

Abstract
Protein–protein interactions lie at the heart of many biological processes and therefore represent promising drug targets. Despite this opportunity, identification of protein–protein interfaces remains challenging. We have previously developed a method that relies on coating protein surfaces with small-molecule dyes to discriminate between solvent-accessible protein surfaces and hidden interface regions. Dye-bound, solvent-accessible protein regions resist trypsin digestion, whereas hidden interface regions are revealed by denaturation and sequenced by MS. The small-molecule dyes bind promiscuously and with high affinity, but their binding mechanism is unknown. Here, we report on the optimization of a novel dye probe used in protein painting, Fast Blue B + naphthionic acid, and show that its affinity for proteins strongly depends on hydrophobic moieties that we call here “hydrophobic clamps.” We demonstrate the utility of this probe by sequencing the protein–protein interaction regions between the Hippo pathway protein Yes-associated protein 2 (YAP2) and tight junction protein 1 (TJP1 or ZO-1), uncovering interactions via the known binding domain as well as ZO-1's MAGUK domain and YAP's N-terminal proline-rich domain. Additionally, we demonstrate how residues predicted by protein painting are present exclusively in the complex interface and how these residues may guide the development of peptide inhibitors using a case study of programmed cell death protein 1 (PD-1) and programmed cell death 1 ligand 1 (PD-L1). Inhibitors designed around the PD-1/PD-L1 interface regions identified via protein painting effectively disrupted complex formation, with the most potent inhibitor having an IC50 of 5 μm.
Keywords: mass spectrometry (MS), cell signaling, protein-protein interaction, Yes-associated protein (YAP), structural biology, immune checkpoint inhibitor, PD-L1, programmed cell death protein 1 (PD-1), protein painting, ZO-1
Introduction
Protein–protein interactions lie at the heart of signaling pathways and are crucial interactions that influence many disease states. However, despite their relevance to disease pathology, protein–protein interactions have historically been infrequent targets of drug discovery efforts due to difficulties in targeting the interaction interface (1). Lack of crystal structures of a protein–protein complex of interest can make identifying precise interface regions and interface topology difficult, not to mention that crystal structures of protein–protein complexes may not accurately depict the biological assembly due to nonphysiological crystallization conditions (2). Additionally, it is often difficult to obtain crystal structures of conformationally flexible or unstructured proteins without modifications to the protein construct or co-crystallization with stabilizing ligands. Furthermore, interface regions of protein complexes as identified by crystallography do not reveal which molecular interactions between two proteins are most responsible for driving affinity. Alanine scanning is the gold standard for determination of residues that contribute to binding affinity, but even combinatorial alanine-scanning methods can be quite time-consuming (3). Other noncrystallographic techniques for identification of protein–protein interfaces, such as hydroxy radical labeling, hydrogen deuterium exchange, or chemical cross-linking, can also be laborious and prone to false positives due to nonphysiological solution conditions (4).
We have previously reported a new technique called protein painting, shown in Fig. 1, that identifies protein–protein interfaces via binding of small-molecule dyes, or “paints” (5). In this technique, preformed protein complexes are pulsed with molecular paints that noncovalently coat the solvent-accessible surface of the complex. The paints remain bound to the protein during denaturation and inhibit digestion via trypsin, allowing the protein–protein interaction fragments to be detected via MS. Peptides that are identified in an unpainted protein sample, yet are absent in a painted sample, represent sequences to which molecular dyes have bound to prevent trypsinization, and are considered the solvent-accessible peptide fragments. A sample consisting of a preformed protein complex can then be painted; peptides identified in the complexed sample, yet absent in the painted protein sample alone, represent peptide fragments that are solvent-accessible when the protein is alone yet are solvent-inaccessible when a protein is complexed. These peptides are the hot spots reported from the analysis. For peptides to be considered absent from the sample, they must be unidentified within a 1% false discovery rate using Proteome Discoverer with Sequest to search a protein database, where mass tolerance for precursor ions is 5 ppm (Orbitrap Fusion) or 10 ppm (LTQ Orbitrap), and mass tolerance for fragment ions is 0.06 Da. The key differentiators of this technique versus other protein MS methods, such as hydroxy radical labeling or hydrogen deuterium exchange, are the native solution conditions employed and small-molecule dyes utilized. These dyes noncovalently bind to native proteins with high affinity (koff ≈ 10−5 s−1, whereas kon ≈ 102 m−1 s−1; see Fig. S4), remain bound to the proteins following denaturation, and block trypsin cleavage of peptide bonds (5). Using this technique, we have been able to identify the unique techniques used by interleukin 1 (IL-1) versus interleukin-33 (IL-33)2 to recruit interleukin-1 receptor accessory protein (ILRAcP) in engaging their respective receptors IL-R1 and ST2 (6). This finding is particularly significant, given the fact that the crystallographic interfaces in the ternary complex between IL-1, IL-R1, and ILRAcP are very similar to those in the ternary complex of IL-33, ST2, and ILRAcP.
Figure 1.

Protein painting methods reveal protein–protein interaction hot spots by blocking tryptic cleavage sites at noninterface regions using molecular dyes. A, preformed protein complexes are pulsed with a great molar excess of small-molecule dyes. These dyes cover solvent-accessible surface regions of the protein complex but cannot bind to the solvent-inaccessible interface regions. After the dye pulse, unbound dye is removed via gel filtration. B, denaturation of the complex reveals the interface regions of the proteins that were previously solvent-inaccessible. Trypsin cleavage of arginine or lysine residues that are not dye-bound (such as Lys-78 of PD-1) yields tryptic peptides that are sequenced by subsequent MS. Those peptides that are exclusively found in samples consisting of a painted complex and absent in samples consisting of a painted individual protein are identified as hot spots of interaction.
However, despite the fact that protein painting has shown great utility in identifying solvent-inaccessible interface regions that are smaller than the crystallographic interfaces, there remain a few unanswered questions about the method. First, the mechanisms of dye-binding are currently unexplored, including how the small-molecule dyes used in this method bind to proteins with such high affinity and what properties allow for the development of a dye that binds universally to all protein complexes. Currently, every prospective complex examined by protein painting is screened against multiple different dyes to optimize coverage, as there is no objective criterion by which a dye can be chosen to ensure best coverage of a specific protein. Obtaining a more complete understanding of the mechanism by which these small-molecule dyes bind to proteins, and how they remain bound during protein denaturation, can lead to improved candidate dye structures with affinity for many different protein surfaces. These new dye candidates could be incorporated into the next generation of protein-painting methodology to allow for more rapid screening of different kinds of protein–protein complexes, enabling identification of additional drug targets.
In this study, we screened small-molecule dyes to identify structural motifs common to those dyes with the highest coverage and then used this information to develop a next-generation molecular dye probe. Using this new probe in our protein-painting protocol, we investigated two clinically relevant protein–protein interactions. First, we investigated the interactions between YAP, a core transcriptional regulator in the Hippo pathway, and ZO-1, a scaffolding protein of the tight junction. Neither of these proteins have proved amenable to crystallography, especially given that YAP is thought to be unstructured in the absence of a binding partner (7), yet the complex could represent an important drug target. YAP has been implicated in some cancers (8), macular degeneration (9), and wound healing (10), such that understanding its interactions could provide new drug targets for many different disease states. YAP may have both pro-apoptotic and pro-proliferative effects; YAP interaction with TEAD is shown to promote expression of growth factors and support proliferation (11), whereas YAP interaction with p73 promotes apoptosis (12). Given YAP's role as a “model of functional dichotomy,” as so deemed by Sudol et al. (12), as well as the fact that YAP is thought to be partially unstructured in its native state (7), drug development for modulation of Hippo pathway signaling is often focused on binding partners of YAP, such as TEAD, rather than on YAP itself (11). YAP also interacts with the zona occludens (ZO) family of proteins consisting of three isoforms, ZO-1, ZO-2, and ZO-3, which form intracellular plaques that join transmembrane proteins with the actin cytoskeleton at tight junctions (13). ZO-2 was found to facilitate nuclear localization of YAP and specifically was involved in regulating the pro-apoptotic function of YAP2 (14).
Second, we chose to investigate the protein–protein interface of PD-1 and PD-L1, the immune checkpoint complex used by some cancer cells to evade immune recognition. Despite the success of immune checkpoint inhibitors, there remain several problems associated with antibody therapeutics. First, the large size of mAb therapeutics can lead to poor tissue penetration, especially considering the high cell densities and constricted blood and lymphatic vessels in many dense tumors. This limits access to the type of PD-L1–expressing tumors that respond well to checkpoint inhibition (15). Additionally, mAb therapeutics cannot be dosed orally (16), and difficulty in manufacturing/validation make mAbs harder to produce than small molecules (17). Side effects can also include detrimental immune responses. Therefore, small molecules or peptides may represent the next generation of checkpoint inhibitors (18). Crystallographic studies have shown that the PD-1/PD-L1 interface is large and featureless, making targeted drug discovery using small molecules relatively difficult (19). Identification of hot spot regions in this interface may help guide further small-molecule drug discovery efforts toward particular regions essential for complex formation. Taken together, these two case studies demonstrate the use of protein painting to define solvent-inaccessible, protein-interaction hot spots in clinically relevant complexes without a dependence on crystallographic structural information.
Results
Development and optimization of azo protein dyes produces a molecular dye, Fast Blue B + naphthionic acid (FBBNA), with high-affinity binding
To identify motifs common to high-affinity dyes, we screened a series of small dyes with the model proteins thyroglobulin and lysozyme to compare the number of bound dye molecules per protein and identified differences in the maximum number of bound molecules at equilibrium between different dye classes. In our previous work, we identified from a screen of small-molecule dyes a total of four compounds with suitably low off-rates and high protein binding that could be used in protein painting: Acid Orange 50 (AO50; CAS 10214-07-0), Reactive Blue 19 (CAS 2580-78-1), Red 49 (CAS 5517-38-4), and Congo Red (CAS 573-58-0) (5). These dyes were validated using the model protein carbonic anhydrase, a small 29-kDa protein, and of these, AO50 proved to be the most practical as its solutions were stable over time at room temperature, and the dye was not subject to self-aggregation like Congo Red. To identify new candidate dye moieties most highly correlated to successful protein binding, an iterative screen of small molecules of different classes was conducted using the model proteins lysozyme and thyroglobulin, representing very small (lysozyme = 14 kDa) and very large (thyroglobulin = 660 kDa) proteins, and these results were compared with our previously identified model dye, AO50. Each small molecule's equilibrium binding was examined under saturating dye conditions (100-fold molar excess) to account for binding to high and low affinity sites on the protein's surface. For the first screen, we choose two different classes of dyes to compare with AO50, a halogenated small-molecule azo dye. Representing the diazo class of molecular dyes is trypan blue (TB; CAS 72-57-1), a well-known dye used in microscopy, and representing the phthalocyanine class of molecular dyes is Direct Blue 199 (DB199, CAS 12222-04-7), a water-soluble dye known to bind carbohydrates, which we have previously validated binds to thyroglobulin via EM (Fig. S1).
As shown in Fig. 2, examination of these three dyes with lysozyme and thyroglobulin revealed a few key insights into optimization of dye binding. First, we examined the metallodye Direct Blue 199 to determine whether metal ions might be an important mediator of protein binding for this dye class. Because the copper moiety of some phthalocyanine-based dyes has been shown to be essential for carbohydrate binding (20), we wondered whether these metal ions also had a strong effect upon protein binding. If so, this could provide a lead into tuning protein binding properties based on incorporated metal and could provide new insights into mechanisms of protein binding. However, when Direct Blue 199 binding was compared both in the absence and presence of copper sulfate, the number of molecules bound per thyroglobulin molecule was unchanged. Because copper sulfate was unable to reduce Direct Blue 199 binding through competition, we propose that Direct Blue 199 binding is not primarily mediated through the copper ion. Additionally, DB199 binding depended on the age of the dye; after 1 week, significantly lower binding was observed. Preparations of DB199 were therefore prepared fresh every morning prior to experimentation. We also were unable to detect significant Direct Blue 199 binding to lysozyme. Acid Orange 50 conversely was able to bind both lysozyme and thyroglobulin, with a comparable number of dye molecules bound to thyroglobulin at equilibrium as DB199. Of the molecules tested in Fig. 2, TB bound with the fewest molecules at equilibrium to thyroglobulin and was unable to bind to lysozyme. Unlike DB199, no reduction in binding over time was noted for either TB or AO50 as the dye aged. TB is the most hydrophilic of the first cohort of commercially available dyes tested, and this hydrophilicity may reduce binding by rendering hydrophobic pockets on the protein inaccessible to the dye.
Figure 2.

Binding of commercially available molecular dyes reveals differences between dye classes. A, structure of commercially available dyes DB199, AO50, and TB. B, DB199 binds to thyroglobulin, but not to lysozyme. Binding to thyroglobulin in the absence of CuSO4 (15 molecules at equilibrium) is not significantly greater than binding in the presence of CuSO4 (16 molecules at equilibrium) in a competition assay, indicating the copper ion of DB199 is not primarily responsible for binding affinity. Because the copper moiety of phthalocyanine dyes has been shown to be essential for binding to carbohydrates, lack of involvement suggests that binding to proteins occurs through different mechanisms than binding to carbohydrates. C, Acid Orange 50 binds to both thyroglobulin (15 molecules at equilibrium) and lysozyme (one molecule at equilibrium). D, trypan blue binds to thyroglobulin, but not to lysozyme. Binding of trypan blue (three molecules at equilibrium) is reduced compared with either DB199 or AO50. All data points were collected in duplicate, and final data were fit to the Michaelis–Menten equation, where Vmax represents the maximum number of bound dye molecules per protein molecule at equilibrium. Error bars, S.D.
To further explore the binding hypotheses raised by examination of DB199, AO50, and TB, a small series of dye analogs were generated, which revealed that dye binding to the model protein thyroglobulin had a strong dependence on both size and hydrophobicity. Because both TB and AO50 contain an azo linkage, further optimization of dyes could utilize azo coupling reactions, where electrophilic aryl diazonium salts couple to activated aromatic compounds, such as anilines or phenols, favoring the para position if available and the ortho position if not. Aryl diazonium compounds stabilized with salts such as ZnCl2 are commercially available as “fast dyes,” such that diazotization does not have to be completed in-house. This chemistry has the advantage of compatibility with aqueous solvents and can be completed in one step. Additionally, reaction progress is easy to monitor as color develops when the dye is synthesized.
Two different fast dyes were used to synthesize new dye candidates. Fast Blue B was chosen to synthesize new diazo dyes, as it is structurally similar to the core of TB and would test “larger” dye molecules, whereas Fast Red TR was halogenated like the core of AO50 and was chosen to synthesize “smaller” dyes. Four diverse coupling agents, 4-amino-1-naphthalenesulfonic acid or naphthionic acid, 4-amino-5-hydroxy-2,7-naphthalenedisulfonic acid or H-acid, 7-amino-1,3-naphthalenedisulfonic acid or Amino G acid, and 2-napthol, were chosen to evaluate the effect of hydrophobicity, primarily through sulfonate groups, on dye binding. These compounds are common dye components, with naphthionic acid a component of Congo Red, H-acid a component of trypan blue and Direct Blue 15, and 2-napthol a component of the Sudan class of lipid-soluble dyes. The dye candidates were synthesized at room temperature in PBS, pH 7.4, with the exception that dyes prepared with H-acid or 2-napthol were synthesized at basic pH to deprotonate the alcohol group. The structures of each of the fast dyes and coupling agents are presented in Table 1.
Table 1.
Structures of candidate dyes based on AO50 and TB
Diazonium coupling agents Fast Blue B and Fast Red TR were each coupled to one of four naphthalene coupling agents. Binding to thyroglobulin was only measurable for Fast Blue B + naphthionic acid (FBBNA) and Fast Blue B + H-acid (FBBHA).
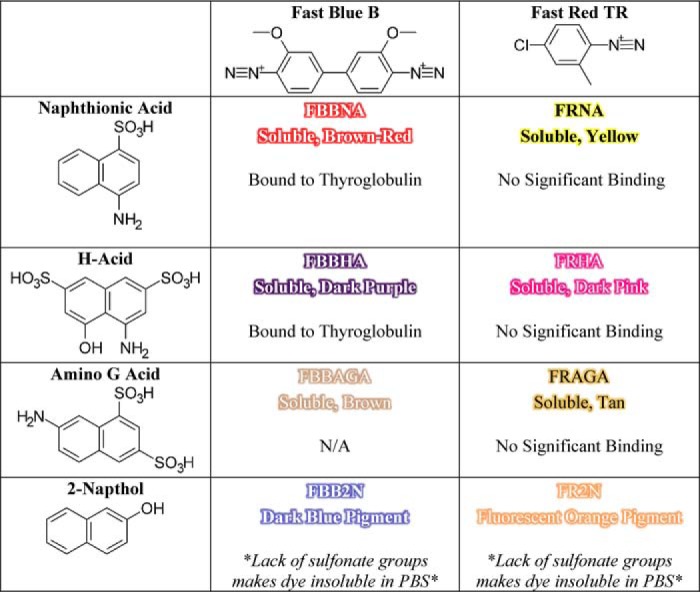
During synthesis, it became clear that the coupling agent lacking a sulfonate moiety, 2-napthol, led to the production of insoluble pigments rather than soluble dyes. Additionally, the unbound dye product of Fast Blue B + Amino G acid could not be removed from a painted protein by Sephadex gel filtration, eliminating it from consideration as a protein-painting dye candidate. The remaining five candidate dyes, FBBNA, Fast Blue B + H-acid (FBBHA), Fast Red TR + naphthionic acid, Fast Red TR + H-acid, and Fast Red TR + Amino G acid, were all evaluated for the number of bound dye molecules per molecule of thyroglobulin as presented in Fig. 3. None of the Fast Red TR-based dyes, including Fast Red TR + naphthionic acid, Fast Red TR + H-acid, and Fast Red TR + Amino G acid, showed any significant binding to thyroglobulin. Both FBBNA (Fig. 3A) and FBBHA (Fig. 3B) were shown to bind to thyroglobulin, where FBBHA had a similar magnitude of bound molecules as TB at equilibrium, which was anticipated based on the structural similarity of TB versus FBBHA. FBBNA, however, had an unexpectedly high number of bound molecules to thyroglobulin at equilibrium, with an almost 10-fold greater number than FBBHA, and was chosen for further investigation. It was hypothesized that the reduced hydrophilicity of naphthionic acid versus H-acid may explain the increased binding of FBBNA versus FBBHA.
Figure 3.

Binding of candidate dyes FBBNA and FBBHA reveals significant difference in binding based on hydrophobicity. A, binding of FBBNA to thyroglobulin shows an average of 57 molecules bound to each thyroglobulin molecule at equilibrium. B, binding of FBBHA to thyroglobulin shows an average of six molecules bound to each thyroglobulin molecule at equilibrium, roughly 10 times fewer than FBBNA. All data points were collected in duplicate and fit to the Michaelis–Menten equation, where Vmax represents the maximum number of bound dye molecules per protein molecule at equilibrium. Error bars, S.D.
To explore this hypothesis, Fast Blue B was coupled to three additional isomers of naphthionic acid to investigate the effect of the position of the sulfonate moiety on binding, which revealed that reducing the hydrophobicity of the “ends” of the dye molecule severely reduced the number of bound molecules. Three additional analogs of FBBNA were prepared using the coupling agents 1-amino-8-naphthalenesulfonic acid (Peri acid), 5-amino-1-naphthalenesulfonic acid (Laurent acid), and 1-naphthylamine-6-sulfonic acid (1,6-Cleve's acid, or Cleve acid) coupled to Fast Blue B. The structures of these coupling agents are shown in Table 2. All of the synthesized dyes were soluble and of similar colors. We hypothesized that reducing the hydrophobic area of the coupling agent, which we called the hydrophobic “clamp” region, would reduce the binding of the dye candidate. The “hydrophobic clamp” region was reduced in Peri acid, Laurent acid, and Cleve acid as compared with naphthionic acid because the sulfonate moiety is coupled to the opposite ring as the amine group. We hypothesized that FBBNA would therefore bind in greater numbers to thyroglobulin than FBBPA, FBBLA, or FBBCA.
Table 2.
Structures of candidate dyes based on FBBNA
Diazonium coupling agent Fast Blue B was coupled to each of the four structural isomers of naphthionic acid. Binding to thyroglobulin was measurable for all four dyes. For each coupling agent, potential sites of coupling to Fast Blue B are marked with a red star, and the “hydrophobic clamp” region is marked with an orange box.
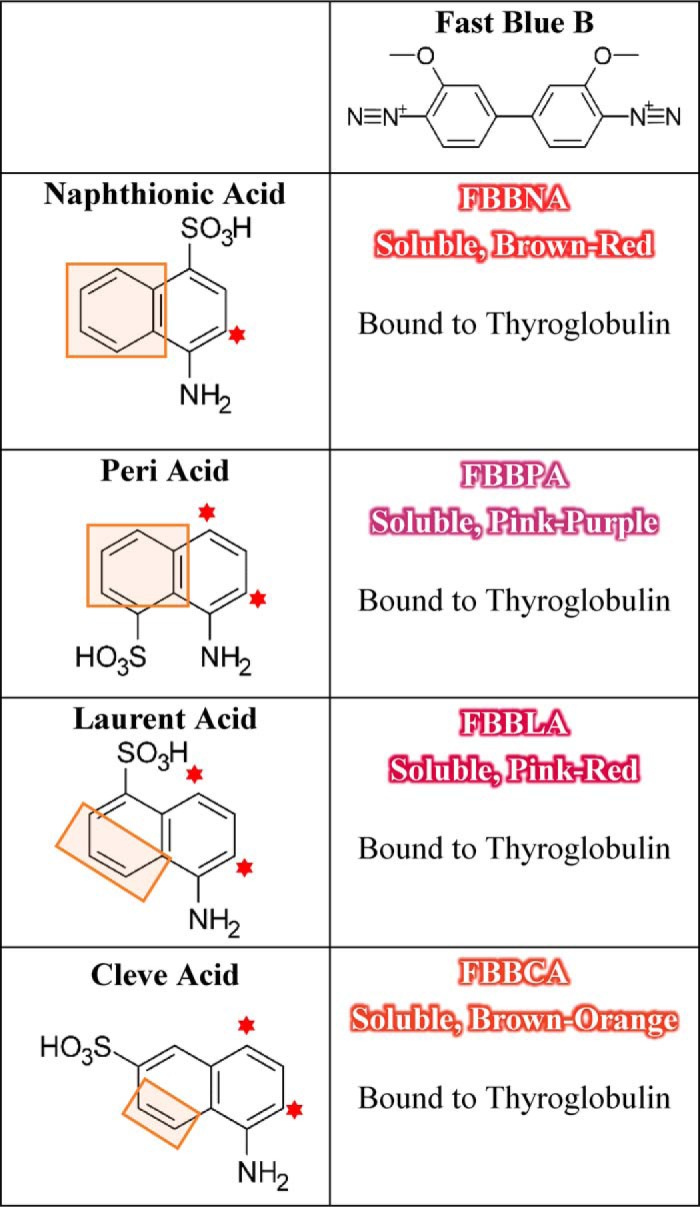
As shown in Fig. 4, FBBNA retained the highest number of bound molecules of the four candidate dyes tested, whereas FBBCA had the lowest number of bound molecules, and FBBPA and FBBLA bound at numbers between FBBNA and FBBCA. This supports the hypothesis that not only is a hydrophobic clamp important for binding to hydrophobic pockets of the protein, but the presence of an unobstructed aryl ring such as is found on FBBNA may allow for tighter π-stacking interactions with aromatic amino acids. This is supported by the reduction in the number of bound molecules seen for FBBPA, because the sulfonate group and amine group of Peri acid are substituted to opposite aryl rings of naphthalene.
Figure 4.

Binding of candidate dye FBBNA compared with FBBPA, FBBLA, and FBBCA reveals the importance of the hydrophobic anchor region. As the hydrophobic portion of each coupling agent is reduced, the number of bound molecules of the corresponding dye is reduced. All data points were collected in duplicate and fit to the Michaelis–Menten equation, where Vmax represents maximum number of bound molecules at equilibrium. FBBNA had the highest number of molecules bound at equilibrium of the four candidate dyes tested. A, binding of FBBNA to thyroglobulin shows an average of 57 molecules bound to each thyroglobulin molecule at equilibrium, as determined previously and shown in Fig. 3A. The graph is reproduced in this figure for direct comparison with B, C, and D. B, binding of FBBPA to thyroglobulin shows an average of 37 molecules bound to each thyroglobulin molecule at equilibrium. C, binding of FBBLA to thyroglobulin shows an average of 32 molecules bound to each thyroglobulin molecule at equilibrium. D, binding of FBBCA to thyroglobulin shows an average of seven molecules bound to each thyroglobulin molecule at equilibrium. Error bars, S.D.
Molecular dye mixture FBBNA binds to proteins of different surface areas and remains significantly bound in the presence of heat treatment
Given the high number of FBBNA molecules bound to thyroglobulin, we investigated the binding of FBBNA to a range of other proteins and found that the number of bound dye molecules was linearly dependent on the surface area of the protein. For additional examination of FBBNA and its binding properties, we assembled a small series of standard proteins to be used in subsequent experiments to better understand FBBNA's properties. Given in Table 3 are the six standard proteins examined with their molecular weight, Stokes radius, and calculated surface area given. Additionally, a short description of the experiments conducted for each protein is given for reference. First, each of the proteins was examined for the number of bound FBBNA or AO50 molecules at equilibrium to evaluate the generalizability of the thyroglobulin-binding analysis. Fig. 5A shows that the number of bound molecules of FBBNA at 2 h for each protein follows a linear surface area–dependent trend. In contrast, whereas the number of bound Acid Orange 50 molecules was also dependent on surface area, this correlation was not exact (R2 = 0.99 for FBBNA and 0.74 for AO50), where BSA was a significant outlier with a higher number of A050 molecules bound than predicted. This confirms that FBBNA binds in higher numbers than AO50 for several given proteins and may be a good candidate as a next-generation protein-painting dye. Because some proteins, such as apoferritin, are particularly stable and may need to be subjected to high temperatures to fully denature prior to trypsinization and MS, it is important that a protein-painting dye remain bound despite such treatment. To test FBBNA's binding properties at high temperature, the number of FBBNA molecules bound to apoferritin was examined following heat denaturation of the protein. FBBNA remains mostly bound to apoferritin following denaturation of the protein at 100 °C for 10 min, as shown in Fig. 5B. Whereas apoferritin has an unusually high denaturation point, greater than 93 °C at pH 7 (21), the residual dye bound after such treatment indicates that significant dye can remain bound upon thermal denaturation.
Table 3.
Standard set of proteins of diverse sizes used to examine candidate dye binding
| Protein name | Uniprot number | Species | Complex mass | pI | Stokes radius | Surface area | Reference | Experiments conducted with protein |
|---|---|---|---|---|---|---|---|---|
| kDa | nm | nm2 | ||||||
| Lysozyme (LY) | B8YK79 | Gallus gallus | 16 (monomer) | 9.36 | 1.89 | 44.89 | 53 | Binding, Docking |
| Smaller size enables docking | ||||||||
| Carbonic anhydrase (CA) | P00921 | Bos taurus | 29 (monomer) | 6.41 | 2.35 | 69.40 | 54 | Binding, Docking |
| Smaller size enables docking | ||||||||
| BSA | P02769 | B. taurus | 69 (monomer) | 5.82 | 3.55 | 158.37 | 55 | Binding, CD, Docking |
| Smaller size enables docking, standard protein used for CD | ||||||||
| Catalase (CT) | P00432 | B. taurus | 240 (tetramer) | 6.79 | 5.20 | 339.79 | 55 | Binding |
| Apoferritin (AF) | P02791 Q8MIP0 | Equus caballus | 479 (24-mer) | 5.37 | 6.10 | 467.59 | 55 | Binding, Denaturation |
| Denatures at very high temperatures | ||||||||
| Thyroglobulin (TG) | P01267 | B. taurus | 660 (dimer) | 5.48 | 8.58 | 925.09 | 22 | Binding |
| Large size results in good signal/noise ratio for binding experiments |
Figure 5.

FBBNA binds to multiple different proteins in a surface area–dependent manner, remains significantly bound even after heat treatment, and binds to tyrosine residues in the absence of other positively charged residues. A, FBBNA binds to multiple proteins in a surface area–dependent manner (R2 = 0.99). Proteins examined include lysozyme (LY), carbonic anhydrase (CA), BSA, catalase (CT), apoferritin (AF), and thyroglobulin (TG) as described in Table 3. Number of bound FBBNA molecules is not significantly affected by the presence of glycosylation (thyroglobulin) or metallo-binding prosthetic groups (catalase) for this sample set. The number of bound AO50 molecules was not as highly correlated with surface area (R2 = 0.74) as FBBNA, with a particularly high number of AO50 molecules binding to BSA. Except for BSA and lysozyme, FBBNA bound in higher numbers to the proteins tested than did AO50. B, the percentage of FBBNA dye bound to apoferritin decreased only slightly upon heating at 100 °C for 10 min. Dye was allowed to bind to apoferritin, excess dye was removed by gel filtration, and then native samples were incubated at room temperature for 10 min, whereas heat denatured samples were incubated at 100 °C for 10 min. Samples were subsequently passed through a second gel filtration column, and binding was compared. C, FBBNA dot-blot dye binding to poly-amino acids reveals that FBBNA has affinity not only for positively charged samples, but also for poly-Tyr as well as poly-Tyr/Glu, emphasizing the importance of non-salt-bridge interactions. Each bar represents the spot intensity for 12 individual spots as calculated by ImageJ, with one sample dot blot shown for reference. D, AO50 dot-blot dye binding to poly-amino acids reveals that AO50 has affinity only for poly-amino acid samples that contain positively charged residues, suggesting that salt bridge formation is essential for AO50 binding. Each bar represents the spot intensity for 12 individual spots as calculated by ImageJ, with one sample dot blot shown for reference. Error bars, S.D.
Amino acid affinity of FBBNA versus AO50
We further supported the hypothesis that FBBNA binds to proteins partially through interactions with aryl residues by examining the amino acid affinity of FBBNA and found that it has affinity for tyrosine residues. Based on the affinity of FBBNA for a wide variety of proteins and the evidence that the hydrophobic component of FBBNA is important for binding, we examined the affinity for both FBBNA and AO50 for different poly-amino acids. We hypothesized that both FBBNA and AO50 would have affinity for positively charged amino acids such as lysine through salt bridge interactions between the amino groups of polylysine and the sulfonate groups of FBBNA or AO50. If the hydrophobic clamp of FBBNA is important for binding to proteins, it was hypothesized that the dye must have affinity for aryl residues as well as positively charged ones. To test this hypothesis, we compared the binding affinity of both FBBNA and AO50 to both positively charged polymers (polylysine), as well as neutral poly-glutamic acid and aromatic polytyrosine and polymers of lysine with either tyrosine or phenylalanine. As shown in Fig. 5C, FBBNA bound not only to polylysine, but also to polytyrosine. Whereas FBBNA did not bind to poly-glutamic acid, it did bind to a 1:1 polymer of glutamic acid to tyrosine, showing the importance of tyrosine binding. Moreover, a 1:1 polymer of lysine and tyrosine showed the highest binding capacity of the polymers tested with FBBNA. Interestingly, the 1:1 polymer of lysine to phenylalanine did not show increased binding over lysine alone, indicating that whereas FBBNA has affinity for tyrosine residues, it does not have similar affinity for phenylalanine residues. These data suggest that additional interactions, such as hydrogen bonding to tyrosine hydroxyl groups, may be important to explain these interactions. Conversely, AO50 was unable to bind to any polymer that did not contain lysine, as shown in Fig. 5D, suggesting that salt bridge interactions are potentially required for significant AO50 binding. The differences between these two binding mechanisms suggest that there may be cases where the two dyes could act in conjunction to bind different kinds of protein topologies and could potentially be used in tandem to increase protein dye coverage.
Based on the estimated importance of positive residues and tyrosine residues for FBBNA or AO50 binding, we examined the frequency of positive amino acids and tyrosine residues in our set of six standard proteins and compared this with the distribution of these amino acids in vertebrate proteins. As given in Fig. S2, we see that percentages of positive residues and tyrosine residues are fairly consistent between proteins, ranging from 9.21 to 14.7% positive residues and from 2.04 to 3.64% tyrosine residues. This falls well in line with the expected values from vertebrate proteins, which average 11.4% positive residues and 3.3% tyrosine residues (22).
Determination of the structure and binding properties of the component compounds of FBBNA reveals the importance of hydrophobic clamp for protein binding
We determined the structure of FBBNA to verify the expected coupling of Fast Blue B to naphthionic acid. We identified a side product consisting of one molecule of Fast Blue B coupled to only one naphthionic acid group, which displayed severely reduced binding to thyroglobulin. Naphthionic acid can be represented by several resonance structures that lead to a partial negative charge on three positions of the naphthalene ring. The classical “activated” position is the ortho position to the primary amine, which is where naphthionic acid is assumed to couple to an aryl diazonium compound as in the case of the dye Congo Red (23). However, side products consisting of compounds coupling at alternative positions may also represent unique, high-affinity dyes; they also provide a way to compare small changes in structure to protein-binding properties. Additionally, partial products or residual starting material may interfere with binding analysis. Therefore, we purified FBBNA dye and determined its structure via NMR.
To isolate products of the reaction of Fast Blue B with naphthionic acid, synthesized FBBNA dye was examined via reverse-phase TLC. Using a mobile phase of 60% MeOH, two primary bands were observed at Rf = 0.32 (orange-colored band, FBBNA-ORANGE) and Rf = 0.61 (pink-colored band, FBBNA-PINK). These bands were separated via reverse-phase flash chromatography, and each of the two compounds was examined for its protein binding capability as shown in Fig. 6. The number of FBBNA-PINK molecules bound to thyroglobulin was twice that of FBBNA-ORANGE to thyroglobulin, indicating that the pink band was the higher-affinity species in the FBBNA dye. However, as the total binding of the two compounds was roughly equal to the number of bound molecules of the complex unpurified FBBNA dye to thyroglobulin, we can hypothesize that these two species bind to unique sites on thyroglobulin. The structures of both the pink band and the orange band were determined via proton NMR, shown in Fig. 6C. The FBBNA-PINK band was the expected primary product, in which Fast Blue B couples to two molecules of naphthionic acid at the ortho position to the activating amine group. FBBNA-ORANGE was an incomplete reaction in which Fast Blue B only coupled to one molecule of naphthionic acid, although coupling still took place at the ortho position to the activating amine on naphthionic acid. The most common decomposition reaction of Fast Blue B is loss of a diazonium group as N2, which indicates that FBBNA-ORANGE is likely the dye product of naphthionic acid and a partially degraded Fast Blue B molecule. What is particularly notable is the severe reduction in binding when Fast Blue B is coupled to only one molecule of naphthionic acid as compared with two. The number of bound molecules was reduced by roughly 50% when thyroglobulin was painted with FBBNA-ORANGE versus FBBNA-PINK. This again highlights the extreme importance of the hydrophobic clamp region of the dye, where the naphthionic acid molecule can be thought to “dig into” the protein structure to interact with hydrophobic or aryl residues and keep the dye bound to the protein.
Figure 6.

Binding of each component of candidate dye FBBNA reveals that >50% of binding is attributable to the hydrophobic anchor region. A, a total of 48 molecules of the high-retention factor component of FBBNA, pink in color, bound to each molecule of thyroglobulin. B, a total of 14 molecules of the low-retention factor component of FBBNA, orange in color, bound to each molecule of thyroglobulin. C, structures of FBBNA-PINK and FBBNA-ORANGE reveal that FBBNA-ORANGE has one fewer coupling agent than FBBNA-PINK. Error bars, S.D.
We docked the structures of FBBNA-PINK and AO50 into the structures of lysozyme, carbonic anhydrase, and BSA to compare predicted binding locations and found that both dyes were predicted to bind in similar sites for each protein but differed in their binding affinity. To examine predicted binding affinity and binding locations, we used the SwissDock web server (24, 25). As shown in Fig. S3, both AO50 and FBBNA are predicted to bind to similar locations on lysozyme, carbonic anhydrase, or BSA based on the top docking positions. Each docked position of the dye was associated with a separate change in predicted Gibbs free energy of binding to the protein. Whereas individual dye molecules binding to a protein will all have different binding affinities, with high-affinity sites being occupied prior to low-affinity sites, we decided to compare the average Gibbs free energy change for a given molecule to a protein using the predictions from our docking studies. When we averaged the predicted change in Gibbs free energy of binding for each of the top 250 docked positions of a dye to a protein, we found that the predicted average change in Gibbs free energy was significantly different (p < 0.0001) for FBBNA binding versus AO50 binding for each protein. Lysozyme (average ΔG = −8.06 kcal/mol for FBBNA binding versus ΔG = −6.97 kcal/mol for AO50 binding), carbonic anhydrase (average ΔG = −9.13 kcal/mol for FBBNA binding versus ΔG = −8.03 kcal/mol for AO50 binding), and BSA (average ΔG = −9.50 kcal/mol for FBBNA binding versus ΔG = −8.33 kcal/mol for AO50 binding) all showed significant differences. These data suggest that whereas there may be individual AO50 molecules that bind to high-affinity sites on a protein versus individual FBBNA molecules that bind in lower-affinity sites, the binding affinity of FBBNA on average is higher than that of AO50.
For comparison, we also determined the average ΔG value for FBBNA binding to thyroglobulin by experimentally determining the average dissociation constant of FBBNA binding to thyroglobulin and using this experimental value to calculate an average ΔG for FBBNA binding to thyroglobulin. Whereas this average dissociation constant does not describe the dissociation of any one particular molecule of FBBNA from thyroglobulin, it represents an estimated affinity if all bound FBBNA molecules are treated as a cohort. As given in Fig. S4, the average Kd value for FBBNA dissociation from thyroglobulin is 1.36 × 10−7 m, giving an experimentally determined ΔG = −9.42 kcal/mol. This value is similar to the average ΔG values predicted from the top docked positions using SwissDock for lysozyme, carbonic anhydrase, and BSA.
Binding of molecular dyes AO50 and FBBNA protects bound regions of proteins from denaturation by chemotropic agent urea
To further characterize whether FBBNA is a suitable next-generation protein-painting dye, we investigated FBBNA's effect on protein structure in the presence and absence of urea and found that it stabilizes protein secondary structure. We performed CD spectroscopy on native and denatured samples of BSA in the presence and absence of FBBNA to determine the extent of residual secondary structure. BSA has been used as a standard protein in CD because of its strong signal in the far ultraviolet due to its α-helical nature combined with its availability (26). In Fig. 7A, we observed that upon treatment with FBBNA for 10 min, BSA was partially stabilized against denaturation at 2, 4, and 6 m urea. Additionally, the presence of FBBNA did not affect the secondary structure of BSA, confirming its utility for protein-painting analysis. We additionally examined the effect of FBBNA binding on protein denaturation compared with AO50 as well as in conjunction with AO50. Shown in Fig. 7B, both AO50 and FBBNA both were able to partially stabilize BSA against denaturation and were able to do so in conjunction as well. Based on Fig. 5 (C and D), suggesting that FBBNA and AO50 bind with different amino acid specificities, and based on Fig. 7B, suggesting that the two dyes can be used in conjunction without interfering with the protection provided by either, it may be prudent to combine the two dyes for protein painting. We have previously observed that coverage from multiple dyes can be additive (5). Furthermore, the discovery that both dyes offer some protection at 4 m urea in addition to 2 m urea, the concentration employed in our protein-painting protocol, suggests that there may be situations in which the protocol could be modified to utilize a 4 m denaturation step. Both a 4 m denaturation step or a high-heat denaturation step (Fig. 5B) could represent future alternatives to expand protein painting to difficult-to-denature proteins. Additionally, these data shed light on the question of how the protein-painting dyes are able to remain bound to proteins during denaturation; they selectively protect regions of the protein from denaturation.
Figure 7.

CD spectroscopy of BSA in the presence of molecular dyes shows stabilizing effect of FBBNA and AO50 in the presence of chaotropic agent urea. A, native secondary structure of BSA (black solid line) does not change in the presence of FBBNA dye (gray dashed line). Denaturation at 2 m (green solid line), 4 m (red solid line), and 6 m (blue solid line) is attenuated in the presence of FBBNA at 2 m (green dashed line), 4 m (red dashed line), and 6 m (blue dashed line) as measured by an increase in helical content of the CD spectra. B, native secondary structure of BSA (black solid line) does not change in the presence of AO50 dye (orange dotted line) or the combination of AO50 and FBBNA (purple dotted line). Denaturation by 4 m urea is attenuated by FBBNA (red dashed line), AO50 (orange dashed line), and FBBNA + AO50 (purple dashed line) as compared with BSA denatured without dye (red solid line). C, mean residue ellipticity at 222 nm was greater for BSA in the presence of FBBNA dye as compared with BSA alone at all urea concentrations tested. The greatest difference in magnitude was observed at 4 m urea.
Protein painting of Hippo signaling pathway proteins using optimized dye FBBNA predicts hot spots for further probe development
We validated our next-generation protein-painting dye, FBBNA, by painting the complex of YAP2/ZO-1 and identifying not only the canonical binding site, but also additional unreported interactions. Interactions between ZO proteins and YAP2 are facilitated by interaction between the first PDZ domain of ZO proteins and the C-terminal PDZ-binding domain of YAP2, the pentapeptide -FLTWL (14, 27). Given the paucity of crystal structures of YAP with protein-binding partners (28), likely due in part to YAP's unstructured behavior in solution, and the utility of protein painting in the absence of crystallographic information, we utilized protein painting to probe the YAP2/ZO-1 interface. We utilized FBBNA in conjunction with AO50 as the protein-painting dye of choice.
We found two hot spots of ZO-1 (isoform a, NP_003248.3), in complex with YAP2 (YAP 1–2 γ, NP_001123617.1), shown in Fig. 8. Tandem mass spectra of each hot spot of ZO-1 are given in Fig. S5, whereas mass spectra of control peptides are given in Fig. S6. To identify hot spot peptides, the identified peptides from an unpainted, painted, and painted complexed sample are compared; those peptides found in both the unpainted cohort and the complexed cohort, but absent in the painted cohort, constitute hot spot peptide hits. This analysis is shown for one trial in Fig. S7A; each experiment was conducted in triplicate with two technical replicates per trial. Fisher's exact test (Freeman–Halton extension; Fig. S7B) was used to determine whether identification of the hot spot peptides was statistically significant. Both identified hot spot peptides are highly conserved in evolution (Fig. S8). The first hot spot, Arg-42, was found within the first PDZ domain of ZO-1, the canonical YAP recognition site. Whereas the first PDZ domain of ZO-1 has not been crystalized with the C-terminal binding domain of YAP, structures of the first PDZ domain of ZO-1 in complex with other C-terminal peptides are available. In Fig. S9, ZO-1 PDZ-1 bound to heptapeptide ligand WRRTTYL (PDB code 2H2B (29)) shows that Arg-42 is bound in close proximity to the peptide-binding cleft. In addition to this hot spot in the known binding domain, we found one additional hot spot at Lys-592 directly between the SH3 domain and guanylate kinase–like domain of ZO-1, within the documented calmodulin-binding site. Due to the large number of binding partners of tight junction proteins, it is conceivable that multiple binding partners of ZO proteins could have overlapping binding sites, such that binding of two partners to ZO proteins is mutually exclusive. Based on early protein-painting data, it is possible that YAP binding and calmodulin binding to ZO-1 are mutually exclusive; additional research into these regulatory questions is ongoing.
Figure 8.

Hot spots identified in the ZO-1/YAP2 complex by protein painting include sites both within the canonical binding site and in novel interacting domains. A, ZO-1 is shown in light gray with PDZ domains highlighted red, SH3 domain highlighted orange, guanylate kinase-like domain highlighted in yellow, and ZU5 domain highlighted in brown. A selection of known binding partners are shown as tabs above the structure, with their binding regions given as amino acid positions in ZO-1. All hot spots, given in red, were identified in three independent protein-painting experiments. Arg-42 is a hot spot in the known binding site. B, YAP2 is shown in gray with WW domains highlighted red, SH3-binding domain highlighted orange, transcriptional activation domain highlighted in yellow, and PDZ-binding domain highlighted in green. A selection of known binding partners are shown as tabs above the structure with their binding regions given as amino acid positions in YAP2. All hot spots were identified in three independent protein-painting experiments and were found in the N-terminal proline-rich domain.
In conjunction with the two hot spots identified on ZO-1, three hot spots were identified on YAP2, as shown in Fig. 8. Sequence coverage of the C-terminal region of YAP via MS was suboptimal; no coverage was obtained from the region of the protein containing the canonical ZO-1–binding domain, residues 441–504, due to lack of tryptic cleavage sites. Tandem mass spectra of each hot spot of YAP2 are given in Fig. S10, whereas mass spectra of control peptides are given in Fig. S11. Analysis for one trial is given in Fig. S12A; each experiment was conducted in triplicate with two technical replicates per trial. Fisher's exact test (Freeman–Halton extension; Fig. S12B) was used to determine whether identification of the hot spot peptides was statistically significant. Interestingly, the three hot spots obtained, Arg-89, Arg-161, and Arg-187, were not in highly evolutionarily conserved regions. The first two hot spots were found in the relatively unconserved, proline-rich N-terminal region of the protein known to bind TEAD. Arg-187 is found in the first WW domain of the protein known to bind LATS as well as Patched (7, 30). Sequence alignment given in Fig. S13 shows the unconserved N-terminal region as well as the location of the three identified hot spot peptides. It is tempting to suppose that the Lys-592 hot spot identified near the SH3 domain of ZO-1 and the Arg-89 hot spot identified in YAP (hot spot peptide KLPDSFFKPPEPK) could indicate some interaction between the SH3 domain and its recognition motif PXXP in the Arg-89 peptide, although the presence of a glutamic acid residue within the PXXP motif is unusual, as the SH3-binding cavity is fairly hydrophobic (31). Regardless, the presence of multiple hot spots within the N-terminal region of YAP is intriguing and suggests that there may be more to this region of the protein than previously supposed based on the limited conservation.
Painting of the PD-1/PD-L1 complex correctly identifies the known binding interface, and interface peptides prohibit formation of the complex
In addition to the ZO-1/YAP2 complex, protein painting was also used as a tool to investigate the binding of human PD-1 and PD-L1, and peptides based on the identified protein-painting hit successfully disrupted complex formation. PD-1 and PD-L1 are two targets of immune checkpoint inhibitors, an overwhelmingly successful class of antibody cancer therapies. There have been very few reports of small-molecule inhibitors targeting the PD-1/PD-L1 interface; reports of Bristol Meyers Squibb compounds targeting the interface showed high cytotoxicity and poor efficacy (32). We proposed the use of new protein-painting dyes to first identify hot spot regions within the crystallographic PD-1/PD-L1 interface and then use the identified peptide regions as probe molecules to disrupt complex formation. Peptides that effectively disrupted complex formation could be useful lead molecules for further optimization and subsequent peptidomimetic generation to aid in development of small-molecule inhibitors of PD-1/PD-L1. As with the Hippo pathway proteins YAP and ZO, painting of PD-L1 and PD-1 was conducted with a mixture of FBBNA and AO50. However, despite confirmation of FBBNA binding to PD-1 (three molecules of FBBNA bound to PD-1 at equilibrium), no difference was observed in the MS coverage between PD-1 painted with FBBNA and AO50 and a sample of unpainted PD-1 alone. In addition to protein surfaces, we have seen that FBBNA binds tightly to some carbohydrates as well. In Fig. S14, we show that FBBNA shows a stronger affinity for cotton and rayon than silk, suggesting that interactions with sugars could be significant, whereas AO50 displayed no such affinity. We hypothesized that the glycosylation of PD-1 could have an effect on the protein-painting procedure, yet pretreatment of PD-1 with peptide:N-glycosidase F did not facilitate significant reduction in sequence coverage in the presence of FBBNA and AO50 in tandem. However, painting PD-1 with AO50 alone, led to a reduction in sequence coverage via MS as expected and allowed for painting of the PD-1/PD-L1 complex. In Fig. 9, we show that the identified hot spot region, Lys-78 of PD-1, falls within the Ig-like V-type fold of PD-1, the known binding site of PD-L1 (19, 33). Tandem mass spectra of the hot spot of PD-1 are given in Fig. S15, whereas mass spectra of control peptides are given in Fig. S16. Analysis for one trial is given in Fig. S17A.
Figure 9.

Hot spot in the PD-1/PD-L1 complex as identified by protein painting falls in the crystallographic binding site, and inhibitors designed around this hot spot disrupt complex formation. A, PD-1 is shown in light gray at the top, whereas PD-L1 is shown in dark gray at the bottom. Domains are marked directly on the protein. Known binding partners are shown as tabs above the structure with their binding regions given as amino acid positions. All hot spots were identified in three independent protein-painting experiments. B, inhibitors 1–8 were tested over a concentration range of 10 nm to 100 μm for inhibition of protein–protein interactions between PD-1 and PD-L1. The most effective inhibitors were 1 and 2, designed to mimic PD-L1, and had IC50 values < 10 μm. All data points were collected in duplicate, and data were fit to a sigmoidal dose–response curve with the following constraint: baseline ≥ 0. C, crystal structure of human PD-1/PD-L1 complex, PDB 4ZQK, where PD-L1 is shown using a space-filling model in yellow and PD-1 is shown using a ribbon model in red. The residues of Inhibitor 1, based on the PD-L1 interface sequence, are shown in blue at the interface of the two proteins. Error bars, S.D.
To test the hypothesis that elimination of interactions primarily between the Lys-78 region of PD-1 and PD-L1 could disrupt complex formation, we designed eight unique peptides targeting the interface based on the Lys-78 protein-painting hit. Shown in Table 4, one inhibitor was designed based on the N-terminal side of the Lys-78 hot spot region on PD-1 (Inhibitor 5), and one inhibitor was designed around a more C-terminal region of PD-1 found in the crystallographic interface (Inhibitor 7). These inhibitors were designed to disrupt the PD-1/PD-L1 complex based on the crystallographic interface but were not centered on inhibiting the Lys-78 interaction identified via protein painting with a PD-L1–derived sequence. Additionally, we designed one inhibitor as a combination of PD-L1 amino acids near the protein–protein interface that are not adjacent in the linear sequence as a scrambled peptide (Inhibitor 3). Finally, we designed one peptide based directly on the region of PD-1 that interacts with our protein-painting hit Lys-78 (Inhibitor 1). Sequences of these inhibitors are shown in the PD-1/PD-L1 cocrystal structure in Fig. S17 (B–D). Sequences were compared in both linear (Inhibitors 1, 3, 5, and 7) and cyclized (Inhibitors 2, 4, 6, and 8) conformations; cyclized peptides were prepared by adding cysteine residues to the N and C terminus of the four parent sequences and subsequently stapling the peptide ends via disulfide bond formation. These cyclized peptides may be more stable in solution and have more rigidity than their uncyclized counterparts. The sequences of each peptide inhibitor and their specific design parameters are given in Table 4.
Table 4.
Sequence and design of eight peptide inhibitors of the PD-1/PD-L1 complex
| Inhibitor no. | Sequence/Design |
|---|---|
| 1 | YRCMISYGGADYKRITV |
| PD-L1: residues 112–128 | |
| 2 | CYRAMISYGGADYKRITC |
| Similar to Inhibitor 1, cyclized via Cys–Cys bond | |
| 3 | LKYDAPAFTVT |
| PD-L1: Leu-15, Lys-124 to Ala-121, Pro-24, Ala-18 to Thr-22 | |
| 4 | CLKYDAPAFTVTC |
| Similar to Inhibitor 3 cyclized via Cys–Cys bond | |
| 5 | LNWYRMSPSNQTDKLAA |
| PD-1: residues 65–81 | |
| 6 | CLNWYRMSPSNQTDKLAC |
| Similar to Inhibitor 5, cyclized via Cys–Cys bond | |
| 7 | AISLAPKAQIK |
| PD-1: residues 125–135 | |
| 8 | CAISLAPKAQIKC |
| Similar to Inhibitor 7, cyclized via Cys–Cys bond |
To determine the ability of each of these peptide inhibitors to disrupt the PD-1/PD-L1 complex, concentrations up to 100 μm were tested in a PD-1/PD-L1 interaction kit available from BPS Biosciences. As shown in Fig. 9B, inhibitors designed with scrambled PD-L1 interface sequences (Inhibitors 3 and 4) were not effective. However, all remaining inhibitors were effective at 100 μm, and Inhibitors 1 and 2 had IC50 values under 10 μm. By developing an inhibitor mimicking the region directly interacting with the protein-painting hit rather than one derived from other regions of the crystallographic interface, higher levels of inhibition of complex formation could be achieved. Shown in Fig. 9C is the crystal structure of the regions of PD-L1 comprising Inhibitor 1 bound to the structure of PD-1. The portion of PD-L1 directly interacting with PD-1 Lys-78 is part of a B-loop motif that could be used for further small-molecule inhibitor development. No significant improvement was seen between Inhibitor 1 and Inhibitor 2, the cyclized version of Inhibitor 1. Further testing on the stability of Inhibitor 1 will indicate whether cyclization is a valuable improvement on the structure of Inhibitor 1. Additional optimization, including peptidomimetic compound generation or peptide stabilization, can be conducted to develop more potent and more stable compounds to use in the development of small-molecule inhibitors of PD-1/PD-L1. These peptides demonstrate the utility of the new protein-painting method in leading from a MS hit directly to a molecule that disrupts protein complex formation.
Commercially available antibodies targeting PD-1, including pembrolizumab and nivolumab, have been crystalized with PD-1 to identify their binding sites. We compared the binding site of Inhibitor 1 with the binding of commercially available antibodies. As shown in Fig. S18, the binding site of pembrolizumab directly overlaps with Inhibitor 1. The crystal structure of pembrolizumab shows multiple interactions directly with Lys-78 of PD-1, our protein-painting hot spot (34). Efforts to develop small-peptide inhibitors retaining the binding site of pembrolizumab can use Inhibitor 1 as a starting point for further optimizations.
Discussion
In this study, we answered remaining questions about how small dyes bind to proteins by comparing structurally similar dyes, which revealed the importance of a hydrophobic clamp region for achieving high numbers of bound dye molecules. By understanding more about the mechanism of dye binding, we were able to develop a novel dye probe, FBBNA, that binds to multiple different proteins with high affinity. To test the power of this new chemical probe, we applied our optimized protocol utilizing FBBNA to two highly challenging but clinically relevant complexes, YAP2/ZO-1 and PD-1/PD-L1.
Azo dye binding to proteins is optimized by balancing hydrophobic, hydrophilic, and aryl moieties in the candidate dye
In general, mechanisms of dye binding to proteins have not been exhaustively explored. Two exceptions are Coomassie Brilliant Blue, reported to bind primarily through salt-bridge interactions between the negatively charged sulfonate groups and positively charged basic amino acids (35), and Congo Red, thought to bind through both hydrophobic and electrostatic interactions (36, 37). To elucidate the importance of hydrophobic versus hydrophilic interactions, we investigated the mechanism of binding of FBBNA. To our knowledge, there has been no literature examining the systematic change of aryl regions of dyes to ascertain their effect on binding. We present evidence of the extreme importance of these hydrophobic aryl moieties for dye binding, as reduction in the number of hydrophobic aryl moieties at the edges of the molecule (Fig. 5) reduces the number of bound dye molecules by 50%. We propose, as with protein folding, that dye binding is composed of multiple weak interactions between the dye and the protein, such that elimination of several weak interactions leads to failure to bind. This is supported by our investigation showing that all dye candidates synthesized from Fast Red TR (including only one coupling group at maximum) were unable to bind significantly to thyroglobulin, whereas those dyes synthesized from Fast Blue B (two coupling groups at maximum) demonstrated detectable protein binding, as shown in in Table 1. To maximize the number and type of weak interactions, we proposed that a dye would bind more tightly to proteins when it balances opportunities to make hydrophobic, π-stacking, and ionic interactions. We show that FBBNA best balances these interactions by maintaining a hydrophobic clamp on either end of the molecule, balanced by sulfonate groups on one “side” of the molecule and π-stacking interactions between the multiple aryl rings. Affinity is reduced when substitution with a sulfonate group takes place on a separate ring from the amino group of the amino naphthalenesulfonic acid coupling group, eliminating the hydrophobic clamp of FBBNA. The highest binding dye component of FBBNA retained two hydrophobic clamps on either end of the molecule. Overall, these data emphasize the importance of the aryl hydrophobic contributions to binding.
Small-molecule dyes such as FBBNA and AO50 stabilize bound regions in the presence of chaotropic agent urea
By demonstrating that FBBNA and AO50 in combination do not influence secondary structure of BSA, but in fact stabilize secondary structure in the presence of urea, we decided to use both dyes in combination for protein painting. In this model, both FBBNA and AO50 bind to locations on the surface of the protein. When subjected to chaotropic agents, such as urea, the dye protects the protein backbone from direct interaction with urea, stabilizing the area from denaturation. Regions that are unprotected can be denatured and are more accessible to trypsin during the short digestion step of protein painting. These regions, both denatured and undyed, are preferentially trypsinized over other solvent-accessible areas that remain dye-bound. In this model, residues separated in the primary sequence but adjacent in the tertiary structure are able to remain protected during urea treatment due to the stabilizing effects of the dye. Binding of small ligands has been shown to have the capacity to influence the stability or flexibility of proteins (38); we show how this effect can be effectively utilized for structural analysis.
Additional optimization of protein painting will require accounting for post-translational modifications and lack of tryptic cleavage sites in protein–protein interfaces
Whereas our investigation originally centered around the protein-binding properties of FBBNA, we were surprised to uncover the carbohydrate binding properties of FBBNA in addition. Unlike protein binding, in which the presence of the hydrophobic clamp is of utmost importance, carbohydrate binding is probably utilizing different molecular interactions with FBBNA. We find that whereas FBBNA binds very strongly to cotton, it binds very poorly to filament acetate (Fig. S14), in which most of the hydroxyl groups of the carbohydrate monomers are acetylated. This suggests that there could be some kind of hydrogen bond formation, potentially between the primary amine of FBBNA and the carbohydrate hydroxyl groups. Discovery of FBBNA's affinity for tyrosine residues versus phenylalanine residues supports the hypothesis that hydrogen bonding is important for additional binding affinity of this dye.
Based on studies with FBBNA, we have started to identify key structural moieties that are involved in dye binding and better understand how these dyes might be acting in a protein-painting protocol. In addition to further improvements to the dye panels used in protein painting, we must also consider adapting the protein-painting protocol to utilize proteases beyond trypsin. Protein interaction hot spots have been shown to be enriched in both arginine and tyrosine residues as compared with noninteracting regions (39); while trypsin cleaves peptide bonds C-terminal to arginine residues, additional proteases will be required to identify hot spot tyrosine residues. We are pursuing the adaptation of protein painting to incorporate chymotrypsin to address this challenge and will potentially continue to add more proteases in the future to increase the resolution of the protein-painting technique.
It is worth noting that one limitation in our examination of dye binding is that for each experiment, we ranked dyes based on number of bound dye molecules per protein. It is conceivable that a dye that binds in higher numbers to a target might still be inferior to a dye that binds in lower numbers but has a higher affinity specifically for tryptic cleavage sites. Going forward, we will continue to look for additional classes of small molecules that bind with high affinity to protein targets to not only maximize the number of interactions but also diversify the type of molecular interactions that the dyes are making with the proteins under study. This will increasingly be important as we expand protein painting to proteases beyond trypsin, when it will be critical that protein-painting dyes block different types of recognition sequences. Furthermore, it is worth noting that an additional limitation in our examination of dye binding is that experimental constraints required the use of multiple standard proteins, potentially limiting generalizability of individual experimental results. For example, CD experiments were conducted with BSA due to the intense CD signal from the α-helical content that allows for suitable signal/noise ratios; the experiment was not conducted with carbonic anhydrase due to its greater β-sheet character. Additionally, larger panels of well-characterized proteins would be important going forward to ensure that multiple proteins can be found that are suitable for each technique utilized.
Unique hot spots identified between proteins in the Hippo signaling pathway provide novel targets for anti-cancer or wound-healing therapeutics and sites for probe development
Whereas confirmation of protein–protein interaction hot spots using protein painting is useful to validate interfaces identified using crystallography, additional power of the technique is found in cases where crystallographic structural information is unavailable. In the case of YAP2 and ZO-1, neither protein has been fully crystallized, although there are partial crystal structures and NMR structures of YAP (40, 41) and several crystal structures of various PDZ domains of ZO-1 (29, 42). There are currently no crystallized complexes of full-length YAP2 and ZO-1 or crystallized complexes of YAP2/ZO-1–binding domains. However, even in the absence of tertiary structural information about the proteins, we were able to identify hot spots of interaction between YAP2 and ZO-1, both within the canonical PDZ-binding domain and in additional regions that have not been implicated in YAP/ZO-binding interactions. The Ser-127 residue within the N-terminal region of YAP is the site of phosphorylation by the kinase LATS, allowing for phosphopeptide recognition by 14-3-3 and subsequent cytoplasmic retention of YAP (43). Evidence suggests that ZO family proteins, ZO-2 in particular, can form a complex with YAP that facilitates nuclear translocation (27). The presence of multiple hot spots within the N-terminal region may indicate that in complex with ZO-1, the YAP N-terminal region is somewhat occluded from solvent and may be inaccessible to LATS for phosphorylation on Ser-127. This could segregate the nuclear translocation activity of YAP/ZO complexes from the cytoplasmic retention activities of YAP/14-3-3 complexes by modulating the accessibility of the N-terminal region of YAP. In all, the results of this protein-painting analysis unveil unique sites to target on ZO-1 (Lys-592) as well as the N-terminal region of YAP (Arg-89, Arg-161, and Arg-187) to disrupt complex formation without risking potential cross-reactivity by targeting the PDZ domain directly. Future work to biologically validate each of the peptides as well as to determine minimum inhibitory sequences will form the basis for new peptide probes disrupting the ZO/YAP complex. Given that regulation of the Hippo pathway is complex and members cross-talk with a huge variety of other signaling pathways (44), additional targeted probes to specifically interrogate the role of individual complexes would provide new ways to unravel Hippo pathway regulation. In particular, the hot spots found outside the PDZ domain of ZO-1 may represent probes that can disrupt the YAP/ZO-1 complex with limited interference of other signaling complexes.
There are some potential limitations to the protein-painting analysis of YAP2 and ZO-1. Given that interactions of YAP2 with ZO-1 in vivo would occur in the context of many other signaling or scaffolding proteins, it is plausible that the interactions seen in isolation in vitro may not faithfully recapitulate binding in vivo. Additionally, protein-painting analysis could potentially identify regions that interact solely due to proximity in solution rather than reflecting a biologically relevant interaction. To address these issues, we plan to expand our protein-painting technique to larger complexes to potentially facilitate more accurate representations of protein–protein interactions in the cellular context going forward. However, difficulty in simulating in vivo protein–protein interactions is a problem common to X-ray crystallography as well as other MS techniques. Biological evaluation is of the utmost importance to verify that regions identified in protein painting faithfully represent binding in a cellular context.
Protein painting provides a rational start to development of inhibitors targeting the PD-1/PD-L1 protein–protein interaction
Peptide inhibitors designed directly from the hot spots generated using protein painting will ideally be able to disrupt complex formation and be used as probes or models for future drug design. Using the case study of PD-1/PD-L1, we find that protein painting can both identify the interface region and inform the design of multiple peptide inhibitors that can disrupt complex formation. The interface region between PD-1 and PD-L1 is large, featureless, and unattractive to attempt to target with a small molecule. Our protein-painting experiments have identified one specific region of the interface, Lys-78 on PD-1, that represents a potent hot spot that is solvent-inaccessible and allows for more targeted drug discovery against the complex. Rather than focusing drug discovery efforts on targeting any region within the interface, our results suggest that small molecules will be most efficacious if they specifically disrupt the interactions between PD-L1 and Lys-78 of PD-1. Using small-peptide inhibitors, we found that the most potent inhibitors of the PD-1/PD-L1 complex have IC50 values < 10 μm. This includes stabilized cyclic peptide Inhibitor 2. Macrocyclic peptides have shown greater resistance to proteolysis than acyclic peptides and have been of great interest to communities targeting protein–protein interactions (45). We show that the peptides identified in protein painting were amenable to cyclization through cysteine bond formation, with cyclized peptide Inhibitors 2, 6, and 8 showing potency roughly equal to that of their parent acyclic peptide inhibitors. These peptides, in addition to providing proof-of-concept for our protein-painting technology, can be useful lead compounds for further drug development targeting the PD-1/PD-L1 interface.
In summary, protein–protein interactions remain an incredibly underexplored area of drug discovery due to difficulties in identifying and targeting the specific sites of protein–protein interaction. However, a significant amount of research has been conducted in the past several years in optimizing cell-penetrating peptides, improving oral stability of peptides, and constraining the conformational flexibility of peptides (46). All of these improvements will hopefully continue to push small peptides forward as viable drug candidates in the future to target protein–protein interactions. Protein painting as a technique is unique in that it does not require detailed structural information to identify hot spot regions for further drug development, and it does not require that experimentation be done in nonnative conditions. Further improvements to the method and its application to new complexes will result in additional hot spot lead candidates reported for drug discovery.
Experimental procedures
Production of proteins and dyes
Thyroglobulin, catalase, carbonic anhydrase, and lysozyme standards were purchased from Sigma-Aldrich (catalogue nos. 9010-34-8, C1345, 9001-03-0, and 12650-88-3, respectively). Apoferritin was purchased from Alfa Aesar (catalogue no. AAJ60630MC). Protein stocks were prepared in 1× PBS and stored in single-use aliquots at −80 °C. YAP2, transcript variant 1 (catalogue no. TP325864), and ZO-1 (catalogue no. TP322836) were purchased from Origene. Fast Blue B salt and Fast Red TR salt were purchased from Sigma (catalogue nos. 14263-94-6 and 368881, respectively). Coupling agents naphthionic acid, H-acid, 2-napthol, Peri acid, Cleve acid, and Laurent acid were purchased from TCI Chemicals. Dye stocks were prepared to 1 mg/ml final concentrations. Synthesis was conducted as follows. Fast dyes were mixed in 1× Dulbecco's PBS with an excess of coupling agent (3-fold molar concentration of coupling agent to fast dye) and vortexed. Reactions were conducted at room temperature and proceeded in the first 5 min of mixing with evident color change. Dye stocks were allowed to incubate at room temperature for 1 week prior to use. Dyes were used without further purification and were diluted prior to use in protein painting. Dye reagents were vortexed before each use and were stable for >6 months at room temperature. Reactions were monitored by reverse-phase C18 TLC (mobile phase of 60% methanol, 40% water) to determine number of product bands. For Fast Blue B + naphthionic acid, two major product bands were observed with Rf = 0.32 (orange) and Rf = 0.61 (pink), whereas one minor product band was observed with Rf = 0.51 (light orange). The two major product bands were separated via reverse-phase flash column chromatography conducted essentially as described previously (47) using a 20-mm column, stationary phase of COSMOSIL C18-OPN reverse phase resin (Nacalai USA, catalogue no. 37842-66), and mobile phase of 60% methanol. Fractions containing individual bands were identified via reverse-phase TLC and were pooled and dried. Samples purified by TLC were analyzed by NMR to confirm the structure of both major products.
NMR structure confirmation
Confirmation of coupling positions of naphthionic acid to Fast Blue B for each major product were confirmed via NMR. Samples were isolated via reverse-phase column chromatography as described above, and 5–20 mg of dried sample was resuspended in DMSO-d6 for NMR. 1H NMR was recorded on a Bruker spectrometer at 400 MHz with solvent signal as the internal standard. Chemical shifts are given in ppm, and spin multiplicities are given using the following abbreviations: s (singlet), br s (broad singlet), d (doublet), dd (doublet of doublets), and t (triplet).
Fast Blue B
1H NMR (400 MHz, (CD3)2SO) δ 8.76 (d, J = 8.8 Hz, 2H), 8.14 (s, 2H), 7.97 (dd, J = 8.8, 1.6 Hz, 2H), 4.37 (s, 6H).
Naphthionic acid
1H NMR (400 MHz, (CD3)2SO) δ 8.72 (dd, J = 8.6, 1.0 Hz, 1H), 8.03 (dd, J = 8, 0.8 Hz, 1H), 7.66 (d, J = 8 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 6.52 (d, J = 8 Hz, 1H), 5.83 (br s, 2H).
Orange band
1H NMR (400 MHz, (CD3)2SO) δ 8.76 (d, J = 7.6 Hz, 1 H), 8.42 (d, J = 8.4 Hz, 1H), 8.36 (br s, 2H), 8.28 (s, 1 H), 7.89 (d, J = 8.4 Hz, 1H), 7.58 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.2 Hz, 1H), 7.47 (s, 1 H), 7.41 (d, J = 7.6 Hz, 1H), 7.36 (dd, J = 10.6, 1.8 Hz, 1H), 7.32 (s, 1H), 6.99 (dd, J = 7.2, 1.6 Hz, 1H), 4.087 (s, 3H), 3.861 (s, 3H).
Pink band
1H NMR (400 MHz, (CD3)2SO) δ 8.77 (dd, J = 7.6, 1.2 Hz, 2H), 8.43 (d, J = 8.8 Hz, 2H), 8.42 (br s, 4H), 8.30 (s, 2H), 7.94 (d, J = 8.4, 2H), 7.60 (s, 2H), 7.58 (t, J = 8 Hz, 2H), 7.50 (d, J = 6.8 Hz, 2H). 7.49 (t, J = 8.4, 2H), 4.136 (s, 6H).
Determination of number of bound dye molecules per protein at equilibrium under saturating conditions
To determine the number of bound dye molecules per model protein of each candidate dye, thyroglobulin was primarily used as a model protein. The large surface area of thyroglobulin facilitated more molecules of dye binding per protein and thus higher signal/noise ratios versus smaller proteins. Candidate dyes were examined on a UV-2501PC UV-visible spectrometer to determine λmax values. Standard curves were prepared for quantification of molecules of dye; dyes were eliminated from consideration if sensitivity of detection was not greater than 10 μm in solution. Thyroglobulin (1–5 μm) was mixed with a 100-fold molar excess of candidate dye dissolved in PBS and allowed to associate between 15 s and 2 h at room temperature. After association, 50 μl of the dye/protein mixtures were passed through a mini Quick Spin Oligo Column (Sephadex G-25 resin, Roche Applied Science) and centrifuged at 1000 × g for 1 min to separate unbound dye from the protein–dye complex. The collected flow-through was examined with UV-visible spectroscopy, and the quantity of dye in the flow-through was calculated using the previously developed standard curve for the dye. The number of bound dye molecules per protein molecule was calculated as paint molecules bound/protein. A plot of the number of bound dye molecules per protein molecule versus time was fit with the Michaelis–Menten equation in GraphPad Prism 6 as a model to determine maximum dye binding, where Vmax represents the maximum amount of dye molecules that bind to the protein at equilibrium. All curves are fit to data from two independent experiments. The number of bound dye molecules per molecule of apoferritin and catalase were determined using the same method as thyroglobulin. The number of bound dye molecules per molecule of lysozyme, carbonic anhydrase, and BSA were determined similarly, with the change that the protein (2–10 μm) was mixed with a 50-fold molar excess of dye candidate dissolved in PBS and allowed to associate for the indicated times. To determine the relationship between the number of bound dye molecules per protein and protein surface area, the surface area (SA) of each protein was estimated using Equation 1,
| (Eq. 1) |
where the protein was assumed to be spherical with a radius equal to the published Stokes radius.
Determination of association and dissociation constants of FBBNA to thyroglobulin
To measure dissociation of FBBNA from thyroglobulin, 258 μm FBBNA was incubated with 2.58 μm thyroglobulin for 10 min to reach equilibrium, and unbound dye was removed via a mini Quick Spin Oligo Column (Sephadex G-25 resin, Roche Applied Science) according to the manufacturer's directions. FBBNA-thyroglobulin solutions were allowed to incubate for 0, 30, 60, or 180 min before the solutions were passed through a second mini Quick Spin Oligo Column. Residual bound dye was measured via UV-visible spectroscopy and compared with the zero time point to determine the percentage of dye loss over time. FBBNA dissociation data were plotted after the association of FBBNA to thyroglobulin data to prepare an association and dissociation curve. Association and dissociation constants for FBBNA were determined by fitting the association and dissociation curve of FBBNA in GraphPad Prism 6 to an association and then dissociation model, with HotNM set to 258,881 (concentration of FBBNA in nm) and time 0 set to 120 (the time at which association experiments conclude and dissociation experiments begin).
Determination of amino acid binding specificity of FBBNA and AO50
Poly-Lys (catalog no. 2636); poly-Tyr (P1800); poly-Glu (P4761); poly-Lys,Tyr 1:1 (P4274); poly-Lys,Phe 1:1 (P3150); and poly-Glu,Tyr 1:1 (P0151) were purchased from Sigma, and stock solutions were prepared to 5 mg/ml. Polytyrosine was dissolved in 1 n NaOH, whereas all other poly-amino acids were dissolved in MilliQ water. Gel spotting was conducted essentially as described (48) with the following modifications. Polyacrylamide protein gels were dried for 30 min at ambient temperature in a fume hood and then spotted with 8 μl of a 50:50 poly-amino acid/MilliQ water working solution (20 μg/spot). All poly-amino acids were spotted in triplicate on each gel. Following spotting, gels were subsequently dried for an additional 15–30 min at ambient temperature and then fixed in 40% methanol, 10% acetic acid overnight. Staining with FBBNA was conducted essentially as described (49) with the following modifications. Following fixation, gels were transferred to 0.2 m acetate buffer, pH 3.5, for 30 min. Gels were then stained in 1 mg/ml FBBNA dye in 0.2 m acetate buffer, pH 3.5, for 2 h with shaking and then destained overnight in 0.2 m acetate buffer, pH 3.5. Staining with AO50 was conducted as follows. 1 mg/ml AO50 was dissolved in fixation solution (40% methanol, 10% acetic acid). After spotting and drying, gels were stained in the AO50/fixation solution overnight. Destaining was conducted using 40% methanol, 10% acetic acid. All gels were imaged with an Azure Biosystems C300 imager, and spot intensities were determined using ImageJ. Spot intensities were reported from four independent protein spotting experiments per dye.
Molecular docking
Docking was conducted using the SwissDock web server (24) (25). The coordinates for the hen egg white lysozyme structure (PDB code 6QWY), bovine carbonic anhydrase structure (PDB code 1V9E), and BSA structure (PDB code 2V03) were downloaded, and homodimeric chains, crystallographic waters, and metal ions or salts were removed prior to docking. The ligands FBBNA and AO50 were built with MarvinSketch and converted to mol2 files with three-dimensional coordinates in OpenBabel. Under “Extra Parameters” on the SwissDock submission web page, “Accurate” mode was selected for docking, and flexibility was set to 3 Å for optimization of side chain–ligand interactions. A total of 31 clusters comprising 257 docked molecules for FBBNA and 253 docked molecules for AO50 were generated for each protein. Predicted ΔG values for each docked position of each dye for each protein were averaged, and statistical significance was determined via unpaired Student's t test in GraphPad Prism 6. Differences in predicted Kd values for each dye using the carbonic anhydrase model were compared using Equation 2.
| (Eq. 2) |
CD spectroscopy
CD spectroscopy was conducted using BSA on a Jasco J-1500 CD spectrometer from 190 to 350 nm using a 1-mm cuvette. BSA stock solutions were prepared to 2 mg/ml from lyophilized protein in sterile-filtered 10 mm phosphate buffer, pH 7.0, and centrifuged at 16,000 × g to remove particulates. Concentration was determined via A280 using an extinction coefficient of 43,824 m−1 cm−1 for BSA (50). Stocks were stored at −80 °C and diluted to the appropriate concentration in sterile-filtered MilliQ water for each experiment. Final concentration of BSA in each sample was 2 μm (0.12 mg/ml). Samples were incubated for 10 min with dye, final concentration of 36 μm for both AO50 and FBBNA. Controls were incubated with equivolume amounts of MilliQ water in place of dye. Samples were subsequently either incubated at room temperature or denatured at 37 °C for 1 h in the presence of varying concentrations of urea (2, 4, or 6 m) and 3.5 mm DTT. Each spectrum shown is an average of three scans taken using a bandwidth of 1 nm, a scanning speed of 50 nm/min, and an integration time of 8 s. All samples with dye or urea have CD contributions from these buffers alone subtracted from the final spectrum. Helical content of BSA spectra was determined using the online server DICHROWEB (51, 52) to confirm native structure.
Protein-painting methodology
To prepare protein complexes, 2 μg of the protein with the greatest molecular weight in the complex was mixed with an equimolar concentration of its protein-binding partner (final molar concentration of proteins ranged from 0.1 to 2 μm) in 1× PBS and allowed to incubate for 1 h under rotation at room temperature to facilitate complex formation. After incubation, protein complexes were pulsed with a >100-fold molar excess of dye dissolved in PBS and allowed to associate for 5 min. Dye stock was prepared as a 1:1 ratio of 0.25 mg/ml FBBNA to 0.25 mg/ml AO50 for painting of ZO-1 and YAP2; painting of PD-1 and PD-L1 was conducted with 0.5 mg/ml AO50. After association, samples were passed through a mini Quick Spin Oligo Column (Sephadex G25 resin, Roche Applied Science) and centrifuged at 1000 × g for 1 min at room temperature. The flow-through was collected and denatured with urea (final concentration 2 m), reduced with 10 mm DTT, and incubated at 37 °C for 15 min and then alkylated with 50 mm iodoacetamide and incubated for 15 min in the dark. Samples were digested for 1.5 h at 37 °C with sequencing-grade trypsin (Promega) at a 1:10 (w/w) protease/protein ratio. Digestion was stopped with the addition of 4 μl of glacial acetic acid to a final concentration of 2%, and Hippo pathway samples were split into two technical replicates. Tryptic peptides were purified using Pierce C18 spin columns (Thermo Scientific) according to the manufacturer's directions. Desalted peptides were dried under nitrogen and stored at −20 °C until MS analysis. All protein-painting steps were completed using Eppendorf Lo-Bind microcentrifuge tubes for improved peptide recovery.
Mass spectrometry
LC-MS/MS experiments for the Hippo pathway samples were performed on an Orbitrap Fusion (Thermo Fisher Scientific) equipped with a nanospray EASY-LC 1200 HPLC system (Thermo Fisher Scientific), whereas LC-MS/MS experiments for PD-1/PD-L1 were performed on an LTQ Orbitrap (Thermo Fisher Scientific). Peptides were separated using a reversed-phase PepMap RSLC 75-μm inner diameter × 15 cm long with 2 μm, C18 resin LC column (Thermo Fisher Scientific). The mobile phase consisted of 0.1% aqueous formic acid (mobile phase A) and 0.1% formic acid in 80% acetonitrile (mobile phase B). After sample injection, the peptides were eluted by using a linear gradient from 5 to 50% B over 30 min and ramping to 100% B for an additional 2 min. The flow rate was set at 300 nl/min. The mass spectrometer was operated in a data-dependent mode in which one full MS scan (60,000 resolving power) from 300 Da to 1500 Da using quadrupole isolation was followed by MS/MS scans in which the most abundant molecular ions were dynamically selected by Top Speed and fragmented by collision-induced dissociation using a normalized collision energy of 35%. “Peptide Monoisotopic Precursor Selection” and “Dynamic Exclusion” (8 s duration), were enabled, as was the charge state dependence so that only peptide precursors with charge states from +2 to +4 were selected and fragmented by collision-induced dissociation. Tandem mass spectra were searched using Proteome Discover version 2.1 with SEQUEST against the NCBI Escherichia coli and Saccharomyces cerevisiae databases and a custom database containing sequences of the recombinant proteins using tryptic cleavage constraints. Mass tolerance for precursor ions was 5 ppm for the Hippo samples and 10 ppm for the PD-1/PD-L1 samples, and mass tolerance for fragment ions was 0.06 Da. Data were analyzed with oxidation (+15.9949 Da) on methionine as a variable post-translation modification and carbamidomethyl cysteine (+57.0215) as a fixed modification. A 1% false discovery rate was used as a cut-off value for reporting peptide spectrum matches (PSMs) from the database. A “blank” sample consisting of buffer only was inserted between each true sample and analyzed to evaluate potential carryover of peptides between samples. Any blank containing greater than 10 PSMs of the target proteins was considered to signify significant carryover potential, and samples in that run were not included in protein-painting data analysis.
Protein-painting data analysis and identification of hot spots
Individual PSMs for each protein of interest were aligned with the protein sequence and compared from at least three independent experiments. Unpainted proteins were used as controls to ensure significant sequence coverage of the protein and proper trypsin digestion. Sequence coverage between painted proteins alone and painted proteins in complex were compared; sequences that were present in complexed samples but absent from painted proteins alone were considered hits. Conserved Hippo pathway proteins were identified via protein BLAST and aligned using the Clustal Omega web server.
Generation and evaluation of peptide inhibitors
Peptide inhibitors were synthesized by Peptide 2.0 Inc. (Chantilly, VA) and received lyophilized. Peptides were dissolved to 1 mm stock solutions in MilliQ water with the addition of either acetic acid or acetonitrile (ACN) to promote solubility. Final concentrations of acetic acid or acetonitrile in each 1 mm stock solutions are as follows: Peptide 1, 15.3% acetic acid and 14.2% ACN; Peptide 2, 9.8% acetic acid; Peptide 3, 8.9% ACN; Peptide 4, 10.2% ACN; Peptide 5, 5.1% acetic acid; Peptide 6, 5.2% acetic acid; Peptide 7, 3.0% acetic acid; Peptide 8, 3.4% acetic acid. Peptide inhibitors were evaluated for inhibition of PD-1/PD-L1 binding using the PD-1:PD-L1 (Biotinylated) Inhibitor Screening Assay Kit from BPS Biosciences (catalogue no. 72003) according to the manufacturer's directions. Luminescence was measured using a DTX 880 plate reader with a 100-ms accumulation time. Each data point was collected in duplicate. Curve fitting was conducted using GraphPad Prism 6 to a sigmoidal dose–response curve with the equation, y = Bottom + (Top − Bottom)/(1 + 10(x − logIC50)), where Bottom and Top represent the minimum and maximum plateaus on the sigmoidal curve, respectively. Curve fitting was constrained by setting the parameter Bottom ≥0.
Author contributions
A. H., R. C., A. D., and V. N. performed the protein-painting experiments. A. H., D. D., and P. N. performed the CD experiments. A. H., D. D., and M. P. performed the PD-1/PD-L1 inhibitor design and evaluation experiments. A. H. and S. V. performed the docking studies. A. H. performed the binding and NMR experiments. A. H., M. P., L. L., and A. L. conceived of the study design. A. H. wrote the manuscript with L. L. and A. L.
Supplementary Material
Acknowledgments
We most gratefully acknowledge Dr. Paul Russo and Dr. Weidong Zhou for MS expertise. We also thank Dr. Barney Bishop for assistance with the CD experiments. Protein structure images were generated with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California (San Francisco, CA) with support from National Institutes of Health Grant P41-GM103311.
This work was funded in part by NIAMS, National Institutes of Health, Grant RO1AR068436; NCI, National Institutes of Health, Grant R33CA206937; and Center for Innovative Technology Award MF18-007-LS. The funding agencies had no role in experimental design, data analysis, or manuscript preparation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S18.
- IL
- interleukin
- ILRAcP
- interleukin-1 receptor accessory protein
- YAP
- Yes-associated protein
- ZO
- zona occludens
- FBBNA
- Fast Blue B + naphthionic acid
- FBBHA
- Fast Blue B + H-acid
- FBBLA
- Fast Blue B + Laurent acid
- FBBCA
- Fast Blue B + Cleve acid
- AO50
- Acid Orange 50
- TB
- trypan blue
- DB199
- Direct Blue 199
- Peri acid
- 1-amino-8-naphthalenesulfonic acid
- Laurent acid
- 5-amino-1-naphthalenesulfonic acid
- Cleve acid
- 1-naphthylamine-6-sulfonic acid
- PDB
- Protein Data Bank
- SH3
- Src homology 3
- TLC
- thin-layer chromatography
- PSM
- peptide spectrum match
- ACN
- acetonitrile.
References
- 1. Scott D. E., Bayly A. R., Abell C., and Skidmore J. (2016) Small molecules, big targets: drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 15, 533–550 10.1038/nrd.2016.29 [DOI] [PubMed] [Google Scholar]
- 2. Kobe B., Guncar G., Buchholz R., Huber T., Maco B., Cowieson N., Martin J. L., Marfori M., and Forwood J. K. (2008) Crystallography and protein–protein interactions: biological interfaces and crystal contacts. Biochem. Soc. Trans. 36, 1438–1441 10.1042/BST0361438 [DOI] [PubMed] [Google Scholar]
- 3. Weiss G. A., Watanabe C. K., Zhong A., Goddard A., and Sidhu S. S. (2000) Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. U.S.A. 97, 8950–8954 10.1073/pnas.160252097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dailing A., Luchini A., and Liotta L. (2015) Unlocking the secrets to protein-protein interface drug targets using structural mass spectrometry techniques. Expert Rev. Proteomics 12, 457–467 10.1586/14789450.2015.1079487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luchini A., Espina V., and Liotta L. A. (2014) Protein painting reveals solvent-excluded drug targets hidden within native protein-protein interfaces. Nat. Commun. 5, 4413 10.1038/ncomms5413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Günther S., Deredge D., Bowers A. L., Luchini A., Bonsor D. A., Beadenkopf R., Liotta L., Wintrode P. L., and Sundberg E. J. (2017) IL-1 family cytokines use distinct molecular mechanisms to signal through their shared co-receptor. Immunity 47, 510–523.e4 10.1016/j.immuni.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iglesias-Bexiga M., Castillo F., Cobos E. S., Oka T., Sudol M., and Luque I. (2015) WW domains of the Yes-kinase-associated-protein (YAP) transcriptional regulator behave as independent units with different binding preferences for PPxY motif-containing ligands. PLoS One 10, e0113828 10.1371/journal.pone.0113828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moroishi T., Hansen C. G., and Guan K.-L. (2015) The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 15, 73–79 10.1038/nrc3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yan Z., Shi H., Zhu R., Li L., Qin B., Kang L., Chen H., and Guan H. (2018) Inhibition of YAP ameliorates choroidal neovascularization via inhibiting endothelial cell proliferation. Mol. Vis. 24, 83–93 [PMC free article] [PubMed] [Google Scholar]
- 10. Lee M.-J., Byun M. R., Furutani-Seiki M., Hong J.-H., and Jung H.-S. (2014) YAP and TAZ regulate skin wound healing. J. Invest. Dermatol. 134, 518–525 10.1038/jid.2013.339 [DOI] [PubMed] [Google Scholar]
- 11. Holden J. K., and Cunningham C. N. (2018) Targeting the Hippo pathway and cancer through the TEAD family of transcription factors. Cancers (Basel) 10, E81 10.3390/cancers10030081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oka T., Mazack V., and Sudol M. (2008) Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 283, 27534–27546 10.1074/jbc.M804380200 [DOI] [PubMed] [Google Scholar]
- 13. McNeil E., Capaldo C. T., and Macara I. G. (2006) Zonula occludens-1 function in the assembly of tight junctions in Madin-Darby canine kidney epithelial cells. Mol. Biol. Cell 17, 1922–1932 10.1091/mbc.e05-07-0650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oka T., Remue E., Meerschaert K., Vanloo B., Boucherie C., Gfeller D., Bader G. D., Sidhu S. S., Vandekerckhove J., Gettemans J., and Sudol M. (2010) Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem. J. 432, 461–472 10.1042/BJ20100870 [DOI] [PubMed] [Google Scholar]
- 15. Francis D. M., and Thomas S. N. (2017) Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Adv. Drug Deliv. Rev. 114, 33–42 10.1016/j.addr.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamanth A. V. (2016) Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov. Today Technol. 21, 75–83 10.1016/j.ddtec.2016.09.004 [DOI] [PubMed] [Google Scholar]
- 17. Samaranayake H., Wirth T., Schenkwein D., Räty J. K., and Ylä-Herttuala S. (2009) Challenges in monoclonal antibody-based therapies. Ann. Med. 41, 322–331 10.1080/07853890802698842 [DOI] [PubMed] [Google Scholar]
- 18. Li K., and Tian H. (2019) Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target. 27, 244–256 10.1080/1061186X.2018.1440400 [DOI] [PubMed] [Google Scholar]
- 19. Zak K. M., Kitel R., Przetocka S., Golik P., Guzik K., Musielak B., Dömling A., Dubin G., and Holak T. A. (2015) Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 23, 2341–2348 10.1016/j.str.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paris L., Magni R., Zaidi F., Araujo R., Saini N., Harpole M., Coronel J., Kirwan D. E., Steinberg H., Gilman R. H., Petricoin E. F. 3rd, Nisini R., Luchini A., and Liotta L. (2017) Urine lipoarabinomannan glycan in HIV-negative patients with pulmonary tuberculosis correlates with disease severity. Sci. Transl. Med. 9, eaal2807 10.1126/scitranslmed.aal2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stefanini S., Cavallo S., Wang C. Q., Tataseo P., Vecchini P., Giartosio A., and Chiancone E. (1996) Thermal stability of horse spleen apoferritin and human recombinant H apoferritin. Arch. Biochem. Biophys. 325, 58–64 10.1006/abbi.1996.0007 [DOI] [PubMed] [Google Scholar]
- 22. King J. L., and Jukes T. H. (1969) Non-Darwinian evolution. Science 164, 788–798 [DOI] [PubMed] [Google Scholar]
- 23. Pigorsch E., Elhaddaoui A., and Turrell S. (1994) Spectroscopic study of pH and solvent effects on the structure of Congo red and its binding mechanism to amyloid-like proteins. Spectrochim. Acta A 50, 2145–2152 10.1016/0584-8539(94)00151-0 [DOI] [Google Scholar]
- 24. Grosdidier A., Zoete V., and Michielin O. (2011) Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 32, 2149–2159 10.1002/jcc.21797 [DOI] [PubMed] [Google Scholar]
- 25. Grosdidier A., Zoete V., and Michielin O. (2011) SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277 10.1093/nar/gkr366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 10.1038/nprot.2006.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oka T., and Sudol M. (2009) Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ-binding motif. Genes Cells 14, 607–615 10.1111/j.1365-2443.2009.01292.x [DOI] [PubMed] [Google Scholar]
- 28. Sudol M., Shields D. C., and Farooq A. (2012) Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin. Cell Dev. Biol. 23, 827–833 10.1016/j.semcdb.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Appleton B. A., Zhang Y., Wu P., Yin J. P., Hunziker W., Skelton N. J., Sidhu S. S., and Wiesmann C. (2006) Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1: insights into determinants of PDZ domain specificity. J. Biol. Chem. 281, 22312–22320 10.1074/jbc.M602901200 [DOI] [PubMed] [Google Scholar]
- 30. Hao Y., Chun A., Cheung K., Rashidi B., and Yang X. (2008) Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509 10.1074/jbc.M709037200 [DOI] [PubMed] [Google Scholar]
- 31. Kurochkina N., and Guha U. (2013) SH3 domains: modules of protein–protein interactions. Biophys. Rev. 5, 29–39 10.1007/s12551-012-0081-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Skalniak L., Zak K. M., Guzik K., Magiera K., Musielak B., Pachota M., Szelazek B., Kocik J., Grudnik P., Tomala M., Krzanik S., Pyrc K., Dömling A., Dubin G., and Holak T. A. (2017) Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 8, 72167–72181 10.18632/oncotarget.20050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tan S., Zhang H., Chai Y., Song H., Tong Z., Wang Q., Qi J., Wong G., Zhu X., Liu W. J., Gao S., Wang Z., Shi Y., Yang F., Gao G. F., and Yan J. (2017) An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 8, 14369 10.1038/ncomms14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Horita S., Nomura Y., Sato Y., Shimamura T., Iwata S., and Nomura N. (2016) High-resolution crystal structure of the therapeutic antibody pembrolizumab bound to the human PD-1. Sci. Rep. 6, 35297 10.1038/srep35297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tal M., Silberstein A., and Nusser E. (1985) Why does Coomassie Brilliant Blue R interact differently with different proteins? A partial answer. J. Biol. Chem. 260, 9976–9980 [PubMed] [Google Scholar]
- 36. Khurana R., Uversky V. N., Nielsen L., and Fink A. L. (2001) Is Congo Red an amyloid-specific dye? J. Biol. Chem. 276, 22715–22721 10.1074/jbc.M011499200 [DOI] [PubMed] [Google Scholar]
- 37. Skowronek M., Stopa B., Konieczny L., Rybarska J., Piekarska B., Szneler E., Bakalarski G., and Roterman I. (1998) Self-assembly of Congo Red—a theoretical and experimental approach to identify its supramolecular organization in water and salt solutions. Biopolymers 46, 267–281 [DOI] [Google Scholar]
- 38. Celej M. S., Montich G. G., and Fidelio G. D. (2003) Protein stability induced by ligand binding correlates with changes in protein flexibility. Protein Sci. 12, 1496–1506 10.1110/ps.0240003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ofran Y., and Rost B. (2007) Protein–protein interaction hotspots carved into sequences. PLoS Comput. Biol. 3, e119 10.1371/journal.pcbi.0030119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schumacher B., Skwarczynska M., Rose R., and Ottmann C. (2010) Structure of a 14–3-3σ–YAP phosphopeptide complex at 1.15 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 978–984 10.1107/S1744309110025479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Verma A., Jing-Song F., Finch-Edmondson M. L., Velazquez-Campoy A., Balasegaran S., Sudol M., and Sivaraman J. (2018) Biophysical studies and NMR structure of YAP2 WW domain: LATS1 PPxY motif complexes reveal the basis of their interaction. Oncotarget 9, 8068–8080 10.18632/oncotarget.23909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pan L., Chen J., Yu J., Yu H., and Zhang M. (2011) The structure of the PDZ3-SH3-GuK tandem of ZO-1 protein suggests a supramodular organization of the membrane-associated guanylate kinase (MAGUK) family scaffold protein core. J. Biol. Chem. 286, 40069–40074 10.1074/jbc.C111.293084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu F.-X., and Guan K.-L. (2013) The Hippo pathway: regulators and regulations. Genes Dev. 27, 355–371 10.1101/gad.210773.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meng Z., Moroishi T., and Guan K.-L. (2016) Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 10.1101/gad.274027.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White A. M., and Craik D. J. (2016) Discovery and optimization of peptide macrocycles. Expert Opin. Drug Discov. 11, 1151–1163 10.1080/17460441.2016.1245720 [DOI] [PubMed] [Google Scholar]
- 46. Bruzzoni-Giovanelli H., Alezra V., Wolff N., Dong C.-Z., Tuffery P., and Rebollo A. (2018) Interfering peptides targeting protein–protein interactions: the next generation of drugs? Drug Discov. Today 23, 272–285 10.1016/j.drudis.2017.10.016 [DOI] [PubMed] [Google Scholar]
- 47. Still W. C., Kahn M., and Mitra A. (1978) Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 43, 2923–2925 10.1021/jo00408a041 [DOI] [Google Scholar]
- 48. Zhao Z., Aliwarga Y., and Willcox M. D. (2007) Intrinsic protein fluorescence interferes with detection of tear glycoproteins in SDS-polyacrylamide gels using extrinsic fluorescent dyes. J. Biomol. Tech. 18, 331–335 [PMC free article] [PubMed] [Google Scholar]
- 49. Mehta S., and Rajput Y. S. (1998) A method for staining of proteins in nitrocellulose membrane and acrylamide gel using Congo Red dye. Anal. Biochem. 263, 248–251 10.1006/abio.1998.2805 [DOI] [PubMed] [Google Scholar]
- 50. Thermo Scientific (2010) Protein A280: Thermo Scientific NanoDrop Spectrophotometers, Thermo Fisher Scientific, Wilmington, DE [Google Scholar]
- 51. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 10.1002/bip.20853 [DOI] [PubMed] [Google Scholar]
- 52. Whitmore L., and Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 10.1093/nar/gkh371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parmar A. S., and Muschol M. (2009) Hydration and hydrodynamic interactions of lysozyme: effects of chaotropic versus kosmotropic ions. Biophys. J. 97, 590–598 10.1016/j.bpj.2009.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smilgies D.-M., and Folta-Stogniew E. (2015) Molecular weight–gyration radius relation of globular proteins: a comparison of light scattering, small-angle X-ray scattering and structure-based data. J. Appl. Crystallogr. 48, 1604–1606 10.1107/S1600576715015551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 10.1007/s12575-009-9008-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Edelhoch H. (1960) The properties of thyroglobulin I: the effects of alkali. J. Biol. Chem. 235, 1326–1334 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.