

Abstract
Pompe disease is classified by age of onset, organ involvement, severity, and rate of progression in two main forms: the first one, infantile onset Pompe disease (IOPD), presents before the age of 12 months with generalized muscle weakness, hypotonia, respiratory distress, and hypertrophic cardiomyopathy as main clinical features. The second form, late onset Pompe disease (LOPD), is characterized by an onset at the age of 12 months to adulthood, hyperCKemia, and limb-girdle and axial muscle weakness, often complicated by respiratory muscles degeneration. In the last 10–15 years, an increasing interest in Pompe disease has led to multiple studies in an effort to clarify the emerging clinical aspects, to find out the best diagnostic tools to identify the disease as early as possible, and to offer new therapeutic options apart from enzyme replacement therapy (ERT). Since 2006, ERT—the first treatment for Pompe disease—has been universally accepted in the majority of countries all over the world. Although for years Pompe disease has been primarily considered a muscle disorder, nowadays it is clear that the involvement of several other organs has changed the cultural approach to this entity which is now viewed as a multisystem disorder. The emerging clinical aspects have greatly expanded the spectrum of the disease manifestations. In fact, central, peripheral, and autonomous nervous systems are often involved; vascular malformations and heart involvement are frequently observed; musculoskeletal and bone changes as well as oro-gastrointestinal and urinary tract alterations have been better defined. A great deal of effort has been made to clarify the clinical aspects of Pompe disease, to raise awareness of the LOPD patients’ problems and to improve their quality of life.
Keywords: Late onset Pompe disease presentation (LOPD presentation), myopathy, Pompe disease, aneurysms, hyperCKemia
Introduction
For many years, Pompe disease has been mainly considered a muscle disorder, but several recent reports and studies have demonstrated the involvement of other organs or apparatuses, thus warranting the classification of the disease as a multisystem disorder (Table 1). Pompe disease (Glycogen storage type II, GSDII; OMIM #232300) is a rare autosomal recessive disorder caused by mutations of the acid α-glucosidase (GAA) gene, encoding acid maltase. Deficiency of GAA leads to accumulation of lysosomal glycogen in several body tissues, especially in skeletal and respiratory muscles and heart (1). The severity of the disease is quite variable in terms of age of onset, symptoms progression, and different degrees of involvement of affected organs.
Table 1. LOPD clinical multisystem involvement and related investigations.
| Organ involvement | Clinical manifestations | Investigations |
|---|---|---|
| 1. Skeletal muscle | Exercise intolerance/fatigue | Clinical examination |
| Myalgia/hyperCKemia | EMG/ENG | |
| Axial and proximal muscles weakness (> lower limbs) | Muscle MRI | |
| Scapular winging | ||
| 2. Respiratory | Morning headache and sleepiness | PFT (FVC, MIP and MEP at upright and supine position) |
| Sleep apnoea | Polysomnography | |
| Shortness of breath | Respiratory muscles MRI | |
| Impaired cough | Bronchoscopy | |
| Dyspnea (more at supine position) | ||
| 3. Central nervous system/cerebrovascular system | Vertebrobasilar dolichoectasia | Angio-CT |
| Intracranial aneurysms | Angio-MRI | |
| Stroke | Brain MRI | |
| Cerebral hemorrhages | ||
| Lacunar encephalopathy | ||
| Sensorineural deafness | Audiometry | |
| 4. Cognitive and emotional | Mild cognitive impairment | Neuropsychological battery tests |
| Anxiety/depression | ||
| Executive functions impairment | ||
| 5. Peripheral and autonomic nervous system | Paraesthesia at limb extremities | EMG/ENG |
| Burning feet | LEPS | |
| Skin biopsy | ||
| 6. Vascular system | Dilated arteriopathy | Vessels ultrasound |
| Aortic stiffness | Angio-TC | |
| Thoracic and basilar aortic aneurysms | ||
| 7. Heart | Rhythm disturbances | EKG |
| Cardiac hypertrophy | Heart ultrasound | |
| 8. Musculoskeletal and bone | Osteopenia/osteoporosis | Densitometry |
| Vertebral fractures | X-rays | |
| Rigid/bent spine syndromes | Bone MRI | |
| Scoliosis/kyphosis/hyperlordosis | X-rays | |
| 9. Dental-oro gastrointestinal and urinary | Macroglossia | Tongue ultrasound |
| Dysphagia | Gastrointestinal tests | |
| Early satiety | ||
| Chronic diarrhoea | ||
| Urinary/bowel incontinence | Urodynamic studies | |
| 10. Blood | PAS-positive vacuolated lymphocytes | Blood smear |
EMG, electromyography; ENG, electroneurography; MRI, magnetic resonance imaging; PFT, pulmonary function tests; FVC, forced vital capacity; MIP, maximum inspiratory pressure; MEP, maximum expiratory pressure; LEPS, laser evoked potentials.
Pompe disease presents as two main forms: one with an infantile onset (IOPD), classically characterized by cardiomyopathy, respiratory insufficiency and severe muscle hypotonia, usually starting in the first year or even in the first days of life. The second form is a late onset type, and it may begin any time from one year of age to adulthood [late onset Pompe disease (LOPD)] with a chronic course that may progress to significant motor disability and respiratory insufficiency (2).
Classically, LOPD clinical presentations are represented by presymptomatic hyperCKemia, often with exercise intolerance, fatigue or myalgia progressing to limb-girdle and axial weakness with respiratory failure. Respiratory distress alone, as a starting symptom, seems to be very rare; several cases that have been reported with this onset, when carefully restudied, have shown that hyperCKemia (3), exercise intolerance or mild muscle weakness were already present before respiratory involvement (Figure 1).
Figure 1.
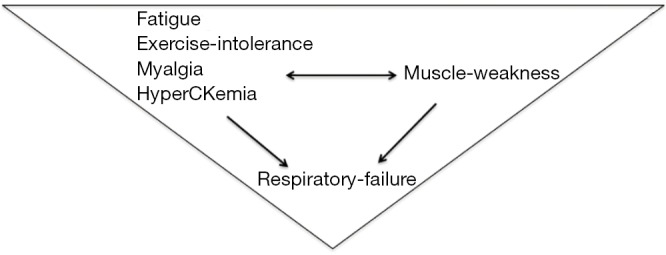
The LOPD “triangle” of clinical presentations. LOPD, late onset Pompe disease.
Epidemiology of Pompe disease is a subject of debate: it is usually reported that the combined frequency of both forms of the disease is 1:40,000/60,000 although several countries have provided a very different account (4).
Skeletal muscle involvement
LOPD is characterized by progressive muscle weakness with respiratory insufficiency that may present at any age greater than 12 months, leading to premature disability, supported ventilation and, eventually, to death (5). Initial symptoms may appear anytime from early infancy to late adulthood and are usually represented by myalgia, exercise intolerance and fatigue that may precede muscle weakness. These symptoms are often ignored and taken as unwillingness of individuals to exercise. These clinical disturbances are usually accompanied by variably increased serum creatine kinase (CK) values. HyperCKemia is a very sensitive but unspecific laboratory parameter; in addition, CK elevation is often complemented by high levels of alanine transaminase (ALT), aspartate transaminase (AST) and lactate dehydrogenase (LDH). When the levels of these three enzymes are high, while CK is not measured at the same time, one can erroneously assume a diagnosis of liver rather than muscle disease. This also may explain a delayed diagnosis of Pompe disease in similar circumstances. In IOPD, CK levels are usually quite high, often reaching over 2,000 U/L, whereas in LOPD, especially in adult cases, the peak level rarely exceeds 1,000–1,500 U/L. In addition, in some cases, adult patients can have normal CK values. Muscle pain is present either at the presymptomatic stage or later, when the disease burden is more heavy. Some authors reported the presence of pain at the early phase of the disease in at least 18% of patients (1). This could also be an overlooked and underappreciated symptom; for example, in a large series of German/Dutch patients, one in two LOPD patients complained of muscle pain. Myalgia can be caused by postural problems due to muscle weakness or by prolonged physical efforts, not sustained by an adequate muscle bioenergetic level (6). Moreover, fatigue and exercise intolerance are highly disabling symptoms which can lead to physical exhaustion caused, again, by muscle weakness or by mental overtiredness or depression in advanced stages of the disease (7). Decreased pulmonary function may also contribute to the level of fatigue.
In conclusion, the combination of muscle aches, fatigue, exercise intolerance and/or hyperCKemia may constitute a presymptomatic stage of LOPD. At advanced phases of the disease, patients complain of difficulties in walking, running, performing sports, climbing stairs or standing up from the floor, bed or chair. Disease progression is quite slow and affects axial, limb-girdle and respiratory muscles, particularly the diaphragm.
Although the regional distribution of skeletal muscle impairment is variable, lower limbs and paraspinal muscles are affected first (5), followed by upper limbs and respiratory muscles. At thigh level, weakness is more evident in hip extensors, adductors and abductors, and then hip flexors; posterior thigh muscles are selectively affected with early involvement of the adductor magnus and semimembranosus and, later, of the long head of the biceps femoris and semitendinosus. Leg and foot muscles are spared or minimally involved till the later stages of the disease.
As for the scapular girdle, weakness is also present at the scapular fixators, particularly at the trapezius inferior, rhomboid, and subscapularis muscles with a later moderate involvement of serratus anterior, deltoid, and supraspinatus muscles. When the fixators of the scapula (lower trapezius, latissimus dorsi and rhomboid), as well as subscapularis and neck flexors (sternocleidomastoid) are involved, a protrusion of the scapula, defined as “winging scapula”, is clearly noticeable in resting position and as soon as the individual lifts the arms anteriorly or laterally.
Progression of motor weakness leads to appearance of scoliosis and then to lumbar hyperlordosis due to reduced strength of truncal muscles. More rarely, patients may present with a “rigid spine syndrome (RSS)”, as in other myopathic disorders.
Facial and bulbar weakness are also present in LOPD patients. It is well known that tongue weakness (often an early sign) as well as bulbar muscles involvement cause swallowing disturbances, dysphagia and dysarthria.
LOPD patients could also present an increased incidence of ophtalmologic abnormalities, such as eyelid ptosis, strabismus and, less frequently, ophthalmoplegia (Figure 2).
Figure 2.

LOPD clinical aspects. (A) Unilateral eyelid ptosis in a 64-year-old LOPD female; (B) scapular winging in a 55-year-old LOPD man; (C) positive Gower’s manoeuvre in a 48-year-old LOPD female. LOPD, late onset Pompe disease.
The clinical picture in the late stages may include progressive inability to walk, a need for mobility assistance devices or a wheelchair and, often, an assisted ventilation. Exitus is usually due to respiratory insufficiency.
Respiratory involvement
LOPD may cause a progressive muscle respiratory impairment characterized by pulmonary symptoms and respiratory failure. In these patients, abdominal muscles and diaphragm are involved resulting in a progressive deterioration of both mobility and pulmonary function. Initial signs of respiratory distress manifest as decreased sleep quality, fatigue, daytime sleepiness, headaches and decreased respiratory capacity. In fact, respiratory and abdominal wall muscle weakness cause ineffective cough and attenuation of airway protection and secretion clearance (8).
Respiratory failure is mainly due to diaphragmatic weakness (9) as well as to the abdominal wall muscles, particularly of internal obliques and rectus abdominis, with sparing of the antero-posterior chest expansion due to the activity of the intercostal muscles. The reduced strength of the diaphragm has also been attributed to the impairment of the respiratory motor neurons in addition to skeletal muscles weakness (10). This specific central nervous system pathology has been observed on autopsy; glycogen accumulation was detected in the ventral region of the spinal cord that houses the phrenic motor neurons innervating the diaphragm. In addition, the atrophy of diaphragm can be accompanied by reduction of lung height and band-like atelectasis (9). To assess respiratory failure, it is necessary to measure forced vital capacity (FVC) both in seated and supine positions, because patients may have a normal seated FVC, and, if supine FVC is not measured, the diaphragm weakness can be missed (11). A postural drop in FVC (usually more than 25% from the sitting to the supine position) is an indicator of reduced diaphragmatic strength. Other useful measurements for inspiratory muscle weakness detection are maximum inspiratory pressure (MIP) and sniff nasal inspiratory pressure (SNIP). Impaired coughing effectiveness can be evaluated by using maximum expiratory pressure (MEP) or peak cough flow (PCF). Respiratory muscle weakness also contributes to sleep-disordered breathing (SDB) and to its progression to nocturnal hypoventilation. These symptoms initially start with hypercapnia at night, eventually followed by daytime respiratory failure. Sleep disturbances and non-restorative sleep contribute to physical and mental exhaustion, causing reduced sleep quality, daytime sleepiness, and fatigue (8).
It is also important to take into consideration glycogen deposition in the lower airway smooth muscles which contributes to LOPD respiratory difficulties (12). In fact, in 2015, Yang et al. reported a 16-year-old girl presenting with progressively altered respiratory function (13). She had a pulmonary hypertension and was already on ERT and nighttime CPAP ventilation for several years. Because of her pulmonary failure, she underwent “flexible bronchoscopy”, followed by an implantation of a bronchial airway stent. After the stent, respiratory function and pulmonary hypertension stabilized and she continued ERT. A similar case was also reported by Brenn et al. in another patient receiving ERT who complained of a progressive obstructive airway and restrictive pulmonary disease: the bronchoscopy revealed a collapse of the lower trachea and bronchi, with a compression of the left main bronchus (14). These findings were likely the result of a large glycogen accumulation in the smooth muscle layers of the trachea and bronchi. Unfortunately, respiratory failure remains a major cause of death in LOPD.
Central nervous system and cerebrovascular involvement
The central and peripheral nervous systems are also involved in LOPD. In fact, several studies have demonstrated accumulation of lysosomal glycogen in brain, anterior horn cells, peripheral nerve and smooth muscles. Some autoptic studies on LOPD patients highlighted the presence of periodic acid-Schiff (PAS)-positive vacuoles within the smooth muscle cells in the tunica media of cerebral arterioles and arteries, together with numerous small aneurysmal dilatations (15,16). Clinically, the vascular smooth muscle pathology may present in brain with a cerebral aneurysm or dilative arteriopathy. These aspects suggest that vacuolar degeneration and glycogen storage alter arterial walls architecture and may contribute to aneurysms formation. In addition, weakened glycogen-filled muscle myocytes may be the origin of dolichoectasia, which reduces blood flow and may cause stenosis of the cerebral artery in some cases (17).
In addition to blood vessel occlusion, the cerebral artery walls may break leading to a fatal rupture of aneurysms, haemorrage, and coma. However, the origins of cerebrovascular abnormalities are not fully understood; inadequate cerebral oxygenation and glycogen accumulation in cerebral arteries may reduce the smooth muscle integrity and alter their anti-thrombotic properties suggesting a hypoxic-ischemic pathogenic mechanism (18). Additional mechanisms have been hypothesized to explain the dilation of resistance vessels: (I) elevated pressure of carbon dioxide can cause vasodilation and (II) glycogen-filled vacuoles in the vessels walls (15) could interfere with the production of extracellular matrix proteins such as collagen and elastin of matrix metalloproteinases or vasoactive substances like nitric oxide. In adult patients with Pompe disease, the abnormalities of cerebral arteries increase with the duration of the disease, resulting in an augmented risk of cerebral ischemic or hemorrhagic stroke.
These cerebrovascular abnormalities have been described as emerging aspects of LOPD, often with adjunctive features such as brain microbleeds or lacunar encephalopathy (18). Vascular alterations are found predominantly at the posterior vascular circulation because of the weakness of intracranial arterial elastic layer which is more vulnerable to the formation of aneurysm expansion and vascular diseases.
Aneurysm pathophysiology is regulated by complex interactions between anatomical (vascular anatomy, aneurysm size, shape, location), structural (arterial walls), and hemodynamic factors (flow patterns, wall shear stress) that play a role in aneurysm formation, growth and rupture.
Vertebrobasilar dolichoectasia is characterized by expansion, elongation and tortuosity of the vertebrobasilar arteries that may become symptomatic due to brainstem compression, obstructive hydrocephalus and/or haemorrhage (Figure 3). The natural histories of more than 200 patients with non-classic IOPD and LOPD showed that cerebral aneurysm rupture caused 3% of deaths, an unexpected high number for an underevaluated symptom (5).
Figure 3.

Brain imaging in LOPD. (A) CT-scan showing a vertebral dolichoectasia in a 64-year-old LOPD female; (B) T2 weighted scan showing a lacunar encephalopathy (Fazekas score 2). LOPD, late onset Pompe disease.
In LOPD patients, magnetic resonance imaging (MRI) and magnetic resonance angiography (MRA) may reveal cerebrovascular abnormalities in vertebrobasilar, cerebral and carotid arteries. In fact, it has been suggested that MRI and MRA should be included in the clinical workup of all adult Pompe disease patients (18).
The incidence of cerebrovascular abnormalities in LOPD patients is higher compared to the healthy, aged-matched populations, suggesting that an early detection of cerebrovascular malformations could avoid life-threatening events such as sub-arachnoid haemorrhage or brainstem compression.
Finally, spinal motor neurons seem to be quite sensitive to the accumulation of an excess of glycogen with a swollen appearance of cells (16), increasing the burden of motor dysfunctions in LOPD patients.
Hearing loss
In LOPD, otoacoustic alterations have been rarely investigated. Evaluation of hearing loss was performed in 24 GSDII patients, mainly with the infantile form, but including juvenile patients: a sensorineural audiological defect was present in almost all infantile patients but rarely in juvenile cases (19). Moreover, in a group of adult Pompe disease patients, hearing dysfunction occurred frequently but no more than in general population (20). Another study investigated more deeply the origin of sensorineural, conductive or mixed hearing impairment. In a cohort of 20 patients, hearing loss was detected in 21/40 ears (52.5%); the affected patients had sensorineural (57%) or conductive (33%) hearing loss, whereas 10% presented with a mixed pattern. This study revealed that the auditory system impairment with prominent cochlear involvement was more recurrently present than previously thought (21).
Cognitive and emotional involvement
Cognitive impairment has been reported in IOPD patients but less frequently in LOPD. Studies in two different small groups of patients found no relevant alterations of the cognitive profile apart from a mild dysfunction in attentive and executive functions (22,23). More recently, a comprehensive study in a larger cohort has been performed to explore the potential impact of brain damage on cognitive functions including a neuropsychological evaluation. Cognitive ability was moderately compromised in 57% of patients with a prevalent impairment of visual-constructive abilities and executive functions. These data have shown a cortical impairment strictly related to cognitive status (18). Depression and anxiety have also been described in LOPD patients (7) due to multiple reasons, not necessarily linked to the specific disease condition (24).
Peripheral nerve system involvement
Peripheral nerve involvement has also been described in LOPD, including the results of autopsies. Glycogen storage has been detected in Schwann cells within the spinal cord and in sural nerve (25). The presence of lysosomal-bound glycogen was described in the axons of intramuscular nerves (26) as well as in Schwann cells of both myelinated and unmyelinated nerve fibers with a large prevalence in myelinated nerve fibers (27).
Some patients complain of painful paresthesias, mainly distally at lower limbs—a condition known as a “small fiber neuropathy (SFN)” that involves myelinated and unmyelinated nerves and accounts for neuropathic pain and temperature alterations due to glycogen deposition in the Schwann cells of peripheral nerves. In fact, SFN was observed in two patients after skin biopsy: one of them showed epidermal nerve fibers swellings, likely related to axonal degeneration, whereas the second patient had a loss of epidermal nerve fiber density and an absence of intraepidermal nerve fibers at the distal calf site. In a larger group, 22 (50%) of 44 individuals with LOPD, showed neuropathic involvement such as SFN (25). The presence of SFN with neuropathic pain could be an important clue to alert physicians of a potential autonomic dysfunction that may also cause orthostasis, gastrointestinal (GI) dysfunction, dry eyes, and sexual dysfunction.
Musculoskeletal and bone involvement
In the last few years, an important group of pathologies, strictly related to the musculoskeletal and bone involvements came to light: osteoporosis, bone fractures, scoliosis, kyphosis, hyperlordosis, RSS or bent spine syndrome (BSS), all likely due to the progressive degeneration of bone and joints related to muscular structures weakness.
RSS is a neuromuscular disorder that affects patients of all ages and is characterized by a very limited flexion of cervical and dorsolumbar spine. This clinical picture can be associated with severe restrictive respiratory changes that, in turn, may lead to respiratory distress. LOPD patients with RSS present with a moderate to severe limbs muscle involvement, and, often, with an extreme difficulty to walk. RSS has been described in about 20% of LOPD patients and is more prevalent in the adult form than in the infantile form (28).
A similar syndrome—BSS, also called “camptocormia”—is characterized by progressive weakness of the extensor muscles of the spine causing an anterior curvature of the thoracolumbar tract. The patient may get some relief with passive extension of the spine itself. In this case, the erector spinae muscles are often massively infiltrated by fat and connective tissue. Other abnormal conditions related to the axial skeleton are represented by scoliosis, kyphosis and lumbar lordosis (29).
In 2011, Roberts et al. (30), analysing the patients data from the international Pompe Registry, found that the prevalence of scoliosis in Pompe patients was as high as about 33% (235 of 711 patients), but it seemed to be more recurrent in patients with onset of symptoms in childhood or adolescence. They also observed that a large percentage of patients later needed a respiratory support because of highly reduced pulmonary function.
Kyphosis and lumbar lordosis may also affect LOPD patients. Kyphosis is characterized by an excessive turning of the back at the thoracic or lumbar region, whereas hyperlordosis shows an exaggerated spinal extension that is more prominent at the lumbar region due to abdominal and hip extensor weakness. Unfortunately, some LOPD patients may be sequentially affected by scoliosis with kyphosis and lordosis.
There is an emerging evidence that bone mass density (BMD), e.g., osteoporosis and fractures are often present in subjects with Pompe disease (31,32). According to the literature data, there is no current evidence suggesting that alpha-glucosidase deficiency has a direct role in bone metabolism. On the other hand, a combination of degenerative myopathy, loss of weight-bearing ability and reduction of force may lead to secondary loss of bone mass (33) and progressive bone degeneration.
In a recent study by Bertoldo et al. (34), a high prevalence of morphometric vertebral fractures was found: in fact, at least one fracture, although asymptomatic, was detected in 17 of the 22 (77%) patients studied. However, the authors emphasize that fracture prevalence was independent of muscular and respiratory functional parameters and of genotype (34). Bone fractures have been reported not only in infants and children, but also in adults (mostly in the long bones such as femur and humerus), mainly when they are nearly immobile or bedridden. In LOPD patients, there is a greater involvement of the femoral neck compared to the lumbar spine. In fact, the “trabecular bone” (e.g., lumbar spine) is mainly influenced by general and systemic factors (e.g., hormone status), whereas the cortical bone (e.g., femur) is more subject to mechanical influences such as gravity, muscle mass, and muscle strength.
Screening for asymptomatic vertebral fractures should be routinely performed in LOPD, irrespective of the disease severity (34). Importantly, physical activity and exercise training may help to reduce bone involvement in these patients.
Cardiac involvement
The first case of Pompe disease was a 7-month-old girl with severe cardiomyopathy, described in 1932 by Dr. JC Pompe. The case was characterized by massive accumulation of glycogen in vacuoles in all the examined tissues, particularly, in the heart. In the infantile classic form, the heart is nearly always severely affected; in contrast, in the adult form only a minority of patients have cardiovascular involvement, including atrio-ventricular conduction abnormalities and myocardial hypertrophy.
In a large group of patients, Soliman et al. (35) reported only one patient with permanent atrial fibrillation and a second one with cardiac hypertrophy. Other studies did not reveal any major cardiac abnormalities; only minor changes were observed, most likely related to other factors such as hypertension and/or advanced age. In a study of 87 patients, Forsha et al. (36) analysed cardiac features before and after ERT; at baseline, a short PR interval was present in 10%, decreased left ventricular systolic function in 7%, and a mildly elevated left ventricular mass in 5%. No abnormalities were detected after ERT.
In 2012, Angelini et al., after having followed 74 patients on ERT over 4 years, found that 13% of them showed a variable degree of heart hypertrophy (37). Rhythm disturbances have been also reported in LOPD, especially as sinus arrhythmias, supraventricular tachycardia, Wolff-Parkinson-White syndrome or atrio-ventricular block that required pacemaker implantation (7,38).
Vascular involvement
In LOPD patients, extracerebral vascular involvement may include cervical arteries, thoracic aorta, iliac and renal arteries, the latter leading to a possible kidney infarct (39). Evidence of dilated arteriopathy, involving primarily the ascending thoracic aorta, was reported in five females (40). One individual had a bicuspid aortic valve and developed dissection, whereas another individual with a juvenile onset of the disease had both thoracic and basilar aortic aneurysms. Furthermore, aortic stiffness leading to increased blood pressure, a predictor of cardiovascular accidents, was observed in a cohort of 17 LOPD patients (41). Aortic pathology from an autopsy of an individual who died because of respiratory failure, showed glycogen accumulation in the vascular smooth muscle cells of the aorta (42), as well as of the carotid artery, coronary arteries and small vessels of numerous organs including heart, skeletal muscle, skin, small intestinal serosa, kidney, liver, diaphragm, lung, and cerebellum (16). Moreover, Nemes et al. reported a significant increase in diastolic aortic diameter and aortic stiffness index in patients with Pompe disease compared to age and gender-matched controls (41).
Peripheral blood involvement
Glycogen storage is found in lysosomes throughout the body, even in the peripheral blood, particularly, in lymphocytes (43). Vacuolated lymphocytes, e.g., lysosomes filled with non-degraded material, may occur in many storage disorders. In a retrospective review of a database, Anderson et al. identified vacuolated lymphocytes in 156 of 2,500 samples of which 23% were detected in patients with Pompe disease—15% in the classic infantile form and 8% in milder forms presenting in childhood, adolescence, or adulthood (44). A unique feature of Pompe disease pathology is that lymphocytes stain positively with PAS, indicating glycogen storage. The test can be performed by using a simple and inexpensive technique such as the Blood Smear Examination (BSE). Consequently, this kind of examination has been proposed as a possible screening tool for Pompe disease. In addition, it has been recently shown that the number of glycogen-filled vacuoles in lymphocytes may decrease after ERT, thus, making BSE a possible biomarker of therapeutic efficiency (45).
Endocrine involvement
Endocrine system has been rarely studied in LOPD. Very few reports tried to explain the possible relationships between this system and LOPD. In some infantile cases, different glands—thyroid, parathyroid, adrenal cortex, and pituitary—have been found infiltrated by glycogen. Most recently, Schneider et al. reported a high prevalence of hypothyroidism in a group of ten patients with LOPD compared to the general adult population, suggesting a link between the enzyme deficiency and thyroid function (46).
Dental-oro-gastrointestinal and urinary tract involvements
Dental abnormalities have been described only in few cases, mainly in children with some developmental abnormalities. Large gingival overgrowth has been reported in a single case of an 8-year-old girl with IOPD, and several comorbidities could have contributed to this dental pathology (47).
In this review (see section “Skeletal muscle involvement”), we have already mentioned the frequent presence of macroglossia and lingual weakness in LOPD. Therefore, the patients need to be carefully examined by physicians to avoid severe consequences such as dysarthria and dysphagia (48). These problems usually stem from bulbar muscle weakness, and, in fact, oropharyngeal dysphagia has been identified in the adult population (49). However, a similar clinical pattern has recently been described in the so-called “long-term survivors”, a group of ERT-treated infants who are still alive after at least 5 years of therapy (50). Although these patients could now be considered as juvenile cases, their clinical condition is quite different from age-matched LOPD patients with a juvenile onset. In fact, the “survivors” exhibit motor weakness, speech deficits, sensorineural and/or conductive hearing loss, osteopenia, gastroesophageal reflux and dysphagia with aspiration risk. However, the majority of them are still independently ambulant (49).
Recent reports have pointed out to the GI symptoms including severe chronic diarrhea, intestinal incontinence, abdominal pain, lack of appetite, early satiety and vomiting that are underevaluated in the late onset form (51). It is not surprising that the GI system is affected, since glycogen storage has been detected in smooth muscle cells—a pathology which can impair peristalsis and intestinal transit (52). The ERT treatment greatly reduced the GI discomfort and allowed to avoid embarrassing social situations. Also, Gesquière-Dando et al. (53) report two sisters with an atypical presentation of “fibromyalgia-like pain” and GI symptoms. ERT improved the GI symptoms but did not decrease pain. Pardo et al. (54) described a similar clinical situation and response to ERT in a patient who presented intense lower GI symptoms that were halted by ERT. Nevertheless, in most patients, urge symptoms persisted and did not seem to respond to ERT.
In the last few years, the urinary tract has also been an object of studies to better understand why and how it is involved in LOPD. According to some authors (55), the prevalence of incontinence in LOPD patients can be as high as 25% assuming no other etiologies or risk factors for this condition. Therefore, the frequent occurrence of bowel incontinence or urinary tract symptoms suggests that there is a common origin of these symptoms due to smooth muscle dysfunction (42). Some authors suggested that urinary incontinence is less frequent than bowel incontinence (55), whereas others (56) reported a higher number of patients with lower urinary tract symptoms (LUTS), with specific differences related to gender.
Conclusions
In the last 10–15 years, several major steps have been accomplished by researchers and physicians in the Pompe disease field. A great deal of effort has been put into the development of new diagnostic tools and new therapeutic approaches. Although ERT is quite efficient (especially for infants), we are still looking for other treatment options to improve patients quality of life. In the meantime, it is very clear that the awareness about “old” and “new” clinical aspects of Pompe disease allows for earlier diagnosis and better management of Pompe disease patients.
Acknowledgments
None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Conflicts of Interest: A Toscano is a member of Global Pompe Registry committee and has received honoraria for speaking engagements and teaching courses from Sanofi Genzyme. O Musumeci received reimbursements for speaking engagements from Sanofi Genzyme. C Rodolico has no conflicts of interest to declare.
References
- 1.Müller-Felber W, Horvath R, Gempel K, et al. Late onset Pompe disease: clinical and neurophysiological spectrum of 38 patients including long-term follow-up in 18 patients, Neuromuscul Disord 2007:17:698-706. 10.1016/j.nmd.2007.06.002 [DOI] [PubMed] [Google Scholar]
- 2.Montagnese F, Barca E, Musumeci O, et al. Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment J Neurol 2015;262:968-78. 10.1007/s00415-015-7664-0 [DOI] [PubMed] [Google Scholar]
- 3.Musumeci O, la Marca G, Spada M. LOPED study: looking for an early diagnosis in a late-onset Pompe disease high-risk population. J Neurol Neurosurg Psychiatry 2016; 87:5-11. [DOI] [PubMed] [Google Scholar]
- 4.Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin 2014;32:751. 10.1016/j.ncl.2014.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winkel LP, Hagemans ML, van Doorn PA, et al. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J Neurol 2005;252:875-84. 10.1007/s00415-005-0922-9 [DOI] [PubMed] [Google Scholar]
- 6.Güngör D, Schober AK, Kruijshaar ME, et al. Pain in adult patients with Pompe disease: a cross-sectional survey. Mol Genet Metab 2013;109:371-6. 10.1016/j.ymgme.2013.05.021 [DOI] [PubMed] [Google Scholar]
- 7.Chan J, Desai AK, Kazi ZB, et al. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol Genet Metab 2017;120:163-72. 10.1016/j.ymgme.2016.12.004 [DOI] [PubMed] [Google Scholar]
- 8.Boentert M, Karabul N, Wenninger S, et al. Sleep-related symptoms and sleep-disordered breathing in adult Pompe disease. Eur J Neurol 2015;22:369-76, e27. [DOI] [PubMed]
- 9.Gaeta M, Barca E, Ruggeri P, et al. Late-onset Pompe disease (LOPD): correlations between respiratory muscles CT and MRI features and pulmonary function. Mol Genet Metab 2013;110:290-6 10.1016/j.ymgme.2013.06.023 [DOI] [PubMed] [Google Scholar]
- 10.DeRuisseau LR, Fuller DD, Qiu K, et al. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci U S A 2009;106:9419-24. 10.1073/pnas.0902534106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger KI, Chan Y, Rom WN, et al. Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul Disord 2016;26:481-9. 10.1016/j.nmd.2016.05.018 [DOI] [PubMed] [Google Scholar]
- 12.McCall AL, ElMallah MK. Macroglossia, Motor Neuron Pathology, and Airway Malacia Contribute to Respiratory Insufficiency in Pompe Disease: A Commentary on Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. Int J Mol Sci 2019. doi: . 10.3390/ijms20030751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang CF, Niu DM, Jeng MJ, et al. Late-Onset Pompe Disease With Left-Sided Bronchomalacia. Respir Care 2015;60:e26-9. 10.4187/respcare.03419 [DOI] [PubMed] [Google Scholar]
- 14.Brenn BR, Theroux MT, Shah SA, et al. Critical Airway Stenosis in an Adolescent Male With Pompe Disease and Thoracic Lordosis: A Case Report. A A Case Rep 2017;9:199-203. 10.1213/XAA.0000000000000564 [DOI] [PubMed] [Google Scholar]
- 15.Kretzschmar HA, Wagner H, Hübner G, et al. Aneurysms and vacuolar degeneration of cerebral arteries in late-onset acid maltase deficiency. J Neurol Sci 1990;98:169-83. 10.1016/0022-510X(90)90258-O [DOI] [PubMed] [Google Scholar]
- 16.Hobson-Webb LD, Proia AD, Thurberg BL, et al. Autopsy findings in late-onset Pompe disease: a case report and systematic review of the literature. Mol Genet Metab 2012;106:462-9. 10.1016/j.ymgme.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 17.Malhotra K, Carrington DC, Liebeskind DS. Restrictive arteriopathy in late-onset Pompe disease: case report and review of the literature. J Stroke Cerebrovasc Dis 2017;26:e172-5. 10.1016/j.jstrokecerebrovasdis.2017.05.032 [DOI] [PubMed] [Google Scholar]
- 18.Musumeci O, Marino S, Granata F, et al. Central nervous system involvement in late-onset Pompe disease: clues from neuroimaging and neuropsychological analysis. Eur J Neurol 2019;26:442-e35. 10.1111/ene.13835 [DOI] [PubMed] [Google Scholar]
- 19.Kamphoven JH, de Ruiter MM, Winkel LP, et al. Hearing loss in infantile Pompe's disease and determination of underlying pathology in the knockout mouse. Neurobiol Dis 2004;16:14-20. 10.1016/j.nbd.2003.12.018 [DOI] [PubMed] [Google Scholar]
- 20.van der Beek NA, Verschuure H, Reuser AJJ, et al. Hearing in adults with Pompe disease. J Inherit Metab Dis 2012;35:335-41. 10.1007/s10545-011-9396-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Musumeci O, Catalano N, Barca E, et al. Auditory system involvement in late onset Pompe disease: a study of 20 Italian patients. Mol Genet Metab 2012;107:480-4. 10.1016/j.ymgme.2012.07.024 [DOI] [PubMed] [Google Scholar]
- 22.Bekircan-Kurt CE, Gunes HN, Yildiz FG, et al. New mutations and genotype-phenotype correlation in late-onset Pompe patients. Acta Neurol Belg 2017;117:269-75. 10.1007/s13760-016-0738-7 [DOI] [PubMed] [Google Scholar]
- 23.Borroni B, Cotelli MS, Premi E, et al. The brain in late onset glycogenosis II: a structural and functional MRI study. J Inherit Metab Dis 2013;36:989-95. 10.1007/s10545-013-9601-7 [DOI] [PubMed] [Google Scholar]
- 24.Schoser B, Bilder DA, Dimmock D, et al. The humanistic burden of Pompe disease: are there still unmet needs? A systematic review. BMC Neurol 2017;17:202. 10.1186/s12883-017-0983-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hobson-Webb LD, Austin SL, Jain S, et al. Small-fiber neuropathy in Pompe disease: first reported cases and prospective screening of a clinic cohort. Am J Case Rep 2015;16:196-201 10.12659/AJCR.893309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fidziańska A, Ługowska A, Tylki-Szymańska A. Late form of Pompe disease with glycogen storage in peripheral nerves axons. J Neurol Sci 2011;301:59-62. 10.1016/j.jns.2010.10.031 [DOI] [PubMed] [Google Scholar]
- 27.Origuchi Y, Itai Y, Matsumoto S, et al. Quantitative histological study of the sural nerve in a child with acid maltase deficiency (glycogenosis type II). Pediatr Neurol 1986;2:346-9. 10.1016/0887-8994(86)90075-5 [DOI] [PubMed] [Google Scholar]
- 28.Laforêt P, Doppler V, Caillaud C, et al. Rigid spine syndrome revealing late-onset Pompe disease. Neuromuscul Disord 2010;20:128-30. 10.1016/j.nmd.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 29.Taisne N, Desnuelle C, Juntas Morales R, et al. Bent spine syndrome as the initial symptom of late-onset Pompe disease. Muscle Nerve 2017;56:167-70. 10.1002/mus.25478 [DOI] [PubMed] [Google Scholar]
- 30.Roberts M, Kishnani PS, van der Ploeg AT, et al. The prevalence and impact of scoliosis in Pompe disease: Lessons learned from the Pompe Registry. Mol Genet Metab 2011;104:574-82. 10.1016/j.ymgme.2011.08.011 [DOI] [PubMed] [Google Scholar]
- 31.Papadimas GK, Terzis G, Methenitis S, et al. Body composition analysis in late-onset Pompe disease. Mol Genet Metab 2011;102:41-3. 10.1016/j.ymgme.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 32.Khan A, Weinstein Z, Hanley DA, et al. In vivo bone architecture in pompe disease using high-resolution peripheral computed tomography. JIMD Rep 2013;7:81-8. 10.1007/8904_2012_146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Berg LE, Zandbergen AA, van Capelle CI, et al. Low bone mass in Pompe disease Muscular strength as a predictor of bone mineral density. Bone 2010;47:643-9. 10.1016/j.bone.2010.06.021 [DOI] [PubMed] [Google Scholar]
- 34.Bertoldo F, Zappini F, Brigo M, et al. Prevalence of Asymptomatic Vertebral Fractures in Late-Onset Pompe Disease. J Clin Endocrinol Metab 2015;100:401-6. 10.1210/jc.2014-2763 [DOI] [PubMed] [Google Scholar]
- 35.Soliman OI, van der Beek NA, van Doorn PA, et al. Cardiac involvement in adults with Pompe disease. J Intern Med 2008;264:333-9. 10.1111/j.1365-2796.2008.01966.x [DOI] [PubMed] [Google Scholar]
- 36.Forsha D, Li JS, Smith PB, et al. Cardiovascular abnormalities in late-onset Pompe disease and response to enzyme replacement therapy. Genet Med 2011;13:625-31. 10.1097/GIM.0b013e3182142966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angelini C, Semplicini C, Ravaglia S, et al. Italian GSDII Group Observational clinical study in juvenileadult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 2012;259:952-8. 10.1007/s00415-011-6293-5 [DOI] [PubMed] [Google Scholar]
- 38.Sacconi S, Wahbi K, Theodore G, et al. Atrio-ventricular block requiring pacemaker in patients with late onset Pompe disease. Neuromuscul Disord 2014;24:648-50. 10.1016/j.nmd.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 39.Quenardelle V, Bataillard M, Bazin D, et al. Pompe disease presenting as an isolated generalized dilative arteriopathy with repeated brain and kidney infarcts J Neurol 2015;262:473-5. 10.1007/s00415-014-7582-6 [DOI] [PubMed] [Google Scholar]
- 40.El-Gharbawy AH, Bhat G, Murillo JE, et al. Expanding the clinical spectrum of late-onset Pompe disease: dilated arteriopathy involving the thoracic aorta, a novel vascular phenotype uncovered. Mol Genet Metab 2011;103:362-6. 10.1016/j.ymgme.2011.04.009 [DOI] [PubMed] [Google Scholar]
- 41.Nemes A, Soliman OI, Geleijnse ML, et al. Increased aortic stiffness in glycogenosis type 2 (Pompe's disease). Int J Cardiol 2007;120:138-41. 10.1016/j.ijcard.2006.07.215 [DOI] [PubMed] [Google Scholar]
- 42.McCall AL, Salemi J, Bhanap P, et al. The impact of Pompe disease on smooth muscle: a review. J Smooth Muscle Res 2018;54:100-18. 10.1540/jsmr.54.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagemans ML, Stigter RL, van Capelle CI, et al. PAS-positive lymphocyte vacuoles can be used as diagnostic screening test for Pompe disease. J Inherit Metab Dis 2010;33:133-9. 10.1007/s10545-009-9027-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderson G, Smith VV, Malone M, et al. Blood film examination for vacuolated lymphocytes in the diagnosis of metabolic disorders; retrospective experience of more than 2500 cases from a single centre. J Clin Pathol 2005;58:1305-10. 10.1136/jcp.2005.027045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parisi D, Musumeci O, Mondello S, et al. Vacuolated PAS-Positive Lymphocytes on Blood Smear: An Easy Screening Tool and a Possible Biomarker for Monitoring Therapeutic Responses in Late Onset Pompe Disease (LOPD). Front Neurol 2018;9:880. 10.3389/fneur.2018.00880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider J, Burmeister LA, Rudser K, et al. Hypothyroidism in late-onset Pompe disease. Mol Genet Metab Rep 2016;8:24-7. 10.1016/j.ymgmr.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Gijt JP, van Capelle CI, Oosterhuis JW, et al. Gingival overgrowth in Pompe disease: a case report. J Oral Maxillofac Surg 2011;69:2186-90. 10.1016/j.joms.2011.03.070 [DOI] [PubMed] [Google Scholar]
- 48.Dubrovsky A, Corderi J, Lin M, et al. Expanding the phenotypeof late onset Pompe disease: Tongue weakness: a new clinical observation. Muscle Nerve 2011;44:897-901. 10.1002/mus.22202 [DOI] [PubMed] [Google Scholar]
- 49.Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord 2013;23:319-23. 10.1016/j.nmd.2012.12.003 [DOI] [PubMed] [Google Scholar]
- 50.Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genet Med 2012;14:800-10. 10.1038/gim.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karabul N, Skudlarek A, Berndt J, et al. Urge incontinence and gastrointestinal symptoms in adult patients with Pompe disease: a cross-sectional survey. JIMD Rep 2014;17:53-61. 10.1007/8904_2014_334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernstein DL, Bialer MG, Mehta L, et al. Pompe disease: dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol Genet Metab 2010;101:130-3. 10.1016/j.ymgme.2010.06.003 [DOI] [PubMed] [Google Scholar]
- 53.Gesquière-Dando A, Attarian S, De Paula AM, et al. Fibromyalgia-like symptoms associated with irritable bowel syndrome: A challenging diagnosis of late-onset Pompe disease. Muscle Nerve 2015;52:300-4. 10.1002/mus.24618 [DOI] [PubMed] [Google Scholar]
- 54.Pardo J, Garcia-Sobrino T, Lopez-Ferreiro A. Gastrointestinal symptoms in late onset Pompe disease: Early response to enzyme replacement therapy. J Neurol Sci 2015;353:181-2. 10.1016/j.jns.2015.04.012 [DOI] [PubMed] [Google Scholar]
- 55.Remiche G, Herbaut AG, Ronchi D, et al. Incontinence in late-onset Pompe disease: an underdiagnosed treatable condition. Eur Neurol 2012;68:75-8. 10.1159/000338776 [DOI] [PubMed] [Google Scholar]
- 56.McNamara ER, Austin S, Case L, et al. Expanding our understanding of lower urinary tract symptoms and incontinence in adults with pompe disease. JIMD Rep 2015;20:5-10. 10.1007/8904_2014_381 [DOI] [PMC free article] [PubMed] [Google Scholar]