

Abstract
Background
Pompe disease is a neuromuscular disease caused by a deficiency of lysosomal acid alpha-glucosidase (GAA) which degrades glycogen, resulting in progressive accumulation of lysosomal glycogen, lysosomal swelling and rupture. In addition, mitochondrial abnormalities have been frequently observed in muscle biopsy specimens of Pompe patients. Enzyme replacement therapy (ERT) using alglucosidase alfa, a recombinant human GAA, is so far the only available therapy. We evaluated glycolysis and basal respiration in primary human myoblasts from patients with Pompe disease and in mouse myoblasts from GAA knockout mice before and after alglucosidase alfa treatment.
Methods
We tested patient-derived primary human myoblasts and immortalized GAA-/- mouse myoblasts for GAA activity, glycolytic activity, and mitochondrial respiration before and after alglucosidase alfa treatment using enzyme activity assays and SeaHorse measurements.
Results
A significant reduction in glycolysis (30%) and in mitochondrial respiration (50%) was observed in both, human and mouse GAA-deficient myoblasts. Treatment with alglucosidase alfa resulted in partial recovery of both metabolic pathways with some variability in human myoblasts.
Conclusions
Future assessments of treatment efficacy should include screening for the metabolic effects on both glycolysis and mitochondrial respiration in order to obtain a better read-out of the cellular energy metabolism.
Keywords: Pompe disease, mitochondria, metabolic measurement, enzyme replacement therapy (ERT)
Introduction
Pompe disease (Glycogen storage disease type II; OMIM #232300) is caused by homozygous or compound heterozygous mutations of the GAA gene resulting in a deficiency of lysosomal acid alpha-glucosidase (GAA) (1,2). GAA degrades lysosomal-bound glycogen and its deficiency causes lysosomal glycogen accumulation in different tissues. These glycogen accumulations result in the swelling and rupture of lysosomes and subsequently cell damage, organ dysfunction, and metabolic crisis (3).
Clinically Pompe disease can be distinguished in the severe infantile form (IOPD) with cardiomyopathy and muscular hypotonia and the later onset form (LOPD) with a predominant involvement of skeletal muscles (4). Since 2006 an enzyme replacement therapy (ERT) using alglucosidase alfa, a recombinant human GAA (rhGAA) carrying an M-6-P glycan, is available for patients. The uptake of alglucosidase alfa occurs via the cation independent mannose 6-phosphate (M6P) receptor followed by delivery of the enzyme to the lysosome by receptor-mediated endocytosis (5). This therapy shows a remarkable amelioration of lysosomal glycogen burden in cardiac muscle but has less effect in other tissues including skeletal muscle. While walking distance and respiratory function are stabilized or partially improve on therapy, weakness of axial skeletal muscles has been shown to persist or decline further (6). Additionally, a high variability of individual response to ERT can be observed in treated patients (7-9).
These limitations of the current ERT make further improvement necessary. Despite the reasonably well understood pathomechanism, many open questions remain. One such a question is the involvement of mitochondria in the disease pathology; morphologically abnormal mitochondria are frequently observed in patients muscle biopsies. These include larger than normal mitochondria and granular or paracrystalline inclusions (10-13). In addition, recent data indicate the presence of bioenergetically compromised mitochondria in GAA-deficient mouse myoblasts (14).
Here we investigate the mitochondrial respiration and glycolysis of primary human myoblasts from LOPD patients before and after alglucosidase alfa treatment in comparison to human myoblast controls using the SeaHorse technology; furthermore, we verified the results using myoblasts derived from a GAA-/- mouse model (15).
Methods
Culture of primary skeletal muscle cell lines
Primary human skeletal muscle cells (myoblasts) from 4 genetically proven patients with late-onset Pompe disease and 2 healthy human subjects were obtained from Muscle Tissue Culture Collection (MTCC) of the Friedrich Baur Institute (Department of Neurology, Ludwig-Maximilians-University, Munich, Germany). All human myoblasts are primary cells (not immortalized) that are capable to differentiate into myotubes. All control and patient materials were obtained with written informed consent of the donor. Ethical approval for this study was obtained from the ethical review committee at the Ludwig-Maximilians-University, Munich, Germany (IRB-No. reference 45-14).
Primary human myoblasts were cultured in Skeletal Muscle Growth Medium supplemented with SkMC Supplement (PELOBiotech), GlutaMax and 40 U/mL Penicillin, 0.04 mg/mL Streptomycin at 37 °C with 5% CO2. Myoblasts were kept from reaching confluence to avoid differentiation. Passage numbers were matched for controls and patients cells for the respective experiments; throughout all experiments passage numbers 8 to 10 have been used.
Culture of immortalized mouse cells
Immortalized GAA-/- mouse myoblasts cell line [clone 6; derived from a GAA-/- mouse (15)] were kindly provided by Nina Raben. The myoblasts were cultured in DMEM supplemented with 20% FCS, 10% Horse serum, GlutaMax, 1% Chick embryo extract, IFN-γ and 40 U/mL penicillin, 0.04 mg/mL Streptomycin at 33 °C with 5% CO2.
GAA uptake assay
Alglucosidase alfa (10 µg/mL) or Na-phosphate-buffer pH 6.2 (25 mM; enzyme diluent) as a negative control was added to myoblasts, grown to 70% confluence. The cells were incubated for 24 h at 37 °C with 5% CO2. Experiments were done in triplicates.
Preparation of cell lysates
After 24 hrs of incubation the medium was aspirated, cells were washed with 1× PBS twice, trypsinized and collected via centrifugation at 1,000 g at room temperature. The pellet was resuspended in 1 mL of 1× PBS followed by centrifugation for 10 min, at 16,100 g, 4 °C. PBS was aspirated completely, and the pellet was resuspended in 100 µL of H2O, sonicated (Sonopuls ultrasonic homogenizer HD2070 with sonotrode MS73, Bandelin; settings: 30 sec; power 40 sec; 5 cycles), and centrifuged for 10 min, at 16,100 g, 4 °C. Supernatant was collected in a fresh tube and protein concentration was determined via Nanodrop 1000. Concentration of cell lysate was adjusted to 4 µg/µL.
GAA activity assay
For GAA activity assay (on black 96 well plates), 20 µL of 4 µg/µL cell lysate/standards were used in triplicates. Eighty µL of reaction buffer (0.5 mM 4-methylumbelliferyl alpha-D-glucopyranoside, 56 mM citric acid, 88 mM Na2HPO4, 0.4% BSA) were added to the samples, mixed for 10 sec at 900 rpm, and incubated for 60 min at 37 °C. Reaction was stopped with Stop buffer (0.1 M glycine, 0.1 M NaOH). Measurements were performed using Tecan Infinite plate reader (orbital mixing step for 5 sec, amplitude 1.5 mm, excitation: 360 nm, emission: 450 nm, gain: 50).
Real time metabolic measurements
Metabolic pathways were assessed using the Seahorse XFp Extracellular Flux Analyzer (Seahorse Bioscience). Myoblasts were seeded in XFp Cell Culture Miniplates (103025-100, Seahorse Bioscience) at a density of 1.5×104 cells per well. Alglucosidase alfa (10 µg/mL) or 25 mM Na-Phosphate-buffer pH 6.2 (enzyme diluent; negative control) was added, and the cells were incubated overnight.
To measure glycolytic function, cells were incubated in unbuffered Basal Assay Medium (Seahorse) pH 7.4 supplemented with 2 mM glutamine at 37 °C without CO2 for 1 h before the assay. Sequential injection of glucose (10 mM), oligomycin (1.0 µM) and 2-deoxy-D-glucose (50 mM) measures glycolysis, glycolytic capacity, glycolytic reserve, and extracellular acidification rate (ECAR).
To measure mitochondrial function, cells were incubated in unbuffered Basal Assay Medium (Seahorse) pH 7.4 supplemented with 2 mM glutamine, 1mM pyruvate and 10 mM glucose at 37 °C without CO2 for 1 h before the assay. Sequential injection of oligomycin (1.0 µM), FCCP (1.0 µM) and rotenone/antimycin A (0.5 µM) measures ATP-linked respiration, maximal respiration, and non-mitochondrial respiration monitoring oxygen consumption rate (OCR). Proton leak and spare respiratory capacity can then be calculated comparing these parameters with basal respiration.
At least four measurements were done per sample; analysis of the data was performed well-wise and data were normalized to the number of cells.
PAS-staining
Cells were fixed with 5% glacial acetic acid in 96% EtOH, rinsed for 1 min in slowly running tap water, and immersed in Periodic Acid solution for 5 min at room temperature. Afterwards, coverslips were rinsed several times with distilled water and immersed in Schiff’s reagent for 15 minutes at room temperature. After washing coverslips in running tap water for 5 minutes, cells were counterstained in hematoxylin solution for 90 sec, and rinsed in running tap water for 5 minutes. Fluorescent mounting medium (DAKO) was used for mounting.
Ultrastructural analysis of human muscle biopsy specimens
Muscle tissue was fixed in ice-cold glutaraldehyde 6.25% buffered with 0.1 M Soerensen phosphate pH 7.3. After thorough rinsing in buffer, samples were fixed in 2% osmium tetroxide. After rapid dehydration in graded series of acetone, tissue blocks were embedded in Epon. Thin sections of the embedded blocks were stained with uranyl acetate and lead citrate and examined by transmission electron microscopy (Philips, EM420, Hamburg, Germany).
Results
Electron microscopy
Electron microscopy of a female LODP patient, aged 58 years, shows aggregated mitochondria with paracrystalline inclusions (parking lot type 1, Figure 1).
Figure 1.

Electron microscopy of mitochondrial alteration in a late-onset Pompe disease. Biopsy was taken from the quadriceps femoris muscle of a 58-year-old female Pompe patient. At a 6,800× magnification paracrystalline inclusions in mitochondria (red arrows) are detectable.
GAA-activity and real time metabolic measurements in myoblasts from Pompe disease patients
To assess the level of GAA activity in our human primary skeletal myoblasts, the cells were harvested by trypsinization, lysed, and the enzyme activity was measured. The GAA activity in all four patients samples was 40% or less compared to the average of the controls (Figure 2A).
Figure 2.
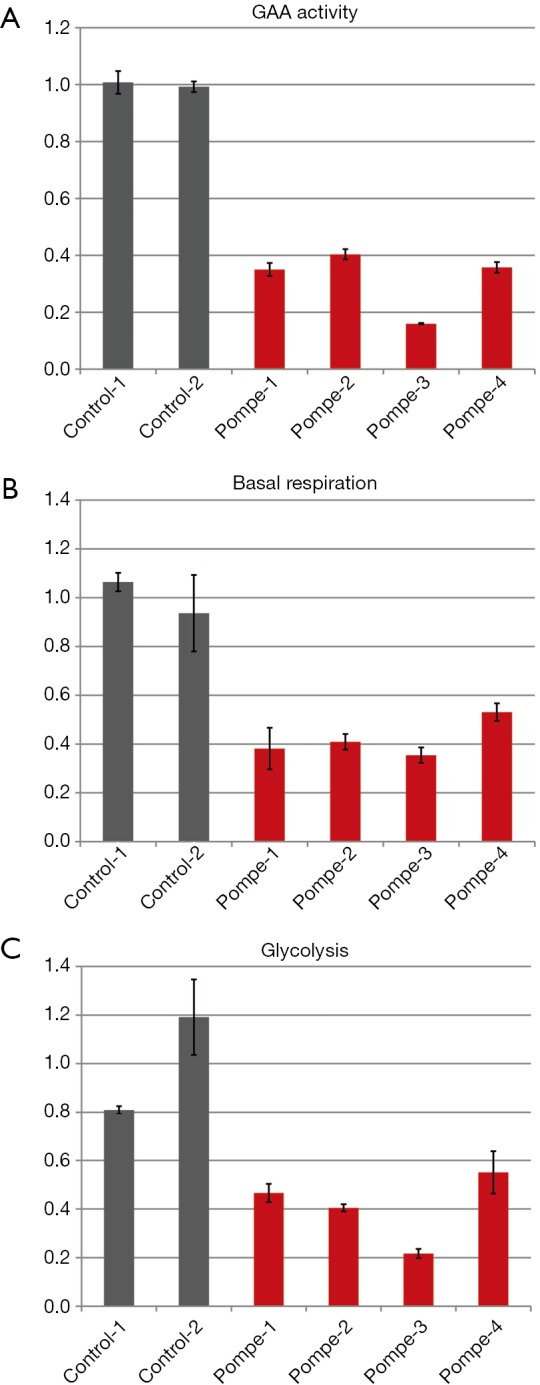
GAA activity and metabolism of primary human myoblasts of a late-onset Pompe patient compared to non-disease human control specimen. (A) GAA activity of Pompe myoblasts (red) compared to controls (grey); in Pompe patients a 15% to 40% GAA activity compared to controls is displayed. (B) Basal respiration of Pompe myoblasts (red) compared to controls (grey) is declined by 30 to 60%; and (C) glycolysis of Pompe myoblasts (red) compared to controls (grey) shows a up to 70% lower level. All graphs with standard deviation and normalized to the mean of controls.
The basal respiration of control and patient myoblasts was evaluated using the Mito Stress Test Kit. Basal respiration is representing the cellular energetic demand under baseline conditions. The two control samples showed some variability; the sample means differed by approximately 20 percent. However, all patient myoblasts had a reduced basal respiration of about 50% or less compared to the controls (Figure 2B).
Glycolysis of the control and patient myoblasts was tested using the Glycolysis Stress Test Kit. Glycolysis is the process of converting glucose to pyruvate. The XF Glycolysis Stress Test measures glycolysis based on the extracellular acidification rate (ECAR) reached after the addition of saturating amounts of glucose. Again, the two controls showed some variability—a higher basal respiration in control 2 was associated with slightly lower glycolysis compared to control 1 but the difference was not dramatic. In contrast, all patients samples did show reduced glycolysis of about 50% or less compared to the mean of the controls (Figure 2C).
GAA-activity and real time metabolic measurements in GAA-/- mouse myoblasts
To verify the results from primary human myoblasts we repeated the experiments in immortalized GAA-/- mouse myoblasts. GAA-activity in these cells was less than 5% of that in immortalized wild type mouse myoblasts (Figure 3A).
Figure 3.

GAA activity and metabolism of immortalized GAA-/- mouse myoblasts compared to immortalized mouse wild type myoblasts. (A) GAA activity of GAA-/- mouse myoblasts (red) compared to wild type (grey) shows an up to 90% decline; (B) basal respiration of GAA-/- mouse myoblasts (red) compared to wild type (grey) displays a 50% reduction of activity; and (C) glycolysis of GAA-/- mouse myoblasts (red) compared to wild type (grey) reveals a 70% reduction. All graphs with standard deviation and normalized to wild type.
As in human primary myoblasts, basal respiration and glycolysis were reduced in immortalized GAA-/- mouse myoblasts by 50% and 30% respectively compared to wild type (Figure 3B,C). The glycolysis of the GAA-/- mouse myoblasts was reduced to ~30% compared to wild type.
The effect of alglucosidase alfa in human myoblasts from Pompe disease patients
Next, we analyzed the effect of alglucosidase alfa treatment on basal respiration and glycolysis. Primary patient myoblasts (Figure 4A) were treated with alglucosidase alfa (final concentration: 10 µg/mL) or buffer (untreated) for 24 hrs. After 24 hrs, cells were harvested by trypsinization, lysed, and the enzyme activity was measured. An increased GAA activity ranging from ~180% to ~950% relative to the control levels was detected in myoblasts from all patients (Figure 4B). This increase was associated with a marked decrease in glycogen level in all patient myoblasts as shown by PAS staining (Figure 4C).
Figure 4.

Alglucosidase alfa uptake and metabolism in primary human myoblasts of Pompe patients. (A) Summary of patient information on GAA-mutations, sex, age at biopsy, and the muscle from which the biopsy was taken. (B) GAA activity of Pompe myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to mean of controls reveals an excellent improvement of GAA activity after treatment. (C) PAS staining showing glycogen accumulations for patient myoblasts before and after treatment, scale bar 40 µm, with a clear reduction of glycogen in treated patient myoblasts. (D) Basal respiration of Pompe myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to untreated. Here a minor increase in basal respiration is found. (E) Glycolysis of Pompe myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to untreated shows an improvement after treatment.
Having demonstrated that alglucosidase alfa is taken up by the cells, we proceeded to test basal respiration and glycolysis. Basal respiration slightly increased in one sample (Pompe-1), did not change in two samples (Pompe-2 and -3), and nearly doubled reaching normal control levels in Pompe-4 (Figure 4D). Glycolysis did not improve after treatment in Pompe-1 but increased in the other three samples by ~30–60% compared to untreated cells (Figure 4E).
The effect of alglucosidase alfa in GAA-/- mouse myoblasts
Again, we verified the results from human primary myoblasts in the GAA-/- mouse myoblasts. GAA activity was increased by 160% compared to the wild type levels following alglucosidase alfa treatment (final concentration: 10 µg/mL) for 24 hrs (Figure 5A).
Figure 5.

Alglucosidase alfa uptake and metabolism in immortalized GAA-/- mouse myoblasts. (A) GAA activity of GAA-/- mouse myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to wild type shows an excellent GAA activity increase after treatment; (B) basal respiration of GAA-/- mouse myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to untreated reveals an up to 20% increase after treatment; (C) glycolysis of GAA-/- mouse myoblasts untreated (grey) and alglucosidase alfa treated (blue) normalized to untreated displays an up to 30% increase in glycolytic activity.
Basal respiration in the mouse GAA-deficient cells improved by 20%, whereas glycolysis increased by ~30% compared to untreated cells (Figure 5B,C).
Discussion
Distinct mitochondrial abnormalities have been observed in muscle tissue of patients with Pompe disease and in a mouse model of the disease (10-13). Our metabolic profiling data confirm these findings. A proposed and likely explanation is defective macroautophagy (autophagy), a mechanism that delivers macromolecules and organelles for degradation and recycling to lysosomes (11). Indeed, defective mitophagy—inefficient elimination of damaged or worn-out mitochondria through the autophagic pathway (16)—may underlie the mitochondrial defects in the diseased muscle cells. Furthermore, abnormal and functionally altered mitochondria are most likely to remain despite the therapy, considering the inability of ERT to fully reverse lysosomal glycogen accumulation and autophagic defect. Therefore, the consequences of these abnormalities on the cellular energy metabolism may represent another hurdle for the long-term treatment efficacy and have to be taken into account. Thus, the need to monitor mitochondrial alterations may become an additional important issue in evaluating the effect of the current and future therapies.
In Pompe patient myoblasts we found reduced basal respiration and reduced glycolysis, pointing to a cellular energy crisis. Of note, the basal respiration was the lowest in myoblasts with the lowermost GAA activity. This suggests that mitochondrial activity might be an additional read-out parameter when evaluating the effects of residual enzyme levels. The mouse GAA knockout myoblasts confirmed the results we obtained in human myoblasts of Pompe patients. These results are consistent with the observation of reduced glycolysis and mitochondrial respiration in human induced pluripotent stem cells from Pompe disease patients (17).
Although the uptake of alglucosidase alfa in patient myoblasts varied, the levels surpassed those in controls in all cases examined. GAA activity was almost twice the control level in myoblasts from one patient, whereas in the others myoblasts it increased ~4- to 9-times. The uptake in GAA-/- mouse myoblasts was more modest - 1.6 times compared to wild type levels.
Overall, alglucosidase alfa treatment resulted in a different degree of improvement of metabolic defects, and, perhaps not surprisingly, the variability was much higher in human samples. Twenty percent increase in basal respiration and 30% increase in glycolysis were observed in mouse GAA-deficient cells. Only one of the four human GAA-deficient samples showed an increased glycolysis (>60%) as well as improved basal respiration (>80%); one cell line had a slight increase (~15%) in basal respiration but no effect on glycolysis, and the remaining two samples showed only increase in glycolysis (30–40%). This variability is related to general characteristics of primary cells, but may also reflect the situation in patients where variability of individual response to ERT is a well-known phenomenon (7-9). One possible explanation might be the presence of additional mutations in mitochondrial genes (18).
Our data indicate that the evaluation of cellular energy metabolism may be an additional read-out of the efficacy of the currently available as well as future therapies for Pompe disease. Further studies are needed for a comprehensive view of the metabolic perturbations in GAA-deficient muscle cells.
Acknowledgments
We thank Nina Raben for sharing with us the immortalized GAA-deficient mouse myoblasts.
Ethical Statement: This study was approved by the Ethical Review Committee at the Ludwig-Maximilians-University, Munich, Germany (IRB-No. reference 45-14); written and informed consent was obtained from all patients.
Footnotes
Conflicts of Interest: B Schoser has received advisory board membership honoraria from Genzyme Sanofi, Amicus therapeutics, Vertex therapeutics, and Valerion therapeutics. B Schoser has an unrestricted grant from Sanofi Genzyme and Greenovation biopharma. P Meinke receives honoraria from Greenovation biopharma. The other authors have no conflicts of interest to declare.
References
- 1.Lam CW, Yuen YP, Chan KY, et al. Juvenile-onset glycogen storage disease type II with novel mutations in acid alpha-glucosidase gene. Neurology 2003;60:715-7. 10.1212/01.WNL.0000048661.95327.BF [DOI] [PubMed] [Google Scholar]
- 2.Martiniuk F, Bodkin M, Tzall S, et al. Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower "affinity" for glycogen (GAA 2) and transient gene expression in deficient cells. Am J Hum Genet 1990;47:440-5. [PMC free article] [PubMed] [Google Scholar]
- 3.Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest 2006;86:1208-20. 10.1038/labinvest.3700484 [DOI] [PubMed] [Google Scholar]
- 4.Matsuishi T, Yoshino M, Terasawa K, et al. Childhood acid maltase deficiency. A clinical, biochemical, and morphologic study of three patients. Arch Neurol 1984;41:47-52. 10.1001/archneur.1984.04050130053022 [DOI] [PubMed] [Google Scholar]
- 5.Van Hove JL, Yang HW, Wu JY, et al. High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci U S A 1996;93:65-70. 10.1073/pnas.93.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 2017;264:621-30. 10.1007/s00415-016-8219-8 [DOI] [PubMed] [Google Scholar]
- 7.Montagnese F, Barca E, Musumeci O, et al. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment. J Neurol 2015;262:968-78. 10.1007/s00415-015-7664-0 [DOI] [PubMed] [Google Scholar]
- 8.Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013;260:951-9. 10.1007/s00415-012-6636-x [DOI] [PubMed] [Google Scholar]
- 9.van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol 2017;24:768-e31. 10.1111/ene.13285 [DOI] [PubMed] [Google Scholar]
- 10.Engel AG, Dale AJ. Autophagic glycogenosis of late onset with mitochondrial abnormalities: light and electron microscopic observations. Mayo Clin Proc 1968;43:233-79. [PubMed] [Google Scholar]
- 11.Raben N, Wong A, Ralston E, et al. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet 2012;160C:13-21. 10.1002/ajmg.c.31317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez R, Fernandez JM, Cervera C, et al. Adult glycogenosis II with paracrystalline mitochondrial inclusions and Hirano bodies in skeletal muscle. Neuromuscul Disord 1999;9:136-43. 10.1016/S0960-8966(98)00117-5 [DOI] [PubMed] [Google Scholar]
- 13.Schoser BG, Muller-Hocker J, Horvath R, et al. Adult-onset glycogen storage disease type 2: clinico-pathological phenotype revisited. Neuropathol Appl Neurobiol 2007;33:544-59. [DOI] [PubMed] [Google Scholar]
- 14.Lim JA, Li L, Kakhlon O, et al. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015;11:385-402. 10.1080/15548627.2015.1009779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raben N, Nagaraju K, Lee E, et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem 1998;273:19086-92. 10.1074/jbc.273.30.19086 [DOI] [PubMed] [Google Scholar]
- 16.Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183:795-803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HP, Chen PH, Hwu WL, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet 2011;20:4851-64. 10.1093/hmg/ddr424 [DOI] [PubMed] [Google Scholar]
- 18.Chamkha I, Alila-Fersi O, Mkaouar-Rebai E, et al. A novel m.12908T>a mutation in the mitochondrial ND5 gene in patient with infantile-onset Pompe disease. Biochem Biophys Res Commun 2012;429:31-8. 10.1016/j.bbrc.2012.10.105 [DOI] [PubMed] [Google Scholar]