

Abstract
Pompe disease (PD) is an autosomal recessive lysosomal disorder caused by the deficient activity of acid alpha-glucosidase (GAA) enzyme due to mutations in the GAA gene. The enzymatic deficiency leads to the accumulation of glycogen within the lysosomes. Clinically, the disease has been classically classified in infantile and childhood/adult forms. The GAA gene has been localized to chromosome 17q25.2-q25.3 and to date, 582 mutations distributed throughout the whole gene have been reported (HGMD: http://www.hgmd.cf.ac.uk/ac/). All types of mutations have been described; missense variants are the most frequent type followed by small deletions. Most GAA mutations are private or found in a small number of families. However, an exception is represented by the c.-32-13T>G splice mutation that is very common in patients of Caucasian origin affected by the childhood/adult form of the disease, with an allelic frequency ranging from 40% to 70%. In this article, we review the spectrum of GAA mutations, their distribution in different populations, and their classification according to their impact on GAA splicing process, protein expression and activity. In addition, whenever possible, we discuss the phenotype/genotype correlation. The information collected in this review provides an overview of the molecular genetics of PD and can be used to facilitate diagnosis and genetic counseling of families affected by this disorder.
Keywords: Pompe disease, GAA mutations, phenotype/genotype correlation
Introduction
Pompe disease (PD-MIM# 232300), is an autosomal recessive lysosomal storage disorder due to mutations in the acid alpha-glucosidase (GAA) gene (MIM#606800) encoding the lysosomal GAA. This genetic defect leads to the deficient activity of GAA resulting in impaired glycogen degradation and accumulation within the lysosomes (1).
Clinically, PD encompasses a highly variable range of phenotypes that differ in the age of onset, extent of organ involvement, and rate of progression (2). Patients with the classic infantile onset form, the most severe and rapidly progressive phenotype, die within the first year of life from cardiorespiratory insufficiency if untreated (3,4). Patients with onset in childhood or adulthood are characterized by the presence of progressive limb-girdle myopathy and respiratory dysfunction. These patients become wheel chair and/or ventilator dependent, and the respiratory insufficiency is the leading cause of death (5-7).
The GAA gene is localized to chromosome 17q25.2-q25.3 (8) and it was cloned and sequenced in 1991 (9). Since then, significant advances have been made in understanding the molecular bases of this disorder, and to date almost 600 mutations have been reported (HGMD: http://www.hgmd.cf.ac.uk/ac/). However, the pathological mechanism by which a mutant enzyme leads to the wide range of phenotypes in patients affected by this disease remains elusive.
The GAA gene is approximately 28 kb long and encompasses 20 exons. The first exon contains the 5' untranslated sequences and is separated from the second one by a large intron of approximately 2.7 kb. The first ATG is located in exon 2, 32 nt downstream from the beginning of the exon. The second and the last exons are quite big (578 and 607 bp, respectively), while the remaining exons length ranges from 85 to 187 bp (9).
The promoter sequence, located upstream of the first GAA exon, has been characterized and, as expected, has the characteristics of housekeeping gene promoters (10).
The GAA cDNA is 3.6 kb long (8) and encodes a precursor peptide of 952 amino acids with a predicted molecular weight of 105 kD. The precursor has an amino-terminal signal peptide for co-translational transport into the lumen of the endoplasmic reticulum, where it is N-glycosylated resulting in a glycosylated precursor with an apparent molecular mass of 110 kDa (11). Seven glycosylation sites at residues 140, 233, 390, 470, 652, 882 and 925 have been predicted (12).
The enzyme is then transferred to the Golgi complex where high-mannose type oligosaccharide side chains are phosphorylated (13-15) and subsequently targeted to the lysosome via the mannose-6-phosphate receptor. Within the late endosomal/lysosomal compartment the enzyme undergoes a series of proteolytic and N-glycan processing events: the 110-kDa precursor is proteolytically processed at the amino terminus, resulting in a 95-kDa intermediate with a sequence beginning at amino acid 122; the 95-kDa intermediate is processed at the carboxy terminal to a 76-kDa form, which is then proteolytically cleaved at the amino terminus to a 70-kDa mature form (13-16). In 2005, Moreland and colleagues demonstrated that the lysosomal human GAA is composed of four different peptides of 70, 19.4, 10.3, and 3.9 kDa, the latter two being disulfide-bonded (17). Recently, the structure of recombinant human GAA (rhGAA) has been solved by X-ray crystallography, using Myozyme® as the source of protein (18). Interestingly, four disordered surface loops have been identified—notably, the same that were absent in the protein complex studied by Moreland et al.—that hampered the formation of productive protein crystals, and a high-resolution diffraction model was obtained only after their proteolytic removal (PDB entry: 5NN3). Scanning the entire length of GAA protein from N-terminus to C-terminus, a trefoil Type-P domain is separated from the catalytic GH31 (β/α)8 barrel domain by a β-sheet domain. A proximal and a distal β-sheet domains constitute the C-terminal end of GAA. Two unstructured inserts arising from the catalytic domain and a loop bearing from the N-terminal β-sheet domain delimit the active site, in which the conserved residues R600 and D282 play a pivotal role in the substrate recognition and stabilization, and residues D518 and D616 are essential for the catalysis. A second substrate-binding pocket, shaped within the N-terminal trefoil Type-P domain, has been identified and could potentially boost the enzyme processivity (18). Finally, of the seven glycosylation sites originally predicted (17), only five of them, namely N140, N233, N390, N470 and N 652, have been experimentally validated to bind M6P glycans (18,19).
The GAA mutations
The mutational spectrum of GAA gene is very heterogeneous. To date 582 mutations distributed throughout the whole gene have been listed at HGMD-http://www.hgmd.cf.ac.uk/ac/. All types of mutations have been described. Missense mutations are most frequent followed by small deletions. Indeed, as shown in Figure 1, 297 (51.0%) of reported mutations are missense, 87 (14.9%) are small deletions, 16 of which are in frame, 74 (12.7%) are splicing variants, 51 (8.8%) are nonsense, 35 (6.0%) are small insertions/duplications, 19 (3.3%) are gross insertions/deletions, 13 (2.2%) are small indels, 5 (0.9%) are complex rearrangements. Only one variant in the regulatory region has been described so far.
Figure 1.
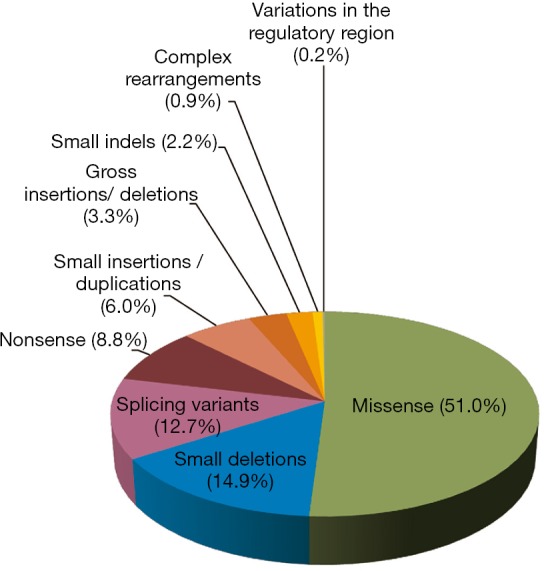
Frequency of GAA mutant alleles reported in the HGMD-http://www.hgmd.cf.ac.uk/ac/ classified by mutation type. GAA, acid alpha-glucosidase.
PD is considered a pan-ethnic disease. However, there are some countries in which the prevalence of the disease is very low. This is the case, for instance, of Finland where only one Pompe patient was definitively diagnosed (20,21).
A number of studies have been conducted aimed at depicting the mutational profile of the GAA gene on a national basis, often distinguishing between the classic severe infantile and the late onset childhood/adult cohorts.
Even though most GAA mutations are private or found in a small number of families, the c.-32-13T>G splice mutation is an exception, since it is very common in patients of Caucasian origin affected by the childhood/adult form of the disease, with an allelic frequency ranging from 40% to 70% in different populations (6,22-36).
The presence of this intronic mutation results in the exclusion of exon 2 from a variable proportion of the expressed GAA mRNA. In other words, by affecting the overall splicing efficiency, it changes the balance between the GAA splicing isoforms towards the non-functional, exon 2-skipped species, yet not completely preventing the expression of the normal transcript that can be translated into an enzymatically active GAA protein. Therefore, the resulting variable levels of GAA residual activity may explain the delay of the phenotypic manifestation of the disease in those who carry the c.-32-13T>G mutation (37-40).
Apart from the common intronic c.-32-13T>G mutation, few variants are overrepresented in particular populations. Perhaps the best examples are the c.525delT and the c.2481+102_2646+31del mutations, the so called “Dutch mutations” due to their high recurrence in Dutch population (41-43). Both variants have been classified as severe mutations. Indeed, the deletion of a single nucleotide (c.525delT) or the entire exon 18 (c.2481+102_2646+31del) cause in both cases a shifting of the open reading frame leading to the generation of a premature stop codon. No transcript from the c.525delT allele is detected in patient’s cells, probably due to the synthesis of an unstable mRNA which would be rapidly degraded (44). As expected, these variants are associated with the infantile-onset form of PD and occur in a cohort of infantile Dutch patients with an allelic frequency of 35% and 31%, respectively (41). The allelic frequencies of both mutations are much lower in Dutch patients with the childhood/adult phenotype, representing only 15% and 8% of the alleles, respectively (41). By comparison, the allelic frequency of the c.525delT in a similar cohort of Italian infantile patients is 13.8% (45).
Geographical differences in the allelic frequency of GAA mutations within the same country have been noted. For instance, the most frequent pathogenic mutation encountered in the infantile subset of patients in the North of China is the c.2662G>T (p.Glu888*) mutation, accounting for 23.1% of total mutant alleles (46). By contrast, the most frequent mutation in a cohort of patients from Southern China is the c.1935C>A (p.Asp645Glu) accounting for the 20–25% of total mutant alleles (46,47). Notably, such a geographical partition is not evident in childhood/adult patients from mainland China in which the most frequent pathogenic GAA mutation is the c.2238G>C (p.W746C) accounting for 27.1% of total mutant alleles (48). Also, in Taiwan the c.1935C>A mutation seems to be the most recurrent mutation in patients affected by the infantile onset form of PD, accounting for 12.2% of total mutant variants. This could be explained by the Chinese origin of the Taiwanese population (49). The importance of historical migratory fluxes in the determination of the mutational profile of specific populations is evidenced by some studies carried out in countries that had experienced a marked immigration in their history, such as Argentina, Brazil, Colombia or Canada. Indeed, a very heterogeneous profile of GAA mutations has been reported in these countries (50-53). However, the severe mutation c.2560C>T (p.R854*) was found to be overrepresented (allele frequency of 16.7%) in a Brazilian cohort of infantile patients (54). This is not an unexpected finding considering the massive migratory flux coming from North Africa, where a high frequency of this mutation is very well documented (55). Not surprisingly, the same c.2560C>T mutation is the most common defect in African-Americans with PD (55).
Recently, Fukuhara and colleagues demonstrated that the mutation c.546G>T occurs with high allelic frequency (22.9%) in patients affected by the childhood/adult phenotype in Japan (56).
Mutations associated with GAA pseudodeficiency
Two sequence variants, c.1726G>A (p.Gly576Ser) and c.2065G>A (p.Glu689Lys), often present in cis on the same allele, are associated with a pseudodeficiency of GAA. The first one, effectively reduces both the amount of expressed GAA protein and its catalytic activity (57-59), while the second one has little effect on GAA functionality (60). Therefore, individuals who are homozygous for this allele have very low GAA enzymatic activity but do not develop PD (49,59,61,62).
This complex allele is quite frequent among people of Asian descent. Indeed, about 4% of subjects in Asian populations carry this allele in homozygosity (62).
The functional impact of GAA mutations
While the possible impact of a nonsense mutation or a deletion/insertion leading to a frameshift on the synthesis and function of the GAA enzyme could be foreseeable, it is much more difficult to predict the effect of missense mutations or variants affecting sequences involved in the splicing process. Indeed, sequence variations leading to the generation of premature stop codons (nonsense of frameshift mutations) would necessarily result in the synthesis of truncated non-functional proteins. Furthermore, in many cases transcripts carrying premature stop codons are quite unstable and degraded via nonsense mediated decay (NMD) with the consequent loss of GAA synthesis. Therefore, in general these types of mutations could be considered as severe ones.
Several in silico prediction tools have been developed to analyze the impact of genetic variants on the mRNA splicing processes. However, although in silico analysis became increasingly promising and trustable in predicting the likely pathogenic effect of most of the analyzed variants (63), this tool cannot substitute functional analysis. Indeed, minigene splicing assays or, even better, direct mRNA analysis should be required for linking variants to a putative splicing defect. Furthermore, functional analysis remains the only reliable tool to determine the severity of a given mutation since in many cases the mutation may not completely abrogate the expression of normally spliced transcript leading to the protein expression and retention of some residual activity (64). The best example of this type of variant is the common c.-32-13T>G mutation (see below).
Similarly, several programs have been developed to predict the potential effect of missense mutations. However, analysis of protein expression and activity in patients’ cells and/or by in vitro expression studies of the mutated proteins remain the gold standard to determine their pathogenetic nature and to gain insights on their severity.
It is worth noting that in vitro expression studies are very useful to test the effect of a given variant when mutations are found in compound heterozygous individuals.
A classification of GAA mutations based on a severity rating system organized in six classes has been proposed by Kroos et al. (58). The severity of a particular mutation is defined based on the results of a transient transfection of an expression construct carrying wild-type or mutated GAA cDNA; each sequence variation is compared to the wild-type GAA in terms of quantity and quality of processed and unprocessed GAA protein, and residual enzymatic activity in cells and media (58).
The six severity classes are identified by letters, whereby A stands for “very severe”, B for “potentially less severe”, C for “less severe”, D for “potentially mild”, E for “presumably nonpathogenic”, and finally, F for “nonpathogenic” (58).
Class A mutations are Cross Reactive Immunologic Material (CRIM)-negative, since no molecular forms of GAA can be detected by immunoblotting, and no residual enzymatic activity can be measured. Even though classes B, C and D mutations are CRIM-positive, the levels of processed and unprocessed forms of GAA are lower than normal ones, leading to a residual enzymatic activity ranging from 0 to 30%. Finally, both classes E and F mutations present quantitatively and qualitatively normal levels of GAA forms, but they differ in residual enzymatic activity, which is higher than 30% in class E and higher than 60% in class F (58,65).
The Pompe Disease Mutation Database, available at http://www.pompecenter.nl has been created with the aim of providing a list of GAA variants and to describe their effect in order to facilitate the diagnosis and genetic counseling.
The database, last updated in May 2016 reports 558 GAA sequence variations: class A and B mutations are the most represented groups with 183 and 139 hits, respectively. Thirty mutations are labeled as “less severe”, 22 as “potentially mild”, and only 6 as “presumably nonpathogenic”. Class F is the third largest group, including 93 sequence variations, and finally, the severity of 85 mutations still remains unknown (Figure 2).
Figure 2.
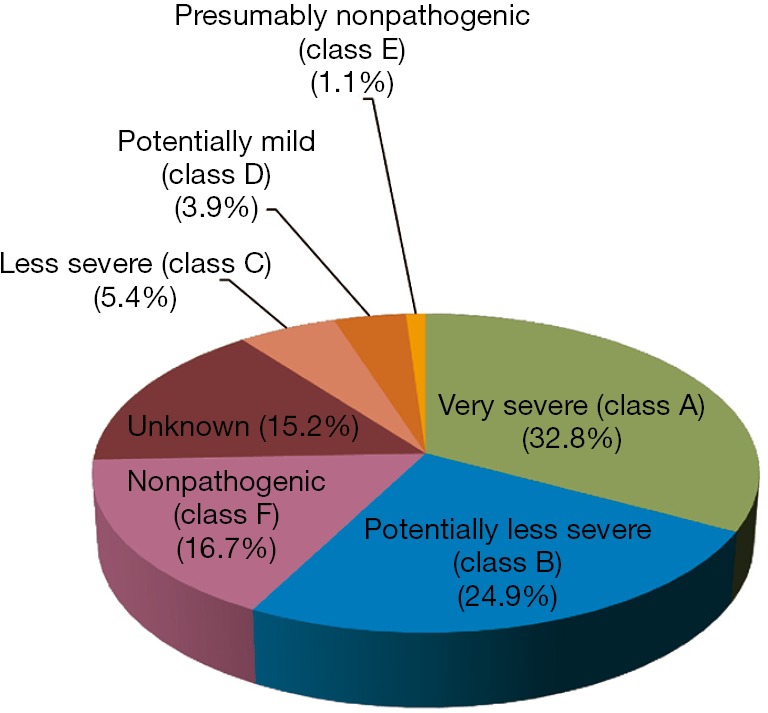
Frequency of GAA mutant alleles reported in the Pompe Disease Mutation Database, available at http://www.pompecenter.nl classified according to their functional impact on GAA. GAA, acid alpha-glucosidase.
Besides being useful for the interpretation of genetic data obtained in PD patients, the knowledge of the functional consequences of a mutant variant might be useful to select patients that could benefit from experimental therapies, such as the use of antisense oligonucleotides able to rescue splicing defects (66,67) or molecular chaperons to improve GAA activity by promoting folding, processing and trafficking to the lysosome of mutated but partially active variants (68).
The common c.-32-13T>G mutation
The c.-32-13T>G variant is the most frequent GAA mutation associated with the childhood/adult phenotype. Indeed, almost 90% of patients affected by this phenotype carry this mutation on at least one allele. This mutation, first described by Huie et al. in a patient affected by the adult onset form of PD (37), is located 13 nucleotides upstream of the canonical acceptor splice site of GAA intron 1 (Figure 3). As supported by studies performed in fibroblasts derived from patients carrying the c.-32-13T>G base change, the main functional consequence of this mutation is the synthesis of different aberrant splicing variants in which the exon 2 is completely or partially spliced out and a limited amount of normally spliced GAA mRNA. Interestingly, the same non-functional splicing isoforms were detected, although in low quantities, in cells from normal subjects (38). These data were then further confirmed in vitro by Raben and colleagues using a minigene assay (39). Therefore, from a clinical point of view, these data strongly support the concept that the production of a certain amount of “normally spliced” wild-type mRNA may represent a general mechanism underlying the delayed symptomatic expression in patients bearing the c.-32-13T>G mutation (39,40).
Figure 3.

GAA mRNA splicing isoforms expressed in cultured fibroblasts. (A) Schematic representation of the 5' region of the GAA gene (exons 1 to 3). The position of the c.-32-13T>G mutation is highlighted in red. The cryptic splice sites, located 35 nt downstream from the normal donor splice site of exon 1 and at 60 nt upstream from the donor site of exon 2, are shown as c1 and c2, respectively. The presence of the c.-32-13T>G mutation abrogates the binding of the U2AF65 splicing factor. (B) Schematic list of the GAA mRNA species expressed in human fibroblasts. Normal spliced GAA mRNA (N) and splicing species SV2 and SV3 are detected both in fibroblasts from patients carrying the c.-32-13T>G variant and healthy controls. In patients vs. controls samples, however, the relative abundance of the various splicing isoforms is different. While N is the main mRNA species expressed in normal cells, SV2 and SV3 are the main species detected in cells from patients bearing the c.-32-13T>G variant. GAA, acid alpha-glucosidase.
From the functional point of view, it has been shown that the mutation interferes with the binding of the splicing factor U2AF65 to the GAA pre-mRNA, almost completely abrogating its interaction with the polypyrimidine tract of exon 2 leading to the general inefficiency of the splicing process (Figure 3). Moreover, it has been demonstrated that the overexpression of specific mRNA binding proteins can modulate the expression of normally spliced GAA mRNA from the c.-32-13T>G mutated allele (40).
Recently, very promising results have been obtained in vitro by targeting a specific silencer located within exon 2 with a combination of antisense oligonucleotides. Indeed, treatment of myotubes of patients carrying the c.-32-13T>G mutation resulted in a significant increase of exon 2 inclusion and GAA activity, and a decrease in lysosomal glycogen accumulation (67). A similar approach supports the possibility of promoting exon 2 inclusion and GAA enzyme activity by targeting inhibitory sequences within intron 1 of GAA (68).
Phenotype-genotype correlation
As for most genetic diseases, a strict correlation between the phenotype and the genotype cannot be established in PD. However, some general conclusions can be drawn from the data obtained in hundreds PD patients.
In general, patients affected by the severe and rapidly progressive infantile form of PD harbour mutations that completely abolish the expression of all forms of GAA protein or lead to a low expression of unprocessed or processed GAA (classified as A or B mutations). In all cases these mutants do not retain significant amount of residual GAA activity (45,69,70). Conversely, the presence of a potentially mild mutation in one allele seems to prevent the occurrence of the severe classic infantile phenotype (22-26,28,30).
Apart from these general considerations, it is difficult to establish a strict correlation between a particular mutation and the clinical presentation and progression of the disease since most GAA mutations are private and found in compound heterozygosity. As mentioned above, the only exception so far is the frequent c.-32-13T>G mutation, which is also often found in compound heterozygosity in patients affected by the childhood/adult phenotype. Therefore, several authors have analyzed the phenotype of patients carrying this mutation in association with a severe “null” variant that is not contributed to GAA activity. Besides the fact that all described patients presented with the childhood/adult form of PD, they demonstrated a wide variability in residual activity, age at onset, and disease progression (21,33,71-73). Furthermore, different phenotypic expression has also been reported in siblings carrying this genotype (74). Considering these observations, it is very likely that secondary factors, genetic and non-genetic, would act as modifiers of the PD phenotype. This hypothesis is further supported by the data collected in patients carrying the c.-32-13T>G mutation in homozygosity who, unexpectedly, presented with the full spectrum of adult PD phenotype (29,75).
In an attempt to explain this wide phenotypic variability, the c.-32-13G>T haplotype was studied in a cohort of 98 compound heterozygous patients carrying the c.-32-13T>G mutation in combination with a null variant. This study failed to demonstrate a correlation between the c.-32-13T>G haplotype and the phenotype. In addition, several authors have explored the possible modifying effect of an angiotensin I-converting enzyme (ACE) polymorphism (76-80). However, the results of these studies do not concur and the role of this polymorphism in the phenotypic expression of PD remains controversial.
Very recently, the c.510C>T variant has been identified as a genetic modifier of the disease onset in compound heterozygous or homozygous for the common c.-32-13T>G variant. Indeed, the c.510C>T when present in cis with the c.-32-13T>G mutation, modulates the splicing pattern of the mutated transcript further reducing the relative amount of correctly spliced mRNA which in turn, resulted in reduced residual GAA activity (81).
Acknowledgments
AFM Telethon Project SPLICESCREENPD.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Hirschhorn R, Reuser AJJ. Glycogen storage disease type II; acid-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Sly W, Valle D. eds. The metabolic and molecular bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001:3389-420. [Google Scholar]
- 2.Güngör D, Reuser AJ. How to describe the clinical spectrum in Pompe disease? Am J Med Genet A 2013;161A:399-400. 10.1002/ajmg.a.35662 [DOI] [PubMed] [Google Scholar]
- 3.van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe's disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332-40. 10.1542/peds.112.2.332 [DOI] [PubMed] [Google Scholar]
- 4.Kishnani PS, Hwu WL, Mandel H, et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr 2006;148:671-6. 10.1016/j.jpeds.2005.11.033 [DOI] [PubMed] [Google Scholar]
- 5.Hagemans ML, Winkel LP, Van Doorn PA, et al. Clinical manifestation and natural course of late-onset Pompe's disease in 54 Dutch patients. Brain 2005;128:671-7. 10.1093/brain/awh384 [DOI] [PubMed] [Google Scholar]
- 6.Herzog A, Hartung R, Reuser AJ, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J Rare Dis 2012;7:35. 10.1186/1750-1172-7-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schüller A, Wenninger S, Strigl-Pill N, et al. Toward deconstructing the phenotype of late-onset Pompe disease. Am J Med Genet C Semin Med Genet 2012;160C:80-8. 10.1002/ajmg.c.31322 [DOI] [PubMed] [Google Scholar]
- 8.Martiniuk F, Mehler M, Pellicer A, et al. Isolation of a cDNA for human acid alpha-glucosidase and detection of genetic heterogeneity for mRNA in three alpha-glucosidase-deficient patients. Proc Natl Acad Sci U S A 1986;83:9641-4. 10.1073/pnas.83.24.9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martiniuk F, Bodkin M, Tzall S, et al. Isolation and partial characterization of the structural gene for human acid alpha glucosidase. DNA Cell Biol 1991;10:283-92. 10.1089/dna.1991.10.283 [DOI] [PubMed] [Google Scholar]
- 10.Tzall S, Martiniuk F. Identification of the promoter region and gene expression for human acid alpha glucosidase. Biochem Biophys Res Commun 1991;176:1509-15. 10.1016/0006-291X(91)90458-J [DOI] [PubMed] [Google Scholar]
- 11.van der Horst GT, Hoefsloot EH, Kroos MA, et al. Cell-free translation of human lysosomal alpha-glucosidase: evidence for reduced precursor synthesis in an adult patient with glycogenosis type II. Biochim Biophys Acta 1987;910:123-9. 10.1016/0167-4781(87)90064-9 [DOI] [PubMed] [Google Scholar]
- 12.Hermans MM, Wisselaar HA, Kroos MA, et al. Human lysosomal alpha glucosidase: functional characterization of glycosylation sites. Biochem J 1993;289:681-6. 10.1042/bj2890681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J Biol Chem 1980;255:4946-50. [PubMed] [Google Scholar]
- 14.Hasilik A, Neufeld EF. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem 1980;255:4937-45. [PubMed] [Google Scholar]
- 15.Reuser AJ, Kroos M, Oude Elferink RP, et al. Defects in synthesis, phosphorylation, and maturation of acid alpha-glucosidase in glycogenosis type II. J Biol Chem 1985;260:8336-41. [PubMed] [Google Scholar]
- 16.Wisselaar HA, Kroos MA, Hermans MM, et al. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J Biol Chem 1993;268:2223-31. [PubMed] [Google Scholar]
- 17.Moreland RJ, Jin X, Zhang XK, et al. Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J Biol Chem 2005;280:6780-91. 10.1074/jbc.M404008200 [DOI] [PubMed] [Google Scholar]
- 18.Roig-Zamboni V, Cobucci-Ponzano B, Iacono R, et al. Structure of human lysosomal acid α-glucosidase-a guide for the treatment of Pompe disease. Nat Commun 2017;8:1111. 10.1038/s41467-017-01263-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park H, Kim J, Lee YK, et al. Four unreported types of glycans containing mannose-6-phosphate are heterogeneously attached at three sites (including newly found Asn 233) to recombinant human acid alpha-glucosidase that is the only approved treatment for Pompe disease. Biochem Biophys Res Commun 2018;495:2418-24. 10.1016/j.bbrc.2017.12.101 [DOI] [PubMed] [Google Scholar]
- 20.Palmio J, Auranen M, Kiuru-Enari S, et al. Screening for late-onset Pompe disease in Finland. Neuromuscul Disord 2014;24:982-5. 10.1016/j.nmd.2014.06.438 [DOI] [PubMed] [Google Scholar]
- 21.Korpela MP, Paetau A, Löfberg MI, et al. A novel mutation of the GAA gene in a Finnish late-onset Pompe disease patient: clinical phenotype and follow-up with enzyme replacement therapy. Muscle Nerve 2009;40:143-8. 10.1002/mus.21291 [DOI] [PubMed] [Google Scholar]
- 22.Montalvo AL, Bembi B, Donnarumma M, et al. Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II. Hum Mutat 2006;27:999-1006. 10.1002/humu.20374 [DOI] [PubMed] [Google Scholar]
- 23.Nascimbeni AC, Fanin M, Tasca E, et al. Molecular pathology and enzyme processing in various phenotypes of acid maltase deficiency. Neurology 2008;70:617-26. 10.1212/01.wnl.0000299892.81127.8e [DOI] [PubMed] [Google Scholar]
- 24.Gort L, Coll MJ, Chabás A. Glycogen storage disease type II in Spanish patients: high frequency of c.1076-1G>C mutation. Mol Genet Metab 2007;92:183-7. 10.1016/j.ymgme.2007.05.011 [DOI] [PubMed] [Google Scholar]
- 25.Wan L, Lee CC, Hsu CM, et al. Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II. J Neurol 2008;255:831-8. 10.1007/s00415-008-0714-0 [DOI] [PubMed] [Google Scholar]
- 26.Joshi PR, Gläser D, Schmidt S, et al. Molecular diagnosis of German patients with late-onset glycogen storage disease type II. J Inherit Metab Dis 2008;31 Suppl 2:S261-5. 10.1007/s10545-008-0820-2 [DOI] [PubMed] [Google Scholar]
- 27.Papadimas GK, Terzis G, Methenitis S, et al. Body composition analysis in late-onset Pompe disease. Mol Genet Metab 2011;102:41-3. 10.1016/j.ymgme.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 28.Byrne BJ, Kishnani PS, Case LE, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab 2011;103:1-11. 10.1016/j.ymgme.2011.02.004 [DOI] [PubMed] [Google Scholar]
- 29.Laforêt P, Laloui K, Granger B, et al. The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease. Rev Neurol (Paris) 2013;169:595-602. 10.1016/j.neurol.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 30.Montagnese F, Barca E, Musumeci O, et al. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment. J Neurol 2015;262:968-78. 10.1007/s00415-015-7664-0 [DOI] [PubMed] [Google Scholar]
- 31.Kuperus E, Kruijshaar ME, Wens SCA, et al. Long-term benefit of enzyme replacement therapy in Pompe disease: A 5-year prospective study. Neurology 2017;89:2365-73 10.1212/WNL.0000000000004711 [DOI] [PubMed] [Google Scholar]
- 32.Mori M, Haskell G, Kazi Z, et al. Sensitivity of whole exome sequencing in detecting infantile- and late-onset Pompe disease. Mol Genet Metab 2017;122:189-97 10.1016/j.ymgme.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semplicini C, Letard P, De Antonio M, et al. Late-onset Pompe disease in France: molecular features and epidemiology from a nationwide study. J Inherit Metab Dis 2018;41:937-46. 10.1007/s10545-018-0243-7 [DOI] [PubMed] [Google Scholar]
- 34.Scheidegger O, Leupold D, Sauter R, et al. 36-Months follow-up assessment after cessation and resuming of enzyme replacement therapy in late onset Pompe disease: data from the Swiss Pompe Registry. J Neurol 2018;265:2783-8. 10.1007/s00415-018-9065-7 [DOI] [PubMed] [Google Scholar]
- 35.Witkowski G, Konopko M, Rola R, et al. Enzymatic replacement therapy in patients with late-onset Pompe disease - 6-Year follow up. Neurol Neurochir Pol 2018;52:465-9. 10.1016/j.pjnns.2018.05.002 [DOI] [PubMed] [Google Scholar]
- 36.Löscher WN, Huemer M, Stulnig TM, et al. Pompe disease in Austria: clinical, genetic and epidemiological aspects. J Neurol 2018;265:159-64. 10.1007/s00415-017-8686-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huie ML, Chen AS, Tsujino S, et al. Aberrant splicing in adult onset glycogen storage disease type II (GSDII): molecular identification of an IVS1 (-13T-->G) mutation in a majority of patients and a novel IVS10 (+1GT-->CT) mutation. Hum Mol Genet 1994;3:2231-6. 10.1093/hmg/3.12.2231 [DOI] [PubMed] [Google Scholar]
- 38.Boerkoel CF, Exelbert R, Nicastri C, et al. Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am J Hum Genet 1995;56:887-97. [PMC free article] [PubMed] [Google Scholar]
- 39.Raben N, Nichols RC, Martiniuk F, et al. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum Mol Genet 1996;5:995-1000. 10.1093/hmg/5.7.995 [DOI] [PubMed] [Google Scholar]
- 40.Dardis A, Zanin I, Zampieri S, et al. Functional characterization of the common c.-32-13T>G mutation of GAA gene: identification of potential therapeutic agents. Nucleic Acids Res 2014;42:1291-302. 10.1093/nar/gkt987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroos MA, Van der Kraan M, Van Diggelen OP, et al. Glycogen storage disease type II: frequency of three common mutant alleles and their associated clinical phenotypes studied in 121 patients. J Med Genet 1995;32:836-7. 10.1136/jmg.32.10.836-a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999;7:713-6. 10.1038/sj.ejhg.5200367 [DOI] [PubMed] [Google Scholar]
- 43.Ausems MG, ten Berg K, Sandkuijl LA, et al. Dutch patients with glycogen storage disease type II show common ancestry for the 525delT and del exon 18 mutations. J Med Genet 2001;38:527-9. 10.1136/jmg.38.8.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hermans MM, De Graaff E, Kroos MA, et al. The effect of a single base pair deletion (delta T525) and a C1634T missense mutation (pro545leu) on the expression of lysosomal alpha-glucosidase in patients with glycogen storage disease type II. Hum Mol Genet 1994;3:2213-8. 10.1093/hmg/3.12.2213 [DOI] [PubMed] [Google Scholar]
- 45.Pittis MG, Donnarumma M, Montalvo AL, et al. Molecular and functional characterization of eight novel GAA mutations in Italian infants with Pompe disease. Hum Mutat 2008;29:E27-36. 10.1002/humu.20753 [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Liu T, Huang M, et al. Clinical and Molecular Characterization of Infantile-Onset Pompe Disease in Mainland Chinese Patients: Identification of Two Common Mutations. Genet Test Mol Biomarkers 2017;21:391-6. 10.1089/gtmb.2016.0424 [DOI] [PubMed] [Google Scholar]
- 47.Fu L, Qiu W, Yu Y, et al. Clinical and molecular genetic study of infantile-onset Pompe disease in Chinese patients: identification of 6 novel mutations. Gene 2014;535:53-9. 10.1016/j.gene.2013.10.066 [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Wang Z, Jin W, et al. Clinical and GAA gene mutation analysis in mainland Chinese patients with late-onset Pompe disease: identifying c.2238G > C as the most common mutation. BMC Med Genet 2014;15:141. 10.1186/s12881-014-0141-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Labrousse P, Chien YH, Pomponio RJ, et al. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol Genet Metab 2010;99:379-83. 10.1016/j.ymgme.2009.12.014 [DOI] [PubMed] [Google Scholar]
- 50.Palmer RE, Amartino HM, Niizawa G, et al. Pompe disease (glycogen storage disease type II) in Argentineans: clinical manifestations and identification of 9 novel mutations. Neuromuscul Disord 2007;17:16-22. 10.1016/j.nmd.2006.09.004 [DOI] [PubMed] [Google Scholar]
- 51.Turaça LT, de Faria DO, Kyosen SO, et al. Novel GAA mutations in patients with Pompe disease. Gene 2015;561:124-31. 10.1016/j.gene.2015.02.023 [DOI] [PubMed] [Google Scholar]
- 52.Niño MY, Mateus HE, Fonseca DJ, et al. Identification and Functional Characterization of GAA Mutations in Colombian Patients Affected by Pompe Disease. JIMD Rep 2013;7:39-48. 10.1007/8904_2012_138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCready ME, Carson NL, Chakraborty P, et al. Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II. Mol Genet Metab 2007;92:325-35. 10.1016/j.ymgme.2007.07.006 [DOI] [PubMed] [Google Scholar]
- 54.Oba-Shinjo SM, da Silva R, Andrade FG, et al. Pompe disease in a Brazilian series: clinical and molecular analyses with identification of nine new mutations. J Neurol 2009;256:1881-90. 10.1007/s00415-009-5219-y [DOI] [PubMed] [Google Scholar]
- 55.Becker JA, Vlach J, Raben N, et al. The African origin of the common mutation in African American patients with glycogen-storage disease type II. Am J Hum Genet 1998;62:991-4. 10.1086/301788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fukuhara Y, Fuji N, Yamazaki N, et al. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol Genet Metab Rep 2017;14:3-9. 10.1016/j.ymgmr.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tajima Y, Matsuzawa F, Aikawa S, et al. Structural and biochemical studies on Pompe disease and a "pseudodeficiency of acid alpha-glucosidase". J Hum Genet 2007;52:898-906. 10.1007/s10038-007-0191-9 [DOI] [PubMed] [Google Scholar]
- 58.Kroos M, Pomponio RJ, van Vliet L, et al. Update of the Pompe disease mutation database with 107 sequence variants and a format for severity rating. Hum Mutat 2008;29:E13-26. 10.1002/humu.20745 [DOI] [PubMed] [Google Scholar]
- 59.Kumamoto S, Katafuchi T, Nakamura K, et al. High frequency of acid alpha-glucosidase pseudodeficiency complicates newborn screening for glycogen storage disease type II in the Japanese population. Mol Genet Metab 2009;97:190-5. 10.1016/j.ymgme.2009.03.004 [DOI] [PubMed] [Google Scholar]
- 60.Huie ML, Menaker M, Mcalpine PJ, et al. Identification of an E689K substitution as the molecular basis of the human acid alpha-glucosidase type 4 allozyme (GAA*4). Ann Hum Genet 1996;60:365-8. 10.1111/j.1469-1809.1996.tb00433.x [DOI] [PubMed] [Google Scholar]
- 61.Chien YH, Chiang SC, Zhang XK, et al. Early detection of Pompe disease by newborn screening is feasible: results from the Taiwan screening program. Pediatrics 2008;122:e39-45. 10.1542/peds.2007-2222 [DOI] [PubMed] [Google Scholar]
- 62.Shigeto S, Katafuchi T, Okada Y, et al. Improved assay for differential diagnosis between Pompe disease and acid α-glucosidase pseudodeficiency on dried blood spots. Mol Genet Metab 2011;103:12-7. 10.1016/j.ymgme.2011.01.006 [DOI] [PubMed] [Google Scholar]
- 63.Grodecká L, Buratti E, Freiberger T. Mutations of Pre-mRNA Splicing Regulatory Elements: Are Predictions Moving Forward to Clinical Diagnostics? Int J Mol Sci 2017;18. doi: . 10.3390/ijms18081668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zampieri S, Buratti E, Dominissini S, et al. Splicing mutations in glycogen-storage disease type II: evaluation of the full spectrum of mutations and their relation to patients' phenotypes. Eur J Hum Genet 2011;19:422-31. 10.1038/ejhg.2010.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kroos M, Hoogeveen-Westerveld M, Michelakakis H, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat 2012;33:1161-5. 10.1002/humu.22108 [DOI] [PubMed] [Google Scholar]
- 66.van der Wal E, Bergsma AJ, Pijnenburg JM, et al. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol Ther Nucleic Acids 2017;7:90-100. 10.1016/j.omtn.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goina E, Peruzzo P, Bembi B, et al. Glycogen Reduction in Myotubes of Late-Onset Pompe Disease Patients Using Antisense Technology. Mol Ther 2017;25:2117-28. 10.1016/j.ymthe.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parenti G, Zuppaldi A, Gabriela Pittis M, et al. Pharmacological Enhancement of Mutated α-Glucosidase Activity in Fibroblasts from Patients with Pompe Disease. Mol Ther 2007;15:508-14. 10.1038/sj.mt.6300074 [DOI] [PubMed] [Google Scholar]
- 69.van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, et al. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis 2015;38:305-14. 10.1007/s10545-014-9707-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Parini R, De Lorenzo P, Dardis A, et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J Rare Dis 2018;13:32. 10.1186/s13023-018-0771-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kroos MA, Pomponio RJ, Hagemans ML, et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007;68:110-5. 10.1212/01.wnl.0000252798.25690.76 [DOI] [PubMed] [Google Scholar]
- 72.van Capelle CI, van der Meijden JC, van den Hout JM, et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J Rare Dis 2016;11:65. 10.1186/s13023-016-0442-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, et al. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet 2012;160C:59-68. 10.1002/ajmg.c.31318 [DOI] [PubMed] [Google Scholar]
- 74.Wens SC, van Gelder CM, Kruijshaar ME, et al. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis 2013;8:182. 10.1186/1750-1172-8-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Musumeci O, Thieme A, Claeys KG, et al. Homozygosity for the common GAA gene splice site mutation c.-32-13T>G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul Disord 2015;25:719-24. 10.1016/j.nmd.2015.07.002 [DOI] [PubMed] [Google Scholar]
- 76.de Filippi P, Ravaglia S, Bembi B, et al. The angiotensin-converting enzyme insertion/deletion polymorphism modifies the clinical outcome in patients with Pompe disease. Genet Med 2010;12:206-11. 10.1097/GIM.0b013e3181d2900e [DOI] [PubMed] [Google Scholar]
- 77.Ravaglia S, De Filippi P, Pichiecchio A, et al. Can genes influencing muscle function affect the therapeutic response to enzyme replacement therapy (ERT) in late-onset type II glycogenosis? Mol Genet Metab 2012;107:104-10. 10.1016/j.ymgme.2012.05.016 [DOI] [PubMed] [Google Scholar]
- 78.De Filippi P, Saeidi K, Ravaglia S, et al. Genotype-phenotype correlation in Pompe disease, a step forward. Orphanet J Rare Dis 2014;9:102. 10.1186/s13023-014-0102-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baek RC, Palmer R, Pomponio RJ, et al. The influence of a polymorphism in the gene encoding angiotensin converting enzyme (ACE) on treatment outcomes in late-onset Pompe patients receiving alglucosidase alfa. Mol Genet Metab Rep 2016;8:48-50. 10.1016/j.ymgmr.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuperus E, van der Meijden JC, In't Groen SLM, et al. The ACE I/D polymorphism does not explain heterogeneity of natural course and response to enzyme replacement therapy in Pompe disease. PLoS One 2018;13:e0208854. 10.1371/journal.pone.0208854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bergsma AJ, In 't Groen SLM, van den Dorpel JJA, et al. A genetic modifier of symptom onset in Pompe disease. EBioMedicine 2019;43:553-61. [DOI] [PMC free article] [PubMed] [Google Scholar]