

Abstract
Purpose
To review the clinical and laboratory spectrum of RAG gene defects in humans, and discuss the mechanisms underlying phenotypic heterogeneity, the basis of immune dysregulation, and the current and perspective treatment modalities.
Methods
Literature review and analysis of medical records
Results
RAG gene defects in humans are associated with a surprisingly broad spectrum of clinical and immunological phenotypes. Correlation between in vitro recombination activity of the mutant RAG proteins and the clinical phenotype has been observed. Altered T and B cell development in this disease is associated with defects of immune tolerance. Hematopoietic cell transplantation is the treatment of choice for the most severe forms of the disease, but a high rate of graft failure has been observed.
Conclusions
Phenotypic heterogeneity of RAG gene defects in humans may represent a diagnostic challenge. There is a need to improve treatment for severe, early-onset forms of the disease. Optimal treatment modalities for patients with delayed-onset disease presenting with autoimmunity and/or inflammation remain to be defined.
Keywords: Recombinase-activating genes, Immunodeficiency, autoimmunity, immunological tolerance, hematopoietic stem cell transplantation
Introduction
The recombination-activating genes (RAG1 and 2) encode for lymphoid-specific proteins that play a crucial role in the early stages of T and B cell development. RAG proteins initiate recombination of the variable (V), diversity (D), and joining (J) genes at the T cell receptor (TCR) and immunoglobulin loci (V(D)J recombination process), allowing for the generation of T and B cells with a broad antigen recognition specificity [1, 2]. In particular, in developing T and B cells, RAG1 and 2 proteins form a tetrameric complex that recognizes the recombination signal sequences (RSSs) that flank coding V, D, and J genes [3]. The RAG complex introduces DNA double-strand breaks (DSBs) at the junction between the RSS and a coding element [4]. Joining of targeted coding genes (D-J, V-DJ, and V-J joining) is then accomplished by proteins of the nonhomologous end-joining (NHEJ) DNA repair machinery.
In mice, targeted biallelic disruption of either one of the Rag genes results in the arrest of T and B cell development due to the inability to initiate V(D)J rearrangements. In particular, T cell maturation is arrested at the CD4− CD8− (double-negative [DN]) stage prior to expression of pre-TCR and TCR complex [5, 6]. B cell development in the marrow is also blocked at the pro-B cell stage, when DNA rearrangement is initiated at the immunoglobulin heavy chain locus. Consistent with these defects of T and B cell development, animals with null Rag mutations have exceptionally small to absent thymuses and hypocellular spleens [5].
Biallelic RAG1 and RAG2 mutations in humans have been associated with a wide range of clinical and immunological phenotypes [7]. While null mutations result in severe combined immunodeficiency with the absence of T and B lymphocytes (T− B− NK+ SCID) [ 8 ], hypomorphic mutations allowing for residual protein function are associated with Omenn syndrome (OS) [9–11], atypical SCID (AS) [8, 12], delayed onset combined immunodeficiency with granulomas and/or autoimmunity (CID-G/AI) [13, 14], and other delayed-onset atypical presentations (Table 1).
Table 1.
Classification criteria
| SCID: absence or very low number of T cells (CD3+ T cells < 300/mL) and no or very low T cell function (< 10% of lower limit of normal) as measured by response to PHA or maternal T cell engraftement. |
| Omenn syndrome (OS): generalized skin rash, absence of maternal engraftment detectable CD3+ T cells, ≥ 300/mL. Absent or low (≤ 30% of normal) T cell proliferation to antigens to which the patient had been exposed. |
| Atypical SCID (AS): reduced number of CD3+ T cells for age; up to 2 years < 1000/mL, for > 2 up to 4 years < 800/mL, for > 4 years < 600/mL. Absence of maternal engraftment and < 30% of lower limit of normal T cell function (as measured by response to PHA). |
| CID-G/AI: delayed age of onset with CD3+ T cell numbers that follow atypical SCID criteria and/or the presence of AI phenomena other that isolated cytopenia. |
Here, we review clinical, molecular, and immunological features of 429 patients [8, 12, 15–21] with defined RAG mutations (RAG1, N = 303; RAG2, N = 126) that have been reported in the literature or are known to us, and whose phenotypic distribution according to criteria reported in Table 1 was as follows: SCID, N = 140; OS, N = 155; AS, N = 66; and CID-G/AI and other delayed-onset forms of the disease, N = 68. We also discuss the mechanisms underlying phenotypical variability and the immune dysregulation of human RAG deficiency and review current and perspective therapeutic options.
T− B− NK+ SCID, Omenn Syndrome, and Atypical SCID: Clinical Features
SCID includes a heterogeneous group of disorders that are characterized by the profound impairment in T and B cell development, and that are associated with life-threatening infections since early in infancy [22]. According to criteria elaborated by the Primary Immune Deficiency Treatment Consortium (PIDTC), SCID is defined by the absence or very low number of T cells (CD3+ T cells < 300/mL) and no or very low T cell function (< 10% of lower limit of normal) as measured by response to PHA, or by the demonstration of maternal T cell engraftment.
In 1996, RAG mutations were identified as the main cause of T− B− NK+ SCID with normal cellular radiosensitivity [8]. In contrast, mutations in genes encoding various components of the NHEJ pathway cause T− B− NK+ SCID with increased cellular radiosensitivity [23].
OS denominates a different phenotype that was first described in 1965 [24], and hypomorphic RAG mutations were identified as the leading cause of OS in 1998 [12]. Patients with OS present in the first weeks of life with generalized erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia, and severe hypogammaglobulinemia but increased IgE levels. These patients typically have absent B cells and expanded, activated, and oligoclonal autologous T cells that infiltrate the skin, gut, liver, and other organs [10, 11].
OS must be distinguished from “Omenn-like syndrome,” a condition in which transplacental passage of maternal T cells into a SCID fetus causes a graft-versus-host (GvH) reaction, with expansion of maternally derived T cells that infiltrate target organs. However, while maternal T cell engraftment is very common in SCID babies [25], Omenn-like disease is rarely observed in these circumstances.
Although T− B− NK+ SCID and OS represent two distinct phenotypes associated with severe RAG mutations, they may both occur in affected members from the same family [10], suggesting different expressivity of the disease. Furthermore, evolution of SCID into OS has been reported following viral infections [26] or upon emergence of T cell clones carrying somatic reversion of RAG gene mutations [27, 28].
Finally, according to PIDTC diagnostic criteria, “atypical SCID” (AS) refers to a condition presenting in infancy in which the count of circulating autologous T cells, albeit lower than normal, is higher than 300 cells/μL, and their function is reduced but not absent (Table 1). Patients with AS may share some features of OS, including skin rash; however, they lack the severe lymphoproliferation that accounts for lymphadenopathy, and hepatosplenomegaly of OS [12]. A distinctive form of AS, associated with expansion of T cells expressing the γδ form of the TCR and the frequent occurrence of autoimmune cytopenias, has been reported in RAG-mutated infants following disseminated cytomegalovirus (CMV) infection [29, 30]. This condition is also characterized by a high risk for Epstein–Barr virus-driven lymphoproliferation [29, 30].
From a clinical standpoint, patients with typical and atypical SCID or OS are highly susceptible to life-threatening viral, fungal, parasitic, and bacterial infections since early after birth. Chronic diarrhea with failure to thrive, Pneumocystis jiroveci pneumonia, respiratory or systemic infections due to cytomegalovirus, adenovirus, and parainfluenza virus type 3, and muco-cutaneous candidiasis are particularly common. In patients with OS in particular, loss of the skin and gut barrier is associated with a higher risk of bacterial infections. In RAG-mutated patients presenting with SCID, OS, or AS, administration of live vaccines may cause serious complications, including severe diarrhea after rotavirus vaccine and disseminated infection after Bacillus Calmette–Guerin (BCG) inoculation.
Autoimmune manifestations are rare in RAG-deficient babies presenting with typical SCID. However, cytopenias, and autoimmune hemolytic anemia (AIHA) in particular, have been reported in more than half of the patients with AS (Table 2). Vasculitis may also occur in patients presenting with AS and may lead to serious consequences such as digital necrosis [31], whereas organ-specific autoimmunity is more rare (Table 2). Because of the severity of the disease, RAG mutations presenting with SCID, OS, or AS lead inevitably to early death, unless patients receive hematopoietic stem cell transplantation (HSCT) [23, 32].
Table 2.
Autoimmune manifestations in 134 patients with CID-G/AI or with AS
| Manifestation | CID-G/AI (n = 68) N (%) | AS (n = 66) N (%) |
|---|---|---|
| Cytopenias | 36 (53) | 38 (57.6) |
| AIHA | 25 (36.7) | 33 (50) |
| ITP | 17 (25) | 11 (16.6) |
| AIN | 15 (22.1) | 8 (12.2) |
| IBD, enteropathy | 9 (13.2) | – |
| Vitiligo | 8 (11.7) | – |
| Hypo/hyper-thyrodism | 7 (10.3) | 1 (1.5) |
| Vasculitis | 6 (8.8) | 3 (4.5) |
| Alopecia | 5 (7.4) | 1 (1.5) |
| Liver disease | 4 (5.9) | 1 (1.5) |
| Autoimmune neuropathies | 4 (5.9) | – |
| Autoimmune myopathies | 4 (5.9) | – |
| Nephritis | 3 (4.4) | 1 (1.5) |
AIHA, autoimmune hemolytic anemia; AIN, autoimmune neutropenia; AS, atypical SCID; CID-G/AI, combined immune deficiency with granuloma and/or autoimmunity; IBD, inflammatory bowel disease; ITP, immune thrombocytopenia
With the availability of newborn screening, it has become possible to establish more precisely how frequently RAG mutations cause SCID, OS, or AS. In particular, in the USA, where SCID and related disorders have a frequency of 1 in 58,000 live births [33], RAG deficiency accounts for only 11.5% of cases of typical SCID, but as many as 41.8% of all cases of OS and AS [34]. A much higher frequency of RAG deficiencies has been reported in countries with increased rate of parental consanguinity [35].
RAG Deficiency Manifesting as Combined Immunodeficiency with Granuloma and/or Autoimmunity (CID-G/AI) or Other Delayed-Onset Phenotypes
In 2008, Schuetz et al. reported on three unrelated girls presenting with excessive granuloma formation in the skin, mucous membranes, and internal organs, and who suffered from severe viral infections and B cell lymphoma. Despite low numbers of T and B cells, surprisingly, they had a diverse TCR repertoire and were able to respond to vaccinations [13]. Since then, several other RAG-mutated patients have been reported with clinical onset later in childhood or even in adulthood, and mainly characterized by autoimmunity (including vasculitis, nephritis, cytopenias, vitiligo, psoriasis, myasthenia gravis, and Guillain–Barré syndrome) and/or inflammatory manifestations, such as granulomatous lesions that may affect various organs [14, 31, 36–41], and sterile chronic multifocal osteomyelitis (CRMO) [42]. Because of these characteristics, this condition has been also referred to as “combined immunodeficiency with granuloma and/or autoimmunity” (CID-G/AI) [37]. In a series of 68 patients with CID-G/AI reviewed here, autoimmune cytopenias, variably affecting various lineages, were seen in 53% of the patients (Table 2). However, manifestations of organ-specific autoimmunity were also common. Granulomatous lesions were noticed in 24–68 patients (35.3%). The skin and lungs were more frequently involved; however, granulomas can affect various tissues, and often more than one in the same patient (Fig. 1).
Fig. 1.
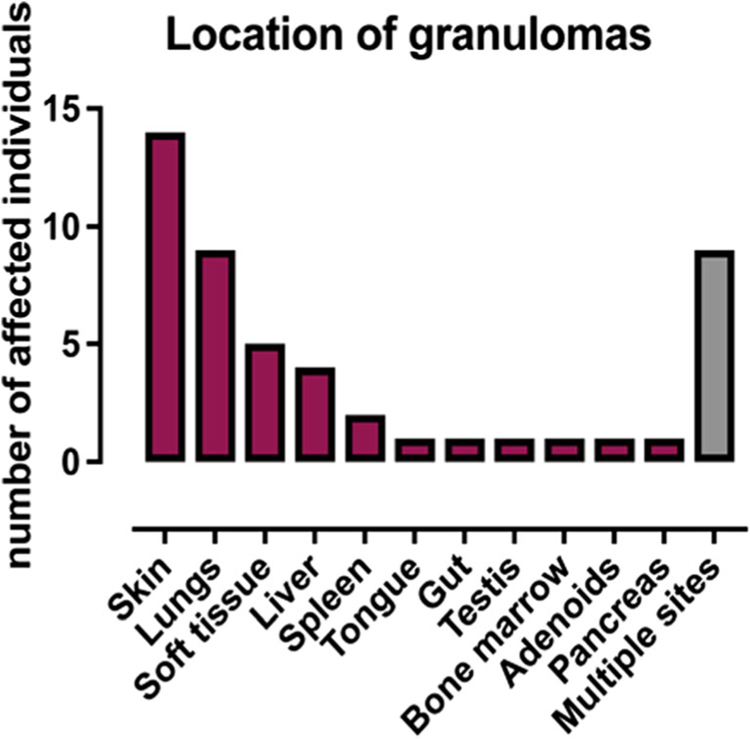
Location of granulomas in 68 patients with combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI)
Life-threatening infections early in childhood are infrequent in patients with CID-G/AI. However, they frequently have a history of recurrent sinopulmonary infections, often leading to bronchiectasis. In addition, severe herpesviridae infections (especially VZV, CMV, and EBV), molluscum, and warts due to human papillomavirus infection (HPV) are also common.
Furthermore, biallelic RAG mutations have been identified in patients with other delayed-onset and atypical presentations, including idiopathic CD4+ T cell lymphopenia [43], common variable immunodeficiency [41, 44], IgA deficiency [45, 46], selective deficiency of polysaccharide-specific antibody responses [46], and hyper-IgM syndrome [47]. In this group of patients, the clinical phenotype is dominated by recurrent sinopulmonary infections.
Overall, these observations have substantially broadened the clinical and immunological spectrum of human RAG deficiency and have identified immune dysregulation as a prominent manifestation of impaired RAG function. Besides delayed clinical onset, CID-G/AI and other atypical presentations of RAG deficiency are often characterized by significant diagnostic delay, which may also lead to organ damage and overall poor prognosis.
Immunological Spectrum of RAG Deficiency
Circulating T cells are absent or very low in patients with RAG deficiency manifesting as typical SCID (Fig. 2). An exception is represented by infants with maternal T cell engraftment, in whom the absolute T cell count can be variable. However, in these infants, the majority of circulating T cells have an activated (CD45R0+) phenotype, which contrasts with the large excess of naïve (CD45RA+) Tcells in peripheral blood from healthy infants.
Fig. 2.

Absolute count of lymphocyte subsets in patients with RAG gene defects presenting as combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI), atypical SCID (AS), Omenn syndrome (OS), and severe combined immune deficiency (SCID). Shown are mean values ± SEM
Patients with OS have a variable, often increased number of peripheral blood autologous Tcells (Fig. 2). However, these T cells are oligoclonal, have an activated phenotype, and infiltrate target organs. Interestingly, within the same patient with OS, the T cells that infiltrate distinct organs carry distinct TCR specificities, suggesting tissue-specific, self-antigen-driven expansion of T cell populations [48]. It is commonly thought that the presence of elevated levels of eosinophils and IgE in these patients is due to skewing of CD4+ T cells to a T helper 2 (Th2) cell phenotype, as indicated by production of higher amounts of IL-4, IL-5, and IL-13 by freshly isolated peripheral blood mononuclear cells and in vitro expanded T cell clones derived from patients with OS [49–52]. However, the mechanisms underlying this skewing remain to be elucidated.
Finally, circulating T cells are present, albeit in reduced number, in patients with AS and in those with CID-G/AI or other delayed forms of disease; however, also in these patients, the proportion of naïve T cells is reduced (Fig. 2).
In vitro T cell proliferation to mitogens is absent in patients with SCID, but is more variable in those with OS and AS, and often normal or only modestly reduced in patients with delayed-onset disease.
Circulating B cells are typically absent in RAG-deficient patients presenting with SCID or OS, whereas they are detected in variable number in those with AS and CID-G/AI (Fig. 2). Consistent with the lack of B cells, serums IgA and IgM are very low to undetectable in patients with SCID and OS (Fig. 3). When present, serum IgG in these infants reflect transplacental passage; however, levels of maternally derived IgG are typically very low to undetectable in patients with OS, due to loss through the skin and gut. By contrast, IgG, IgA, and IgM serum levels are often detected at normal levels in patients with AS and CID-G/AI. Serum IgE are typically elevated in patients with OS and often also in those with AS (Fig. 3). Specific antibody production is absent in patients with SCID and OS and is most often compromised also in patients with AS. By contrast, specific antibody production is variable and may be normal, in patients with CID-G/AI and other forms of delayed-onset disease.
Fig. 3.
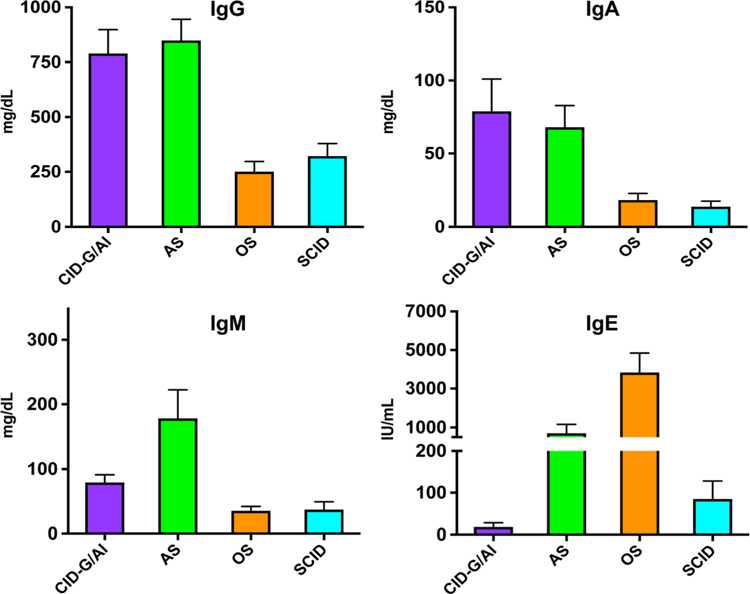
Immunoglobulin serum levels in patients with RAG gene defects presenting as combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI, n = 44), atypical SCID (AS, n = 49), Omenn syndrome (OS, n = 32), and severe combined immune deficiency (SCID, n = 82). Shown are mean values ± SEM
Natural killer (NK) cells are presented in normal or increased numbers in patients with RAG mutations, irrespective of their clinical phenotype (Fig. 2); however, they include a higher than normal proportion of immature (CD56bright) cells and yet display increased perforin content and enhanced degranulation activity [53]. A similar increased cytotoxic potential has been observed also in NK lymphocytes from Rag−/− [54], which however—in contrast to what observed in RAG-mutated patients—display a mature and activated phenotype [54]. Enhanced NK cell-mediated cytotoxicity may contribute to the high rate of graft rejection that has been observed especially after unconditioned haploidentical HSCT in patients with RAG-deficient SCID [55].
Molecular Mechanisms Underlying Phenotypical Variability
Recent knowledge on the molecular structure of the RAG complex [56], development of in vitro assays to evaluate recombination activity of mutant RAG proteins [57, 58], and high throughput sequencing (HTS) of T and B cell repertoire in patients with RAG gene defects [59] have led to improved understanding of the phenotypic variability of the disease.
The nature (nonsense, frameshift, splice site, or missense) of the mutation and its location along the protein may provide important information to predict severity of the clinical phenotype, although variability has been observed even within the same family [47, 60]. More than 200 pathogenic mutations of RAG1 and 2 genes have been documented, with almost 70% of them being missense. Around 80% of the mutations described in the literature fall in RAG1 and RAG2 core domains, affecting catalytic activity [1]. RAG noncore domains are not strictly necessary for the recombinase activity, but mutations in those areas may still cause disease by affecting protein subcellular localization or decreasing chromatin accessibility of the RAG complex [61–63]. Missense mutations associated with CID-G/AI are often located in the coding flank-sensitive region of RAG1 [57, 64]. In vitro and animal studies suggest that these mutations may affect quality of the recombination process by favoring targeting of certain V, D, and J genes [65, 66].
Using in vitro recombination assays [57, 58], correlation has been observed between mean level of recombination activity of the mutant RAG proteins and the clinical phenotype (Fig. 4). However, this correlation is not absolute, as other genetic and environmental factors may modulate the clinical and immunologic phenotype.
Fig. 4.
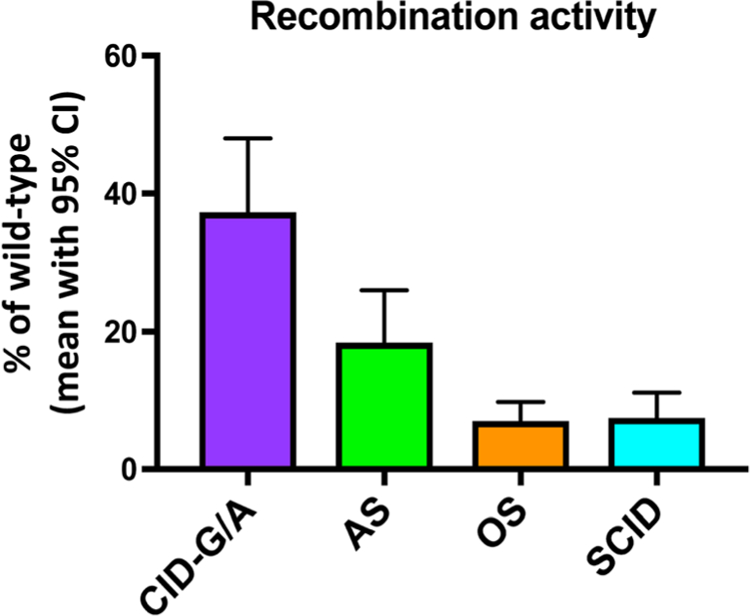
Recombination activity of mutant RAG proteins in patients with combined immune deficiency with granulomas and/or autoimmunity (CID-G/AI; n = 44), atypical SCID (AS, n = 44), Omenn syndrome (OS, n = 103), and severe combined immune deficiency (SCID, n = 68). Shown are mean values (as percentage of wild-type RAG protein) with 95% confidence interval (CI). In each patient, recombination activity was expressed as mean in vitro activity level of the two mutant alleles
Analysis of T and B cell repertoire diversity and composition has also provided important information. CDR3 spectratyping permits to analyze the length of CDR3 products for each TCR and immunoglobulin gene family. This technique can document oligoclonal T cell expansions in patients with OS and AS, but is less informative in those with CID-G/AI [11, 29, 48]. More recently, HTS has been used to study in detail T and B cell repertoire diversity and composition [60, 65]. Restriction of TCRβ repertoire diversity, skewed usage of V, D, and J segment genes, and abnormalities of CDR3 length are more prominent in patients with a more severe phenotype [59]. Within the group of patients with CID-G/AI, restriction of TCRβ repertoire diversity is especially prominent in Treg cells [67], which may have implications for the autoimmunity of this condition. Furthermore, abnormalities of TCRα repertoire have also been described in patients with hypomorphic RAG mutations and other leaky defects of V(D)J recombination, and include decreased usage of TCR-Vα7.2 gene (which can be detected by flow cytometry), and abnormal usage of TRAV and TRAJ genes, with predominant usage of more proximal elements [68]. In general, RAG mutations supporting lower levels of recombination activities lead to the generation of a more restricted TCR and BCR repertoire and are associated with a severe and early-onset clinical phenotype, while higher levels of protein function may lead to a broader repertoire, albeit with qualitative differences of individual gene usage and enrichment in self-reactive clones [59].
Pathophysiology of Immune Dysregulation
The pathophysiology of immune dysregulation observed in patients with CID-G/AI and AS is only partially understood. Consistent with the broad range of autoimmune phenomena of CID-G/AI, a wide spectrum of serum autoantibodies have been reported in these patients [37]. These also include anti-cytokine antibodies; in particular, neutralizing antibodies against interferon-α and interferon-ω have been detected in patients with a history of severe viral infections, especially varicella [37]. In a single case, anticytokine antibodies were demonstrated only after prolonged varicella, suggesting that the production of neutralizing anti-cytokine autoantibodies may be a manifestation of immune dysregulation driven by severe viral infections rather than a factor predisposing to them [69].
While granulomas identified in RAG-deficient patients often have no identifiable infectious or systemic cause [70], it has been shown that rubella virus vaccine strain can establish chronic infection in M2 macrophages and keratinocytes within granulomas of patients with various T cell defects, including RAG deficiency [71], suggesting a role for chronic viral infection and immune disfunction in granuloma formation.
The notion that environmental factors may act as disease modifiers and contribute to immune dysregulation in RAG-mutated hosts is also supported by observations in animal models. In particular, Rigoni et al. have shown that the microbiota is a crucial driver of autoimmunity in a mouse model of OS [72]. In these mice, lack of B cells in the mucosa affects microbiota composition and leads to trans-epithelial bacterial translocation. Furthermore, impaired Tcell tolerance to the gut flora and expansion of T effector cells contribute to gut inflammation. Administration of antibiotics is able to reverse the inflammation and is also associated with a decrease of serum IgE levels, a hallmark of immune dysregulation of OS [72].
Abnormalities of central and peripheral T and B cell tolerance play a major role in causing immune dysregulation in patients with hypomorphic RAG mutations. In the thymus, sustained generation of single positive CD4+ thymocytes expressing CD40 ligand and RANK ligand is required to provide instructive signals to medullary thymic epithelial cells (mTEC) expressing the cognate receptors CD40 and RANK [73]. In response to these signals, mTECs mature and express the autoimmune regulator (AIRE), a transcriptional regulator that allows for the expression of tissue-restricted antigens (TRAs). Presentation of TRAs in the context of MHC molecules on the surface of mTECs and of dendritic cells allows negative selection of self-reactive T cells and generation of Treg lymphocytes [74, 75]. In both patients and animal models with RAG mutations, altered thymopoiesis is associated with abnormalities of thymic architecture, impaired maturation of mTECs, defective expression of AIRE, reduced generation of Treg cells [66, 76–78], and enrichment of CD4+ conventional T cells with molecular signatures of self-reactivity in their TCR repertoire [67].
B cell tolerance is also impaired in RAG deficiency as supported by the presence of a wide spectrum of autoantibodies both in patients [37] and in animal models [79, 80]. Re-expression of RAG proteins allows receptor editing in bone marrow immature B cells, whereby secondary rearrangements in the IGK and IGL loci reduce the frequency of self-reactive cells [81]. Reduced usage of downstream IGK J genes and decreased frequency of peripheral Igλ+ B cells, two biomarkers of receptor editing, have been documented in patients and mice with hypomorphic RAG mutations [60, 66].
Peripheral B cell tolerance is also affected in patients with RAG deficiency. B cell activating factor (BAFF) is a cytokine that plays an important role in B cell survival. Anergic self-reactive B cells express lower amounts of BAFF receptor (BAFF-R) than mature naïve B cells [82]. In steady-state conditions, this results in depletion of self-reactive specificities from the pool of mature naïve B cells. However, peripheral B cell lymphopenia in RAG-mutated patients and mice is associated with higher levels of BAFF, thereby allowing survival of immature self-reactive B cells [79, 80]. Administration of anti-BAFF-R antibody was able to rescue some of the immune dysregulation in a mouse model of OS, further suggesting an important role of BAFF in sustaining B cell-driven autoimmunity in patients with hypomorphic RAG defects [79].
Therapy for RAG Deficiencies
In patients with typical and atypical SCID and OS, supportive measures such as antibiotic prophylaxis, regular immunoglobulin substitution therapy, nutritional support, and contact barrier may delay, but not prevent the invariably fatal outcome. Curative treatment is possible with HSCT. The best results are obtained with transplantation from a matched sibling donor. When this is not available, HSCT from other matched or haploidentical family donors and from unrelated donors should be considered. Because SCID patients lack T cells and therefore do not have the ability to reject allogeneic stem cells, HSCT without chemotherapy-based conditioning regimen should be able to restore a T cell system into adulthood. By contrast, use of conditioning regimen (CR), including serotherapy, is required for patients with OS. Controversial results of overall survival after HSCT for severe RAG deficiency have been reported in the literature [55, 83]. However, there is consensus that the best results are obtained when the transplant is performed early in life (at < 3.5 months of age) in the absence of infections [32, 83]. In a comprehensive multicenter study across 33 pediatric North American centers, Haddad et al. compared the outcome between classical SCID patients and atypical forms including Omenn syndrome: interestingly, no difference was found for survival, need of second procedure, GvHD, or immune reconstitution [83].
Importantly, in the absence of conditioning, HSCT for severe RAG deficiency is associated with a particularly high rate (up to 25%) of graft failure [55, 83]. This likely reflects the notion that the bone marrow and thymus niches are not empty in these patients, but rather are occupied by RAG-mutated progenitor cells that compete with donor-derived cells. NK cells may also play a role in inducing graft rejection. In the absence of true and durable stem cell engraftment, immune reconstitution after non-conditioned HSCT is provided by common lymphoid progenitor cells. However, because of the limited self-renewing capacity of donor-derived thymocytes, late exhaustion of thymopoiesis may ensue years after non-conditioned HSCT. Such a decline of T cell number and function and persistently impaired B cell function have been frequently reported (especially after non-conditioned HSCT from haploidentical donors) and are a major risk factor for increased susceptibility to infections and autoimmunity [55, 83, 84]. Use of chemotherapy can promote donor stem cell engraftment and allow more robust and durable immune reconstitution. However, use of preparative conditioning has to be weighed against the toxicities of chemotherapy, especially if myeloablative regimens are used. A current trial in North America is aimed at exploring the minimal dose of busulfan required to achieve sufficient and stable stem cell engraftment and immune reconstitution in babies identified with RAG deficiency at birth. Another trial (ClinicalTrials.gov ) is exploring the safety and efficacy of conditioning with anti-CD117 (anti-c-kit) monoclonal antibody in patients with SCID, with the aim of depleting recipient stem cells and thereby favoring engraftment of donor-derived hematopoietic stem cells. Whether this approach alone is sufficient in patients with RAG mutations, and especially in those with hypomorphic mutations that allow partial T and B cell development, remains to be seen.
Whereas patients with typical and atypical SCID and OS due to RAG defects need to be treated with HSCT, the situation is more complex in older individuals with CID-G/AI or other delayed-onset and atypical forms of the disease. Immunosuppressive or immunomodulatory drugs are often needed to control autoimmune problems, but they may also increase susceptibility to chronic and opportunistic infections. Limited information is available on the outcome of HSCT for these patients [13, 14, 37, 40, 41, 55, 69, 70, 85–87]. Twenty-six patients were transplanted at the average age of 5.2 years (range 1.5–19 years). Donor type was reported for 11 patients, 9 of which received HSCT from a matched unrelated donor. In most cases, myeloablative CR was used. Eighteen patients (69.2%) were reported to be alive at a median follow-up of 9 months. Causes of death were known for 6 patients and included infections (n = 3), chronic GvHD (n = 1), idiopathic pulmonary syndrome ( n = 1 ), and graft failure ( n = 1 ) [ 31, 44, 88 ]. Unfortunately, patients with CID-G/AI are often diagnosed too late, when organ damage or malignancies have already developed. Overall, management of RAG-deficient patients with immune dysregulation and chronic infections remains a challenge.
Finally, several groups are working at the development of gene therapy for RAG deficiency. Controversial results have been recently obtained after lentivirus-mediated gene therapy in Rag1−/− mice, with one group documenting insufficient reconstitution and conversion of SCID into an OS phenotype [89], whereas another group has reported more successful, albeit incomplete immune reconstitution [90]. More promising results have been achieved after gene therapy in a mouse model of RAG2 deficiency [91–93]. However, the amount of chemotherapy required to enable stable and durable engraftment, especially in hosts carrying hypomorphic mutations, remains to be determined.
Conclusions
The clinical and immunological heterogeneity of RAG deficiency in humans may represent an important diagnostic challenge, especially in patients with atypical forms of the disease. Correlation has been observed between in vitro recombination activity of the mutant RAG proteins and the clinical phenotype; however, stochastic and environmental factors may also modify severity of the disease phenotype. Altered T and B cell development in patients with RAG gene defects is associated with impairment of immunological tolerance. Ultimately, the degree in the severity of T and B cell lymphopenia, breakage of immune tolerance, and exposure to environmental triggers are the main factors that shape the clinical phenotype. While hematopoietic stem cell transplantation represents the mainstay of treatment for the most severe forms of the disease, there is need to develop novel strategies to improve stem cell engraftment and permit durable and robust immune reconstitution, while minimizing the risk of transplant-related toxicity. Such strategies may include in the future also gene therapy. Optimal therapeutic modalities for patients with CID-G/AI remain yet to be defined.
Acknowledgements
For unpublished data, clinical, immunological, and molecular data were obtained according to NIH IRB approved protocols 93-I-0119, 05-I-0213, and 16-I-N139.
Funding Information This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Compliance with Ethical Standards
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol 2016;16(4):234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fugmann SD, Lee AI, Shockett PE, Villey IJ, Schatz DG. The RAG proteins and V(D)J recombination: complexes, ends, and transposition. Annu Rev Immunol 2000;18:495–527. [DOI] [PubMed] [Google Scholar]
- 3.Grawunder U, Harfst E. How to make ends meet in V(D)J recombination. Curr Opin Immunol 2001;13(2):186–94. [DOI] [PubMed] [Google Scholar]
- 4.Feeney AJ, Goebel P, Espinoza CR. Many levels of control of V gene rearrangement frequency. Immunol Rev 2004;200:44–56. [DOI] [PubMed] [Google Scholar]
- 5.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992;68(5):869–77. [DOI] [PubMed] [Google Scholar]
- 6.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992;68(5):855–67. [DOI] [PubMed] [Google Scholar]
- 7.Niehues T, Perez-Becker R, Schuetz C. More than just SCID–the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clin Immunol 2010;135(2):183–92. [DOI] [PubMed] [Google Scholar]
- 8.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science 1996;274(5284):97–9. [DOI] [PubMed] [Google Scholar]
- 9.Villa A, Santagata S, Bozzi F, Giliani S, Frattini A, Imberti L, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell 1998;93(5):885–96. [DOI] [PubMed] [Google Scholar]
- 10.de Saint-Basile G, Le Deist F, de Villartay JP, Cerf-Bensussan N, Journet O, Brousse N, et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome). J Clin Invest 1991;87(4):1352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rieux-Laucat F, Bahadoran P, Brousse N, Selz F, Fischer A, Le Deist F, et al. Highly restricted human T cell repertoire in peripheral blood and tissue-infiltrating lymphocytes in Omenn’s syndrome. J Clin Invest 1998;102(2):312–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Villa A, Sobacchi C, Notarangelo LD, Bozzi F, Abinun M, Abrahamsen TG, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 2001;97(1):81–8. [DOI] [PubMed] [Google Scholar]
- 13.Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med 2008;358(19):2030–8. [DOI] [PubMed] [Google Scholar]
- 14.De Ravin SS, Cowen EW, Zarember KA, Whiting-Theobald NL, Kuhns DB, Sandler NG, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood 2010;116(8):1263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corneo B, Moshous D, Gungor T, Wulffraat N, Philippet P, Le Deist FL, et al. Identical mutations in RAG1 or RAG2 genes leading to defective V(D)J recombinase activity can cause either T-B-severe combined immune deficiency or Omenn syndrome. Blood 2001;97(9):2772–6. [DOI] [PubMed] [Google Scholar]
- 16.Noordzij JG, de Bruin-Versteeg S, Verkaik NS, Vossen JM, de Groot R, Bernatowska E, et al. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood 2002;100(6):2145–52. [PubMed] [Google Scholar]
- 17.Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Hum Mutat 2006;27(12):1174–84. [DOI] [PubMed] [Google Scholar]
- 18.Tabori U, Mark Z, Amariglio N, Etzioni A, Golan H, Biloray B, et al. Detection of RAG mutations and prenatal diagnosis in families presenting with either T-B-severe combined immunodeficiency or Omenn’s syndrome. Clin Genet 2004;65(4):322–6. [DOI] [PubMed] [Google Scholar]
- 19.Alsmadi O, Al-Ghonaium A, Al-Muhsen S, Arnaout R, Al-Dhekri H, Al-Saud B, et al. Molecular analysis of T-B-NK+ severe combined immunodeficiency and Omenn syndrome cases in Saudi Arabia. BMC Med Genet 2009;10:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalal I, Tasher D, Somech R, Etzioni A, Garti BZ, Lev D, et al. Novel mutations in RAG½ and ADA genes in Israeli patients presenting with T-B-SCID or Omenn syndrome. Clin Immunol 2011;140(3):284–90. [DOI] [PubMed] [Google Scholar]
- 21.Meshaal S, El Hawary R, Elsharkawy M, Mousa RK, Farid RJ, Abd Elaziz D, et al. Mutations in recombination activating gene 1 and 2 in patients with severe combined immunodeficiency disorders in Egypt. Clin Immunol 2015;158(2):167–73. [DOI] [PubMed] [Google Scholar]
- 22.Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers 2015;1:15061. [DOI] [PubMed] [Google Scholar]
- 23.Woodbine L, Grigoriadou S, Goodarzi AA, Riballo E, Tape C, Oliver AW, et al. An Artemis polymorphic variant reduces Artemis activity and confers cellular radiosensitivity. DNA Repair (Amst) 2010;9(9):1003–10. [DOI] [PubMed] [Google Scholar]
- 24.Omenn GS. Familial Reticuloendotheliosis with eosinophilia. N Engl J Med 1965;273:427–32. [DOI] [PubMed] [Google Scholar]
- 25.Muller SM, Ege M, Pottharst A, Schulz AS, Schwarz K, Friedrich W. Transplacentally acquired maternal T lymphocytes in severe combined immunodeficiency: a study of 121 patients. Blood 2001;98(6):1847–51. [DOI] [PubMed] [Google Scholar]
- 26.Dalal I, Tabori U, Bielorai B, Golan H, Rosenthal E, Amariglio N, et al. Evolution of a T-B-SCID into an Omenn syndrome phenotype follow- ing parainfluenza 3 virus infection. Clin Immunol 2005;115(1):70–3. [DOI] [PubMed] [Google Scholar]
- 27.Wada T, Toma T, Okamoto H, Kasahara Y, Koizumi S, Agematsu K, et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood 2005;106(6):2099–101. [DOI] [PubMed] [Google Scholar]
- 28.Crestani E, Choo S, Frugoni F, Lee YN, Richards S, Smart J, et al. RAG1 reversion mosaicism in a patient with Omenn syndrome. J Clin Immunol 2014;34(5):551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Villartay JP, Lim A, Al-Mousa H, Dupont S, Dechanet-Merville J, Coumau-Gatbois E, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest 2005;115(11):3291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehl S, Schwarz K, Enders A, Duffner U, Pannicke U, Kuhr J, et al. A variant of SCID with specific immune responses and predominance of gamma delta T cells. J Clin Invest 2005;115(11):3140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henderson LA, Frugoni F, Hopkins G, de Boer H, Pai SY, Lee YN, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol 2013;132(4):969–71 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014;371(5):434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014;312(7):729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dvorak CHE, Buckley RH, Cowan M, Logan B, Griffith LM, Kohn DB, et al. The genetic landscape of SCID in the current era. J Allergy Clin Immunol 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Herz W, Al-Mousa H. Combined immunodeficiency: the Middle East experience. J Allergy Clin Immunol 2013;131(3):658–60. [DOI] [PubMed] [Google Scholar]
- 36.Avila EM, Uzel G, Hsu A, Milner JD, Turner ML, Pittaluga S, et al. Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics 2010;126(5):e1248–52. [DOI] [PubMed] [Google Scholar]
- 37.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest 2015;125(11):4135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharapova SO, Migas A, Guryanova I, Aleshkevich S, Kletski S, Durandy A, et al. Late-onset combined immune deficiency associated to skin granuloma due to heterozygous compound mutations in RAG1 gene in a 14 years old male. Hum Immunol 2013;74(1):18–22. [DOI] [PubMed] [Google Scholar]
- 39.Patiroglu T, Akar HH, Gilmour K, Ozdemir MA, Bibi S, Henriquez F, et al. Atypical severe combined immunodeficiency caused by a novel homozygous mutation in Rag1 gene in a girl who presented with pyoderma gangrenosum: a case report and literature review. J Clin Immunol 2014;34(7):792–5. [DOI] [PubMed] [Google Scholar]
- 40.Chen K, Wu W, Mathew D, Zhang Y, Browne SK, Rosen LB, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG½ mutations. J Allergy Clin Immunol 2014;133(3): 880–2 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buchbinder D, Baker R, Lee YN, Ravell J, Zhang Y, McElwee J, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol 2015;35(2):119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reiff A, Bassuk AG, Church JA, Campbell E, Bing X, Ferguson PJ. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J Clin Immunol 2013;33(8): 1289–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuijpers TW, Ijspeert H, van Leeuwen EM, Jansen MH, Hazenberg MD, Weijer KC, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood 2011;117(22):5892–6. [DOI] [PubMed] [Google Scholar]
- 44.Abolhassani H, Wang N, Aghamohammadi A, Rezaei N, Lee YN, Frugoni F, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol 2014;134(6):1375–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato T, Crestani E, Kamae C, Honma K, Yokosuka T, Ikegawa T, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol 2015;35(3):280–8. [DOI] [PubMed] [Google Scholar]
- 46.Geier CB, Piller A, Linder A, Sauerwein KM, Eibl MM, Wolf HM. Leaky RAG deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PLoS One 2015;10(7):e0133220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol 2012;130(6):1414–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Signorini S, Imberti L, Pirovano S, Villa A, Facchetti F, Ungari M, et al. Intrathymic restriction and peripheral expansion of the T-cell repertoire in Omenn syndrome. Blood 1999;94(10):3468–78. [PubMed] [Google Scholar]
- 49.Schandene L, Ferster A, Mascart-Lemone F, Crusiaux A, Gerard C, Marchant A, et al. T helper type 2-like cells and therapeutic effects of interferon-gamma in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome). Eur J Immunol 1993;23(1):56–60. [DOI] [PubMed] [Google Scholar]
- 50.Melamed I, Cohen A, Roifman CM. Expansion of CD3+CD4-CD8- T cell population expressing high levels of IL-5 in Omenn’s syndrome. Clin Exp Immunol 1994;95(1):14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chilosi M, Facchetti F, Notarangelo LD, Romagnani S, Del Prete G, Almerigogna F, et al. CD30 cell expression and abnormal soluble CD30 serum accumulation in Omenn’s syndrome: evidence for a T helper 2-mediated condition. Eur J Immunol 1996;26(2):329–34. [DOI] [PubMed] [Google Scholar]
- 52.Wada T, Takei K, Kudo M, Shimura S, Kasahara Y, Koizumi S, et al. Characterization of immune function and analysis of RAG gene mutations in Omenn syndrome and related disorders. Clin Exp Immunol 2000;119(1):148–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobbs K, Tabellini G, Calzoni E, Patrizi O, Martinez P, Giliani SC, et al. Natural killer cells from patients with recombinase-activating gene and non-homologous end joining gene defects comprise a higher frequency of CD56(bright) NKG2A(+++) cells, and yet display increased degranulation and higher Perforin content. Front Immunol 2017;8:798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell 2014;159(1):94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014;123(2):281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1-RAG2. Nature 2015;518(7540):507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotypephenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol 2014;133(4):1099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tirosh I, Yamazaki Y, Frugoni F, Ververs FA, Allenspach EJ, Zhang Y, et al. Recombination activity of human RAG2 mutations and correlation with the clinical phenotype. J Allergy Clin Immunol 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee YN, Frugoni F, Dobbs K, Tirosh I, Du L, Ververs FA, et al. (2016) Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci Immunol 1(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.H IJ, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol 2014;133(4):1124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santagata S, Gomez CA, Sobacchi C, Bozzi F, Abinun M, Pasic S, et al. N-terminal RAG1 frameshift mutations in Omenn’s syndrome: internal methionine usage leads to partial V(D)J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc Natl Acad Sci U S A 2000;97(26):14572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simkus C, Anand P, Bhattacharyya A, Jones JM. Biochemical and folding defects in a RAG1 variant associated with Omenn syndrome. J Immunol 2007;179(12):8332–40. [DOI] [PubMed] [Google Scholar]
- 63.Couedel C, Roman C, Jones A, Vezzoni P, Villa A, Cortes P. Analysis of mutations from SCID and Omenn syndrome patients reveals the central role of the Rag2 PHD domain in regulating V(D)J recombination. J Clin Invest 2010;120(4):1337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mo X, Bailin T, Sadofsky MJ. A C-terminal region of RAG1 contacts the coding DNA during V(D)J recombination. Mol Cell Biol 2001;21(6):2038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong SY, Lu CP, Roth DB. A RAG1 mutation found in Omenn syndrome causes coding flank hypersensitivity: a novel mechanism for antigen receptor repertoire restriction. J Immunol 2008;181(6): 4124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ott de Bruin LM, Bosticardo M, Barbieri A, Lin SG, Rowe JH, Poliani PL, et al. (2018) Hypomorphic Rag1 mutations alter the preimmune repertoire at early stages of lymphoid development. Blood [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rowe JH, Stadinski BD, Henderson LA, Ott de Bruin L, Delmonte O, Lee YN, et al. Abnormalities of T-cell receptor repertoire in CD4(+) regulatory and conventional T cells in patients with RAG mutations: implications for autoimmunity. J Allergy Clin Immunol 2017;140(6):1739–43 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I, et al. (2018) PROMIDISα: a TCR α signature associated with immunodeficiencies caused by V(D)J recombination defects. J Allergy Clin Immunol; June 12 [Epub ahed of print]. [DOI] [PubMed] [Google Scholar]
- 69.Goda V, Malik A, Kalmar T, Maroti Z, Patel B, Ujhazi B, et al. Partial RAG deficiency in a patient with varicella infection, autoimmune cytopenia, and anticytokine antibodies. J Allergy Clin Immunol Pract 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mitra A, Pollock B, Gooi J, Darling JC, Boon A, Newton-Bishop JA. Cutaneous granulomas associated with primary immunodeficiency disorders. Br J Dermatol 2005;153(1):194–9. [DOI] [PubMed] [Google Scholar]
- 71.Neven B, Perot P, Bruneau J, Pasquet M, Ramirez M, Diana JS, et al. Cutaneous and visceral chronic granulomatous disease triggered by a rubella virus vaccine strain in children with primary immunodeficiencies. Clin Infect Dis 2017;64(1):83–6. [DOI] [PubMed] [Google Scholar]
- 72.Rigoni R, Fontana E, Guglielmetti S, Fosso B, D’Erchia AM, Maina V, et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med 2016;213(3):355–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Akiyama T, Tateishi R, Akiyama N, Yoshinaga R, Kobayashi TJ. Positive and negative regulatory mechanisms for fine-tuning cellularity and functions of medullary thymic epithelial cells. Front Immunol 2015;6:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002;298(5597):1395–401. [DOI] [PubMed] [Google Scholar]
- 75.Watanabe N, Wang YH, Lee HK, Ito T, Wang YH, Cao W, et al. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature 2005;436(7054):1181–5. [DOI] [PubMed] [Google Scholar]
- 76.Cavadini P, Vermi W, Facchetti F, Fontana S, Nagafuchi S, Mazzolari E, et al. AIRE deficiency in thymus of 2 patients with Omenn syndrome. J Clin Invest 2005;115(3):728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood 2009;114(1):105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rucci F, Poliani PL, Caraffi S, Paganini T, Fontana E, Giliani S, et al. (2011) Abnormalities of thymic stroma may contribute to immune dysregulation in murine models of leaky severe combined immunodeficiency. Front Immunol 2(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cassani B, Poliani PL, Marrella V, Schena F, Sauer AV, Ravanini M, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med 2010;207(7):1525–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walter JE, Rucci F, Patrizi L, Recher M, Regenass S, Paganini T, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med 2010;207(7):1541–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annu Rev Immunol 2004;22:485–501. [DOI] [PubMed] [Google Scholar]
- 82.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 2004;20(4):441–53. [DOI] [PubMed] [Google Scholar]
- 83.Haddad ELBR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month post-transplant CD4 count predict survival and immune recovery: a PIDTC retrospective study. Blood 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Neven B, Leroy S, Decaluwe H, Le Deist F, Picard C, Moshous D, et al. Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency. Blood 2009;113(17):4114–24. [DOI] [PubMed] [Google Scholar]
- 85.John T, Walter JE, Schuetz C, Chen K, Abraham RS, Bonfim C, et al. Unrelated hematopoietic cell transplantation in a patient with combined immunodeficiency with granulomatous disease and autoimmunity secondary to RAG deficiency. J Clin Immunol 2016;36(7):725–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khan TA, Iqbal A, Rahman H, Cabral-Marques O, Ishfaq M, Muhammad N. Novel RAG1 mutation and the occurrence of mycobacterial and Chromobacterium violaceum infections in a case of leaky SCID. Microb Pathog 2017;109:114–9. [DOI] [PubMed] [Google Scholar]
- 87.Lawless D, Geier CB, Farmer JR, Lango Allen H, Thwaites D, Atschekzei F, et al. Prevalence and clinical challenges among adults with primary immunodeficiency and recombination-activating gene deficiency. J Allergy Clin Immunol 2018;141(6):2303–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dutmer CM, Asturias EJ, Smith C, Dishop MK, Schmid DS, Bellini WJ, et al. Late onset hypomorphic RAG2 deficiency presentation with fatal vaccine-strain VZV infection. J Clin Immunol 2015;35(8):754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Til NP, Sarwari R, Visser TP, Hauer J, Lagresle-Peyrou C, van der Velden G, et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol 2014;133(4):1116–23. [DOI] [PubMed] [Google Scholar]
- 90.Pike-Overzet K, Rodijk M, Ng YY, Baert MR, Lagresle-Peyrou C, Schambach A, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia 2011;25(9):1471–83. [DOI] [PubMed] [Google Scholar]
- 91.Yates F, Malassis-Seris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P, et al. Gene therapy of RAG-2−/− mice: sustained correction of the immunodeficiency. Blood 2002;100(12):3942–9. [DOI] [PubMed] [Google Scholar]
- 92.van Til NP, de Boer H, Mashamba N, Wabik A, Huston M, Visser TP, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther 2012;20(10):1968–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Capo V, Castiello MC, Fontana E, Penna S, Bosticardo M, Draghici E, et al. Efficacy of lentivirus-mediated gene therapy in an Omenn syndrome recombination-activating gene 2 mouse model is not hindered by inflammation and immune dysregulation. J Allergy Clin Immunol 2017. 10.1016/j.jaci.2017.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]