

Abstract
Homocysteine, a metabolite of the methionine cycle, is a known agonist of N-methyl-d-aspartate receptor (NMDAR), a glutamate receptor subtype and is involved in NMDAR-mediated neurotoxicity. Our previous findings have shown that homocysteine-induced, NMDAR-mediated neurotoxicity is facilitated by a sustained increase in phosphorylation and activation of extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK MAPK). In the current study, we investigated the role GluN1/GluN2A-containing functional NMDAR (GluN2A-NMDAR) and GluN1/GluN2B-containing functional NMDAR (GluN2B-NMDAR) in homocysteine-induced neurotoxicity. Our findings revealed that exposing primary cortical neuronal cultures to homocysteine leads to a sustained low-level increase in intracellular Ca2+. We also showed that pharmacological inhibition of GluN2A-NMDAR or genetic deletion of the GluN2A subunit attenuates homocysteine-induced increase in intracellular Ca2+. Our results further established the role of GluN2A-NMDAR in homocysteine-mediated sustained ERK MAPK phosphorylation and neuronal cell death. Of note, the preferential role of GluN2A-NMDAR in homocysteine-induced neurotoxicity was distinctly different from glutamate-NMDAR–induced excitotoxic cell death that involves overactivation of GluN2B-NMDAR and is independent of ERK MAPK activation. These findings indicate a critical role of GluN2A-NMDAR–mediated signaling in homocysteine-induced neurotoxicity.
Keywords: homocysteine; N-methyl-D-aspartate receptor (NMDA receptor, NMDAR); glutamate; calcium; extracellular-signal-regulated kinase (ERK); calcium homeostasis; GluN2A-NMDA receptor subunit; hyperhomocysteinemia; kinase signaling; neurotoxicity
Introduction
Homocysteine, a thiol-containing nonessential amino acid, is formed as an intermediate of the methionine cycle. Normal plasma homocysteine level range between 7 and 14 μm (1). Systemic elevation of plasma homocysteine, also known as hyperhomocysteinemia, may occur due to dietary deficiency of folate, vitamin B12, and vitamin B6; renal impairment; and hypothyroidism resulting in a mild (15–30 μm) or moderate (50–100 μm) increase in plasma homocysteine levels (1–4). Genetic mutations of key enzymes in the homocysteine metabolic pathway may also lead to severe (100–500 μm) hyperhomocysteinemic conditions (1, 3, 5). Hyperhomocysteinemia is a metabolic disorder that has been implicated as an independent risk factor for multiple neurodegenerative diseases (2, 6–10). However, a causal link between the elevated levels of plasma homocysteine and neurodegeneration is still elusive. Previous studies have identified homocysteine as an agonist for NMDAR2 (11). However, homocysteine interacts with NMDARs in a complex way, acting as a partial antagonist of the glycine coagonist site with physiological levels of glycine (∼1 μm) and an agonist at the glutamate-binding site of NMDARs when glycine levels are elevated (≥50 μm) under pathological conditions (11). Consistent with these findings, we and others have established that treatment of neurons with mild to moderate levels of homocysteine in conjunction with elevated levels of glycine leads to NMDAR-mediated neurotoxicity (11–18). Our findings further showed that homocysteine-NMDAR–induced neurotoxicity (16, 17) involves a unique signaling pathway, which is different from the glutamate-NMDAR–mediated excitotoxic signaling cascade (19–22). Although homocysteine-NMDAR–induced intracellular signaling involves sustained phosphorylation and subsequent activation of ERK MAPK (16–18), glutamate-mediated NMDAR stimulation leads to a rapid but transient increase in ERK MAPK phosphorylation (17, 23–26). However, both the homocysteine-induced sustained ERK MAPK phosphorylation and the glutamate-mediated transient ERK MAPK phosphorylation are Ca2+-dependent (16, 17, 23), suggesting that the site of Ca2+ entry is a critical determinant of the differential intracellular signaling observed following NMDAR stimulation by homocysteine and glutamate. In this context, earlier studies showed that Ca2+ influx through GluN2B-NMDAR plays a role in the glutamate-NMDAR–induced neurotoxicity (19–22, 27–32), whereas our findings showed that signaling through GluN2B-NMDAR is not involved in homocysteine-NMDAR–dependent neurotoxicity (16). This raises the possibility that homocysteine-NMDAR–mediated neuronal cell death involves GluN2A-NMDAR signaling. To address this issue, in the current study we utilized pharmacological approaches and GluN2A subunit-knockout (GluN2A-KO) mice to investigate the extent and duration of the intracellular Ca2+ increase in primary cortical neuronal cultures following exposure to homocysteine. The study further evaluated the role of GluN2A-NMDAR in homocysteine-induced ERK MAPK phosphorylation and neuronal cell death.
Results
Homocysteine induces sustained increase in intracellular Ca2+ level in cortical neurons
To examine the changes in intracellular Ca2+ dynamics, rat neuronal cultures were loaded with Fura2 acetooxymethyl ester (Fura2-AM) and treated with l-homocysteine (50 μm; 60 min). The frames in Fig. 1A show the Ca2+ responses (in false-color maps) in a representative group of cells treated with homocysteine. The temporal profile of Fura2 fluorescence ratio measured in the soma of 20 individual neurons (Fig. 1B) and their mean data (Fig. 1C) illustrated a slow and progressive increase over time when compared with the unstimulated cells (control). Quantification of the Ca2+ changes further showed that intracellular Ca2+ concentration ([Ca2+]i) remained unchanged in the control cells, whereas treatment with homocysteine resulted in a significant increase in [Ca2+]i over time with a maximum increase of ∼7.2-fold by 60 min when compared with corresponding control cells (Fig. 1D). In contrast, neurons treated with the NMDAR agonist glutamate (50 μm; 10 min) showed a rapid change in Fura2 fluorescence ratio and a ∼21-fold increase in [Ca2+]i within 2.5 min, which then declined progressively with time (Fig. 1, E and F). Such rapid but transient increase in [Ca2+]i when challenged with glutamate is also in agreement with earlier reports (33–36). In neurons where the soma and its main dendritic process (out to at least 40 μm) were identifiable, we further analyzed the temporal profile of [Ca2+]i increase in the processes following exposure to homocysteine. The findings showed a slow and progressive increase in both the Fura2 fluorescence ratio and the [Ca2+]i in the processes over time (Fig. 1, G and H). A comparison of the progressive change in [Ca2+]i between somata and processes showed a similar profile (Fig. 1, I and J).
Figure 1.

Time course of homocysteine- and glutamate-induced changes in [Ca2+]i neurons. A, representative micrographs of neurons showing [Ca2+]i changes over time measured with Fura2 following exposure to l-homocysteine (50 μm). Both black and white (60 min) and false-color images (0–60 min) are shown. B, individual responses in soma of 20 neurons showing the range of increase in Fura2 fluorescence ratio over time following exposure to l-homocysteine (l-Hcy). C and D, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) (C) and [Ca2+]i (mean ± S.D.) (D) in the somata of l-Hcy–treated and untreated cells (control). For [Ca2+]i measurements (n = 17–20), two-way ANOVA shows significant group difference (F(1,140) = 102.0; p < 0.0001). E and F, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) (E) and [Ca2+]i (mean ± S.D.) (F) in somata of glutamate (50 μm)-treated and control cells. For [Ca2+]i measurements (n = 16–19), two-way ANOVA shows significant group difference (F(1,123) = 116.4.0; p < 0.0001). G and H, time-dependent changes in Fura2 fluorescence ratio (mean ± S.E.) (G) and [Ca2+]i (mean ± S.D.) (H) in dendrites of l-Hcy–treated and control cells. For [Ca2+]i measurements (n = 11–13), two-way ANOVA shows significant group difference (F(1,88) = 73.08; p < 0.0001). I and J, comparison of changes in Fura2 fluorescence ratio (mean ± S.E.) (I) and [Ca2+]i (mean ± S.D.) (J) between somata and dendrites of l-Hcy–treated cells. For [Ca2+]i measurements (n = 11–20), two-way ANOVA shows no significant group difference (F(1,116) = 0.2357; p < 0.6283). Post hoc analysis shows p < 0.05 (*), p < 0.001 (**), and p < 0.0001 (****) between the treatment group (l-Hcy or glutamate) and the control group at the given time point. Error bars represent S.D. or S.E.
Homocysteine-induced increase in intracellular Ca2+ level depends on GluN2A-containing NMDAR
To determine the primary source of [Ca2+]i increase, neurons were exposed to l-homocysteine (50 μm; 60 min) in the presence of the Ca2+ chelator EGTA. Fig. 2, A–D, show that homocysteine-mediated increases in Fura2 fluorescence ratio and [Ca2+]i level were blocked in the presence of EGTA in both somata and processes, suggesting that homocysteine-mediated [Ca2+]i increase is derived from extracellular sources. To examine the role of NMDARs in homocysteine-mediated Ca2+ influx, neurons were treated with l-homocysteine (50 μm; 60 min) in the presence of NMDAR inhibitor dl-2-amino-5-phosphopentanoic acid (dl-AP5). Fig. 2, E–H, show that dl-AP5 attenuated homocysteine-induced increases in Fura2 fluorescence ratio and [Ca2+]i in both the somata and the processes.
Figure 2.

Homocysteine-induced Ca2+ influx is NMDAR-dependent. A–D, changes in Fura2 fluorescence ratio (mean ± S.E.) (A and C) and [Ca2+]i (mean ± S.D.) (B and D) in somata and dendrites of cells treated with l-Hcy (50 μm) in the presence or absence of EGTA (2 mm). For [Ca2+]i in somata (n = 18–20), two-way ANOVA shows significant group difference (F(1,144) = 167.4; p < 0.0001). For [Ca2+]i in dendrites (n = 10–11), two-way ANOVA shows significant group difference (F(1,76) = 46.40; p < 0.0001). E–H, changes in Fura2 fluorescence ratio (mean ± S.E.) (E and G) and [Ca2+]i (mean ± S.D.) (F and H) in somata and dendrites of cells treated with l-Hcy (50 μm) in the presence or absence of dl-AP5 (200 μm). For [Ca2+]i in somata (n = 18–20), two-way ANOVA shows significant group difference (F(1,144) = 144.0; p < 0.0001). For [Ca2+]i in dendrites (n = 10–11), two-way ANOVA shows significant group difference (F(1,76) = 48.78; p < 0.0001). Post hoc analysis shows p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****) between l-Hcy–treated groups in the absence and presence of inhibitor at the given time point. Error bars represent S.D. or S.E.
To evaluate the role of GluN2A- and GluN2B-containing NMDARs in homocysteine-induced Ca2+ influx, rat neuronal cultures were incubated with l-homocysteine (50 μm; 60 min) in the presence of selective antagonists for GluN2A subunit ([(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydro-quinoxalin-5-yl)-methyl]phosphonic acid (NVP-AAM077)) or GluN2B subunit (Ro 25-6981). Fig. 3, A–D, show that coincubation with NVP-AAM077 attenuated the homocysteine-induced increase in Fura2 fluorescence ratio and [Ca2+]i in both the somata and the processes. In contrast, exposure to Ro 25-6981 failed to attenuate homocysteine-induced [Ca2+]i increase in either the somata or processes (Fig. 3, E–H). To establish more directly the role of GluN2A-NMDAR in homocysteine-induced [Ca2+]i, neuronal cultures obtained from wildtype (WT) and GluN2A-KO mice were treated with l-homocysteine (50 μm; 60 min). Fig. 4, A–C, G, and H, show progressive increases in Fura2 fluorescence ratio and [Ca2+]i in both the somata and the processes of neurons from WT mice as compared with control cells. However, treatment of neurons from GluN2A-KO mice with homocysteine failed to increase the Fura2 fluorescence ratio or [Ca2+]i in the somata and the processes (Fig. 4, D–F, I, and J) when compared with control cells. In contrast, treatment with glutamate (50 μm) led to rapid increases in Fura2 fluorescence ratio and [Ca2+]i in neurons from both WT and GluN2A-KO mice (Fig. 5, A–E). Additional studies evaluating the protein expression of GluN1, GluN2A, and GluN2B subunits in neuronal lysates from WT and GluN2A-KO mice showed no change in the protein level of GluN1 subunit (Fig. 5F). The findings also confirmed the loss of expression of GluN2A subunit in the neurons from GluN2A-KO mice. However, neuronal GluN2B subunit expression went up substantially in the GluN2A-KO mice.
Figure 3.
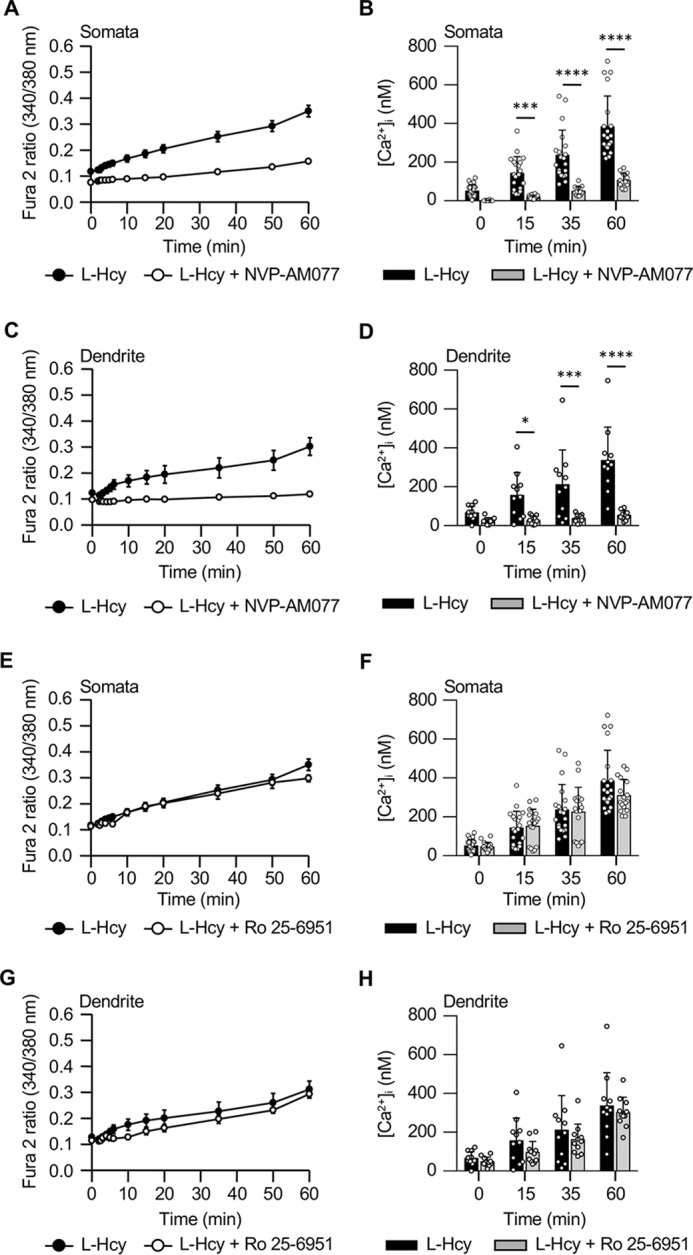
Pharmacological inhibition of GluN2A-NMDAR attenuates homocysteine-induced Ca2+ influx. A–D, changes in Fura2 fluorescence ratio (mean ± S.E.) (A and C) and [Ca2+]i (mean ± S.D.) (B and D) in somata and dendrites of cells treated with l-Hcy (50 μm) in the presence or absence of NVP-AAM077 (30 nm). For [Ca2+]i in somata (n = 14–20), two-way ANOVA shows significant group difference (F(1,128) = 107.3; p < 0.0001). For [Ca2+]i in dendrites (n = 10–11), two-way ANOVA shows significant group difference (F(1,76) = 49.65; p < 0.0001). Post hoc analysis shows p < 0.05 (*), p < 0.001 (***), and p < 0.0001 (****) between l-Hcy–treated groups in the absence and presence of inhibitor at the given time point. E–H, changes in Fura2 fluorescence ratio (mean ± S.E.) (E and G) and [Ca2+]i (mean ± S.D.) (F and H) in somata and dendrites of cells treated with l-Hcy (50 μm) in the presence or absence of Ro 25-6981 (1 μm). For [Ca2+]i in somata (n = 20–17), two-way ANOVA shows no significant group difference (F(1,140) = 1.388; p < 0.2408). For [Ca2+]i in dendrites (n = 11), two-way ANOVA shows no significant group difference (F(1,80) = 3.019; p < 0.0861). Error bars represent S.D. or S.E.
Figure 4.

Knockdown of GluN2A subunit blocks homocysteine-induced Ca2+ influx. A and D, individual responses in soma of 16–18 neurons obtained from WT (A) or GluN2A-KO (D) mice showing the range of increase in Fura2 fluorescence ratio over time following exposure to l-Hcy (50 μm). B, C, E, and F, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) and [Ca2+]i (mean ± S.D.) in the somata of l-Hcy–treated and control cells from WT (B and C) and GluN2A-KO (E and F) mice. For [Ca2+]i in WT mice (n = 16–18), two-way ANOVA shows significant group difference (F(1,128) = 1235; p < 0.0001). Post hoc analysis shows p < 0.0001 (****) between l-Hcy–treated group and control group at the given time point. For [Ca2+]i in GluN2A-KO mice (n = 16), two-way ANOVA shows no significant group difference (F(1,120) = 2.661; p < 0.1055). G–J, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) and [Ca2+]i (mean ± S.D.) in dendrites of l-Hcy–treated and control cells from WT (G and H) and GluN2A-KO (I and J) mice. For [Ca2+]i in WT mice (n = 8–10), two-way ANOVA shows significant group difference (F(1,64) = 603.6; p < 0.0001). Post hoc analysis shows p < 0.0001 (****) between l-Hcy–treated group and control group at the given time point. For [Ca2+]i in GluN2A-KO mice (n = 7–10), two-way ANOVA shows no significant group difference (F(1,60) = 3.975; p < 0.0507). Error bars represent S.D. or S.E.
Figure 5.

Knockdown of GluN2A subunit does not affect glutamate-induced Ca2+ influx. A and B, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) (A) and [Ca2+]i (mean ± S.D.) (B) in the somata of glutamate (50 μm)-treated and untreated (control) cells from WT mice. For [Ca2+]i measurements (n = 13–16), two-way ANOVA shows significant group difference (F(1,108) = 270.1; p < 0.0001). C and D, temporal profile of increase in Fura2 fluorescence ratio (mean ± S.E.) (C) and [Ca2+]i (mean ± S.D.) (D) in the somata of glutamate (50 μm)-treated and untreated (control) cells from GluN2A-KO mice. For [Ca2+]i measurements (n = 12–16), two-way ANOVA shows significant group difference (F(1,104) = 265.4; p < 0.0001). Post hoc analysis shows p < 0.0001 (****) between glutamate-treated group and control group at the given time point. E, immunoblot analysis of neuronal lysates from WT and GluN2A-KO mice with anti-GluN1, -GluN2A, -GluN2B, and -β-tubulin antibodies. Error bars represent S.D. or S.E.
Homocysteine-induced stimulation of GluN2A-containing NMDAR leads to sustained ERK MAPK phosphorylation and neuronal death
Our earlier studies showed that a sustained increase in ERK MAPK phosphorylation plays a crucial role in promoting homocysteine-NMDAR–induced neuronal cell death (17). To evaluate the role of GluN2A-NMDAR in homocysteine-induced ERK MAPK phosphorylation, rat neuronal cultures were treated with l-homocysteine (50 μm) for 30 or 60 min in the presence of dl-AP5 (NMDAR inhibitor) or NVP-AAM077 (GluN2A inhibitor). Fig. 6, A and B, show that pharmacological inhibition with either dl-AP5 or NVP-AAM077 attenuated homocysteine-induced ERK MAPK phosphorylation at both time points. To further confirm the role of GluN2A-NMDAR in homocysteine-induced ERK MAPK phosphorylation, neuron cultures from WT and GluN2A-KO mice were treated with l-homocysteine (50 μm) for 30 or 60 min. Fig. 6C shows that treatment with homocysteine led to a sustained increase in ERK MAPK phosphorylation in neurons obtained from WT mice, whereas it failed to induce ERK MAPK phosphorylation in neurons obtained from GluN2A-KO mice. Subsequent studies evaluated the effect of glutamate (50 μm) on the temporal profile of ERK MAPK phosphorylation (5, 30, or 60 min) in rat neuron cultures. As shown in Fig. 6D, treatment with glutamate led to a rapid but transient increase in ERK MAPK phosphorylation within 5 min of stimulation, which returned to basal level by 30 min. Treatment with dl-AP5 blocked, whereas treatment with NVP-AAM077 failed, to ameliorate the glutamate-induced transient increase in ERK MAPK phosphorylation at 5 min (Fig. 6E), indicating that GluN2A-NMDAR does not play a role in glutamate-induced ERK MAPK phosphorylation. Consistent with this interpretation, studies in neuron cultures from WT and GluN2A-KO mice showed that exposure to glutamate (50 μm) led to a rapid but transient increase in ERK MAPK phosphorylation in both WT and GluN2A-KO mouse cultures (Fig. 6F).
Figure 6.

Homocysteine-induced ERK MAPK phosphorylation is mediated through GluN2A-NMDAR. A and B, tat neuron cultures were exposed to l-Hcy (50 μm) for 30 or 60 min in the absence and presence of dl-AP5 (200 μm) (A) or NVP-AAM077 (30 nm) (B). C, neuronal cultures from WT and GluN2A-KO mice were treated with l-Hcy (50 μm) for 30 or 60 min. D, rat neuron cultures were exposed to glutamate (Glu; 50 μm) for 5, 30, or 60 min. E, rat neuronal cultures were exposed to glutamate for 5 min in the presence or absence of dl-AP5 (200 μm) or NVP-AAM077 (30 nm). F, neuronal cultures from WT and GluN2A-KO mice were treated with glutamate (50 μm) for 5, 30, or 60 min. A–G, immunoblot analysis of cell lysates with anti-phospho-ERK (pERK) (top) and anti-ERK (bottom) antibodies. Values are mean ± S.D. (n = 3–7). Significant differences were assessed by one-way ANOVA: *, p < 0.001 from corresponding 0 min; #, p < 0.001 from 30-min l-Hcy treatment; ¶, p < 0.001 from 60-min l-Hcy treatment; and §, p < 0.0001 from 5-min Glu treatment. Error bars represent S.D.
To evaluate the role of GluN2A-NMDARs in homocysteine-induced neuronal cell death, rat neuronal cultures were treated with l-homocysteine (50 μm; 18 h) in the presence of NVP-AAM077. Cell death was assessed by Hoechst DNA staining, an early indicator of apoptosis (37). The representative photomicrographs and quantitative analysis of pyknotic nuclei showed a significant increase in cell death following exposure to homocysteine (Fig. 7A), which is consistent with our earlier findings (16). Pharmacological treatment with NVP-AAM077 significantly reduced homocysteine-induced neuronal death (Fig. 7A). To assess the role of ERK MAPK activation in homocysteine-GluN2A-NMDAR–induced neurotoxicity, in subsequent studies neurons were treated with l-homocysteine (50 μm; 18 h) in the presence of the ERK MAPK inhibitor PD98059 (15 μm). As shown in Fig. 7A, ERK MAPK inhibition also attenuated homocysteine-induced neurotoxicity. Additional studies in neuron cultures obtained from WT and GluN2A-KO mice showed that exposure to homocysteine (50 μm) significantly increased neuronal death in cultures obtained from WT mice, whereas it failed to induce neurotoxicity in cultures obtained from GluN2A-KO mice (Fig. 7B). The findings also showed that homocysteine-induced neurotoxicity observed in neurons from WT mice was significantly reduced in the presence of ERK MAPK inhibitor (Fig. 7B). In contrast, glutamate (50 μm)-induced neuronal cell death in rat neuron cultures remained unaffected by pharmacological inhibition of either GluN2A-NMDAR or ERK MAPK (Fig. 7C). A comparison of the neurotoxic effects of glutamate in neuron cultures obtained from WT and GluN2A-KO mice further showed that deletion of the GluN2A subunit of NMDAR failed to reduce the neurotoxic effects of glutamate (Fig. 7D). Pharmacological inhibition of ERK MAPK also failed to reduce glutamate-induced neurotoxicity in neurons from either WT or GluN2A-KO mice (Fig. 7D).
Figure 7.

Homocysteine-induced neurotoxicity is mediated through GluN2A-NMDAR–dependent ERK MAPK activation. A, rat neuronal cultures were exposed to l-Hcy (50 μm; 18 h) in the absence and presence of NVP-AAM077 (30 nm) or PD98059 (15 μm). B, neurons from WT and GluN2A-KO mice were exposed to l-Hcy (50 μm; 18 h) in the absence or presence of PD98059 (15 μm). C, rat neuronal cultures were exposed to glutamate (Glu; 50 μm; 1 h) in the absence and presence of NVP-AAM077 (30 nm) or PD98059 (15 μm) and then maintained in original medium for 17 h. D, neurons from WT and GluN2A-KO mice were exposed to glutamate (50 μm; 1 h) in the absence or presence of PD98059 (15 μm) and then maintained in original medium for 17 h. A–D, representative photomicrographs showing pyknotic DNA stained with Hoechst 33342 (indicated with arrows). The percentage of neurons with pyknotic nuclei is represented as mean ± S.D. (n = 13–38 fields with a total of at least 1500 cells/condition from four experiments). Significant differences were assessed by one-way ANOVA: *, p < 0.001 from corresponding control; and #, p < 0.001 from l-Hcy treatment. Error bars represent S.D.
Discussion
Ca2+ is an important intracellular messenger that regulates multiple neuronal functions, including cellular growth, membrane excitability, and synaptic activity. As such, the intracellular Ca2+ level in neurons is tightly regulated to ensure efficient control on downstream signaling cascades involved in maintaining cellular physiology (38). The cellular mechanisms that help maintain Ca2+ homeostasis include the transmembrane Ca2+ gradient, route of Ca2+ entry, and presence of various Ca2+ buffering and extrusion systems (39, 40). Any changes in these homeostatic control mechanisms under a pathological condition leads to an aberrant increase in the intracellular Ca2+ level. Evidences now indicate that overactivation of NMDARs in neurodegenerative disorders involving excessive release of glutamate may lead to an aberrant increase in the [Ca2+]i level (41–44). However, the extent of Ca2+ increase varies depending on the severity of the stimuli and NMDAR subunit composition. A moderate and transient influx of Ca2+ has been coupled to GluN2A-NMDAR, whereas rapid, large Ca2+ overload is associated with GluN2B-NMDAR (41, 45–47). Additional studies using low-affinity Ca2+ indicators have also shown that glutamate-mediated intracellular Ca2+ increases occur in two phases, a rapid but low level of initial increase followed by a delayed, larger increase (47–49). The larger, delayed increase in [Ca2+]i is blocked in the presence of selective antagonists of GluN2B-NMDAR, indicating that the initial smaller Ca2+ influx depends on GluN2A-NMDAR (47).
A particular contribution of our study is the identification of the exclusive role of GluN2A-NMDAR in homocysteine-induced Ca2+ influx. The sole role of GluN2A-NMDAR in the homocysteine-induced low level of Ca2+ influx is not only confirmed using a selective pharmacological inhibitor for GluN2A-NMDAR but also by genetic deletion of GluN2A subunit of NMDAR. The inability of homocysteine to induce [Ca2+]i increase in neurons obtained from GluN2A-KO mice, despite the higher expression of the GluN2B subunit of NMDAR, further emphasizes the contribution of GluN2A-NMDARs in homocysteine-induced [Ca2+]i influx. In this context, a recent study utilizing HEK 293T cells expressing recombinant GluN1/2A and GluN1/2B NMDARs showed that GluN1/2B NMDARs undergo rapid desensitization upon exposure to homocysteine, whereas GluN1/2A receptors do not exhibit desensitization (50).
The distinctly different contributions of GluN2A- and GluN2B-containing NMDARs in homocysteine- and glutamate-mediated [Ca2+]i influx suggest differential roles of these subunits in the regulation of intracellular signaling cascades. This interpretation is substantiated by our findings that homocysteine-mediated NMDAR stimulation leads to a sustained increase in ERK MAPK phosphorylation, which is completely blocked by pharmacological inhibition of GluN2A-NMDAR or genetic deletion of GluN2A subunit. Whereas, in the glutamate treatment paradigm, the increase in ERK MAPK phosphorylation is found to be rapid but transient, and it remains unaffected by pharmacological inhibition or genetic deletion of GluN2A-NMDAR. Our findings also show that this difference in ERK MAPK signaling by the two NMDAR agonists has a considerably different effect on the neurotoxic profile. Pharmacological inhibition of GluN2A-NMDAR or genetic deletion of GluN2A subunit as well as pharmacological inhibition of ERK MAPK activity attenuates homocysteine-induced neuronal death, whereas they fail to attenuate glutamate-induced neuronal cell death. The findings highlight the role of sustained ERK MAPK activation in GluN2A-NMDAR–mediated neurotoxicity. Our earlier studies have shown that such sustained ERK MAPK activation leads to a decrease in surface expression of the GluA2 subunit of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, resulting in Ca2+ influx through GluA2-lacking Ca2+-permeable AMPA receptors, which leads to p38 MAPK activation (18). This novel interplay between ERK and p38 MAPK results in caspase-3–dependent neuronal cell death (17). Whether this detrimental signaling cascade downstream of GluN2A-NMDAR stimulation, observed in our studies, is unique to the homocysteine signaling pathway or could also be triggered by other extracellular stimuli remains to be investigated. The transient increase in ERK MAPK phosphorylation following exposure to glutamate and its inability to attenuate glutamate-induced neurotoxicity further indicate that transient ERK MAPK activation has different consequences as compared with sustained activation (51, 52).
The present study reveals an important functional consequence of GluN2A-NMDAR activation, which is in contrast to the prevailing theory that mainly emphasizes the function of GluN2A-NMDAR in promoting neuronal survival and growth (20, 53–55). Several in vitro and in vivo studies on excitotoxic and ischemic neuronal injury have demonstrated that selective inhibition GluN2A-NMDARs either has no effect or exacerbates neuronal death (20, 27, 56, 57). Also, an elegant study investigating the effect of chimeric constructs of GluN2A/2B subunits expressed in cultured neurons and a knockin mouse model has demonstrated that replacing the C-terminal domain of GluN2B with that of GluN2A lowers the vulnerability of neurons to excitotoxic insult, emphasizing a neuroprotective role of GluN2A-NMDARs (58). However, there are a few studies that have investigated the detrimental role of GluN2A-NMDARs in neurons following ischemic/excitotoxic insult (56, 59, 60), but the interpretation of results obtained from these studies is not straightforward. The in vivo study by Morikawa et al. (56) shows that permanent focal cerebral ischemia in GluN2A-KO mice has no effect on stroke outcome when compared with WT mice, whereas 2 h of focal cerebral ischemia in GluN2A-KO mice leads to significant reduction in infarct size as compared with WT littermates. Similarly, the study by Wang et al. (59) shows involvement of GluN2A-NMDARs in selective degeneration of CA1 hippocampal neurons following stroke only after bilateral vertebral arteries were completely cut and coagulated for 12 h prior to induction of stroke. Furthermore, the in vitro study by Zhou et al. (60) shows that blocking GluN2A-NMDAR leads to partial protection against NMDA-mediated excitotoxicity when the duration and severity of the insult are low. Interestingly, they also demonstrate that blocking GluN2A-NMDAR does not show measurable effects when the duration of the insult is increased from 15 to 30 min or the degree of excitotoxic insult is increased from 50 to 100 μm NMDA. This indicates that blocking GluN2A-NMDAR is not effective in attenuating neuronal death under severe excitotoxic condition. In contrast, significant protection was still achieved by blocking GluN2B-NMDAR at all the above experimental conditions. As such, the issue of whether the GluN2A-NMDAR stimulation influences excitotoxic neuropathology has still remained unresolved. Distinct from these previous studies, our findings provide convincing evidence of intracellular signaling via GluN2A-NMDAR as the primary contributor of homocysteine-NMDAR–induced neurotoxicity.
Although the focus of the present study is on homocysteine-NMDAR–induced neurotoxicity, earlier studies have indicated a possible role of metabotropic glutamate receptors (mGluRs) in homocysteine-induced neurotoxicity (61, 62). However, the involvement of mGluRs in homocysteine-induced neurotoxicity in these studies was observed using very high doses of homocysteine (5–25 mm) typically not observed under pathological conditions (1, 3, 5). In addition, the neurotoxicity observed in these studies does not involve Ca2+ influx in neurons but, on the contrary, leads to efflux of Ca2+ from neurons. In contrast, our study was carried out with a concentration of homocysteine that is generally observed in individuals predisposed to mild to moderate hyperhomocysteinemia (1, 11, 63–65). We also observed that under this condition the increase in [Ca2+]i plays a key role in homocysteine-induced neurotoxicity. As such, the neurotoxicity observed in our study could not be attributed to mGluR activation. This interpretation is further supported by our earlier study in neuron cultures demonstrating that mGluR inhibition fails to block homocysteine-induced ERK MAPK activation and subsequent neurotoxicity (16). In addition, an earlier study that evaluated the relative potency of mGluRs 1–5 to bind to l-homocysteine and its acidic derivatives showed that l-homocysteine by itself is not an agonist of mGluRs (66). Together, these findings present the novel concept that homocysteine, by stimulating GluN2A-NMDARs, may promote neurotoxicity through sustained activation of Ca2+-dependent ERK MAPK signaling.
Experimental procedures
Materials and reagents
Pregnant female Sprague-Dawley rats were purchased from Envigo. GluN2A-KO mice were obtained from Dr. Andrew Holmes, National Institute on Alcohol Abuse and Alcoholism (67), and timed pregnant mice were generated at the animal facility of University of New Mexico. The Institutional Animal Care and Use Committee of University of New Mexico, Health Sciences Center approved all animal procedures. l-Homocysteine thiolactone, glycine, EGTA, and Hoechst 33342 were purchased from Sigma-Aldrich. Fura2-AM and all cell culture reagents were purchased from Invitrogen. Anti-phospho-ERK1/2 (Thr-202/Tyr-204) mAb (pERK) and anti-rabbit and -mouse horseradish peroxidase–conjugated secondary antibodies were purchased from Cell Signaling Technology. Anti-ERK2 (ERK) and anti-β-tubulin polyclonal antibodies were purchased from Santa Cruz Biotechnology. Anti-GluN2A rabbit mAb was purchased from Abcam. Anti-GluN1 mAb and anti-GluN2B polyclonal antibody were purchased from Millipore Sigma. Ionomycin, dl-AP5, Ro 25-6981, PD98059, and NVP-AAM077 were obtained from EMD Biosciences.
Neuron culture, l-homocysteine preparation, and stimulation
Embryos obtained from pregnant Sprague-Dawley rats (16–17-day gestation) or WT and GluN2A-KO mice (15–16-day gestation) were used to establish primary cortical neuronal cultures as described earlier (16–18). Neurons were grown on (a) 35-mm culture dishes (MatTek Corp.) coated overnight with poly-d-lysine (50 μg/ml) and laminin (10 μg/ml) for Ca2+ imaging studies, (b) poly-d-lysine–coated 60-mm dishes (Corning BioCoat) for biochemical studies, and (c) poly-d-lysine–coated 4-well culture slides (Corning BioCoat) for cell-death assay. Neurons were maintained in culture for 12–13 days before experiments. The cells were treated with freshly prepared l-homocysteine (50 μm) in Hank's balanced salt solution containing 50 μm glycine (11, 16–18) for the specified time periods. In a parallel series of experiments, cells were treated with glutamate (50 μm) for the specified time periods. Cells were then processed for live-cell imaging, immunoblotting, or cell-death assay. In some experiments, EGTA, dl-AP5, NVP-AAM077, Ro 25-6981, or PD98059 was added 15 min prior to l-homocysteine or glutamate treatment.
Calcium measurements
[Ca2+]i in neurons was determined using the fluorescent indicator Fura2-AM. Briefly, neurons were loaded with Fura2 (10 μm) in phenol red–free Hank's balanced salt solution for 30 min at 37 °C followed by postincubation for 20 min (47). Time-lapse live-cell imaging was performed following stimulation using a Nikon Ti Eclipse inverted microscope equipped with Tokai Hit stage-top incubator maintained at 37 °C and infused with 95% air, 5% CO2 mixture. Fields of five to eight cells were imaged using a 40× oil immersion objective (Nikon). Fluorescence excitations (340 and 380 nm) were performed using a Sutter LB-LS/30 Lambda xenon arc lamp, and fluorescence emissions (510 nm) were captured using a charge-coupled device (CCD) camera (Photometrics). NIS Elements AR software was used for data acquisition and analysis. Ratiometric data (340/380 nm) from regions of interest were corrected for background and converted into estimates of [Ca2+]i as described previously (47) using the equation developed earlier (68). Maximum and minimum fluorescence ratios were determined from Fura2-loaded cells treated with ionomycin (5 μm) in calcium-containing medium (2 mm) or in calcium-free medium with EGTA (0.5 mm).
Immunoblotting
Rat and mouse neuron cultures were harvested in SDS sample buffer (69). Equal amounts of protein from the cell lysates were resolved by SDS-PAGE (7.5%) and subjected to immunoblotting procedures as described earlier (16, 17, 23). The blots were analyzed with the specified antibodies according to the manufacturer's protocol. Densitometric analyses of the images captured on X-ray films were performed using ImageJ software.
Hoechst DNA staining
Cortical neuron cultures from rats and mice were treated either with l-homocysteine for 18 h or glutamate for 1 h. Glutamate-treated cells were then maintained in their original medium for another 17 h. For some experiments, neurons were treated with l-homocysteine or glutamate in the presence of NVP-AAM077 or PD98059. The neurons were then fixed and stained with Hoechst 33342 dye as described earlier (16). The percentage of pyknotic nuclei was quantitatively assessed by fluorescence microscopy to determine the extent of neuronal death.
Statistical analysis
Statistical analysis was performed using one-way or two-way analysis of variance (ANOVA) followed by post hoc analysis using Bonferroni's multiple comparison test. Differences were considered significant for p < 0.05.
Author contributions
S. N. D. and R. P. data curation; S. N. D., S. P., and R. P. formal analysis; S. N. D., S. P., and R. P. validation; S. N. D., S. M., and S. R. investigation; S. N. D. and R. P. visualization; S. N. D., S. P., and R. P. writing-review and editing; S. M. and S. R. methodology; S. P. and R. P. conceptualization; S. P. and R. P. supervision; S. P. and R. P. funding acquisition; R. P. resources; R. P. writing-original draft; R. P. project administration.
This work was supported by the National Institutes of Health Grants RO1 NS083914 (to R. P.) and RO1 NS059962 (to S. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NMDAR
- N-methyl-d-aspartate receptor
- NMDA
- N-methyl-d-aspartate
- ERK MAPK
- extracellular signal-regulated kinase/mitogen-activated protein kinase
- KO
- knockout
- [Ca2+]i
- intracellular Ca2+ concentration
- dl-AP5
- dl-2-amino-5-phosphopentanoic acid
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- mGluR
- metabotropic glutamate receptor
- NVP-AAM077
- [(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydro-quinoxalin-5-yl)-methyl]phosphonic acid
- AM
- acetooxymethyl ester
- ANOVA
- analysis of variance
- l-Hcy
- l-homocysteine.
References
- 1. Refsum H., Ueland P. M., Nygård O., and Vollset S. E. (1998) Homocysteine and cardiovascular disease. Annu. Rev. Med. 49, 31–62 10.1146/annurev.med.49.1.31 [DOI] [PubMed] [Google Scholar]
- 2. Seshadri S., Beiser A., Selhub J., Jacques P. F., Rosenberg I. H., D'Agostino R. B., Wilson P. W., and Wolf P. A. (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N. Engl. J. Med. 346, 476–483 10.1056/NEJMoa011613 [DOI] [PubMed] [Google Scholar]
- 3. Austin R. C., Lentz S. R., and Werstuck G. H. (2004) Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 11, Suppl. 1, S56–S64 10.1038/sj.cdd.4401451 [DOI] [PubMed] [Google Scholar]
- 4. Kang S. S., Wong P. W., and Malinow M. R. (1992) Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 12, 279–298 10.1146/annurev.nu.12.070192.001431 [DOI] [PubMed] [Google Scholar]
- 5. Gupta S., Kühnisch J., Mustafa A., Lhotak S., Schlachterman A., Slifker M. J., Klein-Szanto A., High K. A., Austin R. C., and Kruger W. D. (2009) Mouse models of cystathionine β-synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J. 23, 883–893 10.1096/fj.08-120584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sharma M., Tiwari M., and Tiwari R. K. (2015) Hyperhomocysteinemia: impact on neurodegenerative diseases. Basic Clin. Pharmacol. Toxicol. 117, 287–296 10.1111/bcpt.12424 [DOI] [PubMed] [Google Scholar]
- 7. Zoccolella S., Martino D., Defazio G., Lamberti P., and Livrea P. (2006) Hyperhomocysteinemia in movement disorders: current evidence and hypotheses. Curr. Vasc. Pharmacol. 4, 237–243 10.2174/157016106777698414 [DOI] [PubMed] [Google Scholar]
- 8. Hankey G. J., and Eikelboom J. W. (2001) Homocysteine and stroke. Curr. Opin. Neurol. 14, 95–102 10.1097/00019052-200102000-00015 [DOI] [PubMed] [Google Scholar]
- 9. Zoccolella S., Bendotti C., Beghi E., and Logroscino G. (2010) Homocysteine levels and amyotrophic lateral sclerosis: a possible link. Amyotroph. Lateral Scler. 11, 140–147 10.3109/17482960902919360 [DOI] [PubMed] [Google Scholar]
- 10. Ansari R., Mahta A., Mallack E., and Luo J. J. (2014) Hyperhomocysteinemia and neurologic disorders: a review. J. Clin. Neurol. 10, 281–288 10.3988/jcn.2014.10.4.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lipton S. A., Kim W. K., Choi Y. B., Kumar S., D'Emilia D. M., Rayudu P. V., Arnelle D. R., and Stamler J. S. (1997) Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. U.S.A. 94, 5923–5928 10.1073/pnas.94.11.5923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kruman I. I., Culmsee C., Chan S. L., Kruman Y., Guo Z., Penix L., and Mattson M. P. (2000) Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 20, 6920–6926 10.1523/JNEUROSCI.20-18-06920.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kruman I. I., Kumaravel T. S., Lohani A., Pedersen W. A., Cutler R. G., Kruman Y., Haughey N., Lee J., Evans M., and Mattson M. P. (2002) Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer's disease. J. Neurosci. 22, 1752–1762 10.1523/JNEUROSCI.22-05-01752.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mattson M. P., and Shea T. B. (2003) Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 26, 137–146 10.1016/S0166-2236(03)00032-8 [DOI] [PubMed] [Google Scholar]
- 15. Jara-Prado A., Ortega-Vazquez A., Martinez-Ruano L., Rios C., and Santamaria A. (2003) Homocysteine-induced brain lipid peroxidation: effects of NMDA receptor blockade, antioxidant treatment, and nitric oxide synthase inhibition. Neurotox. Res. 5, 237–243 10.1007/BF03033381 [DOI] [PubMed] [Google Scholar]
- 16. Poddar R., and Paul S. (2009) Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 110, 1095–1106 10.1111/j.1471-4159.2009.06207.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Poddar R., and Paul S. (2013) Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J. Neurochem. 124, 558–570 10.1111/jnc.12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Poddar R., Chen A., Winter L., Rajagopal S., and Paul S. (2017) Role of AMPA receptors in homocysteine-NMDA receptor-induced crosstalk between ERK and p38 MAPK. J. Neurochem. 142, 560–573 10.1111/jnc.14078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim M. J., Dunah A. W., Wang Y. T., and Sheng M. (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760 10.1016/j.neuron.2005.04.031 [DOI] [PubMed] [Google Scholar]
- 20. Liu Y., Wong T. P., Aarts M., Rooyakkers A., Liu L., Lai T. W., Wu D. C., Lu J., Tymianski M., Craig A. M., and Wang Y. T. (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 27, 2846–2857 10.1523/JNEUROSCI.0116-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardingham G. E., Fukunaga Y., and Bading H. (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414 10.1038/nn835 [DOI] [PubMed] [Google Scholar]
- 22. Zhang S. J., Steijaert M. N., Lau D., Schütz G., Delucinge-Vivier C., Descombes P., and Bading H. (2007) Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron 53, 549–562 10.1016/j.neuron.2007.01.025 [DOI] [PubMed] [Google Scholar]
- 23. Paul S., Nairn A. C., Wang P., and Lombroso P. J. (2003) NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 6, 34–42 10.1038/nn989 [DOI] [PubMed] [Google Scholar]
- 24. Wang J. Q., Tang Q., Parelkar N. K., Liu Z., Samdani S., Choe E. S., Yang L., and Mao L. (2004) Glutamate signaling to Ras-MAPK in striatal neurons: mechanisms for inducible gene expression and plasticity. Mol. Neurobiol. 29, 1–14 10.1385/MN:29:1:01 [DOI] [PubMed] [Google Scholar]
- 25. Ivanov A., Pellegrino C., Rama S., Dumalska I., Salyha Y., Ben-Ari Y., and Medina I. (2006) Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol. 572, 789–798 10.1113/jphysiol.2006.105510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mao L., Tang Q., Samdani S., Liu Z., and Wang J. Q. (2004) Regulation of MAPK/ERK phosphorylation via ionotropic glutamate receptors in cultured rat striatal neurons. Eur. J. Neurosci. 19, 1207–1216 10.1111/j.1460-9568.2004.03223.x [DOI] [PubMed] [Google Scholar]
- 27. Chen M., Lu T. J., Chen X. J., Zhou Y., Chen Q., Feng X. Y., Xu L., Duan W. H., and Xiong Z. Q. (2008) Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke 39, 3042–3048 10.1161/STROKEAHA.108.521898 [DOI] [PubMed] [Google Scholar]
- 28. Riccio A., and Ginty D. D. (2002) What a privilege to reside at the synapse: NMDA receptor signaling to CREB. Nat. Neurosci. 5, 389–390 10.1038/nn0502-389 [DOI] [PubMed] [Google Scholar]
- 29. Li J. H., Wang Y. H., Wolfe B. B., Krueger K. E., Corsi L., Stocca G., and Vicini S. (1998) Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur. J. Neurosci. 10, 1704–1715 10.1046/j.1460-9568.1998.00169.x [DOI] [PubMed] [Google Scholar]
- 30. Stocca G., and Vicini S. (1998) Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J. Physiol. 507, 13–24 10.1111/j.1469-7793.1998.013bu.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tovar K. R., and Westbrook G. L. (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 19, 4180–4188 10.1523/JNEUROSCI.19-10-04180.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li B., Chen N., Luo T., Otsu Y., Murphy T. H., and Raymond L. A. (2002) Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat. Neurosci. 5, 833–834 10.1038/nn912 [DOI] [PubMed] [Google Scholar]
- 33. Connor J. A., Wadman W. J., Hockberger P. E., and Wong R. K. (1988) Sustained dendritic gradients of Ca2+ induced by excitatory amino acids in CA1 hippocampal neurons. Science 240, 649–653 10.1126/science.2452481 [DOI] [PubMed] [Google Scholar]
- 34. de Erausquin G. A., Manev H., Guidotti A., Costa E., and Brooker G. (1990) Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proc. Natl. Acad. Sci. U.S.A. 87, 8017–8021 10.1073/pnas.87.20.8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Glaum S. R., Scholz W. K., and Miller R. J. (1990) Acute- and long-term glutamate-mediated regulation of [Ca++]i in rat hippocampal pyramidal neurons in vitro. The J. Pharmacol. Exp. Ther. 253, 1293–1302 [PubMed] [Google Scholar]
- 36. Randall R. D., and Thayer S. A. (1992) Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J. Neurosci. 12, 1882–1895 10.1523/JNEUROSCI.12-05-01882.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daxhelet G. A., Coene M. M., Hoet P. P., and Cocito C. G. (1989) Spectrofluorometry of dyes with DNAs of different base composition and conformation. Anal. Biochem. 179, 401–403 10.1016/0003-2697(89)90152-8 [DOI] [PubMed] [Google Scholar]
- 38. Arundine M., and Tymianski M. (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34, 325–337 10.1016/S0143-4160(03)00141-6 [DOI] [PubMed] [Google Scholar]
- 39. Tymianski M., and Tator C. H. (1996) Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 38, 1176–1195 10.1097/00006123-199606000-00028 [DOI] [PubMed] [Google Scholar]
- 40. Tymianski M. (1996) Cytosolic calcium concentrations and cell death in vitro. Adv. Neurol. 71, 85–105 [PubMed] [Google Scholar]
- 41. Lynch D. R., and Guttmann R. P. (2002) Excitotoxicity: perspectives based on N-methyl-d-aspartate receptor subtypes. J. Pharmacol. Exp. Ther. 300, 717–723 10.1124/jpet.300.3.717 [DOI] [PubMed] [Google Scholar]
- 42. Dingledine R., Borges K., Bowie D., and Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 43. Lee J. M., Zipfel G. J., and Choi D. W. (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399, A7–A14 10.1038/399a007 [DOI] [PubMed] [Google Scholar]
- 44. Lipton S. A., and Rosenberg P. A. (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622 10.1056/NEJM199403033300907 [DOI] [PubMed] [Google Scholar]
- 45. Scimemi A., Fine A., Kullmann D. M., and Rusakov D. A. (2004) NR2B-containing receptors mediate cross talk among hippocampal synapses. J. Neurosci. 24, 4767–4777 10.1523/JNEUROSCI.0364-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vicini S., Wang J. F., Li J. H., Zhu W. J., Wang Y. H., Luo J. H., Wolfe B. B., and Grayson D. R. (1998) Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J. Neurophysiol. 79, 555–566 10.1152/jn.1998.79.2.555 [DOI] [PubMed] [Google Scholar]
- 47. Paul S., and Connor J. A. (2010) NR2B-NMDA receptor-mediated increases in intracellular Ca2+ concentration regulate the tyrosine phosphatase, STEP, and ERK MAP kinase signaling. J. Neurochem. 114, 1107–1118 10.1111/j.1471-4159.2010.06835.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng C., Fass D. M., and Reynolds I. J. (1999) Emergence of excitotoxicity in cultured forebrain neurons coincides with larger glutamate-stimulated [Ca2+]i increases and NMDA receptor mRNA levels. Brain Res. 849, 97–108 10.1016/S0006-8993(99)01995-2 [DOI] [PubMed] [Google Scholar]
- 49. Hyrc K., Handran S. D., Rothman S. M., and Goldberg M. P. (1997) Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J. Neurosci. 17, 6669–6677 10.1523/JNEUROSCI.17-17-06669.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sibarov D. A., Abushik P. A., Giniatullin R., and Antonov S. M. (2016) GluN2A subunit-containing NMDA receptors are the preferential neuronal targets of homocysteine. Front. Cell. Neurosci. 10, 246 10.3389/fncel.2016.00246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Colucci-D'Amato L., Perrone-Capano C., and di Porzio U. (2003) Chronic activation of ERK and neurodegenerative diseases. BioEssays 25, 1085–1095 10.1002/bies.10355 [DOI] [PubMed] [Google Scholar]
- 52. Zhuang S., and Schnellmann R. G. (2006) A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 319, 991–997 10.1124/jpet.106.107367 [DOI] [PubMed] [Google Scholar]
- 53. Lai T. W., Shyu W. C., and Wang Y. T. (2011) Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 17, 266–275 10.1016/j.molmed.2010.12.008 [DOI] [PubMed] [Google Scholar]
- 54. Martin H. G., and Wang Y. T. (2010) Blocking the deadly effects of the NMDA receptor in stroke. Cell 140, 174–176 10.1016/j.cell.2010.01.014 [DOI] [PubMed] [Google Scholar]
- 55. Terasaki Y., Sasaki T., Yagita Y., Okazaki S., Sugiyama Y., Oyama N., Omura-Matsuoka E., Sakoda S., and Kitagawa K. (2010) Activation of NR2A receptors induces ischemic tolerance through CREB signaling. J. Cereb. Blood Flow Metab. 30, 1441–1449 10.1038/jcbfm.2010.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morikawa E., Mori H., Kiyama Y., Mishina M., Asano T., and Kirino T. (1998) Attenuation of focal ischemic brain injury in mice deficient in the ϵ1 (NR2A) subunit of NMDA receptor. J. Neurosci. 18, 9727–9732 10.1523/JNEUROSCI.18-23-09727.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. DeRidder M. N., Simon M. J., Siman R., Auberson Y. P., Raghupathi R., and Meaney D. F. (2006) Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol. Dis. 22, 165–176 10.1016/j.nbd.2005.10.011 [DOI] [PubMed] [Google Scholar]
- 58. Martel M. A., Ryan T. J., Bell K. F., Fowler J. H., McMahon A., Al-Mubarak B., Komiyama N. H., Horsburgh K., Kind P. C., Grant S. G., Wyllie D. J., and Hardingham G. E. (2012) The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 74, 543–556 10.1016/j.neuron.2012.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J., Liu S., Fu Y., Wang J. H., and Lu Y. (2003) Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat. Neurosci. 6, 1039–1047 10.1038/nn1119 [DOI] [PubMed] [Google Scholar]
- 60. Zhou X., Ding Q., Chen Z., Yun H., and Wang H. (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-d-aspartate receptor function and neuronal excitotoxicity. J. Biol. Chem. 288, 24151–24159 10.1074/jbc.M113.482000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ziemińska E., Stafiej A., and Łazarewicz J. W. (2003) Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 43, 481–492 10.1016/S0197-0186(03)00038-X [DOI] [PubMed] [Google Scholar]
- 62. Lazarewicz J. W., Ziembowicz A., Matyja E., Stafiej A., and Zieminska E. (2003) Homocysteine-evoked 45Ca release in the rabbit hippocampus is mediated by both NMDA and group I metabotropic glutamate receptors: in vivo microdialysis study. Neurochem. Res. 28, 259–269 10.1023/A:1022329317218 [DOI] [PubMed] [Google Scholar]
- 63. Selhub J., Jacques P. F., Bostom A. G., D'Agostino R. B., Wilson P. W., Belanger A. J., O'Leary D. H., Wolf P. A., Schaefer E. J., and Rosenberg I. H. (1995) Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N. Engl. J. Med. 332, 286–291 10.1056/NEJM199502023320502 [DOI] [PubMed] [Google Scholar]
- 64. Perry I. J., Refsum H., Morris R. W., Ebrahim S. B., Ueland P. M., and Shaper A. G. (1995) Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 346, 1395–1398 10.1016/S0140-6736(95)92407-8 [DOI] [PubMed] [Google Scholar]
- 65. Lindgren A., Brattström L., Norrving B., Hultberg B., Andersson A., and Johansson B. B. (1995) Plasma homocysteine in the acute and convalescent phases after stroke. Stroke 26, 795–800 10.1161/01.STR.26.5.795 [DOI] [PubMed] [Google Scholar]
- 66. Shi Q., Savage J. E., Hufeisen S. J., Rauser L., Grajkowska E., Ernsberger P., Wroblewski J. T., Nadeau J. H., and Roth B. L. (2003) l-Homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J. Pharmacol. Exp. Ther. 305, 131–142 10.1124/jpet.102.047092 [DOI] [PubMed] [Google Scholar]
- 67. Sakimura K., Kutsuwada T., Ito I., Manabe T., Takayama C., Kushiya E., Yagi T., Aizawa S., Inoue Y., Sugiyama H., and Mishina M. (1995) Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor ϵ1 subunit. Nature 373, 151–155 10.1038/373151a0 [DOI] [PubMed] [Google Scholar]
- 68. Grynkiewicz G., Poenie M., and Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 69. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]