute to this inefficiency. Some are inherent to the anatomy of the body that cannot be altered, while others may be addressed with better drug design and engineering. The knowledge gained from alglucosidase alfa ERT over the past 2 decades has allowed us to better understand the challenges that hinder its effectiveness. In this review, we detail the problems which must be overcome for improving drug targeting and clinical efficacy. These same issues may also impact therapeutic enzymes derived from gene therapies, and thus, have important implications for the development of next generation therapies for Pompe.
Keywords: Alglucosidase alfa, distribution, enzyme replacement therapy (ERT), gene therapy, M6P
Introduction
Pompe disease, also known as glycogen storage disease type II (GSD II) or acid maltase deficiency, is a rare and typically fatal neuromuscular disease caused by mutations in the GAA gene, which encodes the lysosomal hydrolase acid alpha-glucosidase (GAA) (1,2). GAA functions to hydrolyze glycogen to release free glucose units in lysosomes (3) that are ultimately transported back to the cytosol for use in various cellular pathways. Unlike cytoplasmic glycogen that has a well-defined role for energy production, the biological role of lysosomal glycogen has not yet been elucidated. GAA deficiency results in glycogen accumulation within lysosomes that significantly impairs cellular function, particularly in smooth, cardiac, and skeletal muscle cells. Patients with infantile-onset Pompe disease (IOPD) are most severely affected and usually die by one year of age, primarily due to cardiac or respiratory failure (4,5). Patients with late-onset Pompe disease (LOPD) have varying levels of residual GAA activity that slows progression of disease and present a much wider range of symptoms, age of clinical presentation, and disease severity (2,6). All LOPD patients experience progressive muscle weakness, impaired motor function, and respiratory decline that ultimately leads to their demise (1,7).
Alglucosidase alfa (Myozyme®/Lumizyme®, Sanofi Genzyme, Cambridge, MA, USA) enzyme replacement therapy (ERT) using recombinant human GAA (rhGAA), is currently the only approved treatment for Pompe disease. Alglucosidase alfa irrefutably has provided clinical benefits, particularly for increasing survival in IOPD patients (8-13). The ERT appears to slow disease progression but has not been shown to halt or reverse disease for the majority of patients, and thus, significant unmet medical needs remain (14-18). The most apparent limitation is the poor response of skeletal muscles to alglucosidase alfa treatment (19). The majority of IOPD infants who survive usually develop progressive myopathy in subsequent years (20,21) despite initiation of treatment very early in life (20-22). Accumulation of glycogen-filled autophagosomes due to defective autophagy appears to contribute significantly to disease pathology (3,23,24) and is not resolved by alglucosidase alfa (20,25,26). The humoral immune response to alglucosidase alfa may also interfere with clinical outcomes, especially in cross reactive immunologic material (CRIM)-negative infants (27). CRIM-positive late onset adults can also develop high anti-rhGAA antibody titers (28,29) but the impact of antibodies is not clear since clinical efficacy does not appear to be affected for the majority of LOPD patients (30).
The effectiveness of alglucosidase alfa is limited primarily due to its poor drug targeting to skeletal muscles. There are several main causes for this inefficiency which have been gleaned from nearly three decades of rhGAA ERT development and are discussed in this article. We anticipate that these issues would affect not just ERTs but also gene products from cross-corrective gene therapies, and thus, have important implications for future therapies for Pompe.
Lessons learned
Nonproductive clearance of rhGAA severely reduces amount of ERT actually reaching skeletal muscles
In healthy individuals, GAA is expressed in the endoplasmic reticulum and subsequently transported to lysosomes to perform its vital biological function within most—if not all—cells. In contrast, rhGAA ERT relies on the import of exogenous lysosomal enzyme from the systemic circulation. This process is typically very inefficient and incurs substantial loss of therapeutic enzyme prior to delivery to target muscles. Despite attaining high rhGAA concentrations in blood post intravenous administration, multiple competing pathways seize the vast majority of the ERT such that only a tiny fraction of the total rhGAA administered actually reaches the interstitial space for cellular uptake by skeletal muscles, as depicted in Figure 1.
Figure 1.
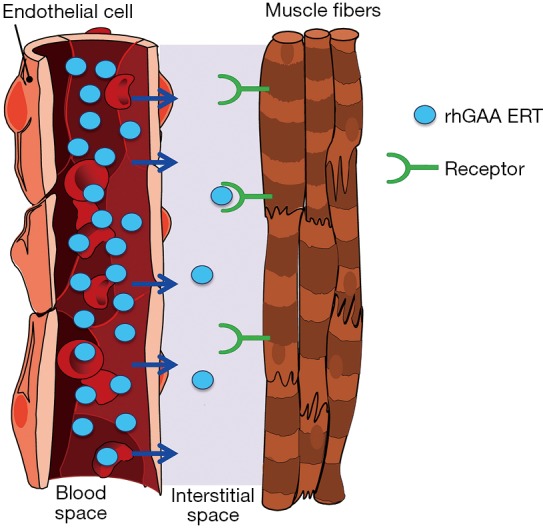
Depiction of rhGAA ERT distribution from the systemic circulation to the interstitial space surrounding muscle fibers. High resultant plasma concentrations of rhGAA (blue circles) can be attained post-intravenous administration but only a very small fraction reaches skeletal muscle. Substantial clearance of rhGAA from the circulation by non-muscle tissues and cells including liver, spleen, fibroblasts, etc. and an inefficient transcytosis process are believed to cause very poor distribution of rhGAA ERT to the interstitia. Receptor-mediated endocytosis is required for efficient cellular uptake of exogenous rhGAA in skeletal muscles (Van der Ploeg et al. 1988; Zhu et al. 2009; Xu et al. 2019). rhGAA, recombinant human acid alpha-glucosidase; ERT, enzyme replacement therapy.
Nonproductive clearance by non-muscle tissues such as liver, spleen, gastrointestinal tract, lymphatic system, etc., removes the vast majority of rhGAA ERT to cause the so called “sink effect” (31-33). rhGAA is a complicated glycoprotein containing various carbohydrate structures that are ligands for several different carbohydrate receptors on cell surfaces of many other cell types throughout body (31-34). These cell types compete with muscles for binding and uptake of ERT. Since there is significant blood flow into these other tissues and the number of competing carbohydrate receptors are numerous, this has been shown to result in nonproductive clearance of ERT to other unintended tissues and substantially decrease the amount of ERT delivered to intended muscles (31,35). The exact mechanism(s) for rhGAA egress out of the vasculature and into the interstitial space are still largely undefined. We are not aware of any documented reports which comprehensively studied rhGAA transport out of the circulation. We speculate that rhGAA is likely internalized in endothelial cells, transported across cell layer within caveolae, and released to the other side via a transcytosis process. This process is known to mediate the transport of albumin, certain hormones, antibodies, and various other glycoproteins from the circulation to tissues (36-39). This is in contrast to the typical endocytosis process that ultimately fuses with lysosomes and retains rhGAA within cells. Transcytosis is thought to be an inefficient process relative to endocytosis and likely accounts for transport of only small fraction of rhGAA from endothelial cells to the interstitial space (40). To understand the relative amounts of rhGAA that is distributed from the systemic circulation to muscles, we measured rhGAA levels in tissue homogenates of Gaa KO mice 24 hrs after administration of 20 mg/kg alglucosidase alfa. Our data indicate that less than 1% of the total rhGAA dose actually reaches muscles 24 hrs after intravenous bolus administration as shown in Table 1. Irrespective of the mechanisms responsible for tissue biodistribution, these data highlight the intrinsic challenges of rhGAA drug targeting from the systemic circulation to muscles that undoubtedly contribute to the suboptimal efficacy for alglucosidase alfa (Supplementary).
Table 1. Tissue distribution of rhGAA in Gaa KO mice post-intravenous administration.
| Variable | Liver | Quad | Tricep | Gastroc | Heart |
|---|---|---|---|---|---|
| Number of animals (n) | 6 | 16 | 10 | 10 | 10 |
| GAA activity in tissue homogenate (nmol 4-MU released/mg protein/hr)a | 776 | 8 | 8 | 11 | 50 |
| mg total protein in homogenate/mg wet tissue | 0.11 | 0.04 | 0.04 | 0.04 | 0.04 |
| GAA activity (nmol/mg wet tissue/hr)b | 85 | 0.32 | 0.32 | 0.44 | 2.0 |
| Total wet tissue weight (mg) | 1,077 | 125 | 81 | 148 | 110 |
| Total GAA activity in tissue (nmol/tissue/hr)c | 9.2×104 | 40 | 26 | 65 | 220 |
| % of rhGAA dose in tissued | 52.57 | <0.03 | <0.02 | <0.04 | <0.13 |
a, amount of GAA enzyme activity as measured by release of 4-MU fluorescence and normalized per milligram of total protein in tissue homogenate; b, amount of GAA activity in 1 mg of wet tissue normalized using determined amount of total protein in tissue homogenate/mg total protein in wet tissue; c, total amount of rhGAA normalized to the entire wet tissue weight; d, fraction of total rhGAA dose in tissue determined by dividing the measured GAA activity in tissue by the total GAA activity from dosing solution (1.75×105 nmol 4-MU released/hr) and expressed as percent of total rhGAA dose. rhGAA, recombinant human acid alpha-glucosidase.
A less appreciated aspect that encumbers ERT delivery to muscles is the instability of rhGAA at the neutral pH of blood. This instability results in a loss of ERT prior to reaching muscles. rhGAA is an acid hydrolase that is most stable and active at acidic pH, but is substantially less stable at the neutral pH environment of blood that ultimately leads to irreversible enzyme inactivation (41). Damaged proteins are thought to be identified and eliminated from the circulation by C3b and other proteins of the complement system (42). Stabilization of rhGAA at neutral pH via small molecule pharmacological chaperones have been shown to reduce irreversible enzyme inactivation for delivery of active ERT in Pompe patient-derived fibroblasts and to muscles of Gaa KO mice (41,43-46).
rhGAA is inherently poorly phosphorylated which severely limits cellular uptake in muscles
For cross-correction of muscles by rhGAA ERT, the exogenous enzyme must be internalized efficiently at clinically relevant doses. Reuser and colleagues conducted the seminal research that conclusively showed exogenous GAA requires the specialized carbohydrate structure mannose 6-phosphate (M6P) for efficient cellular uptake in Pompe patient derived fibroblasts and skeletal muscle cells; GAA lacking M6P or dephosphorylated GAA was not internalized (47,48). Similar results were observed in vivo where only GAA containing M6P was efficiently targeted to cardiac and skeletal muscles in Gaa knockout mice (49), and thus, underscored the requirements and potential for GAA ERT.
M6P is the natural recognition marker that enables newly synthesized soluble lysosomal proteins to bind cation-dependent and cation-independent M6P receptors (CD-MPR and CI-MPR, respectively) for their transport from the trans-Golgi network to lysosomes within cells (50-53). The CI-MPR also cycles to the plasma membrane for facilitating receptor-mediated endocytosis of exogenous rhGAA containing M6P (48,52,54,55). Most soluble lysosomal proteins are modified post-translationally to contain M6P via two resident Golgi enzymes: N-Acetylglucosamine-1-Phosphotransferase transfers an N-acetylglucosamine-linked phosphate group from nucleotide sugar donor UDP-GlcNAc onto certain terminal mannose residues of N-linked high mannose-type oligosaccharides; N-Acetylglucosamine-1-Phosphodiesterase α-N-acetylglucosaminidase removes the “covering” N-acetylglucosamine sugar to expose the M6P targeting moiety (56,57). However, unlike other soluble lysosomal enzymes, GAA is an inherently poor substrate for N-Acetylglucosamine-1-Phosphotransferase such that only a small percentage of the total GAA produced contains M6P (58-61). Affinity chromatography using immobilized CI-MPR revealed that alglucosidase alfa is a mixture of rhGAA comprising both M6P-containing and -lacking fractions (60,61). This is consistent with our internal data as shown in Figure 2A. Since binding the CI-MPR is the mandatory first step for receptor-mediated endocytosis, only the minor rhGAA fraction containing M6P is capable of efficient cellular uptake. Further, the type of M6P-containing N-glycan structure critically impacts receptor binding such that the bis-phosphorylated oligosaccharide (2 M6Ps on same N-glycan) structure has very high affinity for CI-MPR while the mono-phosphorylated oligosaccharide (1 M6P on N-glycan) structure has approximately 3,000-fold lower affinity (62) as highlighted in Figure 2B. This disparity is particularly important at low interstitial enzyme concentrations since only rhGAA with bis-phosphorylated oligosaccharides would be able to bind CI-MPR for cellular uptake in muscle cells under these conditions. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectroscopy analysis of 2-anthranilic acid (2AA)-labeled released N-glycans from alglucosidase alfa revealed that on average, this ERT contained 0.1 mole bis-phosphorylated oligosaccharides per mole rhGAA (63). The total amount of N-glycans from alglucosidase alfa that contained the bis-phosphorylated oligosaccharide structure was empirically determined to be approximately 1% (59). It is therefore expected that only this tiny fraction of alglucosidase alfa would be capable of efficient cellular uptake at the anticipated low interstitial enzyme concentrations. To compensate for low levels of bis-phosphorylated oligosaccharides, significantly higher alglucosidase alfa doses would be needed for optimal in vivo efficacy. This hypothesis is supported by the observation that 100 mg/kg of alglucosidase alfa with low bis-phosphorylated oligosaccharide content was required to obtain the same glycogen substrate reduction in skeletal muscles of Gaa KO mice as was possible using only 20 mg/kg rhGAA ERT with high bis-phosphorylated oligosaccharide content (61). While using 100 mg/kg ERT dose is feasible for establishing proof of concept in preclinical rodent studies, utilizing such high ERT doses for therapeutic treatment in humans is likely not possible due to the limitations caused by significantly higher manufacturing demand, drug costs, and most importantly, to patient safety and tolerability to ERT.
Figure 2.

(A) Alglucosidase alfa was loaded onto a CI-MPR column to assess the relative proportion of the enzyme mixture that contains M6P. rhGAA lacking M6P cannot bind to the CI-MPR and, therefore, flowed through the column, while rhGAA containing M6P binds to the CI-MPR and was retained on the column. The bound rhGAA was then eluted from the column using a linear gradient of increasing free M6P. The beginning of the linear gradient is indicated by the black arrows. Both fractions of rhGAA (unbound and bound/eluted) were collected and assayed for GAA enzyme activity using the fluorogenic substrate 4-methylumbeliferyl-α-glucopyranoside (4-MU-raGlc) to determine the relative percentage of rhGAA in each peak. (B) Representative N-linked oligosaccharide structures and their respective binding affinities for the CI-MPR. The binding affinities for radiolabeled bis- and mono-phosphorylated high mannose oligosaccharides and complex-type oligosaccharides were experimentally determined by equilibrium dialysis as reported by Tong et al. [1989]. Bis-phosphorylated high mannose oligosaccharides have very high affinity while mono-phosphorylated high mannose oligosaccharides have moderate affinity for the CI-MPR. Complex-type oligosaccharides and non-phosphorylated high mannose oligosaccharides (not shown) do not bind CI-MPR. rhGAA has 7 potential N-glycosylation sites (Park et al., 2018) and different enzyme preparations have varying amounts of bis- and mono-phosphorylated high mannose, non-phosphorylated high mannose and complex-type oligosaccharides. rhGAA, recombinant human acid alpha-glucosidase.
Immune response to rhGAA can interfere with clinical efficacy
Nearly all Pompe patients appear to form a humoral immune response to rhGAA ERT and develop anti-rhGAA antibodies (29,64). The most severely affected infantile Pompe patients often do not produce detectable GAA [i.e., cross-reactive immune material (CRIM)-negative] and have robust immune responses to ERT that result in very high, sustained levels of anti-rhGAA antibodies (27). Most late-onset CRIM-positive patients also form antibodies to ERT but often tolerize with a concomitant decline in antibody titres over the course of approximately 2 years (27,65,66). The impact of anti-rhGAA antibodies on clinical efficacy varies among patients. It can negate the benefits of ERT in the following ways: (I) directly, by blocking catalytic activity; or (II) indirectly, by faster clearance of ERT from circulation to reduce muscle targeting; or (III) by blocking receptor binding to prevent cellular uptake of ERT; or (IV) by any combination of the above (27,67). Different approaches—such as antibody-based and chemotherapy regimens to ablate B cells—are being tested as potential ways to address immune response to ERT, particularly for the most vulnerable CRIM-negative patients (65,68). Immune response to ERT undoubtedly complicates treatment and is an enduring problem that needs significant attention and resources to resolve in order to maximize the benefits of rhGAA ERTs and other future treatments.
The cumulative effect of all these factors greatly diminishes rhGAA biodistribution to muscles, particularly skeletal muscles. These issues can be partially mitigated using intravenous (IV) bolus administration of ERT which yields extremely high rhGAA levels in blood that saturates binding of competing pathways in liver, spleen, and other tissues and results in increased delivery of ERT to muscles in preclinical studies involving small rodent models. By comparison, IV infusion of the same dose over 4 hours yields substantially lower rhGAA concentrations in blood that do not saturate binding of competing pathways in non-target tissues and results in significant clearance of ERT prior to reaching muscles (unpublished internal data). Increasing rhGAA distribution to muscles would be highly beneficial but intravenous bolus injection of such high ERT doses in humans is likely not feasible due to safety and tolerability risks. It is therefore challenging to develop an effective dosing strategy in humans to replicate preclinical efficacy based on IV bolus dosing data from rodent studies. Allometric scaling based primarily on metabolic rate and body weight is reasonably effective for small molecule drugs with uniform broad tissue distribution (69) but not accurate for large glycoproteins such as rhGAA because of the aforementioned complexities. It should also be noted that recombinant enzymes with different N-linked carbohydrate structures—particularly incomplete structures with terminal mannose, galactose, or N-acetylglucosamine sugars—are cleared substantially faster by non-muscle tissues and result in very disparate pharmacokinetics (32,33,55) despite having the identical amino acid sequence. Understanding the pharmacokinetics of rhGAA ERT (i.e., Cmax and exposure in blood and in muscle using the intended clinical route and duration time for drug administration) are critical for developing effective dosing strategies in humans.
Future therapies
Next generation ERTs
Alglucosidase alfa is a life-saving drug that extends survival and slows disease progression, but it is not optimally effective for treating Pompe disease. There is need for development of next-generation ERTs that overcome the aforementioned obstacles to improve clinical efficacy. IV infusion of rhGAA ERT yields very low resultant ERT concentrations in the interstitial space and it is unlikely that this can be significantly improved without disrupting the integrity of the vascular system. It is therefore critical that ERT can be internalized by muscle cells at these low enzyme concentrations. Towards this goal, drug companies are developing new ways to increase binding affinities of ERT for cell surface receptors or utilize other transport mechanisms to improve cellular uptake at low enzyme concentrations.
NeoGAA (Avalglucosidase alfa; Sanofi Genzyme, Cambridge, MA, USA) is, in essence, alglucosidase alfa that was chemically modified to attach synthetic bis-phosphorylated oligosaccharides to improve binding to CI-MPR (60,61). The resultant neoGAA glyco-conjugate was shown to have significantly higher binding affinity for CI-MPR and better muscle targeting, resulting in greater glycogen reduction in Gaa KO mice as compared to alglucosidase alfa at equivalent dose (61). NeoGAA was evaluated in human clinical trials and preliminary results after 24 weeks of treatment from a phase 1/2 study showed that naïve patients treated with 20 mg/kg neoGAA increased their mean distance walked in 6-minute walk tests (6MWT) by 24.3±23.0 m over baseline while ERT-switch patients had a decline of −6.2±64.3 m (70). NeoGAA was also shown to slightly improve pulmonary function as measured by upright % predicted forced vital capacity (FVC), maximum expiratory pressure (MEP) and maximum inspiratory pressure (MIP) in naïve patients and stabilized pulmonary function in ERT-switch patients (70). More data are needed to understand the clinical impact of neoGAA, and whether it is a significant improvement over current standard of care. A phase 3 pivotal study has been initiated to evaluate the safety and efficacy of neoGAA for Pompe disease (NCT02782741).
AT-GAA (formerly known as ATB200/AT2221; Amicus Therapeutics, Cranbury, NJ, USA) is a combination product comprised of a novel rhGAA ERT (ATB200) with high M6P content that is co-administered with a small molecule pharmacological chaperone (AT2221) for stabilizing ERT to maintain enzymatic activity at neutral pH during dosing. MALDI-TOF mass spectroscopy revealed that ATB200 contains on average 1.3 moles bis-phosphorylated oligosaccharides per mole of rhGAA indicating that each ATB200 contains at least one bis-phosphorylated oligosaccharide structure for high affinity binding to CI-MPR (63). Importantly, ATB200 was shown to be internalized substantially better in muscle myoblasts than alglucosidase alfa at representative low interstitial enzyme concentrations as shown in Figure 3. The ATB200/AT2221 combination product (AT-GAA) was shown to be better targeted to skeletal muscles and more effective for clearing accumulated lysosomal glycogen in Gaa KO mice than alglucosidase alfa at equivalent dose (41) as shown in Figure 4. AT-GAA was shown to reverse muscle damage and resolve defective autophagy in majority of muscle fibers in this Pompe mouse model as shown in Figure 4C—a feat that had not been achieved with previous rhGAA ERTs or other treatments (26,41). AT-GAA was also shown to increase functional muscle strength and endurance for Gaa KO mice over a 5-month treatment period; no change or a decline was observed with alglucosidase alfa treatment under identical experimental conditions (41). AT-GAA was subsequently evaluated in phase 1/2 clinical studies and shown to be generally well tolerated with a low number of infusion-associated reactions (IARs) (71). AT-GAA improved muscle motor function as evidenced by increased 6MWT distance of 41.8±29.4 m for ambulatory naïve patients and by 23.9±52.2 m for ERT-switch patients after the 24-week treatment phase (72). Further improvements in muscle motor function were observed during the extension phase with a 54.8±34.7 m increase in 6MWT for naïve patients and 53.6±36.4 m for ERT-switch patients after 2 years (72). The magnitude of improvement is surprising, particularly in ERT-switch patients, since these patients had been on alglucosidase alfa for a mean of ~5 years prior to switching to AT-GAA and are typically observed to decline on standard-of-care ERT over this timeframe (15,73,74). AT-GAA improved respiratory function for naïve patients while generally stabilizing pulmonary function in ERT-switch patients in the same study (72). AT-GAA treatment also appeared to improve muscle fatigue and various quality-of-life parameters as monitored by patient-reported outcomes (75). AT-GAA was also evaluated in non-ambulatory ERT-switch patients and was shown to increase upper-body muscle strength in that cohort as measured by various manual muscle and quantitative muscle tests (72). The reported clinical data for AT-GAA thus far are promising but more data are needed to understand whether the observed low rate of IARs and improved muscle function are maintained in the long term. Initiation of a phase 3 pivotal study has commenced for direct comparisons of AT-GAA and alglucosidase alfa for improving muscle motor function and other endpoints in naïve and ERT-experienced late-onset Pompe patients (NCT03729362).
Figure 3.
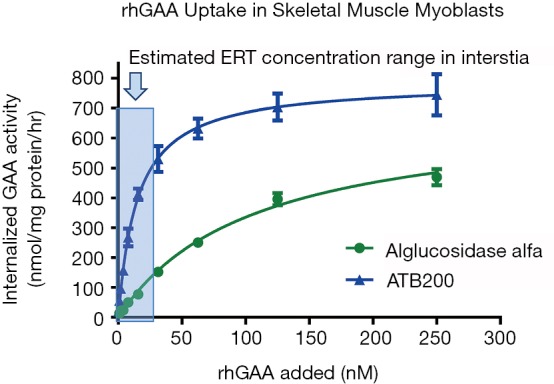
The efficiency of cellular uptake for alglucosidase alfa or ATB200 was evaluated using L6 rat skeletal muscle myoblasts. Varying concentrations of each rhGAA preparation were added to cell culture media and incubated for 16 hours. External rhGAA was inactivated with high pH buffer, cells were then washed and lysed with detergent buffer. The amount of internalized rhGAA within cells was measured by enzyme activity assays using the fluorogenic 4-MU-α-Glc substrate and normalized relative to total cellular protein. The amount of internalized rhGAA (y-axis) was graphed relative to the amount of rhGAA added (x-axis) to correlate the uptake efficiency (i.e., competency) for each rhGAA preparation. Results indicate that high alglucosidase alfa concentrations (i.e., >100 nM) are required for efficient cellular uptake in L6 myoblasts. In contrast, substantially lower ATB200 concentrations are needed for efficient cellular uptake in the same cellular assay. The extrapolated efficiency coefficient for cellular uptake (Kuptake) for ATB200 is approximately 5–15 nM and >125 nM for alglucosidase alfa. It should be noted that saturation of cellular uptake was not achieved for alglucosidase alfa with this concentration range such that an accurate Kuptake for that rhGAA ERT could not be determined. rhGAA, recombinant human acid alpha-glucosidase; ERT, enzyme replacement therapy.
Figure 4.

Approximately 16-week-old male Gaa KO mice received four biweekly intravenous (IV) administrations of vehicle, 20 mg/kg alglucosidase alfa, or 20 mg/kg ATB200/AT2221 (10 mg/kg AT2221 was administered orally 30 minutes prior to ATB200 IV injection). Tissues were collected 14 days after the last administration. (A) Glycogen levels in different skeletal muscles n=6–8 animals per group; ns: not significant (P>0.05); **, 0.001<P<0.01; ***, 0.0001<P<0.001; ****, P<0.0001; Tukey’s multiple comparison under one-way ANOVA. (B) Lamp1 (lysosome-associated membrane protein 1, lysosomal marker, upper panel) and LC3 (microtubule-associated light chain protein, autophagosomal marker, lower panel)-stained sections of skeletal muscle (quadriceps). N=4–5 animals per group (n=2 for the WT), the scale bars: 50 µm. (C) Immunostaining of single fibers from the white part of gastrocnemius with markers for lysosomes (Lamp1; green), autophagosomes (LC3; red), and nuclei (Hoechst dye; blue); the multicolored areas in the core of muscle fibers represent autophagic buildup. n=141 fibers from 4 alglucosidase alfa-treated Gaa-KO mice; n=127 fibers from 4 ATB200/AT2221-treated Gaa-KO mice, the scale bars: 20 µm.
VAL-1221 (Valerion Therapeutics, Concord, MA, USA) is a recombinant fusion protein comprised of rhGAA and a lupus anti-DNA antibody 3E10 that is believed to utilize the nucleoside transporter ENT2 for cellular uptake of ERT in a M6P-independent manner (76,77). VAL-1221 is being developed for hydrolyzing glycogen in cytoplasm and in extra-lysosomal compartments such as in autophagosomes, since 3E10 antibody is known to transport cargo proteins from the outside to cytoplasm and ultimately to nucleus (78). Cytoplasmic glycogen is encapsulated within membranous autophagic vacuoles via autophagy and requires subsequent fusion with lysosomes and GAA hydrolysis for clearing glycogen content. GAA deficiency somehow leads to defective autophagy that impedes fusion of autophagosomes with lysosomes (26,79) and thus, clearance of accumulated glycogen within autophagosomes is a provocative therapeutic approach. This presumably would require capturing both cytosolic VAL-1221 and glycogen for direct substrate degradation within autophagic vacuoles. Would the levels of cytoplasmic VAL-1221 be sufficiently high for capture and distribution in autophagosomes? Is VAL-1221 functionally active in the higher pH environments of autophagosomes and cytoplasm for hydrolyzing glycogen? It should be noted that rhGAA is an acid hydrolase that is most active in the low pH environment of lysosomes but has little to no activity at neutral pH. It will also be important to understand the impact of rhGAA-mediated glycogen hydrolysis in cytoplasm, since phosphorylase b typically performs this function and is not known to be defective in Pompe.
Gene therapies for Pompe
Whether it is the burden of frequent dosing, declining benefit of alglucosidase alfa over time, intolerability to ERT, or other factors, there is longing by the patient community and clinicians for development of a one-time curative treatment. Gene therapy is gaining significant attention as a potential cure for Pompe based on recent successes in other diseases such as spinal muscular atrophy, hemophilia B, various oncology, and ocular diseases among others (80-84). Multiple gene therapy approaches are being contemplated and evaluated for Pompe such as: ex vivo approaches using transduced bone marrow-derived stem cells to produce hGAA for cross-correction of muscles, in vivo gene therapy approaches to transduce muscle tissues for direct correction of muscles, as well as in vivo gene therapy approaches to transduce liver and other tissues for cross-correction of muscles.
AVR-RD-03 (AVROBIO, Cambridge, MA, USA) is an ex vivo gene therapy approach that employs transduced bone marrow-derived hematopoietic progenitor stem cells (HSCs) to secrete a fusion protein comprised of hGAA and an insulin-like growth factor 2 (IGF2) peptide for cross-correction of muscles. IGF2 peptide is a naturally produced peptide growth hormone in humans that has high binding affinity for a distinct domain on CI-MPR (85-87). Therefore it is being utilized for lysosomal targeting of therapeutic protein in lieu of M6P (88,89). AVR-RD-03 is currently in pre-clinical stage of development. Ex vivo gene therapy approaches have been shown to significantly change the course of a disease for the treatment of adenosine deaminase deficiency with severe combined immune deficiency (90) and metachromatic leukodystrophy, particularly when administered early prior to irreversible damage (91,92). Current ex vivo gene therapy approaches require ablation of existing host bone marrow cells via chemotherapy or antibody-based regimens and subsequent engraftment of autologous or allogeneic stem cells that have been transduced with gene therapy (93,94). Modified lentiviruses are typically used for delivery and stable integration of genome into a specific locus within genome of HSCs that is not believed to be oncogenic (95). Transduced bone marrow stem cells ultimately differentiate into various leukocytes in the circulation and tissues, and secrete therapeutic enzyme for cross-correction of distal cells. Proper ablation of bone marrow cells and engraftment of transduced HSCs are critical for maintaining a perpetual population of transduced progenitor stem cells for sustained production of therapeutic protein (96,97). Otherwise, the transduced stem cell population would decline and therefore, gene expression of the therapeutic enzyme would be lost over time.
In vivo gene therapy approaches using recombinant adeno-associated virus (rAAV)-based gene delivery are being evaluated for ability to produce therapeutic levels of hGAA for direct correction of muscles and other key target cells. Keeler and colleagues recently showed that systemic administration of AAVB1-GAA enabled efficient transduction and high-level expression of hGAA resulting in prolonged survival and robust glycogen clearance in heart, gastrocnemius, tongue, and diaphragm in adult Gaa KO mice (98). Further, AAVB1-GAA was also shown to transduce motor neurons to improve respiratory function—a key limitation with current ERT (98). AAV8-LDes (AT845; Audentes Therapeutics, San Francisco, CA, USA) is another in vivo gene therapy that utilizes a novel hybrid liver/desmin promoter that enabled broad, robust hGAA expression in liver, cardiac and skeletal muscles, and the spinal cord in the Gaatm1Rabn mouse model when administered at high doses (99). AAV8-LDes administered at doses ≥3×1013 vg/kg was shown to produce hGAA in muscles that exceeded WT mouse levels and to near-WT levels in spinal cord which normalized glycogen in these tissues after 20 weeks (99). High levels of hGAA derived from liver and presumably secreted into the circulation are believed to help induce immunotolerization as measured by reduced anti-GAA titres (99). To maximize the effectiveness of this in vivo gene therapy approach, transduction of nearly all muscle cells would likely be needed. Is this achievable in humans, and would the high AAV vector doses needed to attain this goal in humans lead to safety issues? Further, it will be important to understand if this approach is able to maintain durable hGAA expression when terminally differentiated myotubes are turned over. Conceivably, transduction of satellite cells with gene therapy could enable continued gene expression when they differentiate into myocytes and myotubes. However, satellite cells are typically quiescent and shown to be difficult to transduce with rAAV gene therapies (100).
In vivo rAAV-based gene therapies for cross-correction of muscles are also being developed and evaluated by the biopharmaceutical industry such as SPK-3006 (Spark Therapeutics, Philadelphia, PA, USA), ACTUS-101 (Actus Therapeutics, Chapel Hill, NC, USA), and yet to be named gene therapies from Sarepta Therapeutics (Cambridge, MA, USA) and Abeona Therapeutics (New York, NY, USA). The main premise of this approach is systemic delivery of rAAV gene therapies for transducing liver and a few other tissues to produce and secrete hGAA into the circulation for cross-correction of muscles. The ACTUS-101 gene therapy is also being developed for immunotolerization to minimize the impact of anti-GAA antibodies in patients receiving ERT. Tropism and transduction efficiency of target tissues are dictated largely by the serotypes of rAAV capsids (101,102) used while promoters and codon optimization can improve transgene expression (103,104). In some cases, the native human GAA signal sequence was replaced with more efficient signal sequences from other human proteins to enable better protein expression and secretion of hGAA (105). In our view, cross-corrective in vivo gene therapies function as constitutive ERT. Thus, the therapeutic enzyme derived from a gene therapy must deal with the same problems of poor biodistribution resulting in low interstitial enzyme levels and inherently inefficient phosphorylation of hGAA that limits its cellular uptake in skeletal muscles. We are not aware of any current in vivo gene therapy approach that can modulate carbohydrate processing in transduced cells to increase levels of bis-phosphorylated oligosaccharides on hGAA. Hence, it is far more likely that hGAA produced by current gene therapies would be very similar to alglucosidase alfa where only a small fraction of the total hGAA contains bis-phosphorylated oligosaccharides (59) for enabling cellular uptake at low interstitial enzyme concentrations as detailed earlier. High rAAV doses are therefore likely needed to produce substantial hGAA levels to compensate for this limitation to enable uptake and glycogen clearance in skeletal muscles of Gaa KO mice (105). Even higher rAAV doses would presumably be needed to produce comparable levels in higher species—such as non-human primates and humans—for replicating in vivo efficacy as observed in mouse models.
Concluding remarks
Alglucosidase alfa has provided undeniable benefits to Pompe patients but is not optimally effective, and unmet medical needs persist. The experience with alglucosidase alfa has led to a better understanding of Pompe disease and the challenges that need to be overcome for developing improved treatments such as next generation ERTs and gene therapies. Next generation ERTs must clear the daunting hurdles of poor distribution from the circulation to muscles, poor phosphorylation of rhGAA that reduce muscle uptake, and interference from anti-GAA antibodies. Systemic administration of ERTs also does not effectively penetrate the blood-brain barrier for treating the neuronal aspects of Pompe. Significant advances have been made in recent years towards development of next-generation ERTs such that both neoGAA and AT-GAA have advanced to the pivotal stage of clinical development. The latter was recently granted breakthrough therapy designation from the U.S. Food & Drug Administration which acknowledges preliminary clinical evidence indicating that the drug may demonstrate substantial improvement on a clinically significant endpoint over other available therapies. These developments suggest that the prospect of improved ERTs for Pompe may be within reach in the not-too-distant future.
The promise of gene therapy as a single-administration curative treatment for Pompe disease is very appealing and seemingly viable based on encouraging preclinical data. However, gene therapies for Pompe are still in a nascent stage of development where enduring challenges and questions must be addressed to realize the full potential and benefits of this approach. Can preclinical efficacy in rodent models be successfully replicated in humans using safe viral doses? How durable is transgene expression? Is gene therapy safe and efficacious in the long term? We anticipate that properly designed clinical studies would be needed to directly compare the effectiveness of gene therapy versus current standard of care to understand if this approach is significantly improved over existing approved therapies. Further, multi-year clinical studies would likely be required to demonstrate long term safety and durable gene expression—neither of which have been established for Pompe. There are also questions about the manufacturability of rAAV gene therapies at large scale to support use of high viral doses. This appears challenging at the moment. In addition to the technical challenges associated with scaling up to large-suspension cell culture processes and downstream purification, there are also major bottlenecks for obtaining needed equipment and high quality plasmids. The analytical assays to support their manufacture will also likely need improvement and standardization to ensure consistent potency of viral batches. We anticipate that substantial time, resources, and effort by both academia and industry will be needed to resolve these issues in the future. These combined factors suggest that gene therapy is not around the corner and will require significant time to fully understand their long term safety and efficacy and for overcoming the manufacturing challenges before it becomes the standard of care for Pompe.
We believe the primary role of the biopharmaceutical industry is to develop efficacious medicines and provide viable treatment options for patients and healthcare providers to effectively manage rare genetic diseases. It is unlikely that one treatment is optimal for all. For example, a patient may opt for gene therapy because of intolerability to ERT or their busy schedules cannot cope with the burden of frequent infusions. Conversely, a patient with pre-existing antibodies against viral capsid proteins may preclude a subject from receiving rAAV gene therapies. Current rAAV gene therapies would also not be appropriate for young children since their organs and tissues are actively growing such that cell turnover would lead to loss of transgene expression. In the future, it may be appropriate to combine therapies for optimal clinical outcomes. For example, one may contemplate next-generation ERT treatments for addressing peripheral symptoms and intrathecal administration of a gene therapy to address the neuronal aspects. Potentially, systemic and intrathecal administration of gene therapy may be utilized to address both. We therefore believe that there is need for continual development of more effective next generation therapies to provide treatment options for proper management of Pompe disease.
Acknowledgments
We wish to thank Dr. Nina Raben for independently testing AT-GAA in Gaa KO mice and for use of the resultant immunostaining data in this manuscript. We also greatly appreciate Dr. Raben’s helpful discussions and expert advice. We wish to thank Drs. Su Xu and Rick Hamler and their respective research teams for conducting animal studies and data analysis. We wish to thank Ms. Heather Ordover for editing and revising manuscript. We also thank Dr. Kenneth Valenzano for contributing to data interpretation, helpful suggestions, and revision of manuscript.
Supplementary
Method for quantifying rhGAA levels in tissues after intravenous bolus dosing (data in Table 1)
Gaa KO mice (~25 g) were dosed with a single intravenous (IV) bolus injection of 20 mg/kg rhGAA via tail vein and tissues were harvested 24 hours post-dosing according to approved IACUC procedures. Individual tissues were weighed and flash frozen. Each tissue sample was minced for homogeneity and approximately 50 mg was homogenized in 0.3 mL deionized H2O on ice and centrifuged at 13,000 ×g at 4 °C for 10 minutes to remove cellular debris. Cleared supernatants were then used to measure GAA levels by enzymatic activity using the fluorogenic substrate 4-methylumbeliferyl-α-glucopyranoside (4-MU-α-Glc) as previously described (Khanna et al., 2012). Total protein concentrations from individual tissue homogenates were determined using the Micro BCA method per manufacturer’s procedures (Thermo Fisher). A 4-methylumbelliferone (4-MU) standard curve ranging from 14 nM to 30 µM was run in parallel for conversion of fluorescence data to absolute GAA enzyme activity and expressed as nmol 4-MU released per mg total protein in tissue homogenate per hour (nmol/mg/hr). The measured total protein in tissue homogenate/mg wet tissue was used to convert the amount of rhGAA from tissue homogenate to the amount of rhGAA in 1 mg of wet tissue. The total amount of rhGAA in entire tissue was then calculated from the wet weight of entire tissue. The amount of rhGAA in each tissue was then divided by the starting amount of rhGAA in dosing solution [1.75×105 nmol 4-MU released/hr (for a 25 g mouse)] and expressed as percent of rhGAA dose.
Footnotes
Conflicts of Interest: Authors are employees and shareholders of Amicus Therapeutics.
References
- 1.van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet 2008;372:1342-53. 10.1016/S0140-6736(08)61555-X [DOI] [PubMed] [Google Scholar]
- 2.Hirschhorn R, Reuser AJ. The Metabolic and Molecular Basis of Inherited Disease. New York, NY: McGraw-Hill, 2001:3389-420. [Google Scholar]
- 3.Hers HG. Alpha-glucosidase deficiency in generalized storage disease (Pompe’s disease). Biochem J 1963;86:11-6. 10.1042/bj0860011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332-40. 10.1542/peds.112.2.332 [DOI] [PubMed] [Google Scholar]
- 5.Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006;148:671-6. 10.1016/j.jpeds.2005.11.033 [DOI] [PubMed] [Google Scholar]
- 6.Reuser AJ, Kroos MA, Hermans MM, et al. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995;3:S61-9. 10.1002/mus.880181414 [DOI] [PubMed] [Google Scholar]
- 7.Parenti G, Andria G. Pompe disease: from new views on pathophysiology to innovative therapeutic strategies. Curr Pharm Biotechnol 2011;12:902-15. 10.2174/138920111795542606 [DOI] [PubMed] [Google Scholar]
- 8.Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr 2006;149:89-97. 10.1016/j.jpeds.2006.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winkel LP, Van den Hout JM, Kamphoven JH, et al. Enzyme replacement therapy in late-onset Pompe's disease: a three-year follow-up. Ann Neurol 2004;55:495-502. 10.1002/ana.20019 [DOI] [PubMed] [Google Scholar]
- 10.Van den Hout JM, Kamphoven JH, Winkel LP, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004;113:e448-57. 10.1542/peds.113.5.e448 [DOI] [PubMed] [Google Scholar]
- 11.Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 2009;11:210-9. 10.1097/GIM.0b013e31819d0996 [DOI] [PubMed] [Google Scholar]
- 12.Chien YH, Lee NC, Thurberg BL, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 2009;124:e1116-25. 10.1542/peds.2008-3667 [DOI] [PubMed] [Google Scholar]
- 13.Rossi M, Parenti G, Della CR, et al. Long-term enzyme replacement therapy for pompe disease with recombinant human alpha-glucosidase derived from chinese hamster ovary cells. J Child Neurol 2007;22:565-73. 10.1177/0883073807302598 [DOI] [PubMed] [Google Scholar]
- 14.van der Ploeg AT, Barohn R, Carlson L, et al. Open-label extension study following the Late-Onset Treatment Study (LOTS) of alglucosidase alfa. Mol Genet Metab 2012;107:456-61. 10.1016/j.ymgme.2012.09.015 [DOI] [PubMed] [Google Scholar]
- 15.Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013;260:951-9. 10.1007/s00415-012-6636-x [DOI] [PubMed] [Google Scholar]
- 16.Prater SN, Patel TT, Buckley AF, et al. Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J Rare Dis 2013;8:90-101. 10.1186/1750-1172-8-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hahn A, Praetorius S, Karabul N, et al. Outcome of Patients with Classical Infantile Pompe Disease Receiving Enzyme Replacement Therapy in Germany. JIMD Rep 2015;20:65-75. 10.1007/8904_2014_392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 2017;264:621-30. 10.1007/s00415-016-8219-8 [DOI] [PubMed] [Google Scholar]
- 19.Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018;15:928-42. 10.1007/s13311-018-0655-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med 2012;14:800-10. 10.1038/gim.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Case LE, Beckemeyer AA, Kishnani PS. Infantile Pompe disease on ERT: update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med Genet 2012;160C:69-79. 10.1002/ajmg.c.31321 [DOI] [PubMed] [Google Scholar]
- 22.Peng SS, Hwu WL, Lee NC, et al. Slow, progressive myopathy in neonatally treated patients with infantile-onset Pompe disease: a muscle magnetic resonance imaging study. Orphanet J Rare Dis 2016;11:63. 10.1186/s13023-016-0446-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raben N, Roberts A, Plotz PH. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol 2007;26:45-8. [PMC free article] [PubMed] [Google Scholar]
- 24.Raben N, Takikita S, Pittis MG, et al. Deconstructing Pompe disease by analyzing single muscle fibers. Autophagy 2007;3:546-52. 10.4161/auto.4591 [DOI] [PubMed] [Google Scholar]
- 25.Raben N, Fukuda T, Gilbert AL, et al. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol Ther 2005;11:48-56. 10.1016/j.ymthe.2004.09.017 [DOI] [PubMed] [Google Scholar]
- 26.Raben N, Ralston E, Chien YH, et al. Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy. Mol Genet Metab 2010;101:324-31. 10.1016/j.ymgme.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729-36. 10.1097/GIM.0b013e3182174703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Vries JM, van der Beek NA, Kroos MA, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab 2010;101:338-45. 10.1016/j.ymgme.2010.08.009 [DOI] [PubMed] [Google Scholar]
- 29.Patel TT, Banugaria SG, Case LE, et al. The impact of antibodies in late-onset Pompe disease: a case series and literature review. Mol Genet Metab 2012;106:301-9. 10.1016/j.ymgme.2012.04.027 [DOI] [PubMed] [Google Scholar]
- 30.de Vries JM, Kuperus E, Hoogeveen-Westerveld M, et al. Pompe disease in adulthood: effects of antibody formation on enzyme replacement therapy. Genet Med 2017;19:90-7. 10.1038/gim.2016.70 [DOI] [PubMed] [Google Scholar]
- 31.Zhang XS, Brondyk W, Lyndon JT, et al. Biotherapeutic target or sink: analysis of the macrophage mannose receptor tissue distribution in murine models of lysosomal storage diseases. J Inherit Metab Dis 2011;34:795-809. 10.1007/s10545-011-9285-9 [DOI] [PubMed] [Google Scholar]
- 32.Park EI, Manzella SM, Baenziger JU. Rapid clearance of sialylated glycoproteins by the asialoglycoprotein receptor. J Biol Chem 2003;278:4597-602. 10.1074/jbc.M210612200 [DOI] [PubMed] [Google Scholar]
- 33.Vugmeyster Y, Xu X, Theil FP, et al. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J Biol Chem 2012;3:73-92. 10.4331/wjbc.v3.i4.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SJ, Evers S, Roeder D, et al. Mannose Receptor-Mediated Regulation of Serum Glycoprotein Homeostasis. Science 2002;295:1898-901. 10.1126/science.1069540 [DOI] [PubMed] [Google Scholar]
- 35.Joyner MJ, Casey DP. Regulation of increased blood flow (hyperemia) to muscles during exercise: a hierarchy of competing physiological needs. Physiol Rev 2015;95:549-601. 10.1152/physrev.00035.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuma P, Hubbard AL. Transcytosis: crossing cellular barriers. Physiol Rev 2003;83:871-932. 10.1152/physrev.00001.2003 [DOI] [PubMed] [Google Scholar]
- 37.Ghitescu L, Bendayan M. Transendothelial transport of serum albumin: a quantitative immunohistochemical study. J Cell Biol 1992;117:745-55. 10.1083/jcb.117.4.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghinea N, Mai TV, Groyer MT, et al. How protein hormones reach their target cells. Receptor-mediated transcytosis of hCG through endothelial cells. J Cell Biol 1994;125:87-97. 10.1083/jcb.125.1.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simionescu M, Popov D, Sima A. Endothelial transcytosis in health and disease. Cell Tissue Res 2009;335:27-40. 10.1007/s00441-008-0688-3 [DOI] [PubMed] [Google Scholar]
- 40.Rodman JS, Mercer RW, Stahl PD. Endocytosis and Transcytosis. Curr Opin Cell Biol 1990;2:664-72 10.1016/0955-0674(90)90108-Q [DOI] [PubMed] [Google Scholar]
- 41.Xu S, Lun Y, Frascella M, et al. Improved efficacy of a next generation ERT in murine Pompe disease. JCI Insight 2019;4:e125358. 10.1172/jci.insight.125358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramadass M, Ghebrehiwet B, Kew RR. Enhanced recognition of plasma proteins in a non-native state by complement C3b. A possible clearance mechanism for damaged proteins in blood. Mol Immunol 2015;64:55-62. 10.1016/j.molimm.2014.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parenti G, Fecarotta S, la Marca G, et al. A chaperone enhances blood alpha-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 2014;22:2004-12. 10.1038/mt.2014.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porto C, Cardone M, Fontana F, et al. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther 2009;17:964-71. 10.1038/mt.2009.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khanna R, Flanagan JJ, Feng J, et al. The Pharmacological Chaperone AT2220 Increases Recombinant Human Acid alpha-Glucosidase Uptake and Glycogen Reduction in a Mouse Model of Pompe Disease. PLoS One 2012;7:e40776. 10.1371/journal.pone.0040776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khanna R, Powe AC, Jr, Lun Y, et al. The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS One 2014;9:e102092. 10.1371/journal.pone.0102092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reuser AJJ, Kroos MA, Ponne NJ, et al. Uptake and stability of human and bovine acid α-glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp Cell Res 1984;155:178-89. 10.1016/0014-4827(84)90779-1 [DOI] [PubMed] [Google Scholar]
- 48.Van der Ploeg AT, Loonen MCM, Bolhuis BA, et al. Receptor-mediated uptake of acid α-glucosidase corrects lysosomal glycogen storage in cultured skeletal muscle. Pediatr Res 1988;24:90-4 10.1203/00006450-198807000-00021 [DOI] [PubMed] [Google Scholar]
- 49.Van der Ploeg AT, Kroos MA, Willemsen RM, et al. Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice. J Clin Invest 1991;87:513-8. 10.1172/JCI115025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kornfeld S. Trafficking of lysosomal enzymes. FASEB J 1987;1:462-8. 10.1096/fasebj.1.6.3315809 [DOI] [PubMed] [Google Scholar]
- 51.Dahms NM, Lobel P, Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem 1989;264:12115-8. [PubMed] [Google Scholar]
- 52.Dahms NM, Kornfeld S. The cation-dependent mannose 6-phosphate receptor. Structural requirements for mannose 6-phosphate binding and oligomerization. J Biol Chem 1989;264:11458-67. [PubMed] [Google Scholar]
- 53.Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochim Biophys Acta 2009;1793:605-14. 10.1016/j.bbamcr.2008.10.016 [DOI] [PubMed] [Google Scholar]
- 54.Wisselaar HA, Kroos MA, Hermans MM, et al. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J Biol Chem 1993;268:2223-31. [PubMed] [Google Scholar]
- 55.McVie-Wylie AJ, Lee KL, Qiu H, et al. Biochemical and pharmacological characterization of different recombinant acid α-glucosidase preparations evaluated for the treatment of Pompe disease. Mol Genet Metab 2008;94:448-55. 10.1016/j.ymgme.2008.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Ann Rev Biochem 1985;54:631-64. 10.1146/annurev.bi.54.070185.003215 [DOI] [PubMed] [Google Scholar]
- 57.Rohrer J, Kornfeld S. Lysosomal hydrolase mannose 6-phosphate uncovering enzyme resides in the trans-Golgi network. Mol Biol Cell 2001;12:1623-31. 10.1091/mbc.12.6.1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu L, Lee WS, Doray B., et al. Enginering of GlcNAc-1-phosphotransferase for production of highly phosphorylated lysosomal enzymes for enzyme replacement therapy. Mol Ther Methods Clin Dev 2017;5:59-65. 10.1016/j.omtm.2017.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park H, Kim J, Lee YK, et al. Four unreported types of glycans containing mannose-6-phosphate are heterogeneously attached at three sites (including newly found Asn 233) to recombinant human acid alpha-glucosidase that is the only approved treatment for Pompe disease. Biochem Biophys Res Commun 2018;495:2418-24. 10.1016/j.bbrc.2017.12.101 [DOI] [PubMed] [Google Scholar]
- 60.Zhu Y, Li X, McVie-Wylie A, et al. Carbohydrate-remodelled acid alpha-glucosidase with higher affinity for the cation-independent mannose 6-phosphate receptor demonstrates improved delivery to muscles of Pompe mice. Biochem J 2005;389:619-28. 10.1042/BJ20050364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther 2009;17:954-63. 10.1038/mt.2009.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tong PY, Gregory W, Kornfeld S. Ligand interactions of the cation-independent mannose 6-phosphate receptor. The stoichiometry of mannose 6-phosphate binding. J Biol Chem 1989;264:7962-69. [PubMed] [Google Scholar]
- 63.Gotschall R, Xu S, Lun Y, et al. Novel recombinant human acid α-glucosidase with optimal glycosylation is significantly better than standard of care enzyme replacement for glycogen clearance in skeletal muscles of GAA knock-out mice. Mol Genet Metab 2015;114:S49 10.1016/j.ymgme.2014.12.096 [DOI] [Google Scholar]
- 64.de Vries JM, van der Beek N. AME, Kroos MA, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab 2010;101:338-45. 10.1016/j.ymgme.2010.08.009 [DOI] [PubMed] [Google Scholar]
- 65.Kazi ZB, Desai AK, Berrier KL, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight 2017;2:e94328. 10.1172/jci.insight.94328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masat E, Laforet P, De Antonio M, et al. Long-term exposure to Myozyme results in a decrease of anti-drug antibodies in late-onset Pompe patients. Sci Rep 2016;6:36182. 10.1038/srep36182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, et al. High Sustained Antibody Titers in Patients with Classic Infantile Pompe Disease Following Immunomodulation at Start of Enzyme Replacement Therapy. J Pediatr 2018;195:236-43.e3. 10.1016/j.jpeds.2017.11.046 [DOI] [PubMed] [Google Scholar]
- 68.Banugaria SG, Prater SN, McGann JK, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: lessons learned from Pompe disease. Genet Med 2013;15:123-31. 10.1038/gim.2012.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27-31. 10.4103/0976-0105.177703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pena LD, Barohn R, Byrne B, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onset Pompe disease: A phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord 2019;29:167-86. 10.1016/j.nmd.2018.12.004 [DOI] [PubMed] [Google Scholar]
- 71.Clemens P, Mozaffar T, Schoser B, et al. Safety and efficacy of advanced and targeted acid α-glucosidase (AT-GAA)(ATB200/AT2221) in ERT-switch nonambulatory patients with Pompe disease: preliminary results from the ATB200-02 trial. Mol Genet Metab 2019;126:S40-1. 10.1016/j.ymgme.2018.12.084 [DOI] [Google Scholar]
- 72.Kishnani P, Schoser B, Bratkovic D, et al. First-in-human study of advanced and targeted acid α-glucosidase (AT-GAA) (ATB200/AT2221) in patients with Pompe disease: preliminary functional assessment results from the ATB200-02 trial. Mol Genet Metab 2019;126:S86 10.1016/j.ymgme.2018.12.212 [DOI] [Google Scholar]
- 73.Wyatt K, Henley W, Anderson L, et al. The effectiveness and cost-effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol Assess 2012;16:1-543. 10.3310/hta16390 [DOI] [PubMed] [Google Scholar]
- 74.van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol 2017;24:768-e31. 10.1111/ene.13285 [DOI] [PubMed] [Google Scholar]
- 75.Schoser B, Bratkovic D, Byrne B, et al. Preliminary patient-reported outcomes and safety of advanced and targeted acid α-glucosidase (AT-GAA (ATB200/AT2221) in patients with Pompe disease from the ATB200-02 trial. Mol Genet Metab 2019;126:S132-3. 10.1016/j.ymgme.2018.12.340 [DOI] [Google Scholar]
- 76.Yi H, Sun T, Armstrong D, et al. Antibody-mediated enzyme replacement therapy targeting both lysosomal and cytoplasmic glycogen in Pompe disease. J Mol Med (Berl) 2017;95:513-21. 10.1007/s00109-017-1505-9 [DOI] [PubMed] [Google Scholar]
- 77.Weisbart RH, Gera JF, Chan G, et al. A cell-penetrating bispecific antibody for therapeutic regulation of intracellular targets. Mol Cancer Ther 2012;11:2169-73. 10.1158/1535-7163.MCT-12-0476-T [DOI] [PubMed] [Google Scholar]
- 78.Weisbart RH, Chan G, Jordaan G, et al. DNA-dependent targeting of cell nuclei by a lupus autoantibody. Sci Rep 2015;5:12022. 10.1038/srep12022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raben N, Wong A, Ralston E, et al. Autophagy and mitochondria in Pompe disease: Nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet 2012;160C:13-21. 10.1002/ajmg.c.31317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hardcastle N, Boulis NM, Federici T. AAV gene delivery to the spinal cord: serotypes, methods, candidate diseases, and clinical trials. Expert Opin Biol Ther 2018;18:293-307. 10.1080/14712598.2018.1416089 [DOI] [PubMed] [Google Scholar]
- 81.Lillicrap D. FIX it in one go: enhanced Factor IX gene therapy for hemophilia B. Cell 2017;171:1478-80. 10.1016/j.cell.2017.11.049 [DOI] [PubMed] [Google Scholar]
- 82.Schambach A, Morgan M. Retroviral Vectors for Cancer Gene Therapy. Recent Results Cancer Res 2016;209:17-35. 10.1007/978-3-319-42934-2_2 [DOI] [PubMed] [Google Scholar]
- 83.Rodrigues GA, Shalaev E, Karami TK. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm Res 2018;36:29. 10.1007/s11095-018-2554-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov 2013;3:388-98. 10.1158/2159-8290.CD-12-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tong PY, Tollefsen SE, Kornfeld S. The cation-independent mannose 6-phosphate receptor binds insulin-like growth factor II. J Biol Chem 1988;263:2585-8. [PubMed] [Google Scholar]
- 86.Kornfeld S. Structure and function of the mannose 6-phosphate/insulin-like growth factor II receptors. Ann Rev Biochem 1992;61:307-30. 10.1146/annurev.bi.61.070192.001515 [DOI] [PubMed] [Google Scholar]
- 87.Schmidt B, Kiecke-Siemsen C, Waheed A, et al. Localization of the Insulin-like Growth Factor II Binding Site to Amino Acids 1508–1566 in Repeat 11 of the Mannose 6-Phosphate/Insulin-like Growth Factor II Receptor. J Biol Chem 1995;270:14975-82. 10.1074/jbc.270.25.14975 [DOI] [PubMed] [Google Scholar]
- 88.Maga JA, Zhou J, Kambampati R, et al. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in Pompe mice. J Biol Chem 2013;288:1428-38. 10.1074/jbc.M112.438663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Peng J, Dalton J, Butt M, et al. Reveglucosidase alfa, an IGF2-tagged rhAcid α-glucosidase improves respiratory functional parameters in a murine model of Pompe disease. J Pharmacol Exp Ther 2017;360:313-23. 10.1124/jpet.116.235952 [DOI] [PubMed] [Google Scholar]
- 90.Aiuti A, Roncarolo MG, Naldini L. Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med 2017;9:737-40. 10.15252/emmm.201707573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013;341:1233158. 10.1126/science.1233158 [DOI] [PubMed] [Google Scholar]
- 92.Sessa M, Lorioli L, Fumagalli F, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016;388:476-87. 10.1016/S0140-6736(16)30374-9 [DOI] [PubMed] [Google Scholar]
- 93.Gowing G, Svendsen S, Svendsen CS. Chapter 4 - Ex vivo gene therapy for the treatment of neurological disorders. Prog Brain Res 2017;230:99-132. 10.1016/bs.pbr.2016.11.003 [DOI] [PubMed] [Google Scholar]
- 94.Brendel C, Rothe M, Santilli G, et al. Non-Clinical Efficacy and Safety Studies on G1XCGD, a Lentiviral Vector for Ex Vivo Gene Therapy of X-Linked Chronic Granulomatous Disease. Hum Gene Ther Clin Dev 2018;29:69-79. 10.1089/humc.2017.245 [DOI] [PubMed] [Google Scholar]
- 95.Montini E, Cesana D, Schmidt M, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol 2006;24:687-96. 10.1038/nbt1216 [DOI] [PubMed] [Google Scholar]
- 96.Eichler F, Duncan C, Musolino PL, et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N Engl J Med 2017;377:1630-8. 10.1056/NEJMoa1700554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morris EC, Fox T, Chakraverty R, et al. Gene therapy for Wiskott-Aldrich in a severely affected adult. Blood 2017;130:1327-35. 10.1182/blood-2017-04-777136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keeler AM, Zieger M, Todeasa SH, et al. Systemic Delivery of AAVB1-GAA Clears Glycogen and Prolongs Survival in a Mouse Model of Pompe Disease. Hum Gene Ther 2019;30:57-68. 10.1089/hum.2018.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heffner G, James L, Chaivorapol C, et al. A novel hybrid promote directing AAV-mediated expression of acid alpha-glucosidase to liver, muscle and CNS yields optimized outcomes in a mouse model of Pompe disease. Neuromuscular Disord 2018;28:S135-6. 10.1016/j.nmd.2018.06.398 [DOI] [Google Scholar]
- 100.Arnett ALH, Konieczny P, Ramos JN, et al. Adeno-associated viruses do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev 2014;1:14038. 10.1038/mtm.2014.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meyer K, Ferraiuolo L, Schmelzer L, et al. Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther 2015;23:477-87. 10.1038/mt.2014.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol 2016;21:75-80. 10.1016/j.coviro.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Powell SK, Rivera-Soto R, Gray SJ. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med 2015;19:49-57. [PMC free article] [PubMed] [Google Scholar]
- 104.Colella P, Sellier P, Costa Verdera H, et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol Ther Methods Clin Dev 2018;12:85-101. 10.1016/j.omtm.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Puzzo F, Colella P, Biferi MG, et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci Transl Med 2017;9:eaam6375. [DOI] [PMC free article] [PubMed]