

Abstract
Infantile-onset Pompe disease (IOPD) is characterized by virtually complete absence of acid alpha-glucosidase (GAA)-activity, resulting in rapidly progressive hypertrophic cardiomyopathy (HCM), profound skeletal muscle weakness, and death usually within the first 12 months of life. Enzyme replacement therapy (ERT) with recombinant GAA in humans started in 1999, and pivotal studies demonstrated that the treatment ameliorated HCM, improved motor function in some patients, and prolonged overall and ventilator-free survival. These outcomes led to the approval of ERT in 2006. Implementation of ERT has uncovered multisystemic character of IOPD, not known in the pre-ERT era. Although ERT has substantially improved the prognosis of IOPD, mortality is still considerable, and decline of motor function with time is frequent in long-term survivors. This review details the new complex IOPD phenotype, outlines problems related to ERT, and highlights unmet needs.
Keywords: Infantile-onset Pompe disease (IOPD), enzyme replacement therapy (ERT), long-term outcome, musculoskeletal dysfunction, neurocognitive impairment
Introduction
Pompe disease is caused by biallelic mutations of the acid α-glucosidase (GAA) gene located on chromosome 17q25, that result in deficiency of the lysosomal enzyme GAA (1). Severity of disease depends on the amount of residual enzyme activity. In classic infantile-onset Pompe disease (IOPD), GAA activity is less than 1% (1). Contrary to milder forms with later onset, this causes marked accumulation of glycogen not only in skeletal muscle, but also in heart and other tissues (1). Affected patients present with creatinine kinase elevation, hypertrophic cardiomyopathy (HCM), failure to thrive, muscular hypotonia and axial muscle weakness during the first six months of life. IOPD is rapidly progressive, and the majority of untreated subjects die within the first year of life due to a combination of ventilatory and cardiac failure without achieving any motor milestones such as turning, sitting, or standing (1,2). Survival beyond the age of 18 months is exceptional (2). Classic IOPD should be distinguished from non-classic or late-infantile Pompe disease that also manifests during infancy but has no or much less severe cardiac hypertrophy (1).
Although GAA activity is <1% in all IOPD patients, two groups have to be differentiated. Patients may synthesize a non-functional form of GAA or are completely unable to form any kind of native enzyme. The former patients are designated as cross-reactive immunological material (CRIM)-positive, whereas the latter are classified as CRIM-negative (3).
Enzyme replacement therapy (ERT) substitutes a deficient enzyme by intravenous infusion of the recombinant human enzyme at regular intervals, which is taken up into the cell via the mannose-6-phosphate receptor and transported to the lysosome. Initial ERT trials with recombinant human GAA (rhGAA) in IOPD started in 1999 (4-6), which means that the first surviving patients treated this way have now reached adulthood. In 2006, a multinational study treating eight infants between 3 and 15 months of age with ERT showed a significant reduction of left ventricular muscle mass in all. Five gained motor functions and three achieved free walking. After 12 months on therapy, two had died and one had become ventilator-dependent. Four patients deceased during the extended follow-up phase. Median age of death in the whole group was 21.7 months, being significantly later than in untreated patients (7). Since this suggested that an early start of ERT may yield better results, a further pivotal study included 18 infants younger than 6 months of age (8) who received 20 or 40 mg/kg rhGAA biweekly. After 12 months there was a significant reduction of cardiac hypertrophy and all patients were still alive. Thirteen subjects made motor progress whereas 5 did not. Seven out of 18 achieved independent walking, 3 were able to stand, and 3 were sitting without support, while 6 patients became ventilator-dependent. Following these 18 patients for up to 3 years demonstrated that all of them had survived until age 18 months, that 5 patients (28%) had died thereafter, and that 4 subjects (22%) had become ventilator-dependent. No significant differences were observed between the groups treated with 20 or 40 mg/kg rhGAA, respectively (8,9). Based on these results, ERT with rhGAA was approved 2006. However, from these early studies, it had become clear that IOPD patients respond differently to ERT and that its efficacy is suboptimal, even when treatment starts early (3,10).
In the years since approval of ERT, it has also become clear that IOPD is a multisystemic disorder, and that individuals receiving this kind of treatment develop clinical symptoms not known in the pre-ERT era. Features of this new IOPD phenotype include cardiac, speech, hearing, musculoskeletal, respiratory, swallowing, and neurocognitive symptoms (11,12).
This review describes the new phenotype of IOPD, summarizes current knowledge regarding therapy, discusses the effects and limitations of ERT, and addresses unmet needs in this specific population.
Long-term overall and ventilator-free survival
Several studies including smaller numbers of patients have assessed the long-term outcome of IOPD in different countries within the last 10 years (13-19). Although these data collections vary substantially in follow-up time, age at start of therapy, assessment of CRIM-status, use of newborn screening, and immunomodulation in CRIM-negative patients, some basic conclusions can be made (Table 1). Notably, the study by Chien et al. following a group of Taiwanese CRIM-positive patients diagnosed through newborn screening for 28–90 months yielded by far the best results: this subset of patients stands a very good chance to remain ventilator-free during early childhood and to achieve independent walking at a near normal age (16). Similarly, Prater and colleagues, who analyzed the outcome of an American cohort by including some individuals participating in the pivotal trials, found that no CRIM-negative subject had reached an age of 5 years, thus, again emphasizing the high impact of CRIM-status on prognosis (14). A comparison of the results from countries without newborn screening such as the UK, Germany, the Netherlands, and Italy, reveals that a substantial number of children die within the first years of life despite ERT (15,17-19). In the German cohort, for example, only 40% of children were alive and not ventilated at last follow-up (17). However, this sobering balance may be flawed due to the fact that all these studies included a substantial number of CRIM-negative subjects who did not receive immunomodulation, which is assumed to improve the outcome (see below).
Table 1. Synopsis of studies assessing outcome in IOPD.
| Region | N | Start of ERT (months) | Deceased | Ventilator-dependent | Walking | Reference |
|---|---|---|---|---|---|---|
| UK | 20 | 6.5 (0.5–32) | 7 (35%) | 6 (30%) | 4 (20%) | (13) |
| US* | 17 | ≤6 | 6 (35%) | 0 (0%) | 7 (41%) | (14) |
| NL | 11 | 3.1 (0.1–8.3) | 3 (27%) | 2 (18%) | 6 (55%) | (15) |
| T** | 10 | <1 | 0 (0%) | 0 (0%) | 10 (100%) | (16) |
| D | 23 | 2.8 (0.1–8.1) | 10 (43%) | 4 (17%) | 9 (39%) | (17) |
| UK | 33 | 0.5 (0.1–32) | 13 (40%) | 8 (24%) | 10 (30%) | (18) |
| I | 28 | 4 (0.2–11) | 9 (32%) | 13 (46%) | 7 (25%) | (19) |
ERT, enzyme replacement therapy; IOPD, infantile-onset Pompe disease; UK, United Kingdom; US, United States; NL, The Netherlands; T, Taiwan; D, Germany; I, Italy; *, age ≥5 years as inclusion criteria; all 6 eligible CRIM-negative patients had died before this age. **, use of newborn screening.
Musculoskeletal dysfunction
IOPD patients receiving ERT display residual axial muscular hypotonia and muscle weakness, ranging from mild (independent walking), moderate (free sitting), or severe (no sitting without support). Distribution of muscle weakness differs from what is seen in patients with milder forms of the disease, since the facial muscles, the neck flexors, the hip extensors, and the foot dorsiflexors are particularly affected (20). After a few years of treatment, even subjects responding initially well to ERT and achieving independent walking show progression of muscle weakness. Older ambulant IOPD patients have a characteristic gait pattern with forward leaning of the trunk and increased lordosis due to pelvic tilt, as well as steppage gait caused by prominent weakness of the foot dorsiflexors (20,21). In many subjects, progressive skeletal muscle weakness results in loss of acquired motor milestones (17-19). Marked lumbar hyperlordosis in conjunction with a progressive scoliosis requires spinal fusion in some subjects during the second decade of life (Figure 1). Although fibrosis does not seem to be a prominent feature of muscle pathology in IOPD (Figure 2), the disproportional strength of foot flexors and extensors may necessitate intermittent use of ankle foot orthoses (11,20). As in other neuromuscular disorders IOPD patients require provision with orthopedic devices and wheelchairs as appropriate, while immobility predisposes to osteopenia (11).
Figure 1.
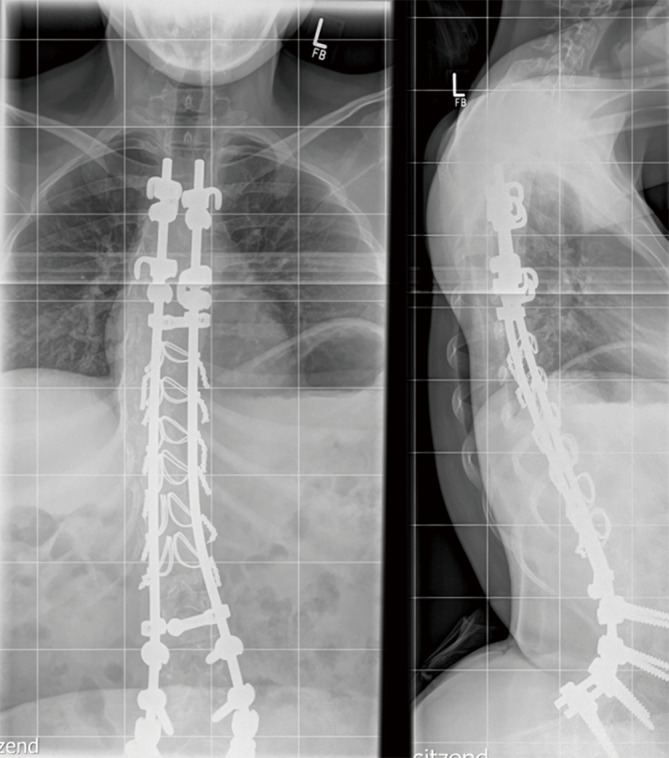
Spinal fusion due to scoliosis and marked hyperlordosis in a 16-year-old girl with IOPD. IOPD, infantile-onset Pompe disease.
Figure 2.

Muscle biopsy findings (right lateral vastus muscle) in a CRIM-positive patient with IOPD achieving free walking at age 19 months and still doing so at age 8 years. Biopsies were taken at age 6 months before start of ERT (left panel) and at age 5 years (right panel). H&E staining shows a progressive vacuolar myopathy, while Desmin staining depicts absent desmin in many fibers, reflecting increasing myofibrillar damage. Resin-PAS semithin (R-PAS) sections show distinct glycogen accumulation (stained red) at age 6 months and persistence of such fibers in conjunction with fibers full of empty vacuoles mirroring abnormal autophagy at age 5 years. Notice that fibers being in different stages of disease pathology are located side by side. Electron microscopy (TEM) depicts a fiber with normal myofibrils (lower part of the image) and a completely destroyed one (upper part of the image) at age 6 months, and a fiber with normal myofibrills, but marked lysosomal (Lys) and extralysosomal (arrow) glycogen deposits. CRIM, cross-reactive immunological material; IOPD, infantile-onset Pompe disease; ERT, enzyme replacement therapy. Length of the scale bar in is 20 µm in all light microscopic images.
The reasons for the variable efficacy of ERT and the progression of disease with time are manifold and only imperfectly understood (12). Known factors include age at start of therapy and pre-treatment muscle pathology, CRIM-status and antibody titers, distribution of type I and II fibers, and altered autophagy (12). Glycogen accumulation and autophagic vacuoles are the most prominent features detected in muscle biopsies from patients with IOPD (22,23). It has been proposed that progression of muscle pathology in IOPD is reflected by incremental enlargement of glycogen-filled lysosomes resulting in lysosomal rupture and release of glycogen and toxic substances into the cytosol, followed by extralysosomal glycogen accumulation and finally complete destruction of the contractile elements and bloating of the cell due to water influx (22). Repeated biopsies during the course of disease have shown a reduction of glycogen storage during ERT in some patients (22,23), but an increase of autophagic vacuoles even despite good response to ERT (20,22) (Figure 2). This corroborates studies by Raben and co-workers demonstrating that abnormal autophagy is a mechanism contributing to progressive muscle pathology in IOPD (24), which is probably not well targeted by ERT (12). Moreover, substantial extralysosomal glycogen deposition before ERT start has also been shown in individuals with IOPD (21,23,25). rhGAA has its maximum activity at the acidic pH of the lysosome. Therefore, it is questionable whether glycogen located outside lysosomes can be degraded by ERT (23).
Cardiac dysfunction
In IOPD, the virtually complete GAA deficiency causes accumulation of glycogen in cardiomyocytes and also in the cardiac conduction system (26). A severe HCM with small ventricular lumen, outflow tract obstruction, and reduced ejection fraction, evolving in the first months of life is a hallmark of IOPD. This causes tachycardia, weak sucking, failure to thrive, and profound sweating and dyspnea during mild efforts. ERT reverses cardiac hypertrophy and improves cardiac function within a few months in the vast majority of patients. However, in some individuals (presumably those who started ERT late), a dilative component of the cardiomyopathy with reduced contractility resulting in reduced exercise tolerance and chronic heart failure may unmask during follow-up (27). Apart from HCM, cardiac arrhythmias have been reported in a substantial number of patients. In the series of McDowell et al. 17% (7 out of 38 subjects) experienced different types of arrhythmias (e.g., nonsustained and sustained supraventricular tachycardia, atrial or ventricular premature beats, and ventricular tachycardia or fibrillation) (28). Similarly, Prater and colleagues reported arrhythmias in 5 out of 11 IOPD long-term survivors (14). These observations demonstrate that careful and regular monitoring of cardiac function is important, even when cardiac hypertrophy has been ameliorated (29).
Respiratory dysfunction
Although ventilatory failure is common in IOPD, lung function data are almost completely lacking. This can be explained by the need for assisted ventilation at a young age and by the concomitant marked orofacial weakness, making measurements of lung function parameters difficult. Patients with Pompe disease have inspiratory and expiratory muscle weakness (30). Expiratory muscle strength can easily be assessed by measuring the peak cough flow (30), or clinically by judging the patient’s cough. Reduced cough predisposes to mucus accumulation, atelectasis and pulmonary infection. Inspiratory muscle strength can be evaluated by determination of maximum inspiratory pressures and sniff nasal inspiratory pressures, or simply by measurement of vital capacity (VC) (30). A distinct decline of VC from the sitting to the supine position is indicative of diaphragm weakness (30). A paradoxical breathing or an abnormal breathing pattern with increased efforts when lying down are clinical signs of reduced inspiratory muscle force. IOPD patients with reduced inspiratory muscle strength spend a higher percentage of their maximum inspiratory muscle force for their resting ventilation and therefore have reduced exercise tolerance. This predisposes to acute ventilatory failure during respiratory tract infections and to chronic ventilatory failure manifesting as nocturnal hypoxemia and hypercapnia (30). Oropharyngeal muscular hypotonia and a large tongue facilitate obstructive sleep apnoea (11). Because of these reasons, respiratory function in IOPD patients should be monitored closely by clinical examination and/or lung function testing, and by polysomnography or measurement of nocturnal SaO2 and etCO2 at regular intervals (11,30). Patients should be supplied with devices supporting coughing, and non-invasive or invasive assisted ventilation should be implemented when necessary (30).
Speech and hearing dysfunction
Glycogen deposition in the inner and outer hair cells of the cochlea has been demonstrated in a knockout mouse model of Pompe disease by Kamphoven et al. (31). In line with this, Capelle et al. found sensorineural hearing loss in 10 out of 11 patients with IOPD (32). Some of them also had additional conductive or retrocochlear hearing loss. Hearing thresholds varied from 10 to 90 dB. Four out of 11 had profound hearing loss (>60 dB) at age 0–4 months, while 8 out of 10 had thresholds greater than 60 dB at age 1–6 years, which suggests that hearing loss increased with time in some and was not improved by ERT (32).
Facial and oropharyngeal muscle weakness is reflected by bilateral ptosis, an expressionless face, and a tent-shaped mouth with dropping of the lower lip (Figure 3). Severity of weakness can also be graded from mild to severe (34). IOPD patients also have reduced tongue and lip mobility in conjunction with velopharyngeal incompetence, resulting in dysfunctional articulation, hypernasal resonance, disordered phonation and prosody, and flaccid dysarthria. Altogether, this leads to a characteristic monotone hoarse and wet voice with substantially reduced speech intelligibility (11,34).
Figure 3.

Photograph of a 15 months old girl with IOPD showing marked facial hypotonia and macroglossia, and demonstrating severe reflux into to the upper third of the oesophagus [from reference (33), used with permission]. IOPD, infantile-onset Pompe disease.
The high risk of progressive hearing impairment necessitates regular assessment of hearing every 6–12 months, and fitting with hearing aids if appropriate. Oro-facial muscle function and speech can be trained by special forms of physiotherapy and language therapy. Speech and hearing problems should be kept in mind when looking for a school best suited for the patient (11,34).
Oropharyngeal and gastrointestinal dysfunction
Oropharyngeal weakness is characterized by oral dysmotility and disorganization with lip incompetence, tongue thrust, and upper esophageal sphincter dysfunction. This predisposes to oral and pharyngeal residues, nasal regurgitation, and delayed initiation of swallow (34-36). Swallowing difficulties can result in insufficient caloric intake and failure to thrive often leading to nutrition with soft foods containing high amounts of sugar. A long retention time in the oral cavity further predisposes to the development of profound caries (Figure 4). This requires monitoring of growth parameters, securing adequate nutrition with proteins, vitamins, and minerals, and caries prophylaxis (11). Dysphagia and gastro-esophageal reflux predispose to increased risk of pneumonia (Figure 3) (33). Silent aspiration should be kept in mind in subjects with recurrent upper respiratory tract infections and frequent coughing. Enteral nutrition via a nasogastric tube or via gastrostomy is important in patients with inadequate caloric intake, and Nissen fundoplication will minimize the risk of aspiration in those with severe gastro-esophageal reflux (33).
Figure 4.
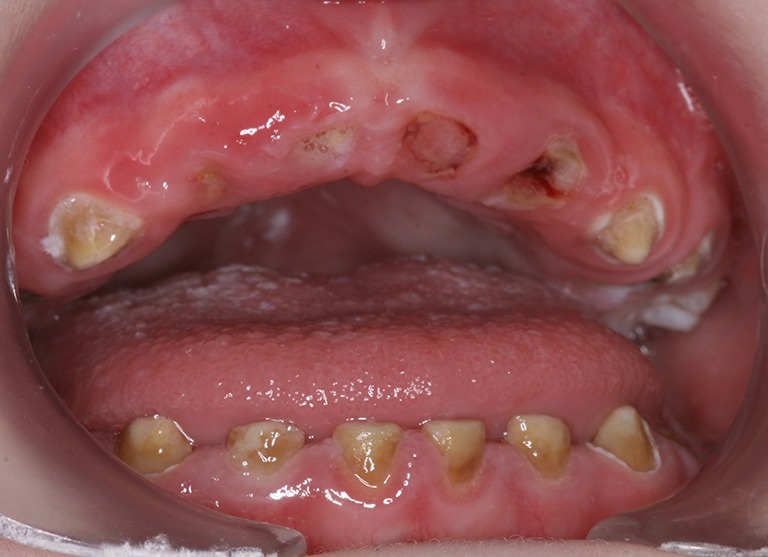
Severe tooth decay in a 4-year-old boy with IOPD. IOPD, infantile-onset Pompe disease.
Neurocognitive dysfunction
Low amounts of glycogen are stored in the brain of patients with IOPD (26). Indeed, signal alterations of the white matter and substantial neurocognitive impairment in individual IOPD patients have been reported by several authors (21,37-39) (Figure 5). However, earlier studies assessing neurocognitive function in small groups of patients showed normal or mildly delayed cognitive development (40) and no evidence of severe neurodevelopmental delay (41). But in a recent study, following prospectively a cohort of 11 IOPD patients up to 17 years of age by neuropsychological testing and brain magnetic resonance imaging, Ebbink and co-workers found slowly progressive white matter abnormalities from approximately age 2 years onwards (42). Although there seemed to be a characteristic pattern of involvement over time, there were considerable variations between patients. In addition, cognitive development ranged from normal to intellectual disabilities (42). Collectively, these studies suggest that potentially progressive neurocognitive impairment is not rare in IOPD. However, due to the limited number of patients and the difficulties in testing children with multiple handicaps, severity and frequency of intellectual disabilities in IOPD are unclear. Moreover, it is not known whether and how white matter alterations and intellectual disabilities are linked to each other.
Figure 5.

Brain magnetic resonance imaging in a 5-year-old girl with IOPD demonstrating signal hyperintensities of the central white matter in T2 and TIRM images. IOPD, infantile-onset Pompe disease.
Problems related to ERT
Based on the results of the 2006 pivotal study leading to approval of ERT, the recommended rhGAA dosage was 20 mg/kg every other week for all patients with Pompe disease. But since this advice depended only on one study comparing two small groups of patients, it has been concluded that there is no robust evidence which dosing schedule is most effective in IOPD (43). Indeed, due to the suboptimal efficacy of ERT, a substantial number of IOPD patients are treated with much higher doses than indicated in the package insert (17-19,44-46). Although there is some evidence that high doses up to 40 mg/kg every week are more effective (44), no larger study has systematically analyzed the efficacy of alternative dosing schedules (43).
In 2010, Kishnani and co-workers showed that the outcome of CRIM-negative patients treated with ERT is similar to that of untreated individuals, and significantly poorer than that of CRIM-positive subjects (3). One year later, the same group analyzed overall and ventilator-free survival in CRIM-negative and CRIM-positive with high and low rhGAA-antibody titers. The authors found that clinical outcome in the high-titer CRIM-positive group was poor relative to the low-titer CRIM-positive group, and that there were no significant differences between the CRIM-negative and high-titer CRIM-positive groups. They concluded that irrespective of CRIM-status, the prognosis of patients with high rhGAA-antibody titers is poor (10). To avoid formation of high antibody titers in CRIM-negative patients, a prophylactic immune tolerance induction with rituximab, methotrexate, and intravenous immunoglobulins (IVIG) was proposed (47) and proved to be effective, feasible, and safe (48). This approach has been adopted by many centers worldwide and can also be applied to CRIM-positive subjects with high antibody titers (15,17-19). CRIM-status can be determined by Western blot analysis using fibroblasts or peripheral mononuclear blood cells (3,49). Alternatively, it can be deduced from the results of genotyping, if the effects of a specific mutation on protein synthesis have been characterized (3,10). Since CRIM-status determination should not delay the start of ERT, this approach requires a fast identification of CRIM-negative subjects, or induction of immune tolerance in all patients with unknown CRIM-status. Antibodies against rhGAA should be regularly controlled in all IOPD patients, and the number of CD19 positive lymphocytes should be determined in those having received immune tolerance induction.
Unmet needs
ERT has transformed IOPD from a rapidly progressive disease fatal in the first year of life into a chronic condition characterized by a new complex phenotype with still high mortality and morbidity. Complex phenotype and sophisticated therapy necessitate a highly specialized multidisciplinary care. Thirteen years after approval of ERT, no international study has assessed the long-term outcome in a large cohort of patients, making it difficult to evaluate the effects of different dosage regimens and immunomodulation on outcome in IOPD. Moreover, international recommendations concerning diagnostics, treatment, and follow-up do not incorporate the recently gained knowledge about the immunological aspects.
Treating skeletal muscle by ERT remains a difficult task. Second generation recombinant enzymes with higher levels of mannose-6-phosphate residues with or without concomitant chaperone therapy are currently in clinical trials and will hopefully allow for more efficient cellular uptake, improved glycogen clearance, and reversal of autophagic defect (50,51).
Involvement of the central nervous system in IOPD seems to be an underestimated challenge (42). Again, studies including a larger number of patients examined in a standardized manner over a longer period of time would be preferable. Neurocognitive problems in IOPD cannot be treated by ERT, since rhGAA does not pass the blood-brain-barrier. Gene therapy using vectors also targeting the brain could be a chance to prevent neurodegeneration in these patients (52).
In conclusion, substantial progress has been made in the treatment of patients with IOPD within the last 10 to 15 years, but the current treatment strategies are far from being perfect and further huge efforts are necessary to improve the outcome of children affected by this most severe form of Pompe disease.
Acknowledgments
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Valle D, et al. editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001:3389-420. [Google Scholar]
- 2.van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332-40. 10.1542/peds.112.2.332 [DOI] [PubMed] [Google Scholar]
- 3.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010;99:26-33. 10.1016/j.ymgme.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van den Hout H, Reuser AJ, Vulto AG, et al. Recombinant human α-glucosi-dase from rabbit milk in Pompe patients. Lancet 2000:356:397-8. 10.1016/S0140-6736(00)02533-2 [DOI] [PubMed] [Google Scholar]
- 5.Van den Hout JM, Reuser AJ, de Klerk JB, et al. Enzyme therapy for Pompe disease with recombinant human α-glucosidase from rabbit milk. J Inherit Metab Dis 2001;24:266-74. 10.1023/A:1010383421286 [DOI] [PubMed] [Google Scholar]
- 6.Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001;3:132-8. [DOI] [PubMed] [Google Scholar]
- 7.Kishnani PS, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human α-glucosidase in infantile-onset Pompe disease. J Pediatr 2006;149:89-97. 10.1016/j.jpeds.2006.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid α-glucosidase. Major clinical benefits in infantile-onset Pompe disease. Neurology 2007:68:99-109. 10.1212/01.wnl.0000251268.41188.04 [DOI] [PubMed] [Google Scholar]
- 9.Kishnani PS, Corzo D, Leslie ND, et al. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr Res 2009;66:329-35. 10.1203/PDR.0b013e3181b24e94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729-36. 10.1097/GIM.0b013e3182174703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267-88. 10.1097/01.gim.0000218152.87434.f3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kishnani PS, Beckemeyer AA, Mendelsohn NJ. The new era of Pompe disease: advances in the detection, understanding of the phenotypic spectrum, pathophysiology, and management. Am J Med Genet C Semin Med Genet 2012;160C:1-7. 10.1002/ajmg.c.31324 [DOI] [PubMed] [Google Scholar]
- 13.Chakrapani A, Vellodi A, Robinson P, et al. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis 2010;33:747-50. 10.1007/s10545-010-9206-3 [DOI] [PubMed] [Google Scholar]
- 14.Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med 2012;14:800-10. 10.1038/gim.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, et al. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis 2015;38:305-14. 10.1007/s10545-014-9707-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr 2015;166:985-91.e1. 10.1016/j.jpeds.2014.10.068 [DOI] [PubMed] [Google Scholar]
- 17.Hahn A, Praetorius S, Karabul N, et al. Outcome of patients with classical infantile pompe disease receiving enzyme replacement therapy in Germany. JIMD Rep 2015;20:65-75. 10.1007/8904_2014_392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broomfield A, Fletcher J, Davison J, et al. Response of 33 UK patients with infantile-onset Pompe disease to enzyme replacement therapy. J Inherit Metab Dis 2016;39:261-71. 10.1007/s10545-015-9898-5 [DOI] [PubMed] [Google Scholar]
- 19.Parini R, De Lorenzo P, Dardis A, et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J Rare Dis 2018;13:32. 10.1186/s13023-018-0771-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Case LE, Beckemeyer AA, Kishnani PS. Infantile Pompe disease on ERT: update on clinical presentation, musculoskeletal management, and exercise considerations. Am J Med Genet C Semin Med Genet 2012;160C:69-79. 10.1002/ajmg.c.31321 [DOI] [PubMed] [Google Scholar]
- 21.Schänzer A, Giese K, Görlach J, et al. Severe distal muscle involvement and mild sensory neuropathy in a boy with infantile onset Pompe disease treated with enzyme replacement therapy for 6 years. Neuromuscul Disord 2019;29:477-82. 10.1016/j.nmd.2019.03.004 [DOI] [PubMed] [Google Scholar]
- 22.Thurberg BL, Lynch Maloney C, Vaccaro C, et al. Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Invest 2006;86:1208-20. 10.1038/labinvest.3700484 [DOI] [PubMed] [Google Scholar]
- 23.Schänzer A, Kaiser AK, Mühlfeld C, et al. Quantification of muscle pathology in infantile Pompe disease. Neuromuscul Disord 2017;27:141-52. 10.1016/j.nmd.2016.10.010 [DOI] [PubMed] [Google Scholar]
- 24.Raben N, Ralston E, Chien YH, et al. Differences in the predominance of lysosomal and autophagic pathologies between infants and adults with Pompe disease: implications for therapy. Mol Genet Metab 2010;101:324-31 10.1016/j.ymgme.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schänzer A, Giese K, Viergutz L, et al. Letter to the Editors: Concerning "Divergent clinical outcomes of alpha-glucosidase enzyme replacement therapy in two siblings with infantile-onset Pompe disease treated in the symptomatic or pre-symptomatic state" by Takashi et al. and Letter to the Editors by Ortolano et al. Mol Genet Metab Rep 2017;12:33-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pena LD, Proia AD, Kishnani PS. Postmortem Findings and Clinical Correlates in Individuals with Infantile-Onset Pompe Disease. JIMD Rep 2015;23:45-54. 10.1007/8904_2015_426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn A, Schmidt D, Hagel KJ, et al. Monitoring cardiac function by B-type natriuretic peptide (BNP) in patients with infantile Pompe's disease treated with recombinant alpha-glucosidase. Clin Lab 2006;52:615-9. [PubMed] [Google Scholar]
- 28.McDowell R, Li JS, Benjamin DK, Jr, et al. Arrhythmias in patients receiving enzyme replacement therapy for infantile Pompe disease. Genet Med 2008;10:758-62. 10.1097/GIM.0b013e318183722f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Capelle CI, Poelman E, Frohn-Mulder IM, et al. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int J Cardiol 2018;269:104-10. 10.1016/j.ijcard.2018.07.091 [DOI] [PubMed] [Google Scholar]
- 30.Mellies U, Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir Med 2009;103:477-84. 10.1016/j.rmed.2008.12.009 [DOI] [PubMed] [Google Scholar]
- 31.Kamphoven JH, de Ruiter MM, Winkel LP, et al. Hearing loss in infantile Pompe's disease and determination of underlying pathology in the knockout mouse. Neurobiol Dis 2004;16:14-20. 10.1016/j.nbd.2003.12.018 [DOI] [PubMed] [Google Scholar]
- 32.van Capelle CI, Goedegebure A, Homans NC, et al. Hearing loss in Pompe disease revisited: results from a study of 24 children. J Inherit Metab Dis 2010;33:597-602. 10.1007/s10545-010-9144-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirschburger M, Hecker A, Padberg W, et al. Treatment of gastroesophageal reflux with nissen fundoplication and gastrostomy tube insertion in infantile pompe's disease. Neuropediatrics 2009;40:28-31. 10.1055/s-0029-1231066 [DOI] [PubMed] [Google Scholar]
- 34.van Gelder CM, van Capelle CI, Ebbink BJ, et al. Facial-muscle weakness, speech disorders and dysphagia are common in patients with classic infantile Pompe disease treated with enzyme therapy. J Inherit Metab Dis 2012;35:505-11. 10.1007/s10545-011-9404-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swift G, Cleary M, Grunewald S, et al. Swallow prognosis and follow-up protocol in Infantile Onset Pompe Disease. JIMD Rep 2017;33:11-17. 10.1007/8904_2016_576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones HN, Muller CW, Lin M, et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 2010;25:277-83. 10.1007/s00455-009-9252-x [DOI] [PubMed] [Google Scholar]
- 37.Rohrbach M, Klein A, Köhli-Wiesner A, et al. CRIM-negative infantile Pompe disease: 42-month treatment outcome. J Inherit Metab Dis 2010;33:751-7. 10.1007/s10545-010-9209-0 [DOI] [PubMed] [Google Scholar]
- 38.Broomfield A, Fletcher J, Hensman P, et al. Rapidly Progressive White Matter Involvement in Early Childhood: The Expanding Phenotype of Infantile Onset Pompe? JIMD Rep 2018;39:55-62. 10.1007/8904_2017_46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebbink BJ, Poelman E, Plug I, et al. Cognitive decline in classic infantile Pompe disease: An underacknowledged challenge. Neurology 2016;86:1260-1. 10.1212/WNL.0000000000002523 [DOI] [PubMed] [Google Scholar]
- 40.Ebbink BJ, Aarsen FK, van Gelder CM, et al. Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology 2012;78:1512-8. 10.1212/WNL.0b013e3182553c11 [DOI] [PubMed] [Google Scholar]
- 41.Spiridigliozzi GA, Heller JH, Case LE, et al. Early cognitive development in children with infantile Pompe disease. Mol Genet Metab 2012;105:428-32. 10.1016/j.ymgme.2011.10.012 [DOI] [PubMed] [Google Scholar]
- 42.Ebbink BJ, Poelman E, Aarsen FK, et al. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Dev Med Child Neurol 2018;60:579-86. 10.1111/dmcn.13740 [DOI] [PubMed] [Google Scholar]
- 43.Chen M, Zhang L, Quan S. Enzyme replacement therapy for infantile-onset Pompe disease. Cochrane Database Syst Rev 2017;11:CD011539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Gelder CM, Poelman E, Plug I, et al. Effects of a higher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompe disease: an open-label single-center study. J Inherit Metab Dis 2016;39:383-90. 10.1007/s10545-015-9912-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Case LE, Bjartmar C, Morgan C, et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul Disord 2015;25:321-32. 10.1016/j.nmd.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 46.Desai AK, Walters CK, Cope HL, et al. Enzyme replacement therapy with alglucosidase alfa in Pompe disease: Clinical experience with rate escalation. Mol Genet Metab 2018;123:92-6. 10.1016/j.ymgme.2017.12.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banugaria SG, Prater SN, Patel TT, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile pompe disease: a step towards improving the efficacy of ERT. PLoS One 2013;8:e67052. 10.1371/journal.pone.0067052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kazi ZB, Desai AK, Berrier KL, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight 2017;2. doi: . 10.1172/jci.insight.94328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bali DS, Goldstein JL, Rehder C, et al. Clinical Laboratory Experience of Blood CRIM Testing in Infantile Pompe Disease. Mol Genet Metab Rep 2015;5:76-9. 10.1016/j.ymgmr.2015.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pena LDM, Barohn RJ, Byrne BJ, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naïve and alglucosidase alfa-treated patients with late-onset Pompe disease: A phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord 2019;29:167-86. 10.1016/j.nmd.2018.12.004 [DOI] [PubMed] [Google Scholar]
- 51.Xu S, Lun Y, Frascella M, et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight 2019;4:e125358. 10.1172/jci.insight.125358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim JA, Yi H, Gao F, et al. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid α-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol Ther Methods Clin Dev 2019;12:233-45. 10.1016/j.omtm.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]