

Abstract
The vacuolar H+-ATPase (V-ATPase) is an ATP-driven proton pump present in various intracellular membranes and at the plasma membrane of specialized cell types. Previous work has reported that plasma membrane V-ATPases are key players in breast cancer cell invasiveness. The two subunit a-isoforms known to target the V-ATPase to the plasma membrane are a3 and a4, and expression of a3 has been shown to correlate with plasma membrane localization of the V-ATPase in various invasive human breast cancer cell lines. Here we analyzed the role of subunit a-isoforms in the invasive mouse breast cancer cell line, 4T1-12B. Quantitation of mRNA levels for each isoform by quantitative RT-PCR revealed that a4 is the dominant isoform expressed in these cells. Using a CRISPR/Cas9-based approach to disrupt the genes encoding each of the four V-ATPase subunit a-isoforms, we found that ablation of only the a4-encoding gene significantly inhibits invasion and migration of 4T1-12B cells. Additionally, cells with disrupted a4 exhibited reduced V-ATPase expression at the leading edge, suggesting that the a4 isoform is primarily responsible for targeting the V-ATPase to the plasma membrane in 4T1-12B cells. These findings suggest that different subunit a-isoforms may direct V-ATPases to the plasma membrane of different invasive breast cancer cell lines. They further suggest that expression of V-ATPases at the cell surface is the primary factor that promotes an invasive cancer cell phenotype.
Keywords: vacuolar ATPase, breast cancer, cell invasion, cell migration, plasma membrane, proton pump, metastasis, a-subunit isoforms, protein trafficking
Introduction
The metastasis of cancer cells from a primary site to secondary sites within the body is the leading cause of cancer mortality. Metastasis is a complex process requiring cancer cells to intravasate from the primary tumor into the circulatory or lymphatic system, followed by extravasation of the cells into secondary sites throughout the body (1, 2). Cancer cells capable of metastasis exhibit an invasive phenotype, meaning they have the ability to penetrate through extracellular matrix and the basement membrane (3). This phenotype is highly dependent on proton extrusion from cancer cells, because proton secretion across the plasma membrane promotes both an alkaline cytosolic pH required for cytoskeletal remodeling and an acidic extracellular space essential for the activity of secreted acid-dependent proteases (cathepsins) that participate in degradation of extracellular matrix (4, 5). This proton transport across the plasma membrane has increasingly been shown to depend upon the activity of the vacuolar H+-ATPase (V-ATPase)3 (6).
V-ATPases are multisubunit, ATP-dependent proton pumps present in both intracellular membranes and the plasma membrane of specialized cell types (7). The 13 subunits that make up the V-ATPase are split into two major domains: the cytosolic V1 domain and the membrane-embedded V0 domain. V1 is responsible for ATP hydrolysis and is composed of subunits A–H, whereas the V0 domain contains subunits a, c, c″, d, and e and transports protons from the cytosol into either intracellular compartments or the extracellular space. Within the cell, V-ATPases localize to the Golgi, secretory vesicles, endosomes, and lysosomes and participate in a variety of processes, including receptor-mediated endocytosis and receptor recycling, zymogen activation, protein trafficking and degradation, and cellular signaling (6–10). The V-ATPase is also present at the cell surface of specialized cell types, such as osteoclasts and renal intercalated cells, where it functions in bone resorption and pH homeostasis, respectively (7, 8, 11–13).
Increasing evidence implicating the V-ATPase in cancer cell invasiveness has emerged in recent years (6). A number of studies have found that there is increased expression of various V-ATPase subunits in many invasive cancers (14–20). Inhibiting V-ATPase activity using either pharmacological inhibitors or genetic approaches significantly reduces the invasiveness and migratory capacity of numerous cancer cell types (5, 18, 21–28). Specifically, plasma membrane V-ATPases appear to play a central role in cancer cell invasiveness (6). Studies looking at localization of the V-ATPase in invasive breast, liver, lung, esophageal, prostate, ovarian, and pancreatic cancers as well as Ewing sarcoma and melanoma all show an up-regulation of the V-ATPase at the plasma membrane (5, 18, 19, 21–25, 29–33). Recent work from our laboratory using an in vitro transwell assay showed that specific inhibition of plasma membrane V-ATPases inhibited the invasion and migration of invasive MDA-MB-231 breast cancer cells to a similar degree as pan-V-ATPase inhibitors (5). This suggests that plasma membrane V-ATPases may play a key role in promoting an invasive phenotype in breast cancer cells.
Different isoforms of subunit V0a are responsible for localizing V-ATPases to various subcellular membranes (34). Mammals express four different subunit a-isoforms, a1–a4, where a3 and a4 are known to target the V-ATPase to the plasma membrane of osteoclasts and renal intercalated cells, respectively (7, 8, 11, 13). Subunit a3 is overexpressed at the mRNA level in a number of cancer types, including melanoma, breast, pancreatic, and ovarian cancers (18, 21, 23–25, 33). Similarly, a4 is overexpressed in glioma (35). Previous work from our laboratory has shown that mRNA levels of a3 and a4 are up-regulated in the invasive MDA-MB-231 breast cancer cell line compared with the noninvasive MCF7 line (24). siRNA-mediated knockdown of either a3 or a4 in MDA-MB-231 cells reduced the migration and invasion of these cells (24). Similarly, the invasive MCF10CA1a breast cancer cell line displays increased levels of a3 mRNA relative to the parental MCF10a cell line, and knockdown of a3 decreased in vitro migration and invasion of MCF10CA1a cells (21). Importantly, overexpression of a3 in noninvasive MCF10a cells increased their invasiveness and localization of the V-ATPase to the plasma membrane (21). Subunit a3 has also been implicated in melanoma, where knockdown of a3 in a melanoma cell line reduced in vivo metastasis in mice (33).
Recently, a3-containing V-ATPases were localized to the plasma membrane of a number of different invasive breast cancer cell lines using isoform-specific antibodies against a3 (25). This was the first study showing that a3-containing V-ATPases localize to the leading edge of highly invasive, migrating breast cancer cells but not of noninvasive breast epithelial cells. Our laboratory also showed that a3 mRNA is overexpressed in 43 of 43 human breast tumor samples relative to normal breast tissue by 2.5–50-fold and that expression of the a3 protein is highest in invasive human breast carcinoma relative to both noninvasive solid tumors and normal breast tissue (25).
Together, this research highlights the importance of plasma membrane V-ATPases in cancer cell invasiveness and the translational relevance of studying these proteins in vitro. Understanding the trafficking and regulation of these pumps in various cancer cell types may provide new therapies against metastatic cancer. Here, we describe the first use of CRISPR/Cas9 to disrupt individual subunit a-isoforms of the V-ATPase in invasive breast cancer cells. Our results suggest that the a4 isoform plays a role in in vitro invasion and migration as well as expression of V-ATPases at the plasma membrane of an invasive mouse breast cancer cell line.
Results
Pharmacological inhibition of the V-ATPase inhibits in vitro migration and invasion of 4T1-12B breast cancer cells
To determine the role of V-ATPases in a mouse model of invasive breast cancer, we examined the 4T1-12B mouse mammary carcinoma cell line. Whereas the V-ATPase has previously been shown to function in in vitro migration and invasion of various human breast cancer cell lines (5, 21, 24, 25), using a breast cancer cell line derived from mouse will be important in future studies employing an animal model of breast cancer in an immunocompetent host. 4T1 cells originate from a spontaneously formed mammary tumor in mouse and are commonly used as a model of breast cancer metastasis (36, 37). When orthotopically injected into mice, these cells metastasize to a number of secondary sites, including liver, lung, brain, and bone (38–43). The 4T1-12B cells used in the current study are a luciferase-expressing cell line derived from 4T1 cells that retain all of the properties of the parental line (44).
To ascertain whether V-ATPase activity is required for in vitro invasion and migration of these cells, an in vitro transwell assay was employed, as described previously (5). Invasion is measured using wells coated with the extracellular matrix–like material Matrigel, whereas migration is measured using uncoated wells. 4T1-12B cells were treated with DMSO or the V-ATPase–specific inhibitor concanamycin A (ConA) at 1 nm, and invasion or migration was induced by the presence of serum in the trans compartment. Treatment with ConA reduced migration by 62% and invasion by 58% relative to control cells (Fig. 1). To validate that the decrease in invasion and migration after treatment with ConA was not due to cell death, cell viability was measured using a trypan blue exclusion assay. No difference in cell viability was detected following ConA treatment (data not shown). These data suggest that V-ATPase activity is important for in vitro invasion and migration by 4T1-12B cells.
Figure 1.
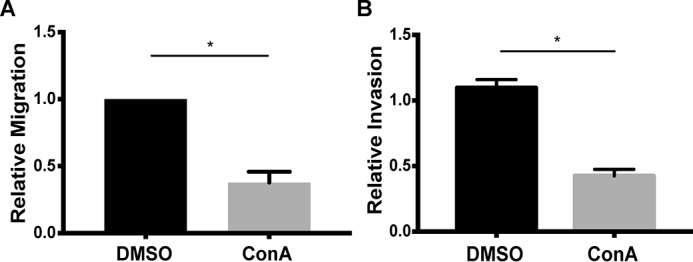
In vitro migration (A) and invasion (B) of 4T1-12B cells after concanamycin A treatment. Transwell migration and invasion assays were performed as described under “Experimental procedures.” Cells were treated with either DMSO or 1 nm ConA, and the assay was initiated. The number of cells that had migrated or invaded to the trans side of the insert were counted in triplicate and normalized to the number of cells in the control condition. Values represent the mean ± S.E. (error bars). A, n = 4 (*, p < 0.05); B, n = 3 (*, p < 0.05).
mRNA levels of subunit a-isoforms in 4T1-12B cells
Previous work has suggested that specific subunit a-isoforms contribute to the invasiveness of breast cancer cells (21, 24, 25). We first wished to determine which subunit a-isoforms are expressed in 4T1-12B cells using quantitative RT-PCR. As shown in Fig. 2, mRNA for a4 was present at the highest levels, followed by a3 and a2. mRNA encoding a1 was barely detectable in samples from 4T1-12B cells. Thus, a4 appears to be the dominant isoform at the mRNA level in these cells.
Figure 2.
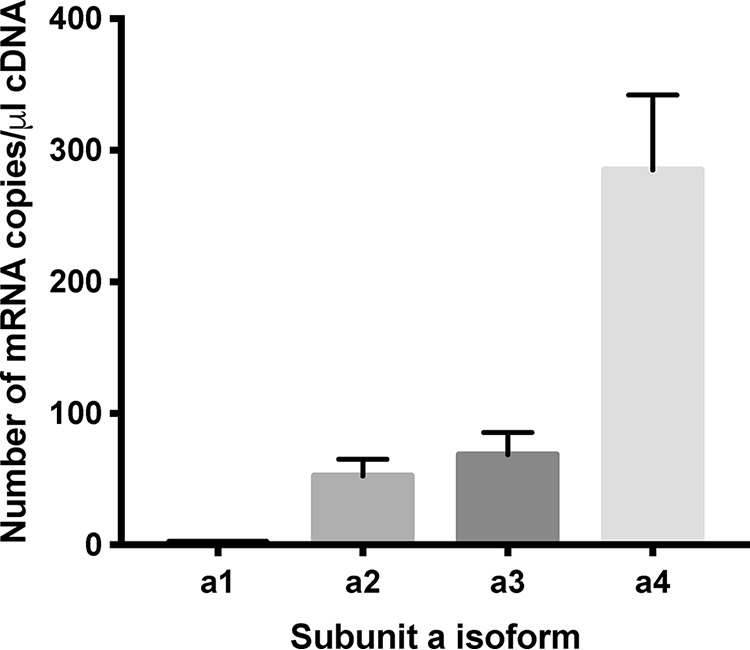
mRNA levels of subunit a-isoforms in 4T1-12B cells. mRNA levels of each a-subunit isoform in 4T1-12B cells were determined using quantitative RT-PCR. Short regions of cDNA of each a-subunit isoform were used to establish a standard curve. The number of mRNA copies present per μg of RNA for each a-isoform is shown. Values represent the mean ± S.E. (error bars), n = 3.
CRISPR/Cas9-mediated knockout of a-subunit isoforms in 4T1-12B cells
Next, to determine whether subunit a-isoforms are important in 4T1-12B cell invasiveness, each of the four a-isoform genes was targeted for disruption using the CRISPR/Cas9 system (45). Cells were transfected with a CRISPR/Cas9 plasmid containing guide RNA (gRNA) sequences specific to each of the four isoforms or an empty plasmid as a control. After clonal populations were isolated, Western blotting was used to determine the level of subunit a-isoform expression. Clones showing the most complete knockout were used for all experiments, and at least two knockout clones for each isoform were tested (Fig. S1). As seen in Fig. 3A, there was successful knockout of each of the targeted subunit a-isoforms without a change in cell viability (Fig. S2). In several cases, partial changes in expression of isoforms not targeted by CRISPR/Cas9 were observed. Thus, for a1-knockout cells, a4 expression is partially reduced, whereas for a4-knockout cells, a1 is partially reduced. Quantitation of expression of a4 in the a1-knockout cells showed that a4 levels were reduced by 30 ± 20% relative to control cells. Similarly, a1 levels were reduced by 30 ± 10% in a4-knockout cells, again relative to control cells. Furthermore, expression of a1 is increased in the a2- and a3-targeted cells, suggesting that a1 expression may increase to compensate for the loss of certain isoforms. Interestingly, only partial knockdown of a3 could be obtained in the cells in which a3 was targeted by CRISPR/Cas9 (Fig. 3A). Quantitation indicates that a3 levels were reduced by 63 ± 7% in a3-knockout cells relative to the control cells. Continued efforts to obtain complete knockout of a3 in 4T1-12B cells were unsuccessful, suggesting that a3 expression may be required for viability of these cells. Nevertheless, previous work using siRNA-mediated knockdown of a3 in breast cancer cell lines revealed significant effects on cell migration and invasion following reduction of expression by a similar magnitude (21, 24, 25).
Figure 3.
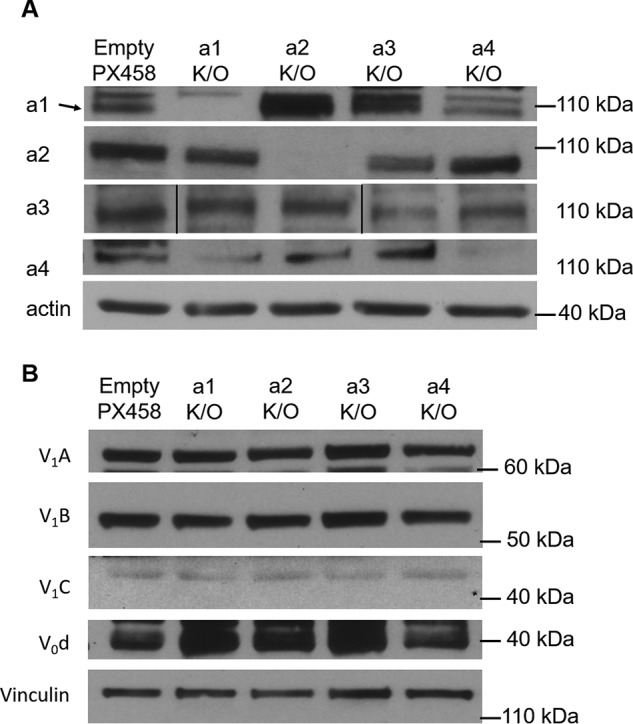
Protein expression of a-subunit isoforms in a-isoform knockout 4T1-12B cells. A, after CRISPR/Cas9 gene editing was performed to knock out individual a-subunit isoforms in 4T1-12B cells, whole-cell lysates were collected and subjected to SDS-PAGE and Western blotting as described under “Experimental procedures.” Antibodies against the individual a-isoforms were used to detect the amount of each a-subunit isoform in all four of the knockout cell lines (a1–a4 K/O) as well as the cell line transfected with the empty CRISPR/Cas9 plasmid (Empty PX458). β-Actin was used as a loading control. Shown is a representative image (n ≥ 3). B, antibodies against V1A, V1B, V1C, and V0d were used to detect the levels of other V-ATPase subunits by Western blotting in all four of the a-subunit isoform knockout cell lines (a1–a4 K/O), as well as the Empty PX458 control cell line. Vinculin was used as a loading control (n = 2).
Western blotting was employed to test whether disruption of any of the subunit a-isoforms affected the expression level of other V-ATPase subunits. Using commercially available antibodies against V1A, V1B, V1C, and V0d, it was found that the levels of these subunits in subunit a-isoform knockout cells did not change relative to control cells (Fig. 3B).
Subunit a4 is required for in vitro migration and invasion of 4T1-12B cells
The subunit a-isoform knockout cells shown in Fig. 3 were used to test whether specific isoforms contribute to the migration and/or invasion of 4T1-12B cells using an in vitro transwell migration assay. As shown in Fig. 4, only knockout of the a4 isoform led to a significant (>90%) reduction in 4T1-12B cell migration. Similar results were obtained using the in vitro transwell invasion assay, where a4 knockout reduced the invasiveness of 4T1-12B cells by ∼80% (Fig. 5). These data suggest that 4T1-12B cell migration and invasion depend upon a4 expression.
Figure 4.
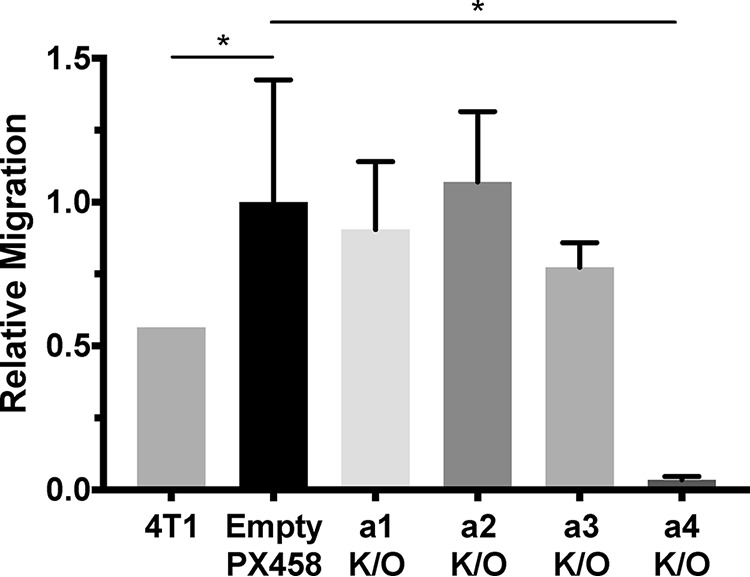
In vitro transwell migration of 4T1-12B a-isoform knockout cell lines. Transwell migration assays were performed as described under “Experimental procedures.” Parental 4T1-12B cells (4T1), cells transfected with a negative control plasmid (Empty PX458), and subunit a-isoform knockout cell lines (a1–a4 K/O) were allowed to migrate through a porous membrane toward a chemoattractant. The number of cells that had migrated to the trans side of the insert were counted in triplicate and normalized to the number of migrating control cells. Knockout of the a4 subunit decreased migration by 97 ± 2% relative to the control. Values represent the mean ± S.E. (error bars), n ≥ 3 (*, p < 0.05).
Figure 5.
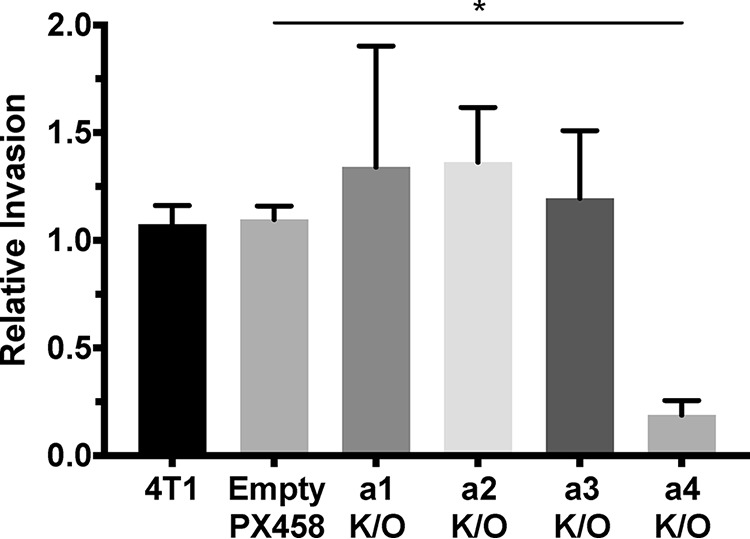
In vitro transwell invasion of 4T1-12B a-isoform knockout cell lines. Transwell invasion assays were performed as described under “Experimental procedures” using MatrigelTM–coated inserts. Parental 4T1-12B cells (4T1), cells transfected with a negative control plasmid (Empty PX458), and subunit a-isoform knockout cell lines (a1–a4 K/O) invaded through Matrigel toward a chemoattractant. The number of cells that had invaded to the trans side of the insert were counted in triplicate, and results are shown relative to the number of invading control cells. Knockout of the a4 subunit decreased invasion by 81 ± 12% relative to the control. Values represent the mean ± S.E. (error bars), n ≥ 3 (*, p < 0.05).
Knockout of the V0a4 subunit reduces plasma membrane V-ATPase staining of migrating 4T1-12B cells
Previous work from our laboratory studying the invasive MDA-MB-231 breast cancer cell line revealed that plasma membrane V-ATPases play a key role in promoting cancer cell invasion (5). The two subunit a-isoforms known to target the V-ATPase to the plasma membrane are a3 and a4, and a3 has been demonstrated to target the pump to the plasma membrane of various invasive breast cancer cell lines (21, 25). We performed immunofluorescence microscopy to determine whether, in 4T1-12B cells, knockout of the various subunit a-isoforms affected V-ATPase localization to the plasma membrane. The a-isoform knockout cells were grown on coverslips and induced to migrate by scratching the monolayer to establish a leading edge in cells. Antibodies against cortactin, a leading edge marker, and the V-ATPase V1A subunit were used to determine the localization of the pump. As seen in Fig. 6, V-ATPases (green) are detected both intracellularly and at the plasma membrane, whereas cortactin (red) localizes to the leading edge of migrating cells. In control, empty plasmid-transfected cells, V1A staining appears in some regions of the plasma membrane, typically colocalized with cortactin (yellow) at the leading edge of the cell. A similar staining pattern was observed for subunit a1–a3-knockout cells (Fig. 6A and Fig. S3). By contrast, V-ATPase staining either does not appear or is highly diminished at the plasma membrane and leading edge of a4-knockout cells (Fig. 6A and Fig. S3). Quantitation of the staining in these cells revealed a 53% decrease in the number of cells expressing any plasma membrane V-ATPase (Fig. 6B). This suggests that in migrating 4T1-12B cells, subunit a4 plays a role in targeting the V-ATPase to the plasma membrane. Importantly, expression of V1A is not affected in any of the V0a-isoform knockout cells (Fig. 3B), indicating that loss of plasma membrane V1A staining is not due to a change in subunit A levels. It should be noted that, because these studies were performed on permeabilized cells, we cannot formally exclude the possibility that what appears to be cell surface staining at the leading edge of cells is, in fact, V-ATPases in close juxtaposition to the plasma membrane. In support of V-ATPases actually localized to the plasma membrane being reduced upon a4 knockout, only a4-knockout cells were found to have a significantly lower extracellular acidification rate (ECAR) relative to control cells (Fig. S4). It should be noted that although the decrease observed in a4-knockout cells is modest (20% relative to parental 4T1 cells and 13% relative to the vector only cells), these experiments were performed under conditions where other cellular mechanisms of pH homeostasis (such as Na/H antiporter) were still functional.
Figure 6.
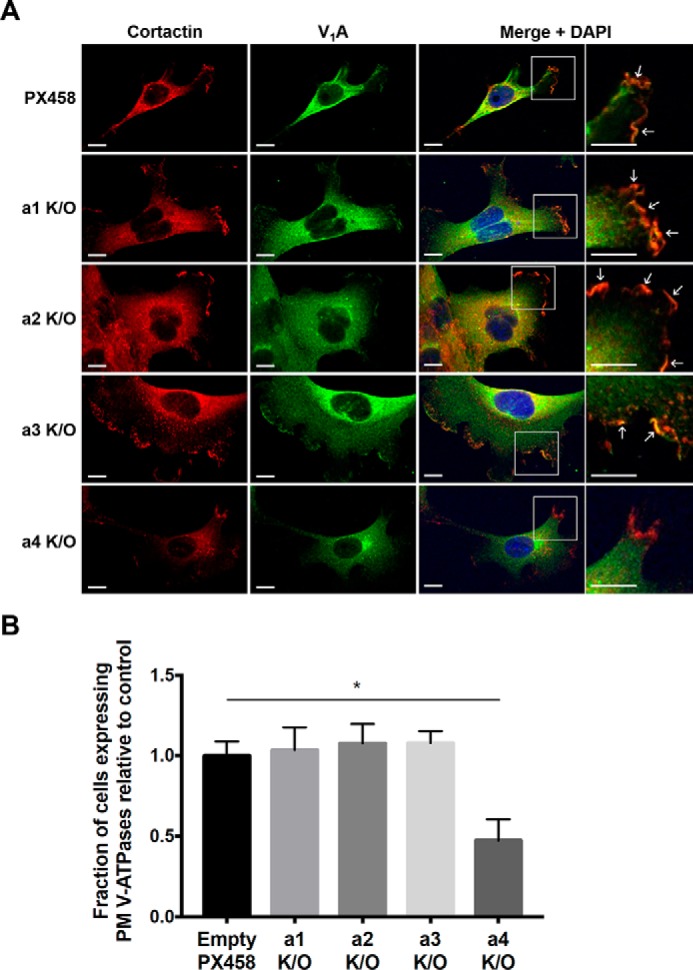
Immunofluorescence of the V-ATPase at the leading edge of migrating 4T1-12B a-isoform knockout cells. 4T1-12B a-isoform knockout cells (a1–a4 K/O) and their corresponding empty plasmid control (PX458) were grown to confluence on coverslips. Migration of the cells was induced as described under “Experimental procedures” by using a pipette tip to scratch the confluent monolayer. Cells were left to migrate for at least 4 h, after which they were fixed and stained for the leading edge marker cortactin (red), the V1A subunit of the V-ATPase (green), and nuclei (DAPI; blue). Plasma membrane V-ATPase localization appears as yellow fluorescence at the leading edge when the red and green channels are merged. The absence of plasma membrane V-ATPase localization appears as red fluorescence at the leading edge in the merged image. A, representative images are shown. Scale bar, 10 μm. White arrowheads, regions of plasma membrane V-ATPase localization. B, a minimum of 60 cells from each cell line were scored for the presence or absence of V-ATPase plasma membrane localization. The fraction of cells with plasma membrane V-ATPase localization was quantified from three independent experiments. Values represent the mean ± S.D. (error bars), n = 3 (*, p < 0.05).
Discussion
The majority of cancer-related deaths occur due to metastasis of primary tumors (46); however, there are no treatments currently available to target this hallmark of cancer. Identifying new anti-metastatic targets would therefore fill an important unmet clinical need. V-ATPases are a family of ATP-dependent proton pumps that have been linked to several hallmarks of cancer, including cancer cell survival, cellular signaling, multidrug resistance, and tumor cell invasiveness (6). Tumor cell invasiveness is essential to the metastasis of cancer cells, as it facilitates both their escape from the site of the primary tumor and their colonization of remote sites. Inhibiting the V-ATPase has been shown to inhibit the invasiveness of a variety of cancer cell types, making it a potential target in the development of anti-metastasis therapies (5, 18, 21–24, 26). Although the mechanism by which inhibiting V-ATPase activity inhibits tumor cell invasiveness remains uncertain, expression of V-ATPases at the plasma membrane of tumor cells may be important for this process (5, 21, 24, 25). Isoforms of subunit a of the V0 domain target V-ATPases to different cellular destinations, with both the a3 and a4 isoforms capable of targeting the V-ATPase to the surface of osteoclasts and renal intercalated cells, respectively (11, 13, 47). Interestingly, both a3 and a4 have been implicated in the invasiveness of human breast cancer cells (21, 24, 25). In the present study, we sought to characterize the role of different isoforms of subunit a in the invasion and migration of a murine breast cancer cell line (4T1-12B).
Previous work characterizing the function of a-subunit isoforms in breast cancer cells has focused on human cancer cell lines (21, 24, 25). Whereas such lines are of direct relevance to human cancer, the study of the in vivo metastasis of these lines must be done in immunocompromised mice to avoid rejection of the human cells by the mouse host. As a result, the role of the immune system in the response of the host to tumor growth and metastasis cannot be studied in such models, despite the importance of the immune system to these processes (48, 49). We thus wished to begin exploring the role of the V-ATPase and a-subunit isoforms in a mouse breast cancer cell line whose in vivo metastasis could subsequently be studied using immunocompetent mice.
We first confirmed the role of V-ATPases in in vitro invasion and migration of 4T1-12B cells using the specific V-ATPase inhibitor concanamycin A (Fig. 1). The observed level of sensitivity is comparable with that previously reported for human breast cancer cell lines (5, 21, 24, 50). We next measured the mRNA levels for the four a-subunit isoforms in these cells using quantitative RT-PCR and found that a4 was the dominant isoform, followed by a2 and a3, with barely detectable levels of a1 present. Interestingly, previous work in MDA-MB-231 cells showed that a3 was the dominant isoform (24), whereas in MCF10CA1a cells, both a2 and a3 were expressed at significantly higher levels than a1 and a4 (21). This is the first cancer cell line of any origin in which a4 mRNA is the dominant species, although previous work from our laboratory has shown that a4 is up-regulated in some human breast cancer samples relative to normal tissue (25).
The differences in expression of a-subunit isoforms between various invasive breast cancer cell lines highlights the importance of characterizing the role of each isoform in the invasive phenotype of these cells. To accomplish this, we performed the first CRISPR/Cas9 genetic knockout of each a-subunit isoform using 4T1-12B cells. Complete knockout of a1, a2, and a4 was achieved, whereas knockdown of somewhat greater than 50% was obtained with a3 (Fig. 3). Despite the use of multiple unique guide sequences targeting a3, no reduction in expression level greater than this was obtained. Our inability to completely disrupt a3 expression using CRISPR/Cas9 may indicate that a3 is a gene essential to the viability of 4T1-12B cells. Previous studies employing CRISPR/Cas9 have resulted in similar incomplete knockdown of the corresponding gene product (51, 52). Nevertheless, because our previous studies using siRNA-mediated knockdown of a-subunit isoforms in breast cancer cell lines showed significant effects on tumor cell invasion and migration following comparable levels of reduction of mRNAs (21, 24, 25), we felt confident in further characterizing cells displaying partial a3 knockdown.
Using an in vitro transwell assay, we found that only cells disrupted in the a4 isoform showed significantly reduced invasion and migration (Figs. 4 and 5). It should be noted that the partial (∼30%) reduction in a4 expression in a1-knockout cells (Fig. 3A) did not result in reduced invasion or migration, suggesting that the remaining level of a4 expression was sufficient to fully support this phenotype. It should also be noted that the partially reduced expression of a1 in the a4-knockout cells (Fig. 3A) is not responsible for the decreased invasion and migration observed for these cells because the a1-knockout cells, in which a1 expression is completely ablated, showed a normal invasive phenotype. Furthermore, compensation by a1 was also observed, with a1 expression increasing in both the a2-knockout and a3 partial knockout cells. Nevertheless, if increased a1 expression accounted for the normal level of migration/invasion in the a2 and a3 knockouts, it might be expected that knockout of a1 would reduce migration/invasion, yet this is not observed.
We previously showed that a3 knockdown had the greatest effect on invasion in MCF10CA1a cells and that ectopic overexpression of a3 (and not the other isoforms) enhanced the invasiveness of the parental MCF10a cells (21, 25). By contrast, a4 knockdown had the greatest effect on invasion in MDA-MB-231 cells, even though it is expressed at the lowest level of any a-isoform in these cells (24). Thus, it is not necessarily knockdown of the most dominantly expressed isoform that affects invasion of breast cancer cells. Nevertheless, we cannot rule out the possibility that the effects on invasion and migration observed with a4 knockout are due, in the case of 4T1-12B cells, to its being the dominantly expressed isoform and hence having the greatest overall effect on V-ATPase activity. By contrast, because a1, a2, and a3 are all expressed at significantly lower levels than a4 (at least at the mRNA level), their disruption would be predicted to have a quantitatively lower effect on overall V-ATPase activity, thus making it difficult to rule out some role of V-ATPases containing these isoforms in invasion and migration. This is particularly true for a3, where knockdown is only partial.
Because we have demonstrated that plasma membrane V-ATPases contribute directly to cancer cell invasiveness (5), and because a4 is known to target the V-ATPase to the plasma membrane in renal intercalated cells (8, 11), we wished to determine whether a4 knockout reduced the level of V-ATPase expressed at the cell surface. We found that only cells disrupted in a4 expression (but not the other isoforms) showed a significant reduction in V-ATPase staining at the plasma membrane, which was localized primarily to the leading edge, as identified by co-localization with cortactin (Fig. 6). This suggests that, although a4 is not located exclusively at the plasma membrane, its expression is required for trafficking of V-ATPases to the cell surface and that it is surface V-ATPases that are contributing to the invasiveness of 4T1-12B cells. Importantly, although 40–50% of a4-knockout cells still show some V-ATPase staining at the cell surface (Fig. 6B), the intensity of staining in knockout cells that do show surface staining is greatly reduced. Thus, our results suggest that 4T1-12B cells target V-ATPases to the cell surface through expression of the a4 isoform, where they serve to enhance cell invasion and migration.
The mechanism by which plasma membrane V-ATPases promote cancer cell invasion and migration is still unclear. Interestingly, knockout of a4 had a greater effect on 4T1-12B cell migration and invasion than pharmacological inhibition of the V-ATPase using concanamycin A (Figs. 1, 4, and 5). One possible explanation for this result is that, in addition to their role in transporting protons out of the cell, plasma membrane V-ATPases play some additional, nonenzymatic role in cell invasion and migration. Because the V-ATPase has been shown to be part of a large complex involved in activation of both mTORC1 and AMPK (albeit on lysosomes) (53, 54) and to directly bind to such kinases as phosphatidylinositol 3-kinase and extracellular signal-regulated kinase (55, 56), it is possible that plasma membrane V-ATPases function in cell surface localization and activation of signaling molecules important in cell invasion or migration.
With respect to the function of the activity of plasma V-ATPases in promoting cell migration and invasion, a number of hypotheses have been proposed. Acidification of the extracellular environment may activate secreted, acid-dependent proteases, such as the cathepsins, that can both degrade extracellular matrix and activate other secreted proteases, such as matrix metalloproteases (57–64). Alternatively, a higher intracellular pH near the cell surface created by plasma membrane V-ATPases may aid in migration through changes in the actin cytoskeleton (4, 65). Interestingly, the V-ATPase is also known to directly interact with F-actin and to promote actin assembly (66–71). Thus, it is possible that activity of V-ATPases at the cell surface creates an acidic extracellular environment that promotes proteolytic degradation of the extracellular matrix and an alkaline intracellular environment that promotes actin polymerization, both of which may aid tumor cell invasion and migration.
V-ATPase activity is also required for proper membrane trafficking within the cell (8). Inhibition of acidification of intracellular compartments may affect the trafficking of signaling molecules involved in cancer cell invasiveness. The peripheral localization and activation of EGFR and Rac1, molecules involved in cell migration, are prevented by V-ATPase inhibition (29). Similarly, inhibition of V-ATPases in secretory vesicles prevents proper trafficking of the Rab27 GTPase that is important for cell movement (72), and work by Matsumoto et al. (73) in osteoclasts found that a3 interacts directly with the inactive, GDP-bound form of Rab27A. Although these findings are not specific to plasma membrane V-ATPases, it is possible that knockdown of a4, in addition to reducing cell surface expression of V-ATPases, may also alter acidification of intracellular compartments.
In summary, this research is the first characterization of the function of V-ATPase a-subunit isoforms in a cancer cell line using CRISPR/Cas9 technology. We have shown that the a4 isoform plays an important role in promoting both the invasion and migration and plasma membrane localization of V-ATPases in 4T1-12B cells. Given our previous results demonstrating a similarly important role for a3 in the invasive MCF10CA1a human breast cancer cell line (21), these results emphasize that different a-subunit isoforms may function to target V-ATPases to the cell surface, where they can promote cell invasion and migration. Thus, isoform-specific therapies to limit breast cancer metastasis may need to be tailored to account for differences in isoform expression of particular tumor types. Future work will be required to understand the mechanism by which plasma membrane V-ATPases contribute to breast cancer cell invasiveness.
Experimental procedures
Materials and equipment
FalconTM T-75 flasks (catalog no. 353136), Corning 96-well plates (catalog no. 3596), Corning Steriflip cups (catalog no. 430758), BioCoatTM MatrigelTM Invasion Chamber with GFR Matrigel Matrix (catalog no. 354483), CorningTM FalconTM FluoroBlokTM 24-well permeable supports (catalog no. 351152), and FalconTM polystyrene 10-cm plates (catalog no. 353003) were purchased from Thermo Fisher Scientific. 4T1-12B cells (44) were a gift from Gary Sahagian (Tufts University). DMSO (D2650), calcein AM (17783), anti-β-actin antibody (A1978; 1:10,000), anti-α-tubulin (T5168; 1:5,000), anti-vinculin (V9131; 1:50,000), WST-1 (catalog no. 5015944001), DMEM without sodium bicarbonate and phenol red (D2902), and PMSF (P7626) were purchased from Sigma. Precast 4–15% Mini-PROTEAN® TGDTM gels (catalog no. 456-1084), iTaqTM Universal SYBR® Green Supermix (catalog no. 172-5121), anti-V1B2 antibody (VMA00190KT; 1:1,000), and peroxidase-conjugated anti-mouse antibodies (catalog no. 170-6516; 1:5,000) were purchased from Bio-Rad. Penicillin/streptomycin (15140-122), DMEM (11885-076), FBS (catalog no. 16140-071), LipofectamineTM 3000 transfection reagent (L300015), SuperScriptTM III First-Strand Synthesis System (catalog no. 18080051), PBS (catalog no. 20012-027), leupeptin (catalog no. 78435), and trypan blue (catalog no. 15250061) were purchased from Thermo Fisher Scientific. Aprotinin (catalog no. 10981532001) and pepstatin (catalog no. 11524488001) were purchased from Roche Applied Science. Poly-d-lysine (A-003-E) was purchased from Millipore. RT Prolong Gold Fixative with DAPI (P36935), Alexa Fluor® 568–conjugated anti-rabbit antibody (A11011; 1:500), and Alexa Fluor® 488–conjugated anti-mouse antibody (A10680; 1:500) were purchased from Life Technologies, Inc. Concanamycin A (BVT-0237) was purchased from BioViotica. The RNeasy® Mini Kit (catalog no. 74104) was purchased from Qiagen. Chemiluminescent substrate (RPN2106) was purchased from GE Healthcare, and autoradiography film (E3012) was purchased from Denville Scientific Inc. Mammalian anti-V0a4 (ab97440; 1:1,000), mammalian anti-V0d (ab56441; 1:1,000), anti-cortactin (ab81208; 1:1,000), peroxidase-conjugated anti-rabbit (ab97051; 1:5,000), and peroxidase-conjugated anti-chick (ab6877; 1:5,000) antibodies were purchased from Abcam, and the mammalian anti-V0a1 antibody (NBP1-89342; 1:1,000) was purchased from Novus. Mammalian anti-V1A antibody that was used for Western blotting was purchased from Abnova (H00000523-A01; 1:1,000), whereas the anti-V1A antibody used for immunocytochemistry was purchased from Sigma (ab1402125; 1:1,000). The chicken monoclonal anti-V0a2 and anti-V0a3 antibodies were described previously by Sun-Wada et al. (74). Mammalian anti-V1C antibody (ARP58589_P050; 1:1,000) was purchased from Aviva. The GFP-expressing CRISPR/Cas9 plasmid, pSpCas9(BB)-2A-GFP (PX458), was purchased from Addgene (catalog no. 48138). The Seahorse XFe96 FluxPak (catalog no. 102601-100) was purchased from Agilent.
Cell culture
4T1-12B cells were maintained in FalconTM T-75 flasks containing DMEM supplemented with 1% penicillin/streptomycin and 10% FBS. Cells were grown at 37 °C with 5% CO2 in a humidified environment. The cells used in these experiments were not passaged more than 10 times, and all experiments were performed using biological replicates.
CRISPR/Cas9
gRNA sequences to target each of the individual V-ATPase subunits were designed using the free online CRISPOR tool (http://crispor.tefor.net/)4 (76) by inputting the region of the mouse V0a1, V0a2, V0a3, or V0a4 gene sequence that we wished to disrupt (usually within the first few exons). Several gRNAs were chosen per a-isoform, but we found that the following sequences produced the best knockout in 4T1-12B cells: a1, 5′-CTCTGGAAAACATTCACATC-3′; a2, 5′-TACGAGTGTCTGAGCGCGCT-3′; a3, 5′-CAGTTGTAAGCAGACCCTGT-3′; a4, 5′-GGCATCTGTGTTTCGAAGTG-3′. These gRNA sequences were cloned into the pSpCas9(BB)-2A-GFP (PX458) plasmid, and 4T1-12B cells were transfected with the empty or a-isoform–targeting plasmid using Lipofectamine 3000 according to the manufacturer's instructions. After 48 h, the BD Biosciences FACS Aria cell sorter was used to plate a single GFP-expressing cell into every well of a 96-well plate to grow clonal a-isoform knockout populations. After clonal populations were grown, their a-isoform protein expression levels were determined using Western blotting. Clones that had full or partial knockout of the targeted a-isoform were used for migration and invasion assays. At least two knockout clones were tested for each a-isoform.
Whole-cell lysis and Western blotting
3 × 106 cells were plated into 10-cm poly-d-lysine–coated plates. The following day, cells were placed on ice and rinsed twice with ice-cold PBS. Cells were scraped into 450 μl of ice-cold lysis buffer (150 mm NaCl, 1% Triton X-100, 50 mm Tris-HCl (pH 7.5), 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml aprotinin, 5 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm NaF, and 1 mm glycerophosphate). Samples were continuously agitated for 30 min to lyse the cells, and then centrifuged at 500 × g for 10 min to clear lysates of unbroken cells and nuclei.
Following whole-cell lysis, protein concentrations were determined using the Lowry method (75). Samples were diluted in SDS-containing sample buffer, and proteins were separated by SDS-PAGE on 4–15% precast gels. Following separation, proteins were transferred to a nitrocellulose membrane. After blocking with 1.5% milk in TBS-T, primary antibodies were added (see “Materials and equipment” for dilution) for at least 1 h. This was followed with TBS-T washes and the appropriate secondary antibodies in milk for 1 h. Protein detection was performed using chemiluminescent substrate and X-ray film. Protein levels in V0a3-knockout cells were quantified using ImageJ version 10.2 software, and the intensity of the a3 band was normalized to the amount of protein in each lane using the β-actin band. This ratio was then normalized to the ratio of a3/actin in empty plasmid control cells (which was set to 1).
Quantitative RT-PCR
5 × 106 4T1-12B cells were harvested, and RNA was isolated using Qiagen's RNeasy® mini kit. cDNA was made from the RNA using Invitrogen's SuperScriptTM III First-Strand Synthesis System, starting with 500 ng of total RNA. Following cDNA synthesis, PCR was performed using Bio-Rad's iTaqTM Universal SYBR® Green Supermix in a Bio-Rad C1000TM thermal cycler according to the manufacturer's instructions. The following primers were designed using NCBI's Primer-BLAST tool and then used in the reaction for detection of each a-isoform: a1, 5′-GATGAGATGGCGGATCCAGA-3′ (forward) and 5′-AGCCACAAAGCCAAGTCGTA-3′ (reverse); a2, 5′-GAGCGGTTACACACTCGTGA-3′ (forward) and 5′-ACTTCTCGGCAGCCTTCTTC-3′ (reverse); a3, 5′-AACTGAGTTTGTCCCTTCTGAGAT-3′ (forward) and 5′-CAGAGACGCAAGTAGGAGGC-3′ (reverse); a4, 5′-CTGTGACGGGTTTCGTGCTA-3′ (forward) and 5′-TCTTCCAGCCTCACGTTGAC-3′ (reverse). To quantify the absolute mRNA levels of each a-isoform, a standard curve was made from serial dilutions of a-isoform cDNA. V0a-isoform–specific cDNA was ordered from Integrated DNA Technologies as gBlocks gene fragments. The region of cDNA amplified by the primers listed above was ordered, and the fragments were used to make the a-isoform–specific standard curves. The specific sequences that were used as the standards for each of the a-isoforms are listed below: a1, 5′-CTGGAACTGACTGAATTAAAATTTATCCTGCGAAAAACCCAGCAGTTTTTCGATGAGATGGCGGATCCAGACCTGTTGGAAGAGTCCTCATCACTCTTGGAGCCAAACGAGATGGGAAGAGGCGCACCCTTACGACTTGGCTTTGTGGCTGGTGTGATTAACCGGGAGCGGATCCCGACCTTTGAGCGCATGCTTT-3′; a2, 5′-TTTTTCTGCTGTGGCTGCACAACGGGCGCAATTGCTTTGGCATGAGCCGGAGCGGTTACACACTCGTGAGGAAGGACAGCGAGGAAGAGGTGTCTCTTCTGGGCAACCAGGACATAGAAGAGGGCAACAGCCGCATGGAAGAAGGCTGCCGAGAAGTGACGTGTGAGGAGTTTAACTTCGGGGAGATCCTGATGACGC-3′; a3, 5′-TGATGAGGAGAAGGCTGGGAGCCCAGGGGATGAAGAAACTGAGTTTGTCCCTTCTGAGATCTTCATGCACCAAGCAATCCACACCATTGAGTTCTGCCTGGGCTGCATCTCCAACACAGCCTCCTACTTGCGTCTCTGGGCCCTGAGCCTGGCCCATGCCCAGCTGTCTGAGGTCCTGTGGGCCATGGTGAT-3′; a4, 5′-CATCATATTTTACCAAGGAGAACAGCTCAGGCTGAAAATCAAGAAGATCTGTGACGGGTTTCGTGCTACCATCTACCCCTGCCCAGAGCATGCAGCAGAGCGCAGAGAGATGCTGACCAGTGTCAACGTGAGGCTGGAAGACTTAATCACCGTCATTACCCAAACAGAGTCTCACCGACAGCGCCTGCTGCAGGAAGCAGC-3′.
Transwell migration assay
In vitro transwell migration assays were performed as described previously (25). Briefly, Fluoroblok inserts with an 8-μm pore size were placed into a 24-well plate containing 500 μl of DMEM with 10% FBS in each well. Cells were trypsinized and diluted to a concentration of 1.5 × 105 cells/ml in filter-sterilized DMEM containing 0.1% BSA. Where necessary, either 1 nm concanamycin A or DMSO was added to the diluted cells. 500 μl of the cell suspension was then seeded onto the membranes of the Fluorblok inserts. Each cell type or treatment was done in triplicate. Cells were then placed in the incubator at 37 °C for an average of 18 h, after which the membrane inserts were placed into wells containing 4 μg/ml calcein AM in PBS and incubated for 5 min at 37 °C in 5% CO2. Cells that had migrated to the trans side of the membrane were imaged using a Zeiss Axiovert 10 fluorescence microscope. An average of 12 images were taken per well, and the number of migrating cells was averaged over three wells. Significant differences in migration for the various a-isoform knockouts were established using a one-way ANOVA combined with a nonparametric post hoc test.
For concanamycin A–treated cells, a trypan blue exclusion assay was performed to asses cell viability after a 24-h treatment.
Transwell invasion assay
In vitro transwell invasion assays were performed as described previously (21). Briefly, Matrigel-coated transwell inserts were placed into a 24-well plate and rehydrated with 200 μl of DMEM (no additives) for 2 h at room temperature. Matrigel-coated inserts were then placed into a new well containing 500 μl of DMEM with 10% FBS. Cells were trypsinized and diluted to a final concentration of 1.5 × 105 cells/ml in filter-sterilized DMEM containing 0.1% BSA. Where necessary, either 1 nm concanamycin A or DMSO was added to the cell suspension. 500 μl of the cell suspension was then seeded onto the Matrigel inserts. Each cell type or treatment was done in triplicate. Cells were then placed in the incubator at 37 °C for an average of 20 h, after which the inserts were placed into wells containing 4 μg/ml calcein AM in PBS and incubated for 5 min at 37 °C in 5% CO2. Importantly, the cis side of the Matrigel insert was gently scrubbed with a cotton swab to remove any cells that did not invade through the Matrigel. Cells that had invaded to the trans side were imaged using a Zeiss Axiovert 10 fluorescence microscope. An average of 12 images were taken per well, and the number of invading cells was averaged over three wells. Significant differences in invasion for the various a-isoform knockouts were established using a one-way analysis of variance combined with a nonparametric post-hoc test. For concanamycin A–treated cells, a trypan blue exclusion assay was performed to assess cell viability after a 24-h treatment.
Immunofluorescence microscopy
1 × 105 cells were plated onto round, poly-d-lysine–coated coverslips in 24-well plates. The following day, the confluent cell monolayer was scratched using a 200-μl pipette tip to create a wound and induce migration. Cells were then incubated in DMEM (no additives) for an average of 5 h to allow for migration at 37 °C in 5% CO2. After the incubation, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for an additional 10 min. Coverslips were placed in a 1% BSA solution for 30 min for blocking. Following blocking, cells were incubated with anti-V1A and anti-cortactin antibodies (both at 1:1,000) in 1% BSA solution overnight at 4 °C. The following day, coverslips were washed with PBS, and a 1% BSA solution containing Alexa Fluor® 568–conjugated anti-rabbit antibody (1:500) and Alexa Fluor® 488–conjugated anti-mouse antibody (1:500) was added for 1 h at room temperature. Coverslips were washed with PBS and then left to cure overnight on slides using the Prolong Gold fixative with DAPI. Images were taken using a Leica TCS SPE confocal microscope with a ×40 objective. A minimum of five unique fields were captured along the wound for each knockout cell line in each experiment.
Quantitation of cells with plasma membrane V-ATPase staining was performed by counting the number of cells with a leading edge and determining how many of those cells had cell surface V-ATPase staining. Over 60 cells were counted for each cell type. An unpaired, two-tailed Student's t test was used to determine significant differences in plasma membrane V-ATPase expression for the various a-isoform knockouts compared with the control.
Cell viability assay
Trypan blue staining was performed according to the manufacturer's instructions. Briefly, 0.4% trypan blue staining solution was mixed with cell suspensions at a 1:1 dilution. The number of cells taking up the dye was counted using a hemocytometer, and the following formula was used to determine cell viability: % viable cells = ((total cells − blue cells)/total cells) × 100.
Cell proliferation assay
Cell proliferation was measured using WST-1 according to the manufacturer's instructions. Briefly, 1 × 104 cells were plated per well in a 96-well plate. The following day, WST-1 was added to each well at a 1:10 dilution. Cells were incubated at 37 °C in 5% CO2 for 1 h, and then the absorbance of each well was measured at 480 nm using a Tecan SpectraFluor Plus plate reader.
Seahorse assay
ECAR in 4T1-12B cells was measured using the Extracellular Flux Assay Kit from Agilent Technologies according to the manufacturer's instructions. Briefly, 4 × 104 cells were seeded in each well of a Seahorse XF96 cell culture microplate, with a minimum of six replicate wells for parental 4T1-12B, negative control PX458, and each a-isoform knockout line. 16 h later, the cells were rinsed once with warm PBS; the medium was replaced with DMEM, pH 7.4, without sodium bicarbonate and phenol red; and the plate was transferred to a 37 °C non-CO2 incubator for 1 h. Five basal measurement cycles (3-min mix, 0-min wait, 3-min measure) were then performed on the Seahorse XFe96 Analyzer. The assay was repeated three times, and the interassay mean ECAR values from the third measurement cycle were compared using an unpaired, two-tailed Student's t test.
Author contributions
C. M. M., M. P. C., and M. F. conceptualization; C. M. M. and M. P. C. data curation; C. M. M. formal analysis; C. M. M. and M. P. C. investigation; C. M. M., M. P. C., G. S.-W., and Y. W. methodology; C. M. M. writing-original draft; C. M. M., M. P. C., G. S.-W., Y. W., and M. F. writing-review and editing; M. P. C. and M. F. funding acquisition; G. S.-W. and Y. W. resources; M. F. supervision.
Supplementary Material
Acknowledgments
We thank Laura Stransky and Kristina Cotter-Cross for many helpful discussions. Additionally, we thank Gary Sahagian of Tufts University for generously providing 4T1-12B cells.
This work was supported by National Institutes of Health Grant R01 GM34478 (to M. F.) and an Exceptional Project Award from the Breast Cancer Alliance (to M. F.) as well as the Sackler Families Collaborative Cancer Biology Award from Tufts University (to M. P. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- V-ATPase
- vacuolar protein-translocating adenosine triphosphatase
- ConA
- concanamycin A
- gRNA
- guide RNA
- ECAR
- extracellular acidification rate
- FBS
- fetal bovine serum
- PMSF
- phenylmethylsulfonyl fluoride
- DMEM
- Dulbecco's modified Eagle's medium
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Gupta G. P., and Massagué J. (2006) Cancer metastasis: building a framework. Cell 127, 679–695 10.1016/j.cell.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 2. Pantel K., and Brakenhoff R. H. (2004) Dissecting the metastatic cascade. Nat. Rev. Cancer 4, 448–456 10.1038/nrc1370 [DOI] [PubMed] [Google Scholar]
- 3. Valastyan S., and Weinberg R. A. (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 10.1016/j.cell.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Webb B. A., Chimenti M., Jacobson M. P., and Barber D. L. (2011) Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer. 11, 671–677 10.1038/nrc3110 [DOI] [PubMed] [Google Scholar]
- 5. Cotter K., Capecci J., Sennoune S., Huss M., Maier M., Martinez-Zaguilan R., and Forgac M. (2015) Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. J. Biol. Chem. 290, 3680–3692 10.1074/jbc.M114.611210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stransky L., Cotter K., and Forgac M. (2016) The function of V-ATPases in cancer. Physiol. Rev. 96, 1071–1091 10.1152/physrev.00035.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cotter K., Stransky L., McGuire C., and Forgac M. (2015) Recent insights into the structure, regulation, and function of the V-ATPases. Trends Biochem. Sci. 40, 611–622 10.1016/j.tibs.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forgac M. (2007) Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929 10.1038/nrm2272 [DOI] [PubMed] [Google Scholar]
- 9. Kane P. M. (2016) Proton transport and pH control in fungi. in Yeast Membrane Transport (Ramos J., Sychrová H., and Kschischo M., eds) pp. 33–68, Springer International Publishing, Cham, Switzerland [Google Scholar]
- 10. Kane P. M. (2012) Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr. Protein Pept. Sci. 13, 117–123 10.2174/138920312800493142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breton S., and Brown D. (2013) Regulation of luminal acidification by the V-ATPase. Physiology (Bethesda) 28, 318–329 10.1152/physiol.00007.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun-Wada G.-H., and Wada Y. (2013) Vacuolar-type proton pump ATPases: acidification and pathological relationships. Histol. Histopathol. 28, 805–815 [DOI] [PubMed] [Google Scholar]
- 13. Toyomura T., Murata Y., Yamamoto A., Oka T., Sun-Wada G.-H., Wada Y., and Futai M. (2003) From lysosomes to the plasma membrane: localization of vacuolar-type H+-ATPase with the a3 isoform during osteoclast differentiation. J. Biol. Chem. 278, 22023–22030 10.1074/jbc.M302436200 [DOI] [PubMed] [Google Scholar]
- 14. García-García A., Pérez-Sayáns García M., Rodríguez M. J., Antúnez-López J., Barros-Angueira F., Somoza-Martín M., Gándara-Rey J. M., and Aguirre-Urízar J. M. (2012) Immunohistochemical localization of C1 subunit of V-ATPase (ATPase C1) in oral squamous cell cancer and normal oral mucosa. Biotech. Histochem. 87, 133–139 10.3109/10520295.2011.574647 [DOI] [PubMed] [Google Scholar]
- 15. Otero-Rey E. M., Somoza-Martín M., Barros-Angueira F., and García-García A. (2008) Intracellular pH regulation in oral squamous cell carcinoma is mediated by increased V-ATPase activity via over-expression of the ATP6V1C1 gene. Oral Oncol. 44, 193–199 10.1016/j.oraloncology.2007.02.011 [DOI] [PubMed] [Google Scholar]
- 16. Pérez-Sayáns M., Reboiras-López M. D., Somoza-Martín J. M., Barros-Angueira F., Diz P. G., Rey J. M. G., and García-García A. (2010) Measurement of ATP6V1C1 expression in brush cytology samples as a diagnostic and prognostic marker in oral squamous cell carcinoma. Cancer Biol. Ther. 9, 1057–1064 10.4161/cbt.9.12.11880 [DOI] [PubMed] [Google Scholar]
- 17. Liu P., Chen H., Han L., Zou X., and Shen W. (2015) Expression and role of V1A subunit of V-ATPases in gastric cancer cells. Int. J. Clin. Oncol. 20, 725–735 10.1007/s10147-015-0782-y [DOI] [PubMed] [Google Scholar]
- 18. Chung C., Mader C. C., Schmitz J. C., Atladottir J., Fitchev P., Cornwell M. L., Koleske A. J., Crawford S. E., and Gorelick F. (2011) The vacuolar-ATPase modulates matrix metalloproteinase isoforms in human pancreatic cancer. Lab. Invest. 91, 732–743 10.1038/labinvest.2011.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu J., Xie R., Liu X., Wen G., Jin H., Yu Z., Jiang Y., Zhao Z., Yang Y., Ji B., Dong H., and Tuo B. (2012) Expression and functional role of vacuolar H+-ATPase in human hepatocellular carcinoma. Carcinogenesis 33, 2432–2440 10.1093/carcin/bgs277 [DOI] [PubMed] [Google Scholar]
- 20. Ohta T., Numata M., Yagishita H., Futagami F., Tsukioka Y., Kitagawa H., Kayahara M., Nagakawa T., Miyazaki I., Yamamoto M., Iseki S., and Ohkuma S. (1996) Expression of 16 kDa proteolipid of vacuolar-type H+-ATPase in human pancreatic cancer. Br. J. Cancer 73, 1511–1517 10.1038/bjc.1996.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Capecci J., and Forgac M. (2013) The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 288, 32731–32741 10.1074/jbc.M113.503771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Michel V., Licon-Munoz Y., Trujillo K., Bisoffi M., and Parra K. J. (2013) Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 132, E1–E10 10.1002/ijc.27811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kulshrestha A., Katara G. K., Ibrahim S., Pamarthy S., Jaiswal M. K., Gilman Sachs A., and Beaman K. D. (2015) Vacuolar ATPase “a2” isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget 6, 3797–3810 10.18632/oncotarget.2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hinton A., Sennoune S. R., Bond S., Fang M., Reuveni M., Sahagian G. G., Jay D., Martinez-Zaguilan R., and Forgac M. (2009) Function of a subunit isoforms of the V-ATPase in pH homeostasis and in vitro invasion of MDA-MB231 human breast cancer cells. J. Biol. Chem. 284, 16400–16408 10.1074/jbc.M901201200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cotter K., Liberman R., Sun-Wada G., Wada Y., Sgroi D., Naber S., Brown D., Breton S., and Forgac M. (2016) The a3 isoform of subunit a of the vacuolar ATPase localizes to the plasma membrane of invasive breast tumor cells and is overexpressed in human breast cancer. Oncotarget 7, 46142–46157 10.18632/oncotarget.10063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu X., Chen L., Chen Y., Shao Q., and Qin W. (2015) Bafilomycin A1 inhibits the growth and metastatic potential of the BEL-7402 liver cancer and HO-8910 ovarian cancer cell lines and induces alterations in their microRNA expression. Exp. Ther. Med. 10, 1829–1834 10.3892/etm.2015.2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang S., Schneider L. S., Vick B., Grunert M., Jeremias I., Menche D., Müller R., Vollmar A. M., and Liebl J. (2015) Anti-leukemic effects of the V-ATPase inhibitor Archazolid A. Oncotarget 6, 43508–43528 10.18632/oncotarget.6180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schempp C. M., von Schwarzenberg K., Schreiner L., Kubisch R., Müller R., Wagner E., and Vollmar A. M. (2014) V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Mol. Cancer Ther. 13, 926–937 10.1158/1535-7163.MCT-13-0484 [DOI] [PubMed] [Google Scholar]
- 29. Wiedmann R. M., von Schwarzenberg K., Palamidessi A., Schreiner L., Kubisch R., Liebl J., Schempp C., Trauner D., Vereb G., Zahler S., Wagner E., Müller R., Scita G., and Vollmar A. M. (2012) The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res. 72, 5976–5987 10.1158/0008-5472.CAN-12-1772 [DOI] [PubMed] [Google Scholar]
- 30. Feng S., Zhu G., McConnell M., Deng L., Zhao Q., Wu M., Zhou Q., Wang J., Qi J., Li Y.-P., and Chen W. (2013) Silencing of atp6v1c1 prevents breast cancer growth and bone metastasis. Int. J. Biol. Sci. 9, 853–862 10.7150/ijbs.6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu X., Qin W., Li J., Tan N., Pan D., Zhang H., Xie L., Yao G., Shu H., Yao M., Wan D., Gu J., and Yang S. (2005) The growth and metastasis of human hepatocellular carcinoma xenografts are inhibited by small interfering RNA targeting to the subunit ATP6L of proton pump. Cancer Res. 65, 6843–6849 10.1158/0008-5472.CAN-04-3822 [DOI] [PubMed] [Google Scholar]
- 32. Avnet S., Di Pompo G., Lemma S., Salerno M., Perut F., Bonuccelli G., Granchi D., Zini N., and Baldini N. (2013) V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim. Biophys. Acta 1832, 1105–1116 10.1016/j.bbadis.2013.04.003 [DOI] [PubMed] [Google Scholar]
- 33. Nishisho T., Hata K., Nakanishi M., Morita Y., Sun-Wada G.-H., Wada Y., Yasui N., and Yoneda T. (2011) The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol. Cancer Res. 9, 845–855 10.1158/1541-7786.MCR-10-0449 [DOI] [PubMed] [Google Scholar]
- 34. Kawasaki-Nishi S., Nishi T., and Forgac M. (2001) Yeast V-ATPase complexes containing different isoforms of the 100-kDa a-subunit differ in coupling efficiency and in vivo dissociation. J. Biol. Chem. 276, 17941–17948 10.1074/jbc.M010790200 [DOI] [PubMed] [Google Scholar]
- 35. Gleize V., Boisselier B., Marie Y., Poëa-Guyon S., Sanson M., and Morel N. (2012) The renal v-ATPase a4 subunit is expressed in specific subtypes of human gliomas. Glia 60, 1004–1012 10.1002/glia.22332 [DOI] [PubMed] [Google Scholar]
- 36. Miller F. R. (1983) Tumor subpopulation interactions in metastasis. Invasion Metastasis 3, 234–242 [PubMed] [Google Scholar]
- 37. Miller F. R., Miller B. E., and Heppner G. H. (1983) Characterization of metastatic heterogeneity among subpopulations of a single mouse mammary tumor: heterogeneity in phenotypic stability. Invasion Metastasis 3, 22–31 [PubMed] [Google Scholar]
- 38. Aslakson C. J., and Miller F. R. (1992) Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 52, 1399–1405 [PubMed] [Google Scholar]
- 39. Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Savagner P., Gitelman I., Richardson A., and Weinberg R. A. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 40. Yoneda T., Michigami T., Yi B., Williams P. J., Niewolna M., and Hiraga T. (2000) Actions of bisphosphonate on bone metastasis in animal models of breast carcinoma. Cancer 88, 2979–2988 [DOI] [PubMed] [Google Scholar]
- 41. Lelekakis M., Moseley J. M., Martin T. J., Hards D., Williams E., Ho P., Lowen D., Javni J., Miller F. R., Slavin J., and Anderson R. L. (1999) A novel orthotopic model of breast cancer metastasis to bone. Clin. Exp. Metastasis 17, 163–170 10.1023/A:1006689719505 [DOI] [PubMed] [Google Scholar]
- 42. Pulaski B. A., and Ostrand-Rosenberg S. (1998) Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 58, 1486–1493 [PubMed] [Google Scholar]
- 43. Eckhardt B. L., Parker B. S., van Laar R. K., Restall C. M., Natoli A. L., Tavaria M. D., Stanley K. L., Sloan E. K., Moseley J. M., and Anderson R. L. (2005) Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol. Cancer Res. 3, 1–13 [PubMed] [Google Scholar]
- 44. Tao K., Fang M., Alroy J., and Sahagian G. G. (2008) Imagable 4T1 model for the study of late stage breast cancer. BMC Cancer 8, 228 10.1186/1471-2407-8-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chaffer C. L., and Weinberg R. A. (2011) A perspective on cancer cell metastasis. Science 331, 1559–1564 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- 47. Toyomura T., Oka T., Yamaguchi C., Wada Y., and Futai M. (2000) Three subunit a isoforms of mouse vacuolar H+-ATPase: preferential expression of the a3 isoform during osteoclast differentiation. J. Biol. Chem. 275, 8760–8765 10.1074/jbc.275.12.8760 [DOI] [PubMed] [Google Scholar]
- 48. Day C.-P., Merlino G., and Van Dyke T. (2015) Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53 10.1016/j.cell.2015.08.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gajewski T. F., Schreiber H., and Fu Y.-X. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sennoune S. R., Bakunts K., Martínez G. M., Chua-Tuan J. L., Kebir Y., Attaya M. N., and Martínez-Zaguilán R. (2004) Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am. J. Physiol. Cell Physiol. 286, C1443–C1452 10.1152/ajpcell.00407.2003 [DOI] [PubMed] [Google Scholar]
- 51. Wang P., Lin M., Pedrosa E., Hrabovsky A., Zhang Z., Guo W., Lachman H. M., and Zheng D. (2015) CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 6, 55 10.1186/s13229-015-0048-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zischewski J., Fischer R., and Bortesi L. (2017) Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 35, 95–104 10.1016/j.biotechadv.2016.12.003 [DOI] [PubMed] [Google Scholar]
- 53. Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., and Sabatini D. M. (2011) mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang C.-S., Jiang B., Li M., Zhu M., Peng Y., Zhang Y.-L., Wu Y.-Q., Li T. Y., Liang Y., Lu Z., Lian G., Liu Q., Guo H., Yin Z., Ye Z., et al. (2014) The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 10.1016/j.cmet.2014.06.014 [DOI] [PubMed] [Google Scholar]
- 55. Marjuki H., Gornitzky A., Marathe B. M., Ilyushina N. A., Aldridge J. R., Desai G., Webby R. J., and Webster R. G. (2011) Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion: ERK and PI3K regulate V-ATPase activity. Cell. Microbiol. 13, 587–601 10.1111/j.1462-5822.2010.01556.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soliman M., Seo J.-Y., Kim D.-S., Kim J.-Y., Park J.-G., Alfajaro M. M., Baek Y.-B., Cho E.-H., Kwon J., Choi J.-S., Kang M.-I., Park S.-I., and Cho K.-O. (2018) Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PLoS Pathog. 14, e1006820 10.1371/journal.ppat.1006820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gocheva V., and Joyce J. A. (2007) Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle 6, 60–64 10.4161/cc.6.1.3669 [DOI] [PubMed] [Google Scholar]
- 58. Victor B. C., Anbalagan A., Mohamed M. M., Sloane B. F., and Cavallo-Medved D. (2011) Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 13, R115 10.1186/bcr3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gondi C. S., and Rao J. S. (2013) Cathepsin B as a cancer target. Expert Opin. Ther. Targets 17, 281–291 10.1517/14728222.2013.740461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Turk B., Turk D., and Turk V. (2000) Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 1477, 98–111 10.1016/S0167-4838(99)00263-0 [DOI] [PubMed] [Google Scholar]
- 61. Withana N. P., Blum G., Sameni M., Slaney C., Anbalagan A., Olive M. B., Bidwell B. N., Edgington L., Wang L., Moin K., Sloane B. F., Anderson R. L., Bogyo M. S., and Parker B. S. (2012) Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 72, 1199–1209 10.1158/0008-5472.CAN-11-2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Premzl A., Zavasnik-Bergant V., Turk V., and Kos J. (2003) Intracellular and extracellular cathepsin B facilitate invasion of MCF-10A neoT cells through reconstituted extracellular matrix in vitro. Exp. Cell Res. 283, 206–214 10.1016/S0014-4827(02)00055-1 [DOI] [PubMed] [Google Scholar]
- 63. Roshy S., Sloane B. F., and Moin K. (2003) Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 22, 271–286 10.1023/A:1023007717757 [DOI] [PubMed] [Google Scholar]
- 64. Nalla A. K., Gorantla B., Gondi C. S., Lakka S. S., and Rao J. S. (2010) Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer Gene Ther. 17, 599–613 10.1038/cgt.2010.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rojas J. D., Sennoune S. R., Maiti D., Bakunts K., Reuveni M., Sanka S. C., Martinez G. M., Seftor E. A., Meininger C. J., Wu G., Wesson D. E., Hendrix M. J. C., and Martínez-Zaguilán R. (2006) Vacuolar-type H+-ATPases at the plasma membrane regulate pH and cell migration in microvascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 291, H1147–H1157 10.1152/ajpheart.00166.2006 [DOI] [PubMed] [Google Scholar]
- 66. Cai M., Liu P., Wei L., Wang J., Qi J., Feng S., and Deng L. (2014) Atp6v1c1 may regulate filament actin arrangement in breast cancer cells. PLoS One 9, e84833 10.1371/journal.pone.0084833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lee B. S., Gluck S. L., and Holliday L. S. (1999) Interaction between vacuolar H+-ATPase and microfilaments during osteoclast activation. J. Biol. Chem. 274, 29164–29171 10.1074/jbc.274.41.29164 [DOI] [PubMed] [Google Scholar]
- 68. Holliday L. S., Lu M., Lee B. S., Nelson R. D., Solivan S., Zhang L., and Gluck S. L. (2000) The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J. Biol. Chem. 275, 32331–32337 10.1074/jbc.M004795200 [DOI] [PubMed] [Google Scholar]
- 69. Vitavska O., Wieczorek H., and Merzendorfer H. (2003) A novel role for subunit C in mediating binding of the H+-V-ATPase to the actin cytoskeleton. J. Biol. Chem. 278, 18499–18505 10.1074/jbc.M212844200 [DOI] [PubMed] [Google Scholar]
- 70. Chen S.-H., Bubb M. R., Yarmola E. G., Zuo J., Jiang J., Lee B. S., Lu M., Gluck S. L., Hurst I. R., and Holliday L. S. (2004) Vacuolar H+-ATPase binding to microfilaments: regulation in response to phosphatidylinositol 3-kinase activity and detailed characterization of the actin-binding site in subunit B. J. Biol. Chem. 279, 7988–7998 10.1074/jbc.M305351200 [DOI] [PubMed] [Google Scholar]
- 71. Zuo J., Vergara S., Kohno S., and Holliday L. S. (2008) Biochemical and functional characterization of the actin-binding activity of the B subunit of yeast vacuolar H+-ATPase. J. Exp. Biol. 211, 1102–1108 10.1242/jeb.013672 [DOI] [PubMed] [Google Scholar]
- 72. Hendrix A., Sormunen R., Westbroek W., Lambein K., Denys H., Sys G., Braems G., Van den Broecke R., Cocquyt V., Gespach C., Bracke M., and De Wever O. (2013) Vacuolar H+ ATPase expression and activity is required for Rab27B-dependent invasive growth and metastasis of breast cancer. Int. J. Cancer 133, 843–854 10.1002/ijc.28079 [DOI] [PubMed] [Google Scholar]
- 73. Matsumoto N., Sekiya M., Tohyama K., Ishiyama-Matsuura E., Sun-Wada G.-H., Wada Y., Futai M., and Nakanishi-Matsui M. (2018) Essential role of the a3 isoform of V-ATPase in secretory lysosome trafficking via Rab7 recruitment. Sci. Rep. 8, 6701 10.1038/s41598-018-24918-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sun-Wada G.-H., Tabata H., Kuhara M., Kitahara I., Takashima Y., and Wada Y. (2011) Generation of chicken monoclonal antibodies against the a1, a2, and a3 subunit isoforms of vacuolar-type proton ATPase. Hybrid (Larchmt.) 30, 199–203 10.1089/hyb.2010.0087 [DOI] [PubMed] [Google Scholar]
- 75. Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 76. Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J. B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J. S., and Concordet J. P. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.