

Abstract
Pompe disease (PD) is a monogenic disorder caused by mutations in the acid alpha-glucosidase gene (Gaa). GAA is a lysosomal enzyme essential for the degradation of glycogen. Deficiency of GAA results in a severe, systemic disorder that, in its most severe form, can be fatal. About a decade ago, the prognosis of PD has changed dramatically with the marketing authorization of an enzyme replacement therapy (ERT) based on recombinant GAA. Despite the breakthrough nature of ERT, long-term follow-up of both infantile and late-onset Pompe disease patients (IOPD and LOPD, respectively), revealed several limitations of the approach. In recent years several investigational therapies for PD have entered preclinical and clinical development, with a few next generation ERTs entering late-stage clinical development. Gene therapy holds the potential to change dramatically the way we treat PD, based on the ability to express the Gaa gene long-term, ideally driving enhanced therapeutic efficacy compared to ERT. Several gene therapy approaches to PD have been tested in preclinical animal models, with a handful of early phase clinical trials started or about to start. The complexity of PD and of the endpoints used to measure efficacy of investigational treatments remains a challenge, however the hope is for a future with more therapeutic options for both IOPD and LOPD patients.
Keywords: Pompe disease (PD), gene therapy, adeno-associated virus vectors (AAV vectors), muscle, liver, tolerance
Introduction
Pompe disease (PD, OMIM #232300) is an autosomal recessive disorder due to mutations in the gene coding for acid alpha-glucosidase (GAA). GAA is the enzyme that degrades lysosomal glycogen to free glucose. Mutations that decrease the activity of this enzyme lead to the accumulation of glycogen in the lysosomal compartment and to the impairment of lysosomal function and autophagy flux (1). In PD, glycogen accumulates virtually in all tissues but the disease manifestation is prominently muscular, with severe hypertrophic cardiomyopathy, respiratory function impairment and proximal muscle weakness (2). Accumulation of lysosomal glycogen in the central nervous system (CNS) is associated with white matter abnormalities and cognitive impairment in particular in patients with no residual GAA activity (3). Additional pathological changes associated with the disease have also been described, i.e., cerebral vessels abnormalities that are relatively frequently found in adult Pompe patients (4). The disease can be broadly classified into (I) infantile-onset PD (IOPD), with no residual GAA activity and associated with generalized hypotonia, and cardio-respiratory failure, leading to death in the first year of life; and (II) late-onset PD (LOPD), characterized by residual levels of GAA activity and a less severe phenotype with progressive limb muscle weakness and respiratory insufficiency (5).
PD is the first and probably the more comprehensively characterized lysosomal storage disease (LSD) that manifests as a metabolic myopathy. For this reason, over the course of the last few decades, several therapeutic approaches aimed at addressing the pathology were explored.
Standard of care and next generation enzyme replacement therapy (ERT)
The discovery of the pathway of uptake of lysosomal enzymes mediated by mannose-6-phophate (M6P) receptor (6-8) allowed the introduction of the concept of cross-correction, i.e., the possibility to replace a lysosomal enzyme by supplementing the enzyme in the extracellular media.
The development of the ERT considerably changed the prognosis of PD and this treatment modality represents the current standard of care for the disease. Indeed, pioneering clinical studies in IOPD patients clearly demonstrated the efficacy of the ERT for the improvement of the cardiac and muscle function (9-11). These results led to the approval in 2006 of the first therapy for IOPD followed in 2010 by the approval of ERT for LOPD (alglucosidase alfa, Lumizyme® within the USA and Myozyme® outside of the USA; Sanofi Genzyme). Recombinant acid alpha-glucosidase (rhGAA) is administered intravenously every other week at a recommended dose of 20 mg/kg, but higher dose regimens (up to 40 mg/kg) are also administered in IOPD patients. These doses are markedly higher than those required in other lysosomal storage disorders, possibly reflecting the higher threshold for correction of GAA deficiency in the skeletal muscle of Pompe patients. In addition, the liver takes up most of the rhGAA (up to 85%) and considerably limits muscle targeting. ERT with recombinant human GAA has clearly demonstrated cardiac and respiratory function improvement and has significantly extended the lifespan of IOPD patients (12). However, the administration of rhGAA in IOPD is frequently associated with the development of neutralizing humoral immune responses against the enzyme and decreased treatment efficacy and survival. This is particularly relevant for cross-reactive immune-material negative (CRIM–) patients with no residual GAA antigen, who therefore completely lack central tolerance to the protein (13). Another important shortcoming of ERT is related to its limited efficacy, for example, respiratory function is only partially rescued by the treatment, and approximately 30% of the rhGAA-treated patients end up requiring assisted ventilation, either invasive or not, over the course of their life (14). Skeletal muscle function is also improved by ERT, although the effect of the treatment is quite variable with some subjects maintaining independent ambulation and others showing only minor improvements and eventually ending up being wheelchair bound (15,16). The majority of reports to date of clinical trials or investigator-initiated clinical studies in LOPD patients are concordant in concluding that, in most patients, ERT leads to an improvement of muscle function as measured by 6-minute walk test whereas long-term studies show that respiratory function is only stabilized (17). Available studies also show that ERT may stabilize or even slightly improve muscle strength and respiratory function among patients at advanced stages of the disease (18,19). In addition to the efficacy limitations, (I) the requirement for frequent intravenous infusions of high doses of rhGAA, (II) the possibility of severe and detrimental immune responses in both CRIM− and CRIM+ patients (9,16,20) and (III) the inability of the recombinant enzyme to cross BBB and correct the nervous system (21), make the development of new therapies for PD an urgent need.
Since the development of ERT for PD, efforts were dedicated to overcoming some of the limitations of the treatment. Two main strategies, both aimed at the enhancement of the enzyme bioavailability in tissues, are now in late-stage clinical testing. The first approach consists in the modification of the recombinant enzyme to increase the M6P residue content (22); the second strategy involves the use of pharmacological adjuvants to enhance ERT efficacy (23,24).
A second generation ERT with rhGAA with higher affinity for the M6P receptors (25) is now under evaluation in a phase III clinical trial (avalglucosidase alfa, Neo-GAA; Sanofi Genzyme; NCT02782741). The study is designed to test doses ranging from 5 to 20 mg/kg of biweekly-administered Neo-GAA with the possibility to switch doses during the study. This phase III clinical trial, aimed at the comparison of the efficacy and safety of bi-weekly infusions of avalglucosidase alfa and alglucosidase alfa in patients with LOPD, is still recruiting. Another experimental rhGAA called ATB200 (Amicus Therapeutics), with a higher content of M6P and bis-M6P glycan residues was developed and is being tested in a clinical trial in association with pharmacological chaperones (vide infra). In preclinical studies, GAA enzymes engineered with synthetic M6P residues improved muscle function in Pompe mice either alone (26) or in combination with chaperones (22) and showed enhanced targeting in Pompe patients fibroblasts (27) when compared to first-generation rhGAA.
Another strategy tested to improve bioavailability of the GAA enzyme in tissues, resulting in enhanced clearance of glycogen, consists in the use of uptake domains. Several chimeric GAA proteins carrying uptake domains were tested in preclinical animal models of PD (28-30). Among these, an engineered form of rhGAA carrying the glycosylation-independent lysosomal targeting (GILT) domain for tissue uptake (29,31) was tested in LOPD patients and reached late-stage clinical development (NCT01924845, BMN 701, BioMarin Pharmaceutical). Unfortunately, concerns over the development of hypoglycemia following enzyme infusion resulted in the discontinuation of the development (32). More recently, a chimeric form of rhGAA containing a humanized Fab fragment derived from the murine 3E10 antibody (30) also entered phase I/II clinical testing (NCT02898753, VAL1221, Valerion Therapeutics, LLC).
The use of pharmacological agents is emerging as a potential strategy to improve the efficacy of ERT in PD. The combination of rhGAA with β2 agonists, e.g., clenbuterol or albuterol mediated increased M6P receptor expression, improved muscle function and reduced glycogen accumulation in muscle and brain when the enzyme is supplied by ERT (33,34) or by liver-mediated gene therapy (vide infra) (35). A phase I/II clinical trial for the evaluation of the combination of albuterol and rhGAA showed increased M6P receptor expression in muscle biopsies and motor function improvement (36). A second clinical trial on the effects of clenbuterol on the ERT efficacy showed improved motor function and the correction of molecular biomarkers of the disease in muscle (37). Both these early-phase trials, realized in LOPD patients, showed signs of improved efficacy in combination with ERT with only mild secondary effects. Larger trials will be required to clearly demonstrate the advantage of the approach compared to ERT alone.
Another combination approach for PD treatment is based on the use of pharmacological chaperone therapy (PCT). Chaperones are small molecules known to promote folding and improve stability of proteins and enzymes (38). In the case of PD, improved rhGAA bioavailability via enhancement of enzyme stability in blood was demonstrated in preclinical models (24,39). Different glucose analogues, acting as allosteric inhibitors of GAA, have been investigated e.g., 1-Deoxynojirimycin (1-DNJ; Duvoglustat®) (40), 1-Deoxynojirimycin-HCl (DNJ-HCl; Duvoglustat®-HCl; AT2220) (41) or N-butyl-deoxynojirimycin (NB-DNJ, Miglustat®) (42) and demonstrated a beneficial effect when combined with ERT. Recent clinical studies sponsored by Amicus Therapeutics tested the combination of the AT2221 chaperone with the engineered enzyme ATB200 in LOPD patients (NCT02675465, AT-GAA, Amicus Therapeutics).
Gene therapy
Given its monogenic origin, PD represents an ideal target for the development of gene replacement strategies. Since 1998, year of the first in vivo gene therapy approach for PD (43), different in vivo and ex vivo approaches (Figure 1) were tested in animal models with the aim of correcting the PD phenotype (51-54). These approaches are reviewed elsewhere (55,56), and this article will mainly focus on the discussion of recent advances in the field of in vivo gene therapy with adeno-associated virus (AAV) vectors.
Figure 1.

Gene therapy modalities. In vivo gene therapy consists in the direct administration of a gene delivery vector (viral or non-viral) directly into the recipient of gene transfer. In Pompe disease, most of the experience to date comes from AAV vector-mediated gene transfer. AAV vectors have been administered either directly into the bloodstream to target the muscle (44), the liver (45,46), or multiple tissues (47), or directly into muscle (48), or intracerebroventricular to target the central nervous system (49). Ex vivo gene therapy uses autologous CD34+ hematopoietic progenitors transduced with integrative vectors [e.g., lentiviral vectors (50)] and re-infused in the recipient following myeloablative bone marrow conditioning. This gene therapy modality has been shown to have the potential to efficiently deliver GAA to the central nervous system. AAV, adeno-associated virus vectors; GAA, acid alpha-glucosidase.
AAV vectors
AAV are small (25 nm) viruses composed by a non-enveloped icosahedral capsid (protein shell) that contains a linear single-stranded DNA genome of about 4.7 Kb. AAV belongs to the family of Parvoviridae, genus Dependovirus, as it can replicate in the nucleus of target cells only in the presence of helper viruses such as adenovirus or herpes virus (57). The AAV genome is flanked by two palindromic inverted terminal repeats (ITR, 145 bp) and includes two open reading frames, rep and cap, which encode proteins involved in the replication and assembly of virions and capsid structural proteins, respectively (57). AAV viruses naturally infect humans; usually exposure to the wild-type virus occurs early in life (58-60) and is not associated with any known disease or illness (61). Importantly, the timing of human exposure to AAV viruses determines the host immunological response to the recombinant AAV vectors [for a comprehensive review, see (62)]. In the genome of recombinant AAV vectors, the only viral sequences that are retained are the two ITRs (cis packaging signals) while the sequences encoding rep and cap are replaced with the exogenous DNA of choice (that is flanked by the ITRs and it is referred to as the transgene expression cassette). Differently from the wild type virus, the genome of the recombinant AAV vectors does not undergo site-specific integration in the host DNA but mainly remains episomal in the nucleus of transduced cells, while random integration events are observed with a low frequency (0.1% to 1% of transduction events) (61,63,64).
Several AAV serotypes have been identified and classified (57,65). The versatility of the AAV production system allows to easily generate pseudotyped AAV vectors composed by the same transgene flanked by the ITRs from serotype 2 (66) (so far the most commonly used) and any of the available AAV capsid (57). AAV vectors can be produced at high yields by transient triple transfection of mammalian cells (67) or infection of packaging eukaryotic (68) and insect cells (69).
Experience with AAV vectors in PD
Intramuscular and systemic gene transfer
Gene therapy holds the potential for improving the standard of care for PD, addressing some of the key limitations of ERT (Table 1). Consistently, in recent years the landscape of gene therapy for PD has dramatically changed, with a pipeline of candidate therapeutics at various stages of development (Table 2).
Table 1. Comparison of enzyme replacement therapy with gene therapy for Pompe disease.
| Features | Enzyme replacement therapy | Investigational gene therapy |
|---|---|---|
| Safety and efficacy in patients | Safe and effective in Pompe patients; long-term efficacy achieved in a subset of patients (5,10-12); hypersensitivity reactions sometimes observed (70) | Safety and efficacy not yet established in Pompe patients |
| GAA immunogenicity | Observed in both IOPD and LOPD patients; associated with treatment failures in IOPD patients (13,71) | Potentially a concern for some gene therapy modalities targeting the muscle (72) |
| Whole-body correction | Not achievable, rhGAA does not cross blood-brain barrier at the doses used | Potential for body-wide correction of the disease (45) |
| Cross-correction | Feasible, highly dependent on levels of expression of mannose 6-phosphate receptor (73) | Feasible (45); potential for expressing GAA directly in target tissues |
| Immune tolerance induction | Achievable but difficult and requires co-administration of immunomodulatory drugs (74) | Potentially achievable with liver gene transfer (45,46,72,75) |
Table 2. Biotechnology companies currently developing gene therapies for Pompe disease.
| Company | Gene therapy vector | Transgene | Target tissue | Development status |
|---|---|---|---|---|
| Actus# | AAV (in vivo) | GAA | Liver | Phase I/II |
| Audentes | AAV (in vivo) | GAA | Muscle and liver | Clinical trial-enabling |
| Sarepta* | AAV (in vivo) | GAA | Central nervous system | Preclinical |
| Spark** | AAV (in vivo) | Secretable GAA | Liver | Clinical trial-enabling |
| Amicus | AAV (in vivo) | Secretable GAA? | Liver | Preclinical |
| Regeneron | AAV (in vivo) | CD63-GAA fusion | Liver | Preclinical |
| AvroBio | Lentivirus (ex vivo) | GILT-GAA fusion | CD34+ HSC | Preclinical |
#, therapeutic candidate inlicensed from Duke University; *, therapeutic candidate inlicensed from Lacerta, Inc.; **, therapeutic candidate inlicensed from Genethon; HSC, hematopoietic stem cells; GILT, glycosylation-independent lysosomal targeting uptake domain; CD63, tetraspanin binding domain
Over the past two decades, the collective experiences across several research groups demonstrated the potential of AAV vectors encoding GAA to rescue PD in animal models, along with the potential for reducing GAA immunogenicity (45,48,76-80). Different AAV serotypes have been explored to develop gene transfer strategies to treat PD in animal models, including AAV1 (81-84), AAV2 (48), AAV5 (85,86), AAV6 (77), AAV8 (45,76,86-88) and AAV9 (49,84,89-93). In these strategies, the muscle (48,76,77,79,81-83,86,90,91,93), the liver (76,87,88), or the CNS (49,84,89,92) were targeted.
First evidences of the efficacy of AAV-based gene transfer efficacy in a PD mouse model were obtained by intramuscular injection of AAV vectors (48,83,84,90). This route of administration was associated with increased GAA transgene immunogenicity and correction of the glycogen accumulation only locally at the level of the injected muscle groups (48). This initial work was followed by studies in which AAV vectors were given systemically or injected directly into the diaphragm. Intra-diaphragmatic injections of AAV vectors led to improvement in respiratory function in Gaa−/− mice. These studies also indicated the ability of AAV vectors to transduce efficiently the phrenic motor neurons that innervates the diaphragm (79,82,84). Based on these results a first-in-human trial of gene therapy for PD with direct injection of an AAV vector expressing GAA in the diaphragm was initiated. This trial, conducted in IOPD patients who required assisted ventilation, established the safety profile of the approach (94). Development of antibodies directed against both the AAV capsid and GAA were observed, in the absence of detectable T cell responses against the vector or the transgene, consistent with the fact that participants were immunosuppressed at the time of vector administration (94). Given the limited size of the trial, no definitive conclusions could be drawn from this study in terms of therapeutic efficacy, aside from a trend toward improved respiratory function (94-97).
Currently, the intramuscular delivery of an AAV9 vector expressing GAA is being tested in a phase I/II trial. In this study, the vector is given concomitantly with an immunosuppressive regimen based on sirolimus (rapamycin) and the B cell-depleting monoclonal antibody rituximab, in an effort to determine if the approach allows for two consecutive intramuscular administrations of AAV vectors while blocking humoral immune responses to the AAV capsid (NCT02240407).
While intramuscular administration of AAV vectors allows to achieve highly efficient tissue transduction, concerns over transgene immunogenicity (98,99), and the systemic nature of the disease, make this route of vector delivery unsuitable to treat effectively PD. The recent advances in the ability to produce AAV at large scale, and the exciting results of clinical trials of systemic delivery of AAV vector to treat neuromuscular diseases (100), resulted in several preclinical studies of systemic delivery of AAV vectors containing muscle-specific expression cassettes for the GAA transgene. These studies demonstrated efficient clearance of glycogen accumulation in muscle and significant improvement of muscle strength as well as cardiac and respiratory function (48,76,77,79,81-83,86,90,91,93). However, one limitation of the approach is that muscle targeting via the systemic route requires extremely high doses of vector [exceeding 1,014 vector genome/kg (101-103)], not easily achievable when addressing the LOPD patient population. Furthermore, muscle-specific expression of GAA is associated with an increased risk of anti-GAA antibody formation and potential immunotoxicities (73,86). One potential solution to the issue of transgene immunogenicity is the use of liver-muscle tandem promoters (47), although this approach is also more suitable for IOPD patients, due to the high vector doses required to transduce muscle in adults with AAV vectors delivered systemically.
Liver gene therapy for PD
The liver is a particularly attractive organ for the development of gene-based therapeutic approaches for PD and other genetic disease for a number of reasons including: (I) it is one of the body’s major biosynthetic organs, is highly vascularized and can efficiently secrete proteins into the bloodstream; (II) studies in small and large animal models and in humans have demonstrated that it is possible to target hepatocytes with high efficiency using AAV vectors administered intravenously (104-109); (III) despite the predominantly non-integrative nature of AAV vectors (64), multi-year transgene expression after gene transfer to the liver has been documented in large animals and humans (110,111); and (IV) expression of a transgene in hepatocytes induces antigen-specific tolerance mediated by regulatory T cells (75,112-114) and other mechanism (Figure 2), which has been shown to prevent and suppress humoral immune responses to GAA in Gaa−/− mice (45-47).
Figure 2.

Induction of hepatic tolerance with AAV vectors [adapted from (115)]. Liver-directed gene transfer with AAV vectors induces immunological tolerance via multiple mechanisms. These include the deletion of reactive CD4+ T helper cells (e.g., by programmed cell death). Induction of FoxP3+ regulatory T cells plays a central role in the induction of hepatic tolerance. Similarly, antigen presentation by resident APCs in the liver draining portal and celiac lymph nodes is key to the induction of hepatic tolerance. MHC II, major histocompatibility complex class II; TCR, T cell receptor; BCR, B cell receptor; AAV, adeno-associated virus vectors; APC, antigen presenting cell.
Liver gene transfer with AAV vectors has been tested in the clinic for a handful of indications, which include hemophilia A and B (105,107-109), acute intermittent porphyria (116), and other indications. A phase I/II trial of liver gene transfer in LOPD patients is currently undergoing (NCT03533673, Actus Therapeutics, Inc.), although results are not available to date. Results gathered thus far in other clinical trials demonstrate that it is possible to target the liver via the systemic administration of AAV vectors. Additionally, they support the favorable safety profile of the approach, as transient increases in liver enzymes, often managed with transient immunosuppression, were the only adverse events reported after hepatic gene transfer with AAV vectors.
Liver gene transfer has been explored as a modality to treat PD. Early studies by Amalfitano and colleagues demonstrated that liver expression of GAA by adenoviral gene transfer mediated cross-correction in the skeletal muscle (53). However, the expression of the transgene in circulation was only transient likely due to the induction of an immune response against the transgene elicited by the vector used in the study.
Stable expression of GAA in the liver was achieved via AAV vector-mediated gene transfer, resulting in cross-correction in peripheral organs with no evident immunogenicity against the transgene (72,78,117). Further studies confirmed the uptake of GAA in heart and skeletal muscle with glycogen clearance and improved muscle function (45,46,118). Collectively, the studies of GAA gene transfer to the liver confirmed the potential efficacy profile and protolerogenic potential of this strategy and, together with toxicology studies in mice (88), supported the initiation of a phase I/II clinical trial sponsored by Actus Therapeutics (NCT03533673, Table 2). The primary objective of this trial is to demonstrate the safety of AAV vector-mediated liver gene transfer for GAA in LOPD patients. Secondary outcome measures aim at the characterization of the biodistribution of the enzyme produced by the liver and at the evaluation of the muscle glycogen accumulation and rescue of muscle and respiratory function.
A limitation of liver gene transfer for GAA is that the supply of the enzyme from the liver to peripheral tissues relies exclusively on cross-correction. Optimization of GAA secretion into the bloodstream therefore could represent an important step toward the development of a safe and efficacious liver-targeted gene therapy for PD. The use of a heterologous signal peptide to improve the secretion of GAA into the bloodstream was previously reported (119). We further improved this strategy by using an optimized signal peptide and a truncated version of the GAA protein with enhanced secretion [a modification determined using bioinformatics prediction (120)]. Using this second generation engineered secretable GAA transgene, we demonstrated a clear dose advantage when compared to the native version of the GAA transgene expressed in the liver (45). In particular, sustained circulating levels of the enzyme resulted in long-term correction of glycogen accumulation in tissues known to be refractory for GAA enzyme uptake (e.g., triceps and quadriceps), or tissue separated from the systemic circulation by physical barriers [spinal cord and brain, which are shielded from most circulating proteins by the blood-brain barrier (BBB)] (Figure 3). We also reported efficient rescue of muscle strength impairment and respiratory function (45). Interestingly, the use of secretable GAA also resulted in a better control of anti-transgene humoral immune responses, possibly via the efficient induction of regulatory T cells in draining lymph nodes (Figures 2,3) (121). Scale-up of this approach in non-human primates demonstrated the uptake of the enzyme secreted by the liver in peripheral tissues, particularly the heart without associated toxicity and supported the translation of this approach into the clinic. Spark Therapeutics is currently engaged in the clinical translation of these proof-of-concept results (Table 2).
Figure 3.
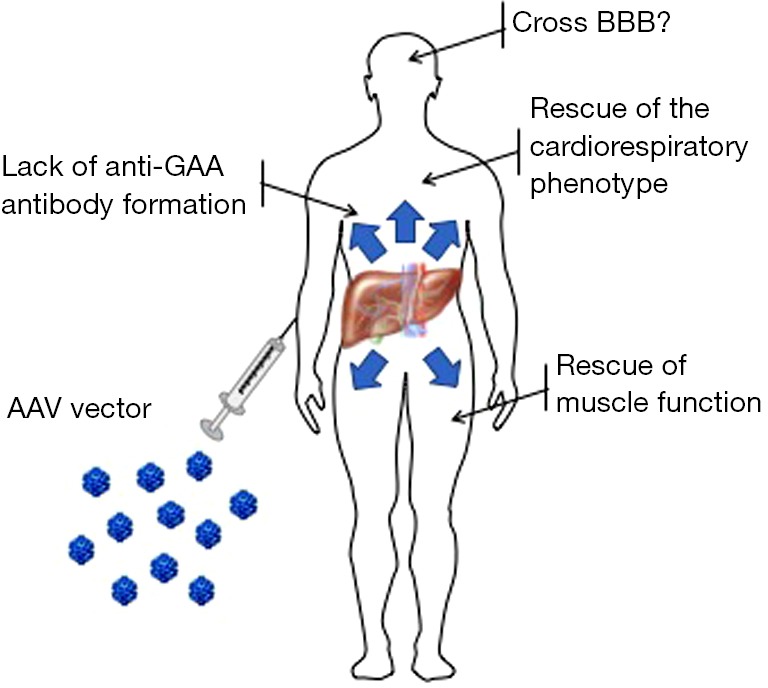
Investigational liver-directed gene therapy for Pompe disease. The graph exemplifies the potential for gene therapy as observed in preclinical studies in Gaa−/− mice (45). Targeting of the liver with AAV vectors expressing secretable forms of GAA can potentially transform the organ in a bio factory of GAA enzyme delivered to the entire body. Experiments in Gaa−/− mice show that the expression of secretable GAA results in the rescue of muscle and cardiorespiratory impairment with reduced GAA transgene immunogenicity. Partial correction of glycogen accumulation in the central nervous system is also observed. BBB, blood-brain barrier; GAA, acid alpha-glucosidase.
Another potential limitation of liver gene transfer for PD is related to the fact that hepatic gene transfer with AAV vectors in neonate animals does not persist at long-term (122). This poses important challenges to the use of this therapeutic strategy in IOPD patients. In pediatric patients, indeed, liver growth is likely to decrease the treatment efficacy due to vector genome dilution, potentially resulting in progressive loss of transgene expression. The formation of a neutralizing antibodies after AAV vector administration would precludes any further administration of the vector. Ongoing work aimed at the identification of immunosuppressive treatments that would allow for vector re-administration yielded promising preclinical results (123-125), and some of them has been translated to the clinic (NCT02240407, University of Florida).
A possible alternative to vector re-administration is based on the use of tandem promoters, which would allow for the induction of GAA transgene immune tolerance through liver expression, while driving persistent transgene expression in non-hepatic tissues (47). This strategy could potentially represent a path forward toward the development of a safe and long-lasting gene transfer approach for IOPD.
Other gene therapy approaches to PD
CNS-targeted delivery of AAV vectors expressing GAA has been recently explored as a possible therapeutic strategy for IOPD, based on the growing evidence that the CNS is significantly affected by glycogen accumulation in this patient population. Results obtained to date demonstrated that the clearance of glycogen in the CNS resulted in the rescue of the functional impairment associated with PD (49,126). However, restricted targeting of CNS is associated with a limited correction of the muscle defect (85,127,128). One possible solution around this is the use of tandem liver-neuron promoters driving simultaneous expression of GAA in liver and CNS after systemic administration of AAV vectors able to cross the BBB (47).
Gene replacement therapy with the GAA transgene is the obvious approach to PD. Nevertheless, given the complexity of the disease, development of alternative therapeutic strategies might result in enhanced efficacy or in the development of adjuvant therapies to be combined with protein- or gene-replacement approaches. Overexpression of the transcription factor EB (TFEB) (129) has been explored as a possible avenue to treat PD. In Gaa−/− mice, the administration of AAV vectors encoding for TFEB in muscle were shown to induce lysosomal exocytosis that was associated with improved muscle performance and delayed disease progression in the absence of reduction of glycogen content in skeletal muscle (130).
Substrate reduction therapy (SRT) has also been proposed for PD (131,132). This strategy is based on the reduction of the activity of the glycogen synthase enzyme acting in muscle (GYS1) to reduce the accumulation of glycogen. Accordingly, using transgenic mouse models, Douillard-Guilloux and colleagues demonstrated that the genetic suppression of the muscle-specific glycogen synthase in Gaa−/− mice was able to reverse cardiac impairment, reduce glycogen accumulation and improve muscle strength (132). Another study, designed to inhibit GYS1 using a phosphorodiamidate morpholino oligonucleotide (PMO) conjugated to a cell penetrating peptide (GS-PPMO), showed decreased glycogen accumulation in heart and skeletal muscle of Pompe mice (131). One caveat related to this approach is that excessive knockdown of GSD1 may lead to toxicities similar to those observed in GSD type 0, which is characterized by and increased risk of cardiac arrest (133). Another approach based on the use of PMO to promote exon inclusion and correct the common mutation c.-32-13T>G has also been proposed (134). Tricyclo-DNA antisense oligonucleotides were also tested as a strategy to correct the aberrant splicing mutation commonly found in LOPD patients (135).
Gene therapy vs. ERT
An intriguing finding coming from the preclinical studies with secretable GAA, which are likely to be generalizable to all gene therapies, is the time-dependent clearance of glycogen in tissues (45). This is likely driven by the steady-state, continuous exposure to the GAA enzyme expressed as a transgene as opposed to the administration of the recombinant form of the enzyme in the setting of ERT, which drives a transient increase in enzyme activity in peripheral tissues (Figure 4). This may provide an advantage for gene therapy, compared to the peak and trough kinetics observed after ERT administration. Notably, results from studies with rhGAA given in combination with pharmacological chaperones (23,24) support this concept, as the enhanced half-life of GAA mediated by the chaperone molecule results in improved efficacy (22). To this aim, gene therapy, when successful in driving sustained levels of GAA expression and uptake into peripheral tissues, is likely to be superior to any ERT, owing its unique pharmacokinetics profile characterized by a greater area under the curve (Figure 4).
Figure 4.
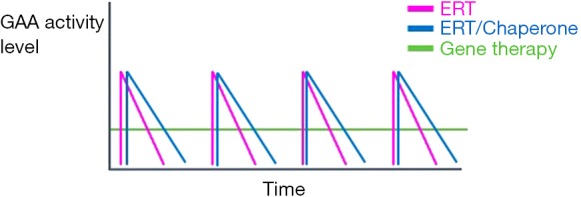
Potential of gene therapy as a treatment modality for Pompe disease. The graph displays the kinetics of GAA activity in tissues following ERT, with rhGAA alone or co-administered with chaperones to extend GAA half-life (ERT/Chaperone), or gene therapy. Both ERT and ERT/Chaperone drive a transient increase in GAA activity with the typical peak and trough kinetics. ERT/Chaperone shows an extended area under the curve vs. ERT only. Investigational gene therapy does not lead to the same peaks of GAA activity, however is associated with steady state levels of enzyme activity, which has the potential to drive stable and efficient glycogen clearance even in tissues naturally refractory to enzyme uptake (45). GAA, acid alpha-glucosidase; ERT, enzyme replacement therapy.
Concluding remarks
PD is a debilitating and potentially fatal disease. The development of ERT for the disease, more than a decade ago (5,10,12), represented a breakthrough in the management of the disease, particularly for IOPD patients. Today, PD remains an unmet medical need, as immunogenicity of recombinant GAA and long-term outcomes of ERT point out to the need for better treatments, both for pediatric and adult patients. Next generation ERTs are in the pipeline (28-30), however, because they mostly rely on the same mechanism of action of the current ERT, they are likely to result in only incremental benefit for patients.
Gene therapy holds the potential to revolutionize the way we treat PD, virtually providing a steady state supply of GAA enzyme to the entire body following a single medical intervention. Promising results obtained in preclinical studies in animal models of PD, along with results from clinical trials for various monogenic diseases, generated a lot of excitement about the prospect of a gene therapy for PD. As for any new investigational therapy, the primary goal of these early gene therapy trials should be focused on safety and on the potential limitations of the current gene transfer technologies (62,136).
Additionally, PD is extremely challenging and diverse, with IOPD patients presenting with clinical features quite different from those in the adult LOPD patient population. Furthermore, even within a category of patients, and even in patients carrying the same GAA genetic background (like most of LOPD patients), inter patient variability is extremely high, making trial design complex, also in view of the fact that endpoints of efficacy have not evolved significantly since the approval of ERT for PD and are far from being sensitive.
These complexities constitute important disease-specific challenges that will play an important role in the development of gene-based approaches for PD. Given the different mechanism of action of investigational gene therapy vs. ERT, future exploratory clinical work will likely help gaining a better understanding of whether the current measures of clinical outcomes used in ERT are best suited to capture the potential benefit of gene therapy.
Acknowledgments
Funding: This work was supported by Genethon and by the European Union H2020 action grant agreement 667751 (MYOCURE).
Footnotes
Conflicts of Interest: F Mingozzi and G Ronzitti are inventors in patent related to the development of gene therapy strategies for Pompe disease. G Ronzitti is recipient of a sponsored research grant from Spark Therapeutics. F Mingozzi is employee and owner of equity of Spark Therapeutics. P Laforet consulted for companies involved in the development of therapeutic approaches for Pompe disease.
References
- 1.Raben N, Fukuda T, Gilbert AL, et al. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol Ther 2005;11:48-56. 10.1016/j.ymthe.2004.09.017 [DOI] [PubMed] [Google Scholar]
- 2.van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet 2008;372:1342-53. 10.1016/S0140-6736(08)61555-X [DOI] [PubMed] [Google Scholar]
- 3.Ebbink BJ, Poelman E, Plug I, et al. Cognitive decline in classic infantile Pompe disease: An underacknowledged challenge. Neurology 2016;86:1260-1. 10.1212/WNL.0000000000002523 [DOI] [PubMed] [Google Scholar]
- 4.Laforet P, Petiot P, Nicolino M, et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology 2008;70:2063-6. 10.1212/01.wnl.0000313367.09469.13 [DOI] [PubMed] [Google Scholar]
- 5.van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe's disease. N Engl J Med 2010;362:1396-406. 10.1056/NEJMoa0909859 [DOI] [PubMed] [Google Scholar]
- 6.Fratantoni JC, Hall CW, Neufeld EF. Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968;162:570-2. 10.1126/science.162.3853.570 [DOI] [PubMed] [Google Scholar]
- 7.Dahms NM, Lobel P, Kornfeld S. Mannose 6-phosphate receptors and lysosomal enzyme targeting. J Biol Chem 1989;264:12115-8. [PubMed] [Google Scholar]
- 8.Neufeld EF. From serendipity to therapy. Annu Rev Biochem 2011;80:1-15. 10.1146/annurev.biochem.031209.093756 [DOI] [PubMed] [Google Scholar]
- 9.Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001;3:132-8. [DOI] [PubMed] [Google Scholar]
- 10.Van den Hout H, Reuser AJ, Vulto AG, et al. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 2000;356:397-8. 10.1016/S0140-6736(00)02533-2 [DOI] [PubMed] [Google Scholar]
- 11.Van den Hout JM, Kamphoven JH, Winkel LP, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004;113:e448-57. 10.1542/peds.113.5.e448 [DOI] [PubMed] [Google Scholar]
- 12.Kishnani PS, Corzo D, Nicolino M, et al. Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 2007;68:99-109. 10.1212/01.wnl.0000251268.41188.04 [DOI] [PubMed] [Google Scholar]
- 13.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010;99:26-33. 10.1016/j.ymgme.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn SH, Kronn D, Leslie ND, et al. Efficacy, safety profile, and immunogenicity of alglucosidase alfa produced at the 4,000-liter scale in US children and adolescents with Pompe disease: ADVANCE, a phase IV, open-label, prospective study. Genet Med 2018;20:1284-94. 10.1038/gim.2018.2 [DOI] [PubMed] [Google Scholar]
- 15.Chakrapani A, Vellodi A, Robinson P, et al. Treatment of infantile Pompe disease with alglucosidase alpha: the UK experience. J Inherit Metab Dis 2010;33:747-50. 10.1007/s10545-010-9206-3 [DOI] [PubMed] [Google Scholar]
- 16.van Gelder CM, Hoogeveen-Westerveld M, Kroos MA, et al. Enzyme therapy and immune response in relation to CRIM status: the Dutch experience in classic infantile Pompe disease. J Inherit Metab Dis 2015;38:305-14. 10.1007/s10545-014-9707-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol 2017;264:621-30. 10.1007/s00415-016-8219-8 [DOI] [PubMed] [Google Scholar]
- 18.Orlikowski D, Pellegrini N, Prigent H, et al. Recombinant human acid alpha-glucosidase (rhGAA) in adult patients with severe respiratory failure due to Pompe disease. Neuromuscul Disord 2011;21:477-82. 10.1016/j.nmd.2011.04.001 [DOI] [PubMed] [Google Scholar]
- 19.Papadopoulos C, Orlikowski D, Prigent H, et al. Effect of enzyme replacement therapy with alglucosidase alfa (Myozyme(R)) in 12 patients with advanced late-onset Pompe disease. Mol Genet Metab 2017;122:80-5. 10.1016/j.ymgme.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 20.Banugaria SG, Prater SN, Ng YK, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med 2011;13:729-36. 10.1097/GIM.0b013e3182174703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebbink BJ, Poelman E, Aarsen FK, et al. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Dev Med Child Neurol 2018;60:579-86. 10.1111/dmcn.13740 [DOI] [PubMed] [Google Scholar]
- 22.Xu S, Lun Y, Frascella M, et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight 2019. doi: . 10.1172/jci.insight.125358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kishnani P, Tarnopolsky M, Roberts M, et al. Duvoglustat HCl Increases Systemic and Tissue Exposure of Active Acid alpha-Glucosidase in Pompe Patients Co-administered with Alglucosidase alpha. Mol Ther 2017;25:1199-208. 10.1016/j.ymthe.2017.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parenti G, Fecarotta S, la Marca G, et al. A chaperone enhances blood alpha-glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 2014;22:2004-12. 10.1038/mt.2014.138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther 2009;17:954-63. 10.1038/mt.2009.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basile I, Da Silva A, El Cheikh K, et al. Efficient therapy for refractory Pompe disease by mannose 6-phosphate analogue grafting on acid alpha-glucosidase. J Control Release 2018;269:15-23. 10.1016/j.jconrel.2017.10.043 [DOI] [PubMed] [Google Scholar]
- 27.Kang JY, Shin KK, Kim HH, et al. Lysosomal Targeting Enhancement by Conjugation of Glycopeptides Containing Mannose-6-phosphate Glycans Derived from Glyco-engineered Yeast. Sci Rep 2018;8:8730. 10.1038/s41598-018-26913-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baik A, Aaron N, Birnbaum MS, et al. Next-generation antibody-guided enzyme replacement therapy in Pompe disease mice. Mol Genet Metab 2018;123:S21 10.1016/j.ymgme.2017.12.028 [DOI] [Google Scholar]
- 29.Maga JA, Zhou J, Kambampati R, et al. Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. J Biol Chem 2013;288:1428-38. 10.1074/jbc.M112.438663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi H, Sun T, Armstrong D, et al. Antibody-mediated enzyme replacement therapy targeting both lysosomal and cytoplasmic glycogen in Pompe disease. J Mol Med (Berl) 2017;95:513-21. 10.1007/s00109-017-1505-9 [DOI] [PubMed] [Google Scholar]
- 31.Peng J, Dalton J, Butt M, et al. Reveglucosidase alfa (BMN 701), an IGF2-Tagged rhAcid alpha-Glucosidase, Improves Respiratory Functional Parameters in a Murine Model of Pompe Disease. J Pharmacol Exp Ther 2017;360:313-23. 10.1124/jpet.116.235952 [DOI] [PubMed] [Google Scholar]
- 32.Byrne BJ, Geberhiwot T, Barshop BA, et al. A study on the safety and efficacy of reveglucosidase alfa in patients with late-onset Pompe disease. Orphanet J Rare Dis 2017;12:144. 10.1186/s13023-017-0693-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koeberl DD, Luo X, Sun B, et al. Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol Genet Metab 2011;103:107-12. 10.1016/j.ymgme.2011.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koeberl DD, Li S, Dai J, et al. beta2 Agonists enhance the efficacy of simultaneous enzyme replacement therapy in murine Pompe disease. Mol Genet Metab 2012;105:221-7. 10.1016/j.ymgme.2011.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li S, Sun B, Nilsson MI, et al. Adjunctive beta2-agonists reverse neuromuscular involvement in murine Pompe disease. FASEB J 2013;27:34-44. 10.1096/fj.12-207472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koeberl DD, Austin S, Case LE, et al. Adjunctive albuterol enhances the response to enzyme replacement therapy in late-onset Pompe disease. FASEB J 2014;28:2171-6. 10.1096/fj.13-241893 [DOI] [PubMed] [Google Scholar]
- 37.Koeberl DD, Case LE, Smith EC, et al. Correction of Biochemical Abnormalities and Improved Muscle Function in a Phase I/II Clinical Trial of Clenbuterol in Pompe Disease. Mol Ther 2018;26:2304-14. 10.1016/j.ymthe.2018.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fink AL. Chaperone-mediated protein folding. Physiol Rev 1999;79:425-49. 10.1152/physrev.1999.79.2.425 [DOI] [PubMed] [Google Scholar]
- 39.Parenti G, Moracci M, Fecarotta S, et al. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med Chem 2014;6:1031-45. 10.4155/fmc.14.40 [DOI] [PubMed] [Google Scholar]
- 40.Flanagan JJ, Rossi B, Tang K, et al. The pharmacological chaperone 1-deoxynojirimycin increases the activity and lysosomal trafficking of multiple mutant forms of acid alpha-glucosidase. Hum Mutat 2009;30:1683-92. 10.1002/humu.21121 [DOI] [PubMed] [Google Scholar]
- 41.Khanna R, Powe AC, Jr, Lun Y, et al. The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS One 2014;9:e102092. 10.1371/journal.pone.0102092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porto C, Cardone M, Fontana F, et al. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther 2009;17:964-71. 10.1038/mt.2009.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsujino S, Kinoshita N, Tashiro T, et al. Adenovirus-mediated transfer of human acid maltase gene reduces glycogen accumulation in skeletal muscle of Japanese quail with acid maltase deficiency. Hum Gene Ther 1998;9:1609-16. 10.1089/hum.1998.9.11-1609 [DOI] [PubMed] [Google Scholar]
- 44.Sun B, Young SP, Li P, et al. Correction of multiple striated muscles in murine Pompe disease through adeno-associated virus-mediated gene therapy. Mol Ther 2008;16:1366-71. 10.1038/mt.2008.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puzzo F, Colella P, Biferi MG, et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci Transl Med 2017. doi: . 10.1126/scitranslmed.aam6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han SO, Ronzitti G, Arnson B, et al. Low-Dose Liver-Targeted Gene Therapy for Pompe Disease Enhances Therapeutic Efficacy of ERT via Immune Tolerance Induction. Mol Ther Methods Clin Dev 2017;4:126-36. 10.1016/j.omtm.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colella P, Sellier P, Costa Verdera H, et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol Ther Methods Clin Dev 2018;12:85-101. 10.1016/j.omtm.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fraites TJ, Jr, Schleissing MR, Shanely RA, et al. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther 2002;5:571-8. 10.1006/mthe.2002.0580 [DOI] [PubMed] [Google Scholar]
- 49.Schuster DJ, Dykstra JA, Riedl MS, et al. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front Neuroanat 2014;8:42. 10.3389/fnana.2014.00042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Til NP, Stok M, Aerts Kaya FS, et al. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood 2010;115:5329-37. 10.1182/blood-2009-11-252874 [DOI] [PubMed] [Google Scholar]
- 51.Zaretsky JZ, Candotti F, Boerkoel C, et al. Retroviral transfer of acid alpha-glucosidase cDNA to enzyme-deficient myoblasts results in phenotypic spread of the genotypic correction by both secretion and fusion. Hum Gene Ther 1997;8:1555-63. 10.1089/hum.1997.8.13-1555 [DOI] [PubMed] [Google Scholar]
- 52.Pauly DF, Johns DC, Matelis LA, et al. Complete correction of acid alpha-glucosidase deficiency in Pompe disease fibroblasts in vitro, and lysosomally targeted expression in neonatal rat cardiac and skeletal muscle. Gene Ther 1998;5:473-80. 10.1038/sj.gt.3300609 [DOI] [PubMed] [Google Scholar]
- 53.Amalfitano A, McVie-Wylie AJ, Hu H, et al. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci U S A 1999;96:8861-6. 10.1073/pnas.96.16.8861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kyosen SO, Iizuka S, Kobayashi H, et al. Neonatal gene transfer using lentiviral vector for murine Pompe disease: long-term expression and glycogen reduction. Gene Ther 2010;17:521-30. 10.1038/gt.2009.160 [DOI] [PubMed] [Google Scholar]
- 55.Kohler L, Puertollano R, Raben N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018;15:928-42. 10.1007/s13311-018-0655-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doerfler PA, Nayak S, Corti M, et al. Targeted approaches to induce immune tolerance for Pompe disease therapy. Mol Ther Methods Clin Dev 2016;3:15053. 10.1038/mtm.2015.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Balakrishnan B, Jayandharan GR. Basic biology of adeno-associated virus (AAV) vectors used in gene therapy. Curr Gene Ther 2014;14:86-100. 10.2174/1566523214666140302193709 [DOI] [PubMed] [Google Scholar]
- 58.Calcedo R, Morizono H, Wang L, et al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin Vaccine Immunol 2011;18:1586-8. 10.1128/CVI.05107-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Erles K, Sebokova P, Schlehofer JR. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J Med Virol 1999;59:406-11. [DOI] [PubMed] [Google Scholar]
- 60.Li C, Narkbunnam N, Samulski RJ, et al. Neutralizing antibodies against adeno-associated virus examined prospectively in pediatric patients with hemophilia. Gene Ther 2012;19:288-94. 10.1038/gt.2011.90 [DOI] [PubMed] [Google Scholar]
- 61.Smith RH. Adeno-associated virus integration: virus versus vector. Gene Ther 2008;15:817-22. 10.1038/gt.2008.55 [DOI] [PubMed] [Google Scholar]
- 62.Mingozzi F, High KA. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu Rev Virol 2017;4:511-34. 10.1146/annurev-virology-101416-041936 [DOI] [PubMed] [Google Scholar]
- 63.Valdmanis PN, Lisowski L, Kay MA. rAAV-mediated tumorigenesis: still unresolved after an AAV assault. Mol Ther 2012;20:2014-7. 10.1038/mt.2012.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, Malani N, Hamilton SR, et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 2011;117:3311-9. 10.1182/blood-2010-08-302729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther 2005;5:285-97. 10.2174/1566523054065057 [DOI] [PubMed] [Google Scholar]
- 66.Rabinowitz JE, Rolling F, Li C, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol 2002;76:791-801. 10.1128/JVI.76.2.791-801.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsushita T, Elliger S, Elliger C, et al. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 1998;5:938-45. 10.1038/sj.gt.3300680 [DOI] [PubMed] [Google Scholar]
- 68.Gao GP, Qu G, Faust LZ, et al. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum Gene Ther 1998;9:2353-62. 10.1089/hum.1998.9.16-2353 [DOI] [PubMed] [Google Scholar]
- 69.Marek M, van Oers MM, Devaraj FF, et al. Engineering of baculovirus vectors for the manufacture of virion-free biopharmaceuticals. Biotechnol Bioeng 2011;108:1056-67. 10.1002/bit.23028 [DOI] [PubMed] [Google Scholar]
- 70.El-Gharbawy AH, Mackey J, DeArmey S, et al. An individually, modified approach to desensitize infants and young children with Pompe disease, and significant reactions to alglucosidase alfa infusions. Mol Genet Metab 2011;104:118-22. 10.1016/j.ymgme.2011.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masat E, Laforet P, De Antonio M, et al. Long-term exposure to Myozyme results in a decrease of anti-drug antibodies in late-onset Pompe disease patients. Sci Rep 2016;6:36182. 10.1038/srep36182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang P, Sun B, Osada T, et al. Immunodominant liver-specific expression suppresses transgene-directed immune responses in murine pompe disease. Hum Gene Ther 2012;23:460-72. 10.1089/hum.2011.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun B, Li S, Bird A, et al. Antibody formation and mannose-6-phosphate receptor expression impact the efficacy of muscle-specific transgene expression in murine Pompe disease. J Gene Med 2010;12:881-91. 10.1002/jgm.1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kazi ZB, Desai AK, Troxler RB, et al. An immune tolerance approach using transient low-dose methotrexate in the ERT-naive setting of patients treated with a therapeutic protein: experience in infantile-onset Pompe disease. Genet Med 2019;21:887-95. 10.1038/s41436-018-0270-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mingozzi F, Liu YL, Dobrzynski E, et al. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J Clin Invest 2003;111:1347-56. 10.1172/JCI200316887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun B, Zhang H, Franco LM, et al. Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther 2005;11:57-65. 10.1016/j.ymthe.2004.10.004 [DOI] [PubMed] [Google Scholar]
- 77.Sun B, Zhang H, Franco LM, et al. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther 2005;11:889-98. 10.1016/j.ymthe.2005.01.012 [DOI] [PubMed] [Google Scholar]
- 78.Franco LM, Sun B, Yang X, et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther 2005;12:876-84. 10.1016/j.ymthe.2005.04.024 [DOI] [PubMed] [Google Scholar]
- 79.Mah C, Pacak CA, Cresawn KO, et al. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol Ther 2007;15:501-7. 10.1038/sj.mt.6300100 [DOI] [PubMed] [Google Scholar]
- 80.Koeberl DD, Kishnani PS. Immunomodulatory gene therapy in lysosomal storage disorders. Curr Gene Ther 2009;9:503-10. 10.2174/156652309790031094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mah C, Cresawn KO, Fraites TJ, Jr, et al. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther 2005;12:1405-9. 10.1038/sj.gt.3302550 [DOI] [PubMed] [Google Scholar]
- 82.Mah CS, Falk DJ, Germain SA, et al. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol Ther 2010;18:502-10. 10.1038/mt.2009.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rucker M, Fraites TJ, Jr, Porvasnik SL, et al. Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development 2004;131:3007-19. 10.1242/dev.01169 [DOI] [PubMed] [Google Scholar]
- 84.Elmallah MK, Falk DJ, Nayak S, et al. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol Ther 2014;22:702-12. 10.1038/mt.2013.282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qiu K, Falk DJ, Reier PJ, et al. Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther 2012;20:21-7. 10.1038/mt.2011.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cresawn KO, Fraites TJ, Wasserfall C, et al. Impact of humoral immune response on distribution and efficacy of recombinant adeno-associated virus-derived acid alpha-glucosidase in a model of glycogen storage disease type II. Hum Gene Ther 2005;16:68-80. 10.1089/hum.2005.16.68 [DOI] [PubMed] [Google Scholar]
- 87.Ziegler RJ, Bercury SD, Fidler J, et al. Ability of adeno-associated virus serotype 8-mediated hepatic expression of acid alpha-glucosidase to correct the biochemical and motor function deficits of presymptomatic and symptomatic Pompe mice. Hum Gene Ther 2008;19:609-21. 10.1089/hum.2008.010 [DOI] [PubMed] [Google Scholar]
- 88.Wang G, Young SP, Bali D, et al. Assessment of toxicity and biodistribution of recombinant AAV8 vector-mediated immunomodulatory gene therapy in mice with Pompe disease. Mol Ther Methods Clin Dev 2014;1:14018. 10.1038/mtm.2014.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Falk DJ, Todd AG, Lee S, et al. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet 2015;24:625-36. 10.1093/hmg/ddu476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Todd AG, McElroy JA, Grange RW, et al. Correcting Neuromuscular Deficits With Gene Therapy in Pompe Disease. Ann Neurol 2015;78:222-34. 10.1002/ana.24433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Falk DJ, Mah CS, Soustek MS, et al. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther 2013;21:1661-7. 10.1038/mt.2013.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.ElMallah MK, Falk DJ, Lane MA, et al. Retrograde gene delivery to hypoglossal motoneurons using adeno-associated virus serotype 9. Hum Gene Ther Methods 2012;23:148-56. 10.1089/hgtb.2012.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Falk DJ, Soustek MS, Todd AG, et al. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol Ther Methods Clin Dev 2015;2:15007. 10.1038/mtm.2015.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Corti M, Liberati C, Smith BK, et al. Safety of Intradiaphragmatic Delivery of Adeno-Associated Virus-Mediated Alpha-Glucosidase (rAAV1-CMV-hGAA) Gene Therapy in Children Affected by Pompe Disease. Hum Gene Ther Clin Dev 2017;28:208-18. 10.1089/humc.2017.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith BK, Collins SW, Conlon TJ, et al. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther 2013;24:630-40. 10.1089/hum.2012.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Byrne PI, Collins S, Mah CC, et al. Phase I/II trial of diaphragm delivery of recombinant adeno-associated virus acid alpha-glucosidase (rAAaV1-CMV-GAA) gene vector in patients with Pompe disease. Hum Gene Ther Clin Dev 2014;25:134-63. 10.1089/humc.2014.2514 [DOI] [PubMed] [Google Scholar]
- 97.Smith BK, Martin AD, Lawson LA, et al. Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: Clinical evidence of respiratory plasticity. Exp Neurol 2017;287:216-24. 10.1016/j.expneurol.2016.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Calcedo R, Somanathan S, Qin Q, et al. Class I-restricted T-cell responses to a polymorphic peptide in a gene therapy clinical trial for alpha-1-antitrypsin deficiency. Proc Natl Acad Sci U S A 2017;114:1655-9. 10.1073/pnas.1617726114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mendell JR, Campbell K, Rodino-Klapac L, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med 2010;363:1429-37. 10.1056/NEJMoa1000228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mendell JR, Al-Zaidy S, Shell R, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 2017;377:1713-22. 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 101.Childers MK, Joubert R, Poulard K, et al. Gene therapy prolongs survival and restores function in murine and canine models of myotubular myopathy. Sci Transl Med 2014;6:220ra10. 10.1126/scitranslmed.3007523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Le Guiner C, Servais L, Montus M, et al. Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat Commun 2017;8:16105. 10.1038/ncomms16105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mack DL, Poulard K, Goddard MA, et al. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Mol Ther 2017;25:839-54. 10.1016/j.ymthe.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nathwani AC, Gray JT, Ng CY, et al. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood 2006;107:2653-61. 10.1182/blood-2005-10-4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nathwani AC, Reiss UM, Tuddenham EG, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 2014;371:1994-2004. 10.1056/NEJMoa1407309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011;365:2357-65. 10.1056/NEJMoa1108046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.George LA, Sullivan SK, Giermasz A, et al. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med 2017;377:2215-27. 10.1056/NEJMoa1708538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rangarajan S, Walsh L, Lester W, et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med 2017;377:2519-30. 10.1056/NEJMoa1708483 [DOI] [PubMed] [Google Scholar]
- 109.Miesbach W, Sawyer EK. Practical Implications of Factor IX Gene Transfer for Individuals with Hemophilia B: A Clinical Perspective. Hum Gene Ther Clin Dev 2018;29:80-9. 10.1089/humc.2017.253 [DOI] [PubMed] [Google Scholar]
- 110.Nathwani AC, Rosales C, McIntosh J, et al. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol Ther 2011;19:876-85. 10.1038/mt.2010.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niemeyer GP, Herzog RW, Mount J, et al. Long-term correction of inhibitor-prone hemophilia B dogs treated with liver-directed AAV2-mediated factor IX gene therapy. Blood 2009;113:797-806. 10.1182/blood-2008-10-181479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mingozzi F, Hasbrouck NC, Basner-Tschakarjan E, et al. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood 2007;110:2334-41. 10.1182/blood-2007-03-080093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dobrzynski E, Mingozzi F, Liu YL, et al. Induction of antigen-specific CD4+ T-cell anergy and deletion by in vivo viral gene transfer. Blood 2004;104:969-77. 10.1182/blood-2004-03-0847 [DOI] [PubMed] [Google Scholar]
- 114.Cao TM, Thomas A, Wang Y, et al. A chromosome 16 quantitative trait locus regulates allogeneic bone marrow engraftment in nonmyeloablated mice. Blood 2009;114:202-10. 10.1182/blood-2009-03-208801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sherman A, Biswas M, Herzog RW. Innovative Approaches for Immune Tolerance to Factor VIII in the Treatment of Hemophilia A. Front Immunol 2017;8:1604. 10.3389/fimmu.2017.01604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.D'Avola D, Lopez-Franco E, Sangro B, et al. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J Hepatol 2016;65:776-83. 10.1016/j.jhep.2016.05.012 [DOI] [PubMed] [Google Scholar]
- 117.Sun B, Kulis MD, Young SP, et al. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine pompe disease. Mol Ther 2010;18:353-60. 10.1038/mt.2009.195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bond JE, Kishnani PS, Koeberl DD. Immunomodulatory, liver depot gene therapy for Pompe disease. Cell Immunol 2017. [Epub ahead of print]. 10.1016/j.cellimm.2017.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun B, Zhang H, Benjamin DK, Jr, et al. Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid alpha-glucosidase in glycogen storage disease type II. Mol Ther 2006;14:822-30. 10.1016/j.ymthe.2006.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Petersen TN, Brunak S, von Heijne G, et al. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011;8:785-6. 10.1038/nmeth.1701 [DOI] [PubMed] [Google Scholar]
- 121.Perrin GQ, Zolotukhin I, Sherman A, et al. Dynamics of antigen presentation to transgene product-specific CD4(+) T cells and of Treg induction upon hepatic AAV gene transfer. Mol Ther Methods Clin Dev 2016;3:16083. 10.1038/mtm.2016.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bortolussi G, Zentillin L, Vanikova J, et al. Life-long correction of hyperbilirubinemia with a neonatal liver-specific AAV-mediated gene transfer in a lethal mouse model of Crigler-Najjar Syndrome. Hum Gene Ther 2014;25:844-55. 10.1089/hum.2013.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meliani A, Boisgerault F, Hardet R, et al. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat Commun 2018;9:4098. 10.1038/s41467-018-06621-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kuranda K, Jean-Alphonse P, Leborgne C, et al. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J Clin Invest 2018;128:5267-79. 10.1172/JCI122372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Corti M, Cleaver B, Clement N, et al. Evaluation of Readministration of a Recombinant Adeno-Associated Virus Vector Expressing Acid Alpha-Glucosidase in Pompe Disease: Preclinical to Clinical Planning. Hum Gene Ther Clin Dev 2015;26:185-93. 10.1089/humc.2015.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lim JA, Yi H, Gao F, et al. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid alpha-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol Ther Methods Clin Dev 2019;12:233-45. 10.1016/j.omtm.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lee NC, Hwu WL, Muramatsu SI, et al. A Neuron-Specific Gene Therapy Relieves Motor Deficits in Pompe Disease Mice. Mol Neurobiol 2018;55:5299-309. 10.1007/s12035-017-0763-4 [DOI] [PubMed] [Google Scholar]
- 128.Hordeaux J, Dubreil L, Robveille C, et al. Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease. Acta Neuropathol Commun 2017;5:66. 10.1186/s40478-017-0464-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Spampanato C, Feeney E, Li L, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 2013;5:691-706. 10.1002/emmm.201202176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gatto F, Rossi B, Tarallo A, et al. AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci Rep 2017;7:15089. 10.1038/s41598-017-15352-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Clayton NP, Nelson CA, Weeden T, et al. Antisense Oligonucleotide-mediated Suppression of Muscle Glycogen Synthase 1 Synthesis as an Approach for Substrate Reduction Therapy of Pompe Disease. Mol Ther Nucleic Acids 2014;3:e206. 10.1038/mtna.2014.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Douillard-Guilloux G, Raben N, Takikita S, et al. Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum Mol Genet 2010;19:684-96. 10.1093/hmg/ddp535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sukigara S, Liang WC, Komaki H, et al. Muscle glycogen storage disease 0 presenting recurrent syncope with weakness and myalgia. Neuromuscul Disord 2012;22:162-5. 10.1016/j.nmd.2011.08.008 [DOI] [PubMed] [Google Scholar]
- 134.van der Wal E, Bergsma AJ, Pijnenburg JM, et al. Antisense Oligonucleotides Promote Exon Inclusion and Correct the Common c.-32-13T>G GAA Splicing Variant in Pompe Disease. Mol Ther Nucleic Acids 2017;7:90-100. 10.1016/j.omtn.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Goyenvalle A, Leumann C, Garcia L. Therapeutic Potential of Tricyclo-DNA antisense oligonucleotides. J Neuromuscul Dis 2016;3:157-67. 10.3233/JND-160146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Colella P, Ronzitti G, Mingozzi F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol Ther Methods Clin Dev 2017;8:87-104. 10.1016/j.omtm.2017.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]