

Abstract
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is a long noncoding RNA overexpressed in various cancers that promotes cell growth and metastasis. Although hypoxia has been shown to up-regulate MALAT1, only hypoxia-inducible factors (HIFs) have been implicated in activation of the MALAT1 promoter in specific cell types and other molecular mechanisms associated with hypoxia-mediated MALAT1 up-regulation remain largely unknown. Here, we demonstrate that hypoxia induces cancer cell-specific chromatin–chromatin interactions between newly identified enhancer-like cis-regulatory elements present at the MALAT1 locus. We show that hypoxia-mediated up-regulation of MALAT1 as well as its antisense strand TALAM1 occurs in breast cancer cells, but not in nontumorigenic mammary epithelial cells. Our analyses on the MALAT1 genomic locus discovered three novel putative enhancers that are located upstream and downstream of the MALAT1 gene body. We found that parts of these putative enhancers are epigenetically modified to a more open chromatin state under hypoxia in breast cancer cells. Furthermore, our chromosome conformation capture experiment demonstrated that noncancerous cells and breast cancer cells exhibit different interaction profiles under both normoxia and hypoxia, and only breast cancer cells gain specific chromatin interactions under hypoxia. Although the HIF-2α protein can enhance the interaction between the promoter and the putative 3′ enhancer, the gain of chromatin interactions associated with other upstream elements, such as putative −7 and −20 kb enhancers, were HIF-independent events. Collectively, our study demonstrates that cancer cell-specific chromatin–chromatin interactions are formed at the MALAT1 locus under hypoxia, implicating a novel mechanism of MALAT1 regulation in cancer.
Keywords: hypoxia; long noncoding RNA (long ncRNA, lncRNA); chromatin structure; histone modification; breast cancer; MALAT1
Introduction
Gene expression is a tightly controlled process incorporating numerous regulatory elements in multiple steps. Enhancers are noncoding regions of the genome that increase gene expression in select tissues or in response to various stresses (1, 2). Enhancer sequences contain transcription factor-binding sites and the recruitment of these proteins to enhancers can up-regulate the target gene through close contact with the promoter (1). In addition, chromatin–chromatin interactions are formed within the three-dimensional chromatin structure, connecting enhancers present upstream, downstream, or at a long distance, to their target promoters (3–5). Gene expression can be further fine-tuned through regulation of histone modifications, such as methylation and acetylation (6).
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1)2 is a long noncoding RNA, which is highly expressed in multiple tissues. Through transcriptional regulation and alternative splicing MALAT1 regulates expression of genes involved in proliferation and cellular motility (7–9). MALAT1 is widely overexpressed in solid tumors where it promotes proliferation, motility, angiogenesis, and metastasis (10). MALAT1 overexpression is a poor prognostic factor in cancer and is a strong indicator of metastatic potential. Although the functional role of MALAT1 is well-documented, the upstream factors involved in MALAT up-regulation are poorly understood. A handful of transcription factors, namely β-catenin/TCF7L2 (11), c-Fos (12), NF-κB (13), and Sp1/Sp3 (14, 15) have been shown to activate MALAT1 transcription, whereas p53 represses transcription (16, 17). However, involvement of these proteins in MALAT1 transcription have not been validated in more than one tumor type, and there is a lack of information regarding how MALAT1 is up-regulated in a broad range of cancer types. Moreover, besides the promoter regions, cis-regulatory elements involved in MALAT1 expression are completely unknown.
Hypoxia, caused by low oxygen levels in the tumor microenvironment, up-regulates a set of genes involved in angiogenesis, apoptosis, cell migration, glycolysis, and stemness (18, 19). Many of these genes are targets of the hypoxia-inducible factor (HIF) proteins, a family of transcription factors stabilized by low-oxygen conditions (20, 21). However, mechanisms that cause HIF-independent gene regulation are largely unexplored and demonstration of these pathways and transcription factors relevant to cancer progression remain elusive. Several studies have shown that MALAT1 is up-regulated under hypoxia (9, 22–26). However, its underlying mechanisms remain poorly understood.
In this study, we demonstrate that the MALAT1 genomic locus contains potential distal enhancers upstream and downstream of the MALAT1 gene body and these regions are involved in long-range chromatin interactions in breast cancer. Importantly, hypoxia modifies histone markers resulting in more open chromatin and induces chromatin interactions in the MALAT1 locus in breast cancer cells. Although HIF facilitates the interaction between the MALAT1 promoter and the enhancer located downstream of the gene, other hypoxia-induced chromatin–chromatin interactions between the potential enhancer regions are HIF-independent, they only occur in breast cancer cell lines and they are not observed in nontumorigenic mammary lines. These findings suggest that the MALAT1 enhancers have a hypoxic-specific response in the cancer condition.
Results
Hypoxia up-regulates MALAT1 in breast cancer cell lines
To determine the expression level of MALAT1 in different subtypes of breast cancer cells, we performed Northern blotting using RNA isolated from nine cancer lines encompassing each subtype: luminal A (MCF7, T47D), luminal B (BT474, MDA-MB-361, ZR-75-30), HER2-enriched (MDA-MB-453, SKBR3), and triple-negative (MDA-MB-157, MDA-MB-231). MCF10A was also included as a nontumorigenic mammary epithelial cell line. We observed high levels of MALAT1 expression in luminal A, luminal B, and HER2-enriched breast cancer cells compared with triple-negative breast cancer cell lines and MCF10A. All three cell lines of the luminal B subtype breast cancer cells had the highest expression levels of MALAT1. This result demonstrated that the MALAT1 expression level is indeed higher in breast cancer cell lines compared with that of a nontumorigenic line (Fig. 1A). To examine how hypoxia affects MALAT1 expression in these cells, we exposed nine of these cell lines to either normoxic (21% O2) or hypoxic (1% O2) conditions for 24 h prior to RNA isolation. Our quantitative PCR (qPCR) data demonstrated that under hypoxia all breast cancer lines increased expression of MALAT1 relative to normoxic levels (Fig. 1B). Hypoxia-mediated up-regulation of MALAT1 is more noticeable in the cell lines with low MALAT1 expression under normoxia (MDA-MB-157, MDA-MB-231, MCF7), compared with the cell lines with the highest MALAT1 expression in normoxia (BT474, MDA-MB-361). Surprisingly, MCF10A did not show any changes in MALAT1 expression under hypoxia (Fig. 1B). These results demonstrated that hypoxia strongly induces MALAT1 expression in breast cancer cells, but not in nontumorigenic mammary epithelial cells.
Figure 1.

MALAT1 and its antisense transcript TALAM1 are up-regulated under hypoxia in breast cancer. A, 5 μg of total RNA was probed for MALAT1 expression by Northern blotting. Cell lines are colored according to receptor status and breast cancer subtype: gray, nontumorigenic; red, triple negative; blue, luminal A; green, luminal B; purple, HER2 positive. B, cells were grown in normoxia (21% O2) or hypoxia (1% O2) for 24 h and total RNA was isolated for RT-qPCR. Increase in MALAT1 expression for each cell line under hypoxia is normalized to its normoxic expression levels. Data are represented as mean ± S.D., n = 3–5, *, p < 0.05; **, p < 0.01. C, diagram of the MALAT1 genomic locus and the location of the antisense transcript TALAM1 gene. The locations indicated in kb are distances from the MALAT1 transcription start site. D, strand-specific qPCR assay design developed by Zong et al. (28). A fusion oligo consisting of a linker sequence and a strand-specific primer for reverse transcription (SSP-RT) is used for cDNA synthesis. The 5′ portion of the linker is used for qPCR in conjunction with a SSP forward primer. E, MALAT1 and TALAM1 strand-specific qPCR for MCF10A and MCF7 exposed to normoxic or 24 hypoxic conditions for 24 h. Data are represented as mean ± S.D., n = 3, *, p < 0.05; **, p < 0.01.
The antisense transcript TALAM1 is up-regulated under hypoxia
Increasing evidence has demonstrated that antisense transcripts are lowly expressed noncoding RNAs transcribed from the opposite strand of coding or noncoding RNAs which then, in many cases, regulate expression of the sense transcript (27). The MALAT1 locus contains an antisense transcript called TALAM1 (Fig. 1C), which promotes MALAT1 up-regulation via 3′ end processing (28). Because MALAT1 was overexpressed under hypoxia and these two RNAs engage in a positive feedback loop with each other, we examined if hypoxia could also up-regulate TALAM1. We used a strand-specific primer (SSP) strategy devised by Zong et al. (28) to differentiate MALAT1 and TALAM1 (Fig. 1D), as regular reverse transcription and PCR cannot discriminate between sense and antisense transcripts. We used a linker sequence not found in the human genome fused to an antisense oligo specific for either MALAT1 or TALAM1 for cDNA synthesis. qPCR amplified an SSP forward primer with a reverse primer for the linker sequence. The result demonstrated no hypoxia-mediated up-regulation of MALAT1 in MCF10A and accordingly no change was found in TALAM1 expression under hypoxia (Fig. 1E). In contrast, when we examined expression of MALAT1 and TALAM1 in the MCF7 breast cancer cell line, both transcripts were significantly up-regulated under hypoxia (Fig. 1E). These expression profiles are consistent with increased MALAT1 transcription under hypoxia and provide support for the importance of this locus in cancer cells.
HIF-2α has a minor role in MALAT1 up-regulation
Having determined that hypoxia up-regulates MALAT1, we next sought to find a candidate factor that would function as a transcriptional activator in this process. We considered HIF-1α or HIF-2α as potential candidates, given their regulation of MALAT1 in various tissues under hypoxia (22, 24, 25). To examine whether HIFs are required for MALAT1 up-regulation under hypoxia, we used small interfering RNA (siRNA) to knockdown either HIF protein alone or in combination with each other. Knockdown of HIF-1α and HIF-2α was confirmed at both the mRNA and protein levels in MCF7 cells (Fig. 2A and Fig. S1A). We next measured the effect of HIF knockdown on the well-known HIF target genes CAIX and PAI1 and found both mRNAs were reduced by HIF depletion (Fig. 2B). Under this condition, we observed a slight, yet significant, decrease of MALAT1 by HIF-2α knockdown alone (Fig. 2C). Consistent with the previous observation on HIF-1α function as a repressor of HIF-2α expression in MCF7 cells (29), our result showed that the combinational treatment of HIF-1α siRNA and HIF-2α siRNA abrogated the effect of HIF-2α siRNA on a marginal decrease of MALAT1.
Figure 2.

HIF-2α plays a minor role in hypoxia-induced MALAT1 up-regulation. A, siRNA targeting HIF-1α or HIF-2α was transfected into MCF7 cells for 48 h prior to transfer to hypoxia for 24 h, and Western blotting was performed to detect HIF-1α and HIF-2α. B and C, MCF7 cells transfected with siRNA as described in A were used for RT-qPCR to measure the expression of the known HIF target genes CAIX and PAI1 (B) and MALAT1 (C). D and E, MCF7 cells were transfected with pcDNA-HA-HIF2α-P405A/P531A (HIF2α-CA; +) or an empty vector (−) for 48 h prior to cell lysis and used for Western blots using the antibodies against HIF-2α and HA (tag) (D) and MALAT1 RT-qPCR (E). F, MCF7 cells were exposed to hypoxia or treated with 100 μm CoCl2 for 24 h prior to lysate preparation for an HIF-2α Western blotting. G, RT-qPCR was performed to measure the MALAT1 expression level following MCF7 treatment with 100 μm CoCl2 for 24 h. Actin was examined as a loading control in Western blots. In the bar graphs, data are represented as mean ± S.D., n = 3–5, *, p < 0.05; **, p < 0.01.
We further examined the role of only HIF-2α in MALAT1 up-regulation by transfecting MCF7 cells with a plasmid encoding a constitutively active form of HIF-2α (HIF2α-CA) with P405A and P531A point mutations (pcDNA-HA-HIF2α-P405A/P531A), rendering the protein stable in the presence of oxygen (30). We confirmed HIF2α-CA overexpression at the mRNA and protein levels and up-regulation of PAI1, a HIF-target gene (Fig. 2D and Fig. S1B). Under this condition, we found that MALAT1 was up-regulated about 1.3-fold (Fig. 2E).
To further verify the minor role of HIF-2α in MALAT1, we treated MCF7 cells with cobalt chloride (CoCl2), which prevents the degradation of the α subunits of HIF under normoxic conditions (31), resulting in increased levels of the HIF2α proteins (Fig. 2F). Upon CoCl2 treatment, we observed only a 1.5-fold increase in MALAT1 expression in MCF7 cells (Fig. 2G), indicating that CoCl2-mediated HIF-stabilization is not sufficient to fully recapitulate the effect of hypoxia. Therefore, it is likely that additional, HIF-independent changes are necessary to fully activate MALAT1 expression in breast cancer cells under hypoxia.
Putative enhancers are identified at the MALAT1 locus and active chromatin marks are increased in some of these enhancer regions under hypoxia
To address whether any cis-elements present near the MALAT1 gene are involved in hypoxia-induced MALAT1 expression, we extensively analyzed various chromatin marks and transcription factor binding patterns at the MALAT1 locus using ENCODE chromatin immunoprecipitation (ChIP)-seq data. Interestingly, besides the known promoter area, we found multiple sites containing the active enhancer/promoter marks histone-3 lysine-4 methylation (mono-methylation, H3K4me1; tri-methylation, H3K4me3) and histone-3 lysine-27 acetylation (H3K27Ac) as well as RNA polymerase II occupancy and transcription factor-binding sites in the sequence upstream of the MALAT1 transcription start site (TSS) and downstream of the transcription termination site (Fig. 3A). Based on enrichment levels of these chromatin features, we propose three putative enhancer regions for MALAT1. These enhancer regions are located between −21.4 and −17.6 kb and −11.1 and −2.7 kb upstream of the TSS of the MALAT1 gene, and between +8.4 and +11 kb, which is the region located immediately downstream of the 3′ end of the MALAT1 gene. We named these putative enhancers −20 kb enhancer, −7 kb enhancer, and 3′ enhancer, respectively (Fig. 3A). Examination of the mouse genome for orthologous sequence revealed each of these putative enhancer elements were conserved at the genetic and epigenetic levels (Fig. S2), supporting a potential functional role.
Figure 3.

The MALAT1 locus contains putative enhancers within the sequences upstream and downstream of the MALAT1 gene body and active chromatin marks are increased in some of these enhancer regions under hypoxia. A, location of putative enhancer regions within the MALAT1 genetic locus are indicated as labeled and highlighted in green boxes. The image incorporates ChIP-seq datasets from GSE107176 and ENCODE (58, 59). Loci for ChIP-qPCR primer sets are indicated as P1–P9. Distances for each primer set from the MALAT1 transcription start site are as follow: P1, −20.5 kb; P2, −15.9 kb; P3, −9.2 kb; P4, −4.7 kb; P5, −2.2 kb; P6, −0.8 kb; P7, +0.8 kb; P8, +1.4 kb; P9, +9.2 kb. B–D, ChIP-qPCR results of the log-fold change in enrichment of H3K27Ac (B), H3K4me1 (C), and H3K4me3 (D) levels in MCF10A (white bars) and MCF7 cells (black bars) following 24 h of exposure to normoxia or hypoxia. Data are shown as mean ± S.D. of one representative result from 5 independent biological replicates. *, p < 0.05.
It is known that as a stress response to hypoxia, a selective group of genes are up-regulated, in part due to an increase of active epigenetic markers such as H3K4me3 at their promoters (32, 33). Therefore, we questioned if hypoxia alters the chromatin status of the MALAT1 promoter or these potential enhancers regions. Nine sites throughout the promoter and putative enhancers were selected to determine whether histone modifications occupancy changed under hypoxia by ChIP-qPCR (Fig. 3A). The P1 locus is found approximately −20.5 kb upstream of the MALAT1 TSS and is within the putative −20 kb enhancer. The P2 locus is at −15.9 kb upstream and is not contained within any putative enhancer. The putative −7 kb enhancer contains two primer sets at each end, P3 and P4, at −9.2 and −4.7 kb, respectively. The P5 locus is at −2.2 kb and is not contained within any putative enhancer. We designed three primer sets for the promoter regions; the P6 locus at −0.8 kb for the upstream promoter, and the P7 locus at +0.8 kb and the P8 locus at +1.4 kb for the internal promoter. Finally, the P9 locus is at +9.2 kb and covers the putative 3′ enhancer.
We performed ChIP-qPCR at the genomic loci P1–P9 under normoxia and hypoxia in the nontumorigenic MCF10A and tumorigenic MCF7 cell lines for the histone modifications H3K27Ac, H3K4me1, and H3K4me3, then calculated the log-fold change of the ratio of hypoxic to normoxic enrichment of each histone modification. Our ChIP-qPCR results revealed distinct levels of histone modifications between these cell lines.
In MCF10A cells, the active enhancer/promoter marker H3K27Ac was enriched at P2, P5, and P9 and decreased at the other six loci under hypoxia (Fig. 3B). Active enhancer marker H3K4me1 levels showed fewer changes in MCF10A cells in response to hypoxia, only increasing at P8 and staying unchanged at P5, whereas decreasing at all other loci (Fig. 3C). Active promoter marker H3K4me3 levels were increased at P1, P7, and P8 but sharply decreased at all other loci, including the P6 locus covering the upstream promoter (Fig. 3D).
Interestingly, we found strikingly different patterns of histone modifications in response to hypoxia in MCF7 cells. Changes in H3K27Ac levels were overall similar to MCF10A, with enrichment at P4, P5, and P9 under hypoxia (Fig. 3B). Interestingly, we found significant increases of the H3K4me1 level under hypoxia compared with normoxia at seven of nine sites examined (except for the P6 and P7 sites) (Fig. 3C). In addition, we detected that the level of H3K4me3 increased at both the upstream and internal promoter loci (P6 and P7, respectively), a part of the putative −7 kb enhancer and its downstream region (P4 and P5) as well as the putative 3′ enhancer (P9) under hypoxia in MCF7 cells (Fig. 3D).
In summary, all three histone markers showed enrichment at the loci of P4 (putative −7 kb enhancer), P5 (downstream of the −7kb enhancer), and P9 (putative 3′ enhancer) in MCF7 under hypoxia, whereas only H3K27Ac was enriched at P5 and P9 in MCF10A. These data demonstrate that the −7 kb enhancer and its downstream region as well as the 3′ enhancer are modified to open, active chromatin in breast cancer in response to hypoxia, but these modifications do not occur in nontumorigenic mammary cells.
Long-range chromatin interactions occur at the MALAT1 locus between enhancers and the MALAT1 promoter in a cell-type specific manner
Although H3K4me3 has traditionally been considered as an active promoter mark, a recent study demonstrated that active enhancers are also highly enriched with H3K4me3 modification (34). Because our ChIP-qPCR data demonstrated greater enrichment of H3K27Ac, H3K4me1, and H3K4me3 under hypoxia at multiple loci within the putative MALAT1 enhancers in breast cancer, we hypothesized that these regions may be in a more open chromatin state under hypoxia and become proximal to each other as well as with the MALAT1 promoter region to facilitate transcription. We tested this possibility using chromosome conformation capture (3C) (Fig. 4A). A dual-digest strategy with the restriction enzymes BamHI and BglII was selected given their complementary overhangs, which yielded 12 restriction sites throughout the MALAT1 genomic locus (Fig. 4B), stopping at −24 kb upstream due to the absence of histone modifications at this locus and further upstream (Fig. 3A). Fragments were named for their distance from the MALAT1 3′ end (fragments A–J), with the fragment containing the MALAT1 gene body omitted (Fig. 4B).
Figure 4.

The 3′ enhancer gains extensive interactions with the MALAT1 promoter as well as upstream enhancers under hypoxia in breast cancer cells. A, experimental outline of the 3C assay. B, diagram of the MALAT1 genetic locus with the indicated BamHI (blue line) and BglII (red line) restriction sites for 3C. The position in kb of each restriction site relative to the MALAT1 TSS is listed at the bottom. The resulting 10 fragments were numbered A–J, by increasing distance from the MALAT1 transcription start site. The 3′ end fragment A containing the 3′ enhancer (red line) was used as the anchor for 3C experiments. The locations of the promoter (orange line), −20 kb enhancer (teal line), and −7 kb enhancer (purple line) are indicated. C, representative gel images of 3C-PCR results in MCF10A and MCF7 under normoxia or hypoxia. D and E, qPCR analyses of the relative interaction frequencies of hypoxia to normoxia for MCF10A (D) and MCF7 (E) between the anchor (3′ enhancer) and fragment B (promoter). Data are represented as mean ± S.D., n = 3, **, p < 0.01.
Comparison of the fragments with the loci of our putative enhancers indicated that the putative −20 kb enhancer is contained within fragments H and I. The vast majority of the putative −7 kb enhancer is contained within fragments D and E. The 3′ enhancer falls mostly into fragment A. The promoter region is contained within the B and C fragments (Fig. 4B). We selected the 3′ enhancer as a 3C anchor point as normoxic epigenetic marker levels were low in this region but sharply increased under hypoxia in MCF7 and the very high density of transcription factors and RNA polymerase II bound at this locus (Fig. 4), which implicates its function in transcriptional regulation, potentially by mediating interaction with the promoter (35, 36).
Our 3C and subsequent PCR using MCF10A and MCF7 cell lines demonstrated that the 3′ enhancer (anchor/fragment A) indeed interacts with upstream fragments in varying degrees in these two cell lines (Fig. 4C and Fig. S3). Under the normoxic condition, the 3′ enhancer demonstrates chromatin–chromatin interactions with the promoter (fragment B) in both cell lines, which demonstrates that chromatin looping between the 3′ enhancer and the promoter regions occurs in both normal and breast cancer cells and this interaction may be important in expression of MALAT1 under normoxic condition. In sharp contrast, the interaction of the anchor with the G fragment was detected only in MCF7 cells, but not in MCF10A cells (Fig. 4C), suggesting this locus is important in cancer cells under normal conditions.
Hypoxia alters long-range chromatin interactions at the MALAT1 locus, resulting in extensive chromatin contacts in breast cancer cells
Because hypoxia altered chromatin modifications at multiple sites upstream and downstream of the MALAT1 gene (Fig. 3, B–D), we next questioned whether hypoxia alters chromatin interactions at the MALAT1 locus. From our 3C-PCR, we found that MCF10A cells did not gain any significant interactions within the examined loci in response to hypoxia. The interaction between the anchor (3′ enhancer) and fragment B (promoter) was even weakened under hypoxia in MCF10A (Fig. 4, C and D, and Fig. S3).
In contrast, in MCF7 cells, the interaction between the anchor (3′ enhancer) and fragment B (promoter) is enhanced under hypoxia. The interaction between the anchor and fragment G was still observed under hypoxia. Interestingly, in addition to these interactions, several novel interactions formed in MCF7 cells under hypoxia; interactions between the anchor and fragments C (part of the upstream promoter), D (−7 kb enhancer), and H (−20 kb enhancer) (Fig. 4, C and E, and Fig. S3). Specificity of these interactions were validated with negative control samples (genomic DNA and unligated samples; Fig. S4). Furthermore, we observed no chromatin–chromatin interactions between the anchor and fragments E, F, I, or J in either cell line, which supports the specificity of the detected interactions. These data demonstrate that novel chromatin–chromatin contacts are formed between MALAT1 enhancers at the MALAT1 locus under hypoxia in MCF7 breast cancer cells, but not in nontumorigenic MCF10A cells.
HIF proteins regulate chromatin–chromatin interactions between the MALAT1 promoter and 3′ enhancer, but not the interactions between enhancers
Having observed a minor but measurable role for HIF-2α in MALAT1 up-regulation in MCF7 cells (Fig. 2), we questioned if this protein could regulate the observed hypoxic chromatin–chromatin interactions. We either overexpressed HIF2α-CA (HIF-2α-P405A/P531A) or treated CoCl2 in MCF7 cells under normoxic conditions and performed 3C-PCR. The results revealed that both HIF-2α overexpression or CoCl2-mediated HIF stabilization increased the chromatin interaction between anchor (3′ enhancer) and fragment B (promoter) (Fig. 5, A, B, D, and E). In line with these results, knockdown of HIF-2α abrogated the effect of hypoxia-mediated chromatin interactions of 3′ enhancer with the promoter region found in fragment B (Fig. 5, C and F). In contrast, HIF-2α does not enhance chromatin–chromatin interactions between the anchor (3′ enhancer) and fragment G (Fig. 5, A–F, and Fig. S5). These results are consistent with our results demonstrating constitutive interactions between these loci (Fig. 4C). Although we observed the interaction between the anchor and fragments C, D, and H under hypoxia in MCF7 cells (Fig. 4C), we did not observe any of those interactions under the condition of HIF-2α overexpression or CoCl2-mediated HIF stabilization (Fig. 5, A and B). These data demonstrate that HIF-2α plays a role in mediating chromatin–chromatin interactions between the MALAT1 promoter and the 3′ enhancer, but not in mediating interactions between other putative enhancers.
Figure 5.

HIF-2α expression levels modulate chromatin looping between the MALAT1 promoter and the 3′ enhancer in MCF7 breast cancer cells. A–C, gel images of 3C-PCR from MCF7 cells transfected with pCDNA-HIF2α-P405A/P531A (HIF2α-CA; +) or an empty vector (−) for 48 h (A), treated with 100 μm CoCl2 (+) or vehicle (−) for 24 h (B), or transfected with HIF2A siRNA (+) or control siRNA (−) for 48 h prior to an additional 24-h incubation under normoxia or hypoxia as indicated (C). D–F, qPCR analyses of the relative interaction frequencies between the anchor (3′ enhancer) and fragments B or G following 48 h of HIF-2α overexpression (HIF-CA) (D), 24 h of 100 μm CoCl2 treatment (E) or 72 total hours of HIF-2α knockdown as described in C (F). G, diagram of the MALAT1 promoter and the 3′ enhancer with the location and the sequence of HIF-binding motifs (HREs) labeled in bold. Two HREs are predicted within the MALAT1 promoter (5′-HRE-1 and 5′HRE-2) and two are predicted within the putative 3′ enhancer (3′-HRE-1 and 3′ HRE-2). H, ChIP-qPCR demonstrates enrichment of HIF-2α under normoxic or 24-h hypoxic conditions in MCF7 cells. Data with bar graphs represent mean ± S.D., n = 3, *, p < 0.05; **, p < 0.01.
To support the notion that HIF-2α plays a role in mediating interaction between the MALAT1 promoter and the 3′ enhancer, we examined whether HIF was recruited to either site under hypoxia. We identified two putative HIF-binding sites (hypoxic response elements; HREs) within the MALAT1 promoter and two HREs within the 3′ enhancer (Fig. 5G). In MCF7 cells, our chromatin immunoprecipitation (ChIP) results revealed that, indeed, HIF-2α was enriched at all four HRE loci (Fig. 5H).
Taken together, our data demonstrate that whereas both nontumorigenic mammary cells and breast cancer cells form chromatin loops at the MALAT1 locus by the interaction between the promoter and 3′ end of the gene, hypoxia triggers further gain of chromatin interactions only in breast cancer cells throughout the MALAT1 locus. This renders extensive chromatin contacts among the promoter and multiple enhancer regions, including the −7 kb enhancer and −20 kb enhancer (Fig. 6). Although the HIF-2α protein can enhance the interaction between the promoter and the 3′ enhancer, the gain of chromatin interactions associated with other upstream enhancers is an HIF-independent event and specifically occurs in breast cancer cells. These findings reveal a mechanism inducing cancer cell-specific MALAT1 up-regulation under hypoxia.
Figure 6.
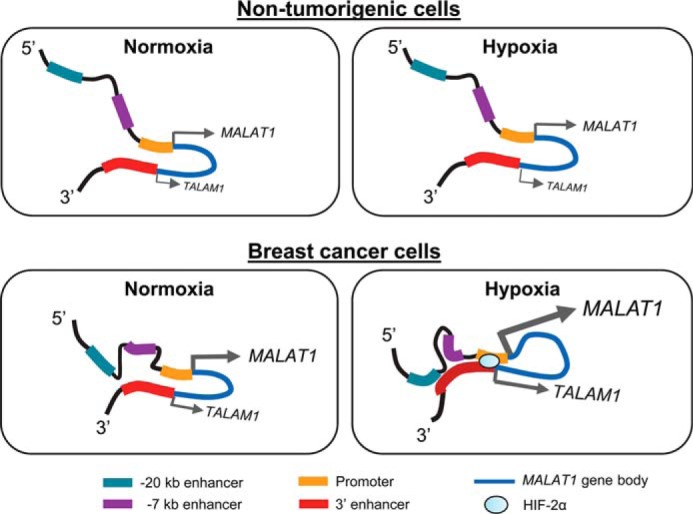
Model of MALAT1 regulation under hypoxia in nontumorigenic cells and breast cancer cells. Top, in nontumorigenic cells, chromatin looping occurs rendering the 3′ enhancer to interact with the MALAT1 promoter under normoxic conditions. Hypoxia does not cause any significant changes in chromatin interaction. MALAT1 expression, as well expression of its antisense transcript TALAM1 is largely unaffected. Bottom, in breast cancer cells, the 3′ enhancer interacts with the MALAT1 promoter as well as the region between −7 and −20 kb enhancers under normoxia. Under hypoxia, the 3′ enhancer gains interactions with multiple loci throughout the MALAT1 locus, including the upstream region of the promoter, −7 kb enhancer, and −20 kb enhancer, which results in extensive chromatin looping. MALAT1 and the antisense transcript TALAM1 are both up-regulated.
Discussion
MALAT1 is an oncogenic lncRNA up-regulated in many solid tumors, which facilitates increased cellular proliferation rates, EMT, and metastasis. Few studies have examined the factors that up-regulate MALAT1 during tumor initiation. Several transcription factors have been shown to regulate MALAT1 transcription (11–17), but these are largely unvalidated in multiple tissues with the exception of p53 and Sp1/Sp3. Environmental stimuli that up-regulate MALAT1 are more well-established, such as transforming growth factor-β signaling in both normal and cancer cells (37–43). Similarly, hypoxia consistently up-regulates MALAT1 in a range of normal and cancerous tissues (9, 22–26); however, the downstream processes from hypoxia that actually result in MALAT1 up-regulation are elusive. In this study, we have demonstrated that hypoxia induces chromatin looping between previously unidentified enhancers located upstream and downstream of the MALAT1 gene in a HIF-independent manner and up-regulates MALAT1 as well as its antisense strand transcript TALAM1 (Fig. 6).
Large scale gene expression changes under hypoxia are regulated through various epigenetic modifying complexes. Interestingly, a large genome-wide increase is consistently noted in the active promoter mark H3K4me3 (32, 44, 45). Our results are consistent with these findings, as we demonstrate hypoxia causes increased H3K4me3 enrichment by ChIP at several enhancer sites throughout the MALAT1 locus in a cancer-specific manner. The active enhancer mark H3K4me1 also increased at a majority of these same loci in cancer cells, supporting the activity of these enhancers under hypoxia.
The most significant findings we present in this study are identification of novel enhancers at the MALAT1 locus and delineation of the chromatin interactions occurring at these potential enhancers under hypoxia. The −20 and −7 kb enhancers appear to be hypoxic-responsive for the cancer condition, as robust chromatin looping was observed between this locus and the 3′ enhancer under hypoxia in breast cancer cells, but not in nontumorigenic mammary epithelial cells. A recent study also identified the downstream region from +8.7 to +17.2 kb relative to the MALAT1 TSS as a putative enhancer, with the 3′ end of this region important for Oct4-mediated up-regulation of MALAT1 in lung cancer (46). This chromatin region largely overlaps our proposed 3′ enhancer, providing support for the importance of this locus in MALAT1 up-regulation in cancer. It would be intriguing to determine whether the −20 and −7 kb enhancers are consistent in MALAT1 regulation in different types of cancer, as well.
The MALAT1 3′ enhancer overlaps the putative promoter of the antisense strand transcript TALAM1, which up-regulates MALAT1 in a positive feedback loop (28). The same genomic regulatory elements are known to have dual functionality, where highly active enhancers can also function as weak promoters (47–49). This model fits well with endogenous expression of TALAM1, which has on average 300-fold lower expression than MALAT1 (28), and our finding of 3′ enhancer and MALAT1 promoter interactions in both nontumorigenic and cancerous cells. This implicates the region downstream of MALAT1 as an important and active enhancer in regulation of this locus.
Although local and global changes to chromatin modifications under hypoxia are well-established, few studies have described changes in chromatin 3D structure and organization under hypoxia. At a genome-wide level, most enhancer–promoter interactions for hypoxic-regulated genes are present in normoxic conditions, with only a handful undergoing compartmental change to the active state upon oxygen deprivation, thereby gaining novel chromatin contacts (50, 51). In a murine cardiac cell model, hypoxia resulted in global chromatin condensation through rapid histone deacetylation compacting nucleosomes, creating novel interactions but largely inactivating chromatin (52). However, these global, average views do not preclude the possibility of local chromatin looping for up-regulation of specific genes under hypoxia, which we observe for MALAT1. For instance, a hypoxic-specific interaction was observed between two enhancers of solute carrier family 2 member 3 (SLC2A3), which increased SLC2A3 gene expression (53). Together, these data indicate that whereas global chromatin structures of hypoxia responsive genes are poised under normoxia, individual gene loci undergo topological changes to facilitate their transcription for hypoxic stress response.
Growing evidence suggests chromatin organization and interactions are altered in cancer and other disease states compared with normal tissue. In breast cancer, recent work has established clear differences in chromatin–chromatin interaction profiles between normal and cancerous cell lines. An enhancer of insulin-like growth factor-binding protein 3 (IGFBP3) had a greater number of long-range chromatin–chromatin interactions in MDA-MB-231 compared with human mammary epithelial cells, particularly with the EGFR gene (54). Our results provide another example of different chromatin interactions in cancer versus noncancerous lines, especially in response to hypoxia.
In this work, we have demonstrated that breast cancer cells undergo specific changes within their chromatin structure to facilitate up-regulation of the lncRNA MALAT1. Hypoxia causes relaxation and opening of chromatin at novel MALAT1 enhancers that specifically interact together in breast cancer to up-regulate MALAT1 in a HIF-independent manner. Our study provides a model for both investigation of genes up-regulated under hypoxia in a HIF-independent manner and changes to chromatin structure. It will be interesting to determine whether this looping occurs in other cancer types or if HIF regulates alternative looping in cell lines such as HeLa where MALAT1 up-regulation is HIF-dependent (23, 25). We predict numerous looping events remain to be discovered between enhancers and promoters of various hypoxic stress response genes and preventing these loops from forming represents a novel therapeutic strategy for treatment of breast cancer.
Experimental procedures
Tissue culture and hypoxia
The nontumorigenic, immortalized mammary cell line MCF10A was obtained from the ATCC and was cultured in MEBM mammary epithelial cell growth basal medium (Lonza) supplemented with 10% FBS, 1% penicillin/streptomycin, 20 ng/ml of epidermal growth factor, 0.5 mg/ml of hydrocortisone, and 10 μg/ml of insulin. Breast cancer cell lines BT474, MCF7, MDA-MB-231, MDA-MB-453, SKBR3, and T47D were obtained from the ATCC (Manassas, VA). MDA-MB-157 and MDA-MB-361 were gifts from Dr. Robert Sobol. All cancer cell lines were maintained in Dulbecco's modified Eagle's medium (Corning, NY) supplemented with 10% FBS + 1% penicillin/streptomycin. All cell lines were maintained in a 37 °C incubator with 5% CO2. Cells were seeded and grown for 48 h under normoxic (21% O2) conditions then either maintained at normoxia or transferred to BD GasPakTM EZ Gas Generating Systems and Supplies:Pouch System (Franklin Lakes, NJ) for hypoxic (∼1% O2) conditions for an additional 24 h.
RNA isolation, cDNA synthesis, and qPCR
Total RNA was extracted using Total RNA Isolation (TRI) reagent, according to the manufacturer's instructions (Sigma). cDNA was generated using the SuperScript III Reverse Transcription Kit (Invitrogen), according to the manufacturer's instructions. The reaction was stopped by incubation at 85 °C for 5 min, then 2 units of RNase H were added for a 20-min incubation at 37 °C. qPCR was performed in triplicate reactions using 2× iTaq Universal SYBR Green Supermix (Bio-Rad) on a Bio-Rad CFX Connect Real-Time System thermocycler. All qPCR were normalized to YWHAZ expression. Primers used are listed in Table S1.
Northern blotting
Five micrograms of total RNA was analyzed by gel electrophoresis in a 1% formaldehyde gel. RNA was transferred to a nylon membrane overnight by capillary action in a 20× SSC buffer. RNA was UV cross-linked and prehybridized with Ultrahyb Hybridization Buffer (Invitrogen). Probes were 3′ labeled with digoxigenin using the DIG Oligonucleotide Labeling Kit (Roche Applied Science), according to the manufacturer's protocol. Probes and RNA were hybridized overnight with gentle rocking at 42 °C. Membranes were washed using the DIG Wash and Block Buffer Set (Roche), according to the manufacturer's instructions. RNA was detected with anti-digoxigen-AP, Fab fragments (Roche), and incubation with disodium 3-(4-methoxyspiro {1,2-dioxetane-3,2′-(5′-chloro)tricyclo [3.3.1.13,7]decan}-4-yl)phenyl phosphate.
Transfection of siRNA and cDNA constructs
Small interfering RNA (siRNA) were purchased from Sigma targeting HIF-1α or HIF-2α and reconstituted in nuclease-free water. The plasmid HA-HIF2α-P405A/P531A-pcDNA3 encoding a constitutively active mutant of HIF-2α was obtained through Addgene (Addgene number 18956) (30). Transfection reactions were prepared in the same way. siRNA was mixed with Opti-MEM (ThermoFisher), combined with Lipofectamine RNAiMax transfection reagent (Invitrogen) and added to the culture media. The HIF-2α construct was mixed with Opti-MEM, combined with Lipofectamine 3000 transfection reagent (Invitrogen), and added to culture media. Both siRNA and construct transfections were incubated at 37 °C for ∼8 h before media change. For HIF knockdown, cultures were grown for 48 h at normoxia, then maintained at 21% O2 or moved to hypoxia for an additional 24 h prior to protein and RNA isolation and 3C analysis. For HIF-2α overexpression, cells were grown for 48 h prior to protein and RNA isolation. siRNA sequences are listed in Table S2.
Protein extraction and Western Blotting
Cells were lysed in an equal volume of 1% Nonidet P-40 buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl, 1% Nonidet P-40) with ×20 PhosphoStop and ×50 protease inhibitor mixture (Roche). Protein was quantified by Pierce Coomassie Protein Assay Reagent (ThermoFisher Scientific), then boiled in a 1× SDS solution. Protein was analyzed by 7–10% SDS-PAGE gel, then transferred to methanol-charged Immobilon PVDF Transfer Membrane (Millipore, Burlington, MA) and blocked in 5% skim milk in 1× TBST prior to overnight incubation with primary antibodies (Table S3). Following washing and secondary antibody incubation at room temperature, protein bands were visualized using Clarity Western ECL Substrate (Bio-Rad).
Chromosome conformation capture (3C)
3C assays were performed as previously described with modifications (55, 56). Cells were fixed in 1% formaldehyde, quenched with glycine, then lysed with lysis buffer (10 mm Tris-HCl, pH 8, 10 mm NaCl, 0.2% Nonidet P-40, protease inhibitor mixture). The lysate solution was homogenized by 15 strokes with a Dounce homogenizer and then centrifuged to collect nuclei. After washing, nuclei were counted and an equal number (0.5–1 × 107) for normoxic and hypoxic conditions were collected by centrifugation. Nuclei were resuspended in 0.5 ml of 1.2× New England Biolabs buffer 3.1 and proteins that are not cross-linked to the DNA were removed by adding 15 μl of 10% SDS and incubated at 37 °C for 1 h followed by quenching by adding 50 μl of 20% Triton X-100. Fifty-five μl was removed and set aside to determine digestion efficiency (no-digest control). Restriction enzymes BamHI (5,000 units) and BglII (1,000 units) were added and then incubated at 37 °C overnight with 950 rpm shaking.
Fifty μl of 1× New England Biolabs buffer 3.1 and 100 μl of 10% SDS were added and incubated at 65 °C for 30 min with 950 rpm shaking. Sixty μl was removed for no-ligation control and kept at −20 °C. Chromatin was combined with 1.1× T4 ligation buffer (550 μl of 10× T4 ligation buffer, 275 μl of 20% Triton X-100, 3.5 ml of water), then incubated at 37 °C for 1 h. For ligation, T4 ligase (800 units) was added and incubated at 16 °C for 4 h. Proteinase K was added to a final concentration of 100 μg/ml and the samples were incubated overnight at 65 °C for reverse cross-linking. After RNase A (40 μg/ml) treatment, DNA was purified using phenol/chloroform, precipitated by adding sodium acetate, pH 5.2, and washed with 70% ethanol.
The no-ligation control was purified by the Qiagen PCR Purification Kit according to the manufacturer's instructions. The concentration of dsDNA was determined by Qubit 3.0 Fluorometer (Invitrogen). Digestion efficiency was determined by PCR using primers listed in Table S1. Successfully digested 3C samples were centrifuged and washed in 70% ethanol, then air-dried and reconstituted in water. 3C sample interaction frequencies were conducted in triplicate by PCR and products were analyzed on a 2% agarose gel.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as previously described (57). Briefly, cells were fixed with 1% formaldehyde, quenched with glycine, then lysed with Farnham buffer (5 mm HEPES, pH 8.0, 85 mm KCl, 20 μl of 50× Protease Inhibitor Mixture, Roche). The lysate solution was centrifuged and the pellet resuspended in RIPA buffer (50 mm Tris, pH 8, 150 mm NaCl, 1% sodium deoxycholate, 1 mm EDTA, 0.1% SDS, 1% Triton X-100) followed by sonication. Successfully sheared chromatin was incubated overnight at 4 °C with rotation with antibodies and Dynabead Protein A or G (Invitrogen) (Table S3). Beads were washed four times in lithium chloride buffer (100 mm Tris-HCl, pH 7.5, 500 mm lithium chloride, 1% Nonidet P-40, 1% sodium deoxycholate) then washed once with TE Buffer (10 mm Tris-HCl, pH 7.5, 0.1 mm EDTA). DNA then was eluted in buffer containing 1% SDS, 0.1 m NaHCO3 with proteinase K, then cleaned up with Qiagen PCR Purification Protocol (Qiagen), according to the manufacturer's instructions.
Statistical analysis
All experiments shown are the result of at least three biological replicates. Data are presented as the mean ± S.D. with p values calculated by 2-tailed t test. All statistics were calculated using GraphPad Prism software version 7.02 (GraphPad).
Author contributions
J. K. S., J.-H. K., M. T., and E.-Y. E. A. conceptualization; J. K. S., J.-H. K., L. V., A. R., H. K. G., and E.-Y. E. A. data curation; J. K. S. and J.-H. K. formal analysis; J. K. S., J.-H. K., L. V., A. R., and E.-Y. E. A. validation; J. K. S., S.-T. S. L., M. T., and E.-Y. E. A. investigation; J. K. S., J.-H. K., and E.-Y. E. A. methodology; J. K. S. and E.-Y. E. A. writing-original draft; J. K. S., J.-H. K., S.-T. S. L., M. T., and E.-Y. E. A. writing-review and editing; S.-T. S. L., M. T., and E.-Y. E. A. funding acquisition; M. T. and E.-Y. E. A. supervision; E.-Y. E. A. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Erik Flemington for critical reading of the manuscript and Dr. Robert Sobol for providing the MDA-MB-157 and MDA-MB-361 cell lines.
This work was supported by the Mitchell Cancer Institute (to E. Y. E. A.) and National Institutes of Health Grants R21 CA185818 (to E. Y. E. A.), R01 CA190688 (to E. Y. E. A.), R01 CA149646 (to M. T.), and R01 HL136432 (to S. T. S. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Tables S1–S3.
- MALAT1
- metastasis-associated lung adenocarcinoma transcript 1
- 3C
- chromosome conformation capture
- SSP
- strand-specific primer
- TSS
- transcription start site
- HRE
- hypoxic response elements
- SLC2A3
- solute carrier family 2 member 3
- qPCR
- quantitative PCR
- lncRNA
- long noncoding RNA
- HIF
- hypoxia-inducible factor
- ChIP
- chromatin immunoprecipitation.
References
- 1. Buecker C., and Wysocka J. (2012) Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 28, 276–284 10.1016/j.tig.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shlyueva D., Stampfel G., and Stark A. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- 3. Allen B. L., and Taatjes D. J. (2015) The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol. 16, 155–166 10.1038/nrm3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., and Young R. A. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 10.1038/nature09380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Merkenschlager M., and Odom D. T. (2013) CTCF and cohesin: linking gene regulatory elements with their targets. Cell 152, 1285–1297 10.1016/j.cell.2013.02.029 [DOI] [PubMed] [Google Scholar]
- 6. Lawrence M., Daujat S., and Schneider R. (2016) Lateral thinking: how histone modifications regulate gene expression. Trends Genet. 32, 42–56 10.1016/j.tig.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 7. Tripathi V., Shen Z., Chakraborty A., Giri S., Freier S. M., Wu X., Zhang Y., Gorospe M., Prasanth S. G., Lal A., and Prasanth K. V. (2013) Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 9, e1003368 10.1371/journal.pgen.1003368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gutschner T., Hämmerle M., Eissmann M., Hsu J., Kim Y., Hung G., Revenko A., Arun G., Stentrup M., Gross M., Zörnig M., MacLeod A. R., Spector D. L., and Diederichs S. (2013) The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 73, 1180–1189 10.1158/0008-5472.CAN-12-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michalik K. M., You X., Manavski Y., Doddaballapur A., Zörnig M., Braun T., John D., Ponomareva Y., Chen W., Uchida S., Boon R. A., and Dimmeler S. (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 114, 1389–1397 10.1161/CIRCRESAHA.114.303265 [DOI] [PubMed] [Google Scholar]
- 10. Yoshimoto R., Mayeda A., Yoshida M., and Nakagawa S. (2016) MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta 1859, 192–199 10.1016/j.bbagrm.2015.09.012 [DOI] [PubMed] [Google Scholar]
- 11. Zhao Y., Yang Y., Trovik J., Sun K., Zhou L., Jiang P., Lau T. S., Hoivik E. A., Salvesen H. B., Sun H., and Wang H. (2014) A novel Wnt regulatory axis in endometrioid endometrial cancer. Cancer Res. 74, 5103–5117 10.1158/0008-5472.CAN-14-0427 [DOI] [PubMed] [Google Scholar]
- 12. Hirata H., Hinoda Y., Shahryari V., Deng G., Nakajima K., Tabatabai Z. L., Ishii N., and Dahiya R. (2015) Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res. 75, 1322–1331 10.1158/0008-5472.CAN-14-2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao G., Su Z., Song D., Mao Y., and Mao X. (2016) The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-κB. FEBS Lett. 590, 2884–2895 10.1002/1873-3468.12315 [DOI] [PubMed] [Google Scholar]
- 14. Huang Z., Huang L., Shen S., Li J., Lu H., Mo W., Dang Y., Luo D., Chen G., and Feng Z. (2015) Sp1 cooperates with Sp3 to upregulate MALAT1 expression in human hepatocellular carcinoma. Oncol. Rep. 34, 2403–2412 10.3892/or.2015.4259 [DOI] [PubMed] [Google Scholar]
- 15. Li S., Wang Q., Qiang Q., Shan H., Shi M., Chen B., Zhao S., and Yuan L. (2015) Sp1-mediated transcriptional regulation of MALAT1 plays a critical role in tumor. J. Cancer Res. Clin. Oncol. 141, 1909–1920 10.1007/s00432-015-1951-0 [DOI] [PubMed] [Google Scholar]
- 16. Jeffers L. K., Duan K., Ellies L. G., Seaman W. T., Burger-Calderon R. A., Diatchenko L. B., and Webster-Cyriaque J. (2013) Correlation of transcription of MALAT-1, a novel noncoding RNA, with deregulated expression of tumor suppressor p53 in small DNA tumor virus models. J. Cancer Ther. 4, 10.4236/jct.2013.43094 10.4236/jct.2013.43094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma X. Y., Wang J. H., Wang J. L., Ma C. X., Wang X. C., and Liu F. S. (2015) Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. BMC Genomics 16, 676 10.1186/s12864-015-1881-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kenneth N. S., and Rocha S. (2008) Regulation of gene expression by hypoxia. Biochem. J. 414, 19–29 10.1042/BJ20081055 [DOI] [PubMed] [Google Scholar]
- 19. Favaro E., Lord S., Harris A. L., and Buffa F. M. (2011) Gene expression and hypoxia in breast cancer. Genome Med. 3, 55 10.1186/gm271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Semenza G. L. (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dengler V. L., Galbraith M., and Espinosa J. M. (2014) Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49, 1–15 10.3109/10409238.2013.838205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choudhry H., Schödel J., Oikonomopoulos S., Camps C., Grampp S., Harris A. L., Ratcliffe P. J., Ragoussis J., and Mole D. R. (2014) Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep. 15, 70–76 10.1002/embr.201337642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lelli A., Nolan K. A., Santambrogio S., Gonçalves A. F., Schönenberger M. J., Guinot A., Frew I. J., Marti H. H., Hoogewijs D., and Wenger R. H. (2015) Induction of long noncoding RNA MALAT1 in hypoxic mice. Hypoxia 3, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luo F., Sun B., Li H., Xu Y., Liu Y., Liu X., Lu L., Li J., Wang Q., Wei S., Shi L., Lu X., Liu Q., and Zhang A. (2016) A MALAT1/HIF-2α feedback loop contributes to arsenite carcinogenesis. Oncotarget 7, 5769–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sallé-Lefort S., Miard S., Nolin M. A., Boivin L., Paré M. È., Debigaré R., Picard F. (2016) Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int. J. Oncol. 49, 1731–1736 10.3892/ijo.2016.3630 [DOI] [PubMed] [Google Scholar]
- 26. Tee A. E., Liu B., Song R., Li J., Pasquier E., Cheung B. B., Jiang C., Marshall G. M., Haber M., Norris M. D., Fletcher J. I., Dinger M. E., and Liu T. (2016) The long noncoding RNA MALAT1 promotes tumor-driven angiogenesis by up-regulating pro-angiogenic gene expression. Oncotarget 7, 8663–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pelechano V., and Steinmetz L. M. (2013) Gene regulation by antisense transcription. Nat. Rev. Genet. 14, 880–893 10.1038/nrg3594 [DOI] [PubMed] [Google Scholar]
- 28. Zong X., Nakagawa S., Freier S. M., Fei J., Ha T., Prasanth S. G., and Prasanth K. V. (2016) Natural antisense RNA promotes 3′ end processing and maturation of MALAT1 lncRNA. Nucleic Acids Res. 44, 2898–2908 10.1093/nar/gkw047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stiehl D. P., Bordoli M. R., Abreu-Rodríguez I., Wollenick K., Schraml P., Gradin K., Poellinger L., Kristiansen G., and Wenger R. H. (2012) Non-canonical HIF-2α function drives autonomous breast cancer cell growth via an AREG–EGFR/ErbB4 autocrine loop. Oncogene 31, 2283–2297 [DOI] [PubMed] [Google Scholar]
- 30. Yan Q., Bartz S., Mao M., Li L., and Kaelin W. G. (2007) The hypoxia-inducible factor 2α N-terminal and C-terminal transactivation domains cooperate to promote renal tumorigenesis in vivo. Mol. Cell Biol. 27, 2092–2102 10.1128/MCB.01514-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yuan Y., Hilliard G., Ferguson T., and Millhorn D. E. (2003) Cobalt inhibits the interaction between hypoxia-inducible factor-α and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-α. J. Biol. Chem. 278, 15911–15916 10.1074/jbc.M300463200 [DOI] [PubMed] [Google Scholar]
- 32. Johnson A. B., Denko N., and Barton M. C. (2008) Hypoxia induces a novel signature of chromatin modifications and global repression of transcription. Mutat. Res. 640, 174–179 10.1016/j.mrfmmm.2008.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou X., Sun H., Chen H., Zavadil J., Kluz T., Arita A., and Costa M. (2010) Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 70, 4214–4221 10.1158/0008-5472.CAN-09-2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pekowska A., Benoukraf T., Zacarias-Cabeza J., Belhocine M., Koch F., Holota H., Imbert J., Andrau J. C., Ferrier P., and Spicuglia S. (2011) H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 30, 4198–4210 10.1038/emboj.2011.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ansari A., and Hampsey M. (2005) A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 19, 2969–2978 10.1101/gad.1362305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. O'Sullivan J. M., Tan-Wong S. M., Morillon A., Lee B., Coles J., Mellor J., and Proudfoot N. J. (2004) Gene loops juxtapose promoters and terminators in yeast. Nat. Genet. 36, 1014–1018 10.1038/ng1411 [DOI] [PubMed] [Google Scholar]
- 37. Wu Y., Liu X., Zhou Q., Huang C., Meng X., Xu F., and Li J. (2015) Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol. Appl. Pharmacol. 289, 163–176 10.1016/j.taap.2015.09.028 [DOI] [PubMed] [Google Scholar]
- 38. Singh K. K., Matkar P. N., Quan A., Mantella L.-E., Teoh H., Al-Omran M., and Verma S. (2016) Investigation of TGF-β-induced long noncoding RNAs in endothelial cells. Int. J. Vasc. Med. 2016, 2459687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fan Y., Shen B., Tan M., Mu X., Qin Y., Zhang F., and Liu Y. (2014) TGF-β–induced upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin. Cancer Res. 20, 1531–1541 10.1158/1078-0432.CCR-13-1455 [DOI] [PubMed] [Google Scholar]
- 40. Zou A., Liu R., and Wu X. (2016) Long non-coding RNA MALAT1 is up-regulated in ovarian cancer tissue and promotes SK-OV-3 cell proliferation and invasion. Neoplasma 63, 865–872 10.4149/neo_2016_605 [DOI] [PubMed] [Google Scholar]
- 41. Wu Y., Zhang L., Zhang L., Wang Y., Li H., Ren X., Wei F., Yu W., Liu T., Wang X., Zhou X., Yu J., and Hao X. (2015) Long non-coding RNA HOTAIR promotes tumor cell invasion and metastasis by recruiting EZH2 and repressing E-cadherin in oral squamous cell carcinoma. Int. J. Oncol. 46, 2586–2594 10.3892/ijo.2015.2976 [DOI] [PubMed] [Google Scholar]
- 42. Jiao F., Hu H., Han T., Yuan C., Wang L., Jin Z., Guo Z., and Wang L. (2015) Long noncoding RNA MALAT-1 enhances stem cell-like phenotypes in pancreatic cancer cells. Int. J. Mol. Sci. 16, 6677–6693 10.3390/ijms16046677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang R., Hardin H., Huang W., Chen J., Asioli S., Righi A., Maletta F., Sapino A., and Lloyd R. V. (2017) MALAT1 long non-coding RNA expression in thyroid tissues: analysis by in situ hybridization and real-time PCR. Endocr. Pathol. 28, 7–12 10.1007/s12022-016-9453-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prickaerts P., Adriaens M. E., van den Beucken T., Koch E., Dubois L., Dahlmans V. E. H., Gits C., Evelo C. T. A., Chan-Seng-Yue M., Wouters B. G., and Voncken J. W. (2016) Hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigenetics Chromatin 9, 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang S., Park B., Choi K., Moon Y., Lee H. Y., and Park H. (2016) Hypoxic reprograming of H3K27me3 and H3K4me3 at the INK4A locus. FEBS Lett. 590, 3407–3415 10.1002/1873-3468.12375 [DOI] [PubMed] [Google Scholar]
- 46. Jen J., Tang Y. A., Lu Y. H., Lin C. C., Lai W. W., and Wang Y. C. (2017) Oct4 transcriptionally regulates the expression of long non-coding RNAs NEAT1 and MALAT1 to promote lung cancer progression. Mol. Cancer 16, 104 10.1186/s12943-017-0674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andersson R., Sandelin A., and Danko C. G. (2015) A unified architecture of transcriptional regulatory elements. Trends Genet. 31, 426–433 10.1016/j.tig.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 48. Mikhaylichenko O., Bondarenko V., Harnett D., Schor I. E., Males M., Viales R. R., and Furlong E. E. M. (2018) The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 32, 42–57 10.1101/gad.308619.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Medina-Rivera A., Santiago-Algarra D., Puthier D., and Spicuglia S. (2018) Widespread enhancer activity from core promoters. Trends Biochem. Sci. 43, 452–468 10.1016/j.tibs.2018.03.004 [DOI] [PubMed] [Google Scholar]
- 50. Platt J. L., Salama R., Smythies J., Choudhry H., Davies J. O., Hughes J. R., Ratcliffe P. J., and Mole D. R. (2016) Capture-C reveals preformed chromatin interactions between HIF-binding sites and distant promoters. EMBO Rep. 17, 1410–1421 10.15252/embr.201642198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Niskanen H., Tuszynska I., Zaborowski R., Heinäniemi M., Ylä-Herttuala S., Wilczynski B., and Kaikkonen M. U. (2018) Endothelial cell differentiation is encompassed by changes in long range interactions between inactive chromatin regions. Nucleic Acids Res. 46, 1724–1740 10.1093/nar/gkx1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kirmes I., Szczurek A., Prakash K., Charapitsa I., Heiser C., Musheev M., Schock F., Fornalczyk K., Ma D., Birk U., Cremer C., and Reid G. (2015) A transient ischemic environment induces reversible compaction of chromatin. Genome Biol. 16, 246 10.1186/s13059-015-0802-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mimura I., Nangaku M., Kanki Y., Tsutsumi S., Inoue T., Kohro T., Yamamoto S., Fujita T., Shimamura T., Suehiro J., Taguchi A, Kobayashi M., Tanimura K., Inagaki T., Tanaka T., Hamakubo T., Sakai J., Aburatani H., Kodama T., and Wada Y. (2012) Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia-inducible factor 1 and KDM3A. Mol. Cell Biol. 32, 3018–3032 10.1128/MCB.06643-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zeitz M. J., Ay F., Heidmann J. D., Lerner P. L., Noble W. S., Steelman B. N., and Hoffman A. R. (2013) Genomic interaction profiles in breast cancer reveal altered chromatin architecture. PLoS ONE 8, e73974 10.1371/journal.pone.0073974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gheldof N., Leleu M., Noordermeer D., Rougemont J., and Reymond A. (2012) Detecting long-range chromatin interactions using the chromosome conformation capture sequencing (4C-seq) method. in Gene Regulatory Networks: Methods and Protocols (Deplancke B., and Gheldof N., eds) pp. 211–225, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 56. Sexton T., Kurukuti S., Mitchell J. A., Umlauf D., Nagano T., and Fraser P. (2012) Sensitive detection of chromatin coassociations using enhanced chromosome conformation capture on chip. Nat. Protocols 7, 1335–1350 10.1038/nprot.2012.071 [DOI] [PubMed] [Google Scholar]
- 57. Kim J. H., Baddoo M. C., Park E. Y., Stone J. K., Park H., Butler T. W., Huang G., Yan X., Pauli-Behn F., Myers R. M., Tan M., Flemington E. K., Lim S. T., and Ahn E. Y. (2016) SON and its alternatively spliced isoforms control MLL complex-mediated H3K4me3 and transcription of leukemia-associated genes. Mol. Cell 61, 859–873 10.1016/j.molcel.2016.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chan H. L., Beckedorff F., Zhang Y., Garcia-Huidobro J., Jiang H., Colaprico A., Bilbao D., Figueroa M. E., LaCava J., Shiekhattar R., and Morey L. (2018) Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 9, 3377 10.1038/s41467-018-05728-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.