

Sedimentary microorganisms in the South China Sea (SCS) remain largely unknown due to the complexity of sediment communities impacted by continent rifting and extension. Distinct geochemical environments may breed special microbial communities including microbes that are still enigmatic. Functional inference of their metabolisms and transcriptional activity provides insight in the ecological roles and substrate-based interactivity of these uncultured Archaea and Bacteria. These microorganisms play different roles in utilizing inorganic carbon and scavenging diverse organic compounds involved in the deep-sea carbon cycle. The genomes recovered here contributed undescribed species to the tree of life and laid the foundation for future study on these novel phyla persisting in marginal sediments of the SCS.
KEYWORDS: carbon fixation, metagenome, metatranscriptome, South China Sea, sulfate reduction
ABSTRACT
Metagenomics of marine sediments has uncovered a broad diversity of new uncultured taxa and provided insights into their metabolic capabilities. Here, we detected microbial lineages from a sediment core near the Jiulong methane reef of the northern South China Sea (at 1,100-m depth). Assembly and binning of the metagenomes resulted in 11 genomes (>85% complete) that represented nine distinct phyla, including candidate phyla TA06 and LCP-89, Lokiarchaeota, Heimdallarchaeota, and a newly described globally distributed phylum (B38). The genome of LCP-89 has pathways for nitrate, selenate, and sulfate reduction, suggesting that they may be involved in mediating these important processes. B38 are able to participate in the cycling of hydrogen and selenocompounds. Many of these uncultured microbes may also be capable of autotrophic CO2 fixation, as exemplified by identification of the Wood-Ljungdahl (W-L) pathway. Genes encoding carbohydrate degradation, W-L pathway, Rnf-dependent energy conservation, and Ni/Fe hydrogenases were detected in the transcriptomes of these novel members. Characterization of these new lineages provides insight to the undescribed branches in the tree of life.
IMPORTANCE Sedimentary microorganisms in the South China Sea (SCS) remain largely unknown due to the complexity of sediment communities impacted by continent rifting and extension. Distinct geochemical environments may breed special microbial communities including microbes that are still enigmatic. Functional inference of their metabolisms and transcriptional activity provides insight in the ecological roles and substrate-based interactivity of these uncultured Archaea and Bacteria. These microorganisms play different roles in utilizing inorganic carbon and scavenging diverse organic compounds involved in the deep-sea carbon cycle. The genomes recovered here contributed undescribed species to the tree of life and laid the foundation for future study on these novel phyla persisting in marginal sediments of the SCS.
INTRODUCTION
Microorganisms are widespread in subseafloor sediments and play a vital role in global geochemical cycles (1–3). It has been estimated that half of prokaryotic cells of the oceans dwell in subseafloor sediments (2). Culture-independent surveys of diversity and transcriptional activity have enhanced our knowledge of marine sedimentary communities (4, 5). Microbial diversity has been studied in a variety marine subseafloor sediments, including estuaries (6), hydrothermal fields (7), basalt rocks (8), and cold seeps (9, 10). A growing number of novel phyla have been discovered and described in terms of metabolisms and adaptive strategies in estuary and marine sediments (11, 12). However, marine sediments are vast diverse habitats, and many niches with special geological settings are left to be explored with newly developed metagenomic and metatranscriptomic approaches.
The South China Sea (SCS) is an oligotrophic marginal sea that is situated in the convergent zone between the Eurasian, the Australian, and the Pacific plates (13). The northern SCS slope was formed by continent rifting and subsequent crustal extension (14). Subseafloor spreading in the SCS began from the northernmost part, followed by southeastern and western parts for more than 40 million years. This resulted in crustal breakup and associated magmatic activities (15), which often left faults beneath the sediments on the northern SCS slope as channels of solutions from magma (16, 17). However, there is no strong evidence of volcanic activities in the northern SCS at present. The northern SCS slope is the major area for exploration of oil and gas hydrates (18). Methane seepage, chemosynthetic fauna, and methane-derived carbonate structures have been observed in the northern SCS slope, such as the Jiulong methane reef (19, 20). Anaerobic methanotrophic archaea (ANME) and associated sulfur-reducing bacteria were identified in the Jiaolong cold seep (21). There are perhaps other novel microorganisms with unique metabolic activities due to the unusual geological settings. In 2008, Han et al. imaged and analyzed large numbers of tubular carbonates and chimneys at sites 1 and 2 near the Jiulong methane reef (Fig. 1) (22), indicating active geochemical activities are ongoing under the marginal slopes.
FIG 1.

Geographic information of the sampling site. Sediment core was obtained at B3 located on a marginal slope at the northern South China Sea. The red dot indicates the sample site. The brown squares represent the sites where carbonates were identified near or within the Jiulong methane reef denoted by a purple circle (22). The yellow star denotes the Jiaolong cold seep (21).
However, our knowledge of the microbial diversity of the cold seep sediments in the north SCS is largely based on culturing and single-gene surveys. In 2015, we obtained a gravity core from the B3 site near sites 1 and 2 (Fig. 1). We employed mineralogical characterization, 16S rRNA gene sequencing, metagenomics, and metatranscriptomics to characterize the sediment samples of the deep-sea site near the Jiulong cold seep reef. The sediment samples exhibited distinct mineralogical components from the seep carbonates, revealing different microbial activities from the cold seep areas. The Lokiarchaeota and Heimdallarchaeota detected here, belonging to Asgard superphylum, are close relatives of eukaryotes and are frequently reported in anoxic marine sediments worldwide (23–25). The acetogenic, strictly anaerobic bacteria Aerophobetes, which may fix CO2 and conserve energy by the Wood-Ljungdahl (W-L) pathway (26, 27), are the most abundant group based on their coverage in metagenomic data. Candidate phyla LCP-89 and B38 (undescribed phyla) were also identified. Interestingly, most of these species are involved in carbon fixation through the W-L pathway or ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO), suggesting an important role for these organisms in carbon cycling.
RESULTS AND DISCUSSION
Mineral composition.
A gravity core of B3 was sampled in the northern SCS near the Jiulong methane reef (Fig. 1). Likely due to the hard bottom at the sampling site, approximately 43 cm of sediment was obtained in the 3-m core length. The major components of layer B3-1 at a depth of 30 cm were quartz (45%), muscovite (32%), and anorthite (18%), while layer B3-2 at a depth of 40 cm differed from B3-1, consisting mainly of quartz (27%), muscovite (24%), and a specific constituent part: clinochlore (ferroan; 34%) (see Fig. S1 in the supplemental material). The presence of clinochlore and muscovite in the sediment suggests geothermal alternation in the B3 sediment (28, 29), but the temperature could not be measured in situ. Since both layers did not contain carbonates, B3 was not a cold seep. We examined the microbial communities and their potential activities in the B3-1 and B3-2 layers.
Microbial community in sediments based on 16S rRNA gene amplicons.
Microbial communities in the samples were examined by sequencing 16S rRNA gene amplicons (see Fig. S2). Taxonomic sorting of 3,197 nonsingleton operational taxonomic units (OTUs) indicated that Proteobacteria (24.9%), Woesearchaeota (24.4%), and Chloroflexi (16.2%) were the major groups in B3-1. Asgard archaea represented by Odinarchaeota (2.3%), Lokiarchaeota (1%), and Heimdallarchaeota (0.1%) were also detected, and candidate phyla LCP-89 (0.6%), TA06 (0.2%), and Aerophobetes (0.1%) were the minor groups in B3-1. For B3-2, 38.4% of the 16S rRNA gene amplicons were assigned to Proteobacteria, followed by 16% to Chloroflexi, 11.1% to Woesearchaeota, 6.4% to Aerophobetes, and 5% to Atribacteria. Atribacteria, potentially living a fermentative, anaerobic, and saccharolytic lifestyle, are frequently detected in anoxic hot environments (12). Our finding of Atribacteria in B3 indicates a potential anoxic condition, although in situ measurement of oxygen flux was not conducted in this study.
The microbial community structure at the B3 site has never been reported in other SCS marine sediments. A recent work revealed that ANME and sulfur-reducing Proteobacteria were the dominant groups in the nearby Jiaolong cold seep (21). However, we did not detect ANME in the 16S rRNA gene amplicons from the B3 community from the SCS. In addition, in the Loki hydrothermal field, Lokiarchaeota and Heimdallarchaeota coexisted with Proteobacteria (23, 25), as we found in B3 layers. However, the other major phyla in B3 samples were not reported in Loki hydrothermal sediments.
Genomic reconstruction and phylogenetic inference.
De novo coassembly of metagenome data from B3-1 (24,446,965 paired reads) and B3-2 (15,401,162 paired reads) yielded a total of 2,175,262 scaffolds with 335,330 reads longer than 1 kbp (see Table S1). Although Proteobacteria dominated the amplicon libraries, each species of Proteobacteria was not the most abundant one in the metagenomes (see Fig. S3). It was difficult to bin a nearly complete Proteobacteria metagenome-assembled genome (MAG) manually; thus, Proteobacteria MAGs were excluded here. Genome binning resulted in 11 archaeal and bacterial genomes (here, MAGs) (86.5% to 97.7% complete) (Fig. S3 and Table 1). Phylogenetic placement of their 16S rRNA genes showed that seven MAGs belonged to Bacteria domain, and the other four were classified as Archaea. The MAGs were derived from nine distinct phyla, Lokiarchaeota (two MAGs), Heimdallarchaeota, Woesearchaeota, TA06, Aerophobetes (two MAGs), Chloroflexi, Planctomycetes, LCP-89, and a novel bacterial phylum (Fig. 2). The taxonomic affiliation of these microorganisms was confirmed by phylogenomic analyses of 24 concatenated ribosomal proteins (Fig. 3 and Table S2).
TABLE 1.
General characteristics of bacterial and archaeal metagenome-assembled genomes (MAGs) from B3
| MAG | Phylum | MAG length (bp) | No. of scaffolds | GC content (%) | No. of genes |
No. of CSCGsa |
No. of CDSs | Coding density (%) | Contamination (%) | Completion (%) | Coverage |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tRNA | rRNA | Total | Unique | B3-1 | B3-2 | |||||||||
| Ae_b3a | Aerophobetes | 2,368,411 | 141 | 41.8 | 36 | 3 | 95 | 93 | 2,306 | 85.3 | 0.9 | 86.5 | 103.3 | 108.3 |
| Ae_b3b | Aerophobetes | 2,041,952 | 86 | 43.1 | 34 | 3 | 94 | 94 | 2,029 | 87.5 | 0.9 | 86.5 | 28.8 | 60.3 |
| B3_TA06 | TA06 | 2,414,740 | 42 | 52.6 | 46 | 3 | 104 | 102 | 2,266 | 87.3 | 0 | 91.9 | 0.8 | 28.3 |
| B3_LCP | LCP-89 | 3,794,239 | 18 | 46.5 | 47 | 3 | 101 | 100 | 2,954 | 90 | 0.9 | 92.8 | 18.3 | 0.2 |
| B3_Pla | Planctomycetes | 5,978,183 | 47 | 52.9 | 46 | 5 | 104 | 100 | 4,605 | 87.3 | 3.6 | 91 | 34.7 | 0.7 |
| B3_B38 | B38 | 2,254,273 | 102 | 49.5 | 37 | 3 | 102 | 99 | 2,015 | 88.6 | 0.9 | 91.9 | 2.3 | 9.0 |
| B3_Chlor | Chloroflexi | 2,817,311 | 73 | 58.2 | 47 | 3 | 100 | 97 | 2,594 | 87.5 | 0.9 | 86.5 | 42.0 | 89.1 |
| Loki_b31b | Lokiarchaeota | 3,518,981 | 99 | 31.5 | 23 | 2 | 77 | 75 | 3,391 | 86.9 | 1.2 | 90.6 | 7.1 | 23.5 |
| Loki_b32b | Lokiarchaeota | 4,129,784 | 71 | 29.9 | 24 | 10 | 84 | 80 | 4,068 | 86 | 3.5 | 92.9 | 17.8 | 4.1 |
| B3_Woesb | Woesearchaeota | 1,021,708 | 4 | 30.2 | 29 | 4 | 85 | 83 | 1,106 | 92.5 | 1.2 | 97.7 | 5.2 | 11.0 |
| B3_Heimb | Heimdallarchaeota | 3,948,260 | 103 | 36.1 | 27 | 8 | 85 | 82 | 3,576 | 86.6 | 3.5 | 97.7 | 2.0 | 10.2 |
Total CSCGs and unique CSCGs refer to the number of all and unique conserved single-copy genes, respectively.
Archaea.
FIG 2.

Maximum likelihood phylogenetic tree based on 16S rRNA genes. Bootstrap support is indicated by the size of the black dots, and only those for values >50% are shown. 16S rRNA genes for these candidate phyla were derived from MAGs and collected from NCBI (accession numbers in Table S6 in the supplemental material).
FIG 3.

Phylogenetic analysis of microorganisms based on concatenated conserved proteins. A total of 24 conserved proteins (Table S2) in Archaea and Bacteria were used to generate a maximum likelihood tree by raxmlGUI (PROTCATLG model and 1,000 replicates). Bootstrap support is indicated by the size of the black dots, and those representing >50% are depicted.
Two MAGs, B3_LCP (92.8% complete) and B3_B38 (91.9% complete), were not grouped with any described phyla. The 16S rRNA gene of B3_LCP was within the clade composed of those for the LCP-89 lineage, which was delineated in Silva database and lacked genomic representation. The B3_B38 MAG falls within a monophyletic clade (with strong confidence) consisting of 16S rRNA genes from several environments but has not been defined as a phylum (Fig. 2). This monophyletic clade is a sibling to Acidobacteria (Fig. 3). A MAG (B3_TA06) from candidate phylum TA06 was also obtained.
Another two 16S rRNA sequences from the MAGs, B3_Chlor and B3_Pla, were classified Chloroflexi and Planctomycetes, respectively. However, the similarity of 16S rRNA genes to those of known species was low (85% similar to that of Litorilinea sp. of Chloroflexi; 82% similar to that of Phycisphaera sp. in Planctomycetes). Based on this 16S rRNA similarity, they were unclassified Chloroflexi and Planctomycetes in the B3 samples. The MAGs of Aerophobetes (Ae_b3a and Ae_b3b) have nearly full-length 16S rRNA genes that share a similarity of 92%. According to taxonomic thresholds (30) of 16S rRNA gene sequences, they should be assigned to different families. The average nucleotide identity (ANI) of two Aerophobetes MAGs (Ae_b3a and Ae_b3b) is 72.5%. Four nearly complete archaeal MAGs for Woesearchaeota (B3_Woes), Lokiarchaeota (Loki_b31 and Loki_b32), and Heimdallarchaeota (B3_Heim) were obtained. The ANI of the two Lokiarchaeota MAGs is 69.5%. The related lineages were dwelling in anoxic sediments (23, 25, 31).
The relative abundance of each MAG in the metagenomes of the two layers was estimated from the metagenomic coverage information. Aerophobetes MAG (Ae_b3a) was most abundant, accounting for 2.3% and 3.9% of genomic sequencing reads in B3-1 and B3-2, respectively. The Planctomycetes and Chloroflexi MAGs were also the abundant members in B3-1, accounting for 2.1% and 1.1%, respectively. Other major species in B3-2 harbored B3_Chlor (3.6%), Ae_b3b (1.9%), and Loki_b31 (1.1%) (see Fig. S4). In total, the microbes of these nine phyla in the metagenomes contributed to 7.8% and 12.9% of the sequencing reads for B3-1 and B3-2, respectively. The distribution of these MAGs in different layers is relatively consistent with that based on analysis of the 16S rRNA gene amplicons.
To examine the worldwide distribution of the phyla detected in B3, we collected 16S rRNA genes from NCBI nucleotide databases with query identities higher than 85% and included them in phylogenetic trees. Bacteria of candidate phyla B38, TA06, and LCP-89 generally inhabit anoxic sediments in hydrothermal vents, cold seeps, and methane hydrate-bearing subseafloor sediments worldwide (see Fig. S5). The 16S rRNA-based tree showed that species of candidate phyla B38 and TA06 from B3 were also identified in the clones isolated from nearby subseafloor of the Taiwan Strait. This is also true for Lokiarchaeota and Heimdallarchaeota (see Fig. S6). In the lokiarchaeotal tree, the 16S rRNA genes from MAGs Loki_b31 and Loki_b32 do not neighbor Lokiarchaeum GC14_75 from Arctic Mid-Ocean Ridge reported previously (23) (Fig. S6A). The sedimentary genotype of TA06 was previously obtained from estuary sediments (11). However, the deep-sea type is distinct.
Potential metabolic capabilities of candidate bacterial phyla from SCS.
To begin to understand the physiological capabilities of the uncultured lineages, their predicted proteins were compared to a variety of function databases (see Materials and Methods). This revealed that candidate phylum B38 (MAG B3_B38) appears to be able to utilize a variety of organic compounds. A set of genes encoding a cross-membrane phosphotransferase system (PTS) for the uptake of mannose was identified in B3_B38 as a potential source of organic carbon. B3_B38 also contains several genes for periplasmic dipeptidase/aminopeptidase for the uptake of amino acids. We identified genes related to KDO2 lipid IV A and peptidoglycan synthesis pathways, which are the components of outer membranes and cell walls, respectively. KDO2 lipid IV A may aid bacterial virulence as well. B3_B38 encodes all the steps for the glycolysis pathway, except it lacks a phosphofructokinase gene. It also contains all the steps for the citrate acid cycle (TCA cycle), except for an aconitate hydratase gene. The conversion of pyruvate to acetyl coenzyme A (acetyl-CoA) may be catalyzed by pyruvate:ferredoxin oxidoreductase (PFOR), as genes encoding PFOR subunits were detected in B3_B38. Acetyl-CoA might be reduced to ethanol as indicated by the identification of an alcohol dehydrogenase gene. B3_B38 also contains the genes for NADH respiration complexes I and II.
Genes encoding a sulfate transporter and sulfate adenylyltransferase (SAT), which work at transporting and activating sulfate or selenate, are present in B3_B38 (32). Sulfate and selenate reductase genes were missing in the MAG. However, selenite might be further reduced by thioredoxin reductase to produce hydrogen selenide and was probably integrated into amino acid as selenocysteine (Sec), as the related genes were detected (Fig. 4). B3_B38 harbors genes for Sec protein biosynthesis, including SelA (selenocysteine synthase), SelB (selenocysteine-specific elongation factor), and SelD (selenophosphate synthase). Moreover, a selenocysteine insertion sequence (SECIS) was also identified (see Fig. S7 and Table S3), which is a necessary mRNA motif for selenocysteine cotranslational insertion into selenoproteins (33). Usage of Sec as the 21st amino acid in proteins has been recorded in all three domains of life (34). Most of the selenoproteins in prokaryotic selenoproteomes are redox proteins. Sec in these proteins either coordinates with redox-active metal (nickel, molybdenum, or tungsten) or catalyzes redox in Sec:thiol (35). Sec is superior to Cys in improving the catalytic efficiency of and maintaining stability in the redox reaction (36). However, only one putative selenoprotein-coding gene was located in B3_B38.
FIG 4.


Schematic metabolisms of new phyla. (A) B3_TA06 (TA06); (B) B3_B38 (B38); (C) B3_Heim (Heimdallarchaeota); (D) B3_LCP (LCP-89). The metabolic networks are predicted based on the gene contents of the MAGs. The pathways with transcriptomic evidence are denoted by a red arrow. PTS system, phosphotransferase system; CRISPRS-Cas, prokaryotic defense mechanism against foreign genetic element; Nap, periplasmic nitrate reductase; SAT, sulfate adenylyltransferase; Apr, adenylylsulfate reductase; Qmo, quinone-interacting membrane-bound oxidoreductase complex; Dsr, dissimilatory sulfite reductase; APS, adenosine 5′-phosphosulfate; FDox, oxidized ferredoxin; FDred, reduced ferredoxin; Q/QH, ubiquinone-reduced ubiquinone; NirB, assimilatory nitrite reductase large subunit; Rnf complex, electron transport complex; Mrp, multiple resistance/pH regulation Na+/H+; SDF, sodium dicarboxylate symporter family; THF, tetrahydrofuran; TrxR, thioredoxin reductase; SelA, selenocysteine synthase; SelD, selenophosphate synthase; Mvh, methyl viologen-reducing hydrogenase; hdr, heterodisulfide oxidoreductase. Details about the genome annotations are provided in Table S3.
This bacterium also appears to be able to generate hydrogen. The respiration complexes I and II of B38 presumably accumulate reduced quinol (QH2), which might be used to generate hydrogen via methyl viologen-reducing hydrogenase complex (MvhADG). This process is also perhaps associated with oxidation of coenzyme M (CoM-SH) and coenzyme B (CoB-SH) by HdrABC (37) (Fig. 4). Therefore, the genomics of the candidate phylum B38 indicates that it is a hydrogen producer.
Metabolic inference from the genome of candidate phylum LCP-89 indicates that it may be able to lead a mixotrophic lifestyle. The W-L pathway, except for formate-tetrahydrofolate ligase and formate dehydrogenase, for CO2 fixations is present in the B3_LCP MAG. Genes encoding cellulase, gingipain (extracellular cysteine proteinase), and membrane aminopeptidase were also detected (Table S3). Genes encoding glycolytic and TCA cycle pathways for heterotrophic growth are complete in the B3_LCP MAG (Fig. 4). Acetyl-CoA may be generated from pyruvate by PFOR by LCP-89. Genes encoding phosphate acetyltransferase (PTA) and acetate kinase (ACK) were detected, suggesting that it is capable of acetate production. It has been reported that the PTA-ACK pathway has an impact on ATP levels, stress tolerance, and oxygen tolerance (38). Acetyl-CoA might be turned into butanoate, as the gene encoding butyrate kinase is present. The alcohol dehydrogenase gene was also identified, suggesting that the organisms are also able to produce ethanol. Genes encoding canonical NADH phosphorylation respiration chains, including complexes I, II, and IV, were also identified in the MAG.
B3_LCP encodes a sulfate transporter, SAT, adenylylsulfate reductase (AprAB), and dissimilatory sulfite reductases (DsrABCD) (Fig. 4). Phylogenetic analyses of the DsrAB proteins revealed monophylogeny of the homologs from LCP-89 and other sulfate-reducing organisms (see Fig. S8), suggesting that the LCP-89 bacterium is probably capable of dissimilatory sulfate reduction. Furthermore, the B3_LCP MAG has genes with homology to those involved in the synthesis of siroheme for the six-electron reduction of sulfite to sulfide (39) (Fig. 4). B3_LCP also encodes ABC transporters for uptake of Fe2+ that is requisite to the Fe4S4 center of siroheme complex (40). Similar to the B3_B38 MAG, the B3_LCP MAG might carry genes for selenocompound metabolism. Genes encoding SECIS, SelA, SelB, and SelD are present in this genome as well. The B3_LCP also seems to enable nitrate reduction, since it contains several genes that encode periplasmic subunits of nitrate reductases NapABDGH (Fig. 4 and Table S3).
The ecological roles of the candidate phylum TA06 are still largely unresolved, but draft genomes have been reconstructed from a shallow estuary (11). The candidate phylum TA06 MAG (B3_TA06) (∼91.9% complete) was recovered from B3. B3_TA06 has a phosphoenolpyruvate carboxykinase gene, which suggests its capacity for gluconeogenesis. However, B3_TA06 contains mannose transporter (PTS) genes, indicating that the organisms are able to import external carbohydrates. TA06 might produce or utilize ethanol by using an alcohol dehydrogenase encoded by the MAG.
ATP generation driven by the ion gradient is essential for the survival of organisms. Several cross-membrane transport complexes function to produce the proton gradient. The SCS B3_TA06 genome encodes the Rnf complex (electron transport complex). This process can associate the oxidation of ferredoxin with the reduction of NAD+ to NADH. B3_TA06 encodes the Mrp complex, a primary cross-membrane Na+/H+ antiporter in Bacteria (41). The Mrp complex encoded in the B3_TA06 MAG might help to achieve homeostasis of their cells.
We identified genes encoding one NiFe hydrogenase (αβγδ subunits) in B3_TA06, in which the large subunit was clustered with group 3b tetrameric bifunctional hydrogenases (Fig. 5) (12). Such a group 3b NiFe hydrogenase could carry out a bifurcating reaction for hydrogen production whereby NAD(P)H and protons are consumed simultaneously (42). The NAD(P)H might be supplied from sugar fermentation. Thus, TA06 may be a source of H2, which is an important electron donor in the deep sea (43). The genes encoding accessory proteins that help synthesize the multicomponent active sites of NiFe hydrogenase were also detected in B3_TA06 (Table S3).
FIG 5.

Phylogeny of large subunits of NiFe hydrogenases. Large subunits detected in B3 are highlighted in red. Sequence alignment was performed by MUSCLE (v3.8.31); phylogenetic tree was inferred by raxmlGUI using maximum likelihood method with PROTGAMMABLOSUM62 model and 1,000 replicates. Microorganisms contain NiFe hydrogenases, encoded by Ae_b3a and Ae_b3b (Aerophobetes), B3_Pla (Planctomycetes), B3_TA06 (TA06), Loki_b31 and Loki_b32 (Lokiarchaeota), B3_B38 (B38), and B3_Chlor (Chloroflexi).
Physiological capabilities of Asgard archaea from SCS.
Heimdallarchaeota, affiliated with Asgard archaea, are of interest because they have been shown to possess eukaryotic signature proteins (ESPs) and are the closest relatives of eukaryotes (25). We reconstructed a nearly complete MAG of Heimdallarchaeota (B3_Heim) from the sample B3. The ANI values between B3_Heim and those publicly available are in the range of 61.5% to 63.2%, considerably different from those recently recovered (25). In the B3_Heim MAG, genes encoding the glycolysis and fermentation pathways were present, probably for the production of ethanol, lactate, and acetate. Notably, the Heimdallarchaeota MAG harbored a complete set of genes involved in the TCA cycle and the respiration complexes I, II, and IV. Complex IV was probably centralized on cytochromes c and bd for pumping H+ into the periplasma (Fig. 4). A gene encoding archaeal type III RuBisCO is also present in B3_Heim. CO2 fixation through this archaeal type III RuBisCO may contribute to AMP and central carbon metabolism (44). B3_Heim also contains genes encoding pathways for lipid utilization and the fatty acid degradation (beta-oxidation) pathway. Moreover, it also possesses a relatively large variety of peptidase-coding genes (see Fig. S9). Genes encoding clostripain (extracellular cysteine peptidase) and membrane dipeptidase/aminopeptidase were found in the SCS MAG (Table S3). There are also ABC transporter genes responsible for the uptake of oligopeptide/dipeptide. The results suggest that the B3 Heimdallarchaeota archaea might capture detrital proteins effectively from the environment and influence the marine sedimentary carbon cycle by involvement in detrital protein remineralization (45). The deep-sea Heimdallarchaeota MAG contains the nitrite reductase (NADH) large subunit nirB gene, suggesting that it is able to reduce NO2 to NO. Detection of glutamine synthetase and glutamate synthase genes supported the assimilation of ammonia in Heimdallarchaeota. In addition, undetermined nitrile compounds might be catalyzed by nitrilase to generate ammonia and utilized for amino acid synthesis by Heimdallarchaeota (Fig. 4).
Lokiarchaeota is another member of Asgard archaea. Its two MAGs bear the genes for dipeptidase/aminopeptidase and a large variety of other peptidase-coding genes (Fig. S9). Fatty acid degradation was present in species of Lokiarchaeota, as their MAGs harbor abundant genes that take part in the pathway. Lokiarchaeota may contribute to the reduction of nitrite to nitric oxide, as we detected the genes that encode cytoplasmic nitrite reductase (NirB) (see Fig. S10A). In the Lokiarchaeota MAG Loki_b32, there was a full set of genes involved in serine and glycine degradation with the release of CO2 (Fig. S10A). In addition, genes encoding the W-L pathway were present in the Lokiarchaeota MAGs (Loki_b31 and Loki_b32). Archaeal CO dehydrogenase/CoA synthase (Cdh) is an enzyme functionally similar to bacterial Acs and evolved independently in archaea via convergent evolution (46). Phylogenetic analyses of the deduced Cdh enzymes revealed that those of Lokiarchaeota formed a novel branch (see Fig. S11A). Tetrahydrofolate (THF) and tetrahydromethanopterin (THMPT) are both C1 carriers in the W-L pathway encoded by lokiarchaeotal MAG (CR4) (47). Lokiarchaeota archaea detected here prefer the THMPT W-L pathway, and the related genes were all identified.
Potential metabolisms of other MAGs in SCS.
The Aerophobetes MAGs encode numerous ABC transporters (Fig. S10B), which might help for the import of carbohydrate, peptide, and other nutrient ions from the milieu (48, 49). However, they also encode a complete W-L pathway (except for NADP+-dependent formate dehydrogenase) and group 3b hydrogenases (Fig. 5 and Fig. S12), as has been reported in Aerophobetes obtained from the Red Sea (26). We found that most of the deep-sea genotypes in B3 encode the W-L pathway (Fig. S12). In the W-L pathway, acetyl-CoA synthase (Acs) is a key enzyme that converts CO2 to acetyl-CoA. Phylogenetic analyses of AcsAB proteins encoded in Aerophobetes MAGs (Ae_b3a and Ae_b3b) revealed that they are adjacent to that from the LCP-89 MAG (B3_LCP), while those of Chloroflexi MAG (B3_Chlor) and Planctomycetes MAG (B3_Pla) were closer to the root (Fig. S11B). The discovery of autotrophic CO2 fixation pathways in the B3 organisms indicates that they may be capable of lithoautotrophic growth.
In addition to the capacity to fix inorganic carbon, the microbes in this study are probably able to utilize a variety of organic carbon compounds. The bacterial MAGs in B3 sediments encode a large variety of carbohydrate-active enzymes (CAZys) to degrade organic carbon (Fig. 6). The Planctomycetes MAG (B3_Pla) bears the highest number of CAZy genes encoding glycoside hydrolases (182 genes) and carbohydrate esterases (38 genes), among which, α-N-acetylgalactosaminidase (GH109) is the dominant group. In contrast, the Chloroflexi MAG (B3_Chlor) is enriched with genes encoding glycosyl transferase CAZy families of GT2, GT4, and GT83. In addition, genes involved in fatty acid degradation (beta-oxidation) pathways were identified in the Chloroflexi MAG.
FIG 6.
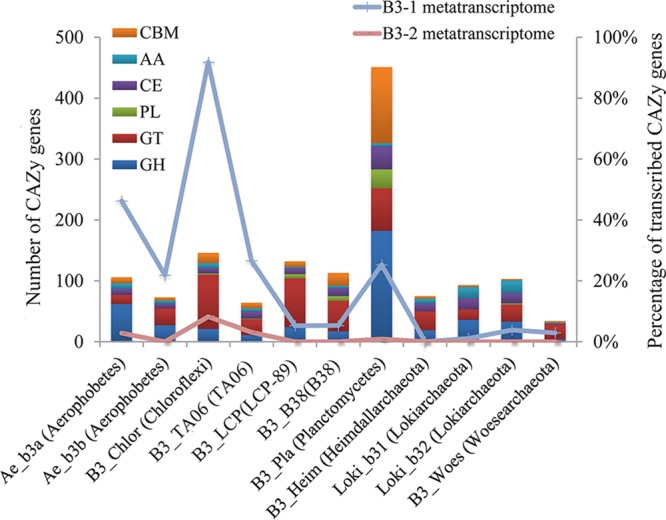
Percentages of CAZy genes in metagenome and metatranscriptome. The bars represent the numbers of CAZy genes. The lines denote the percentages of CAZy transcripts for the organisms. CAZy categories include carbohydrate-binding modules (CBM), auxiliary activities (AA), carbohydrate esterases (CE), polysaccharide lyases (PL), glycosyl transferases (GT), and glycoside hydrolases (GH) (www.cazy.org).
The utilization of organic matter and the W-L pathway coexisted in some of the B3 organisms, indicating flexibility in their abilities to assimilate carbon and energy conservation in the niche (27). Acetyl-CoA produced in the W-L pathway is not only for biomass production but also associated with ATP generation (such as via the PTA-ACK pathway to produce ATP) (38, 50). The presence of genes encoding W-L and PTA-ACK pathways in the Aerophobetes MAG (Ae_b3a) and Planctomycetes MAG (B3_Pla) indicated additional ATP generation in the species. Organic matter degradation may generate CO2, NADH, and reduced ferredoxins. All these could be utilized by the W-L pathway. In addition, the Rnf complex helps to balance NAD+/NADH and ferredoxin (ox/red), as well as to enhance the ion gradient for energy conservation (27). A complete set of Rnf-coding genes was also detected in Aerophobetes MAG (Ae_b3a) (Fig. S10) and Planctomycetes MAG (B3_Pla) (data not shown), while in the Aerophobetes MAG (Ae_b3b), only the genes encoding RnfABC subunits were identified (Ae_b3b) (data not shown). The combination of organic matter fermentation, W-L pathway, and the Rnf complex could balance redox stoichiometry and generate an electrogenic sodium ion potential for ATP production (27), which may help these microorganisms achieve fitness in the niche. The Chloroflexi MAG (B3_Chlor) lacks genes for the PTA-ACK pathway and Rnf complex. Thus, the W-L pathway in Chloroflexi might contribute biomass. We also obtained a small (∼1 Mbp) MAG belonging to Woesearchaeota (B3_Woes). Although the completeness of the Woesearchaeota MAG is 97.7%, it lacks most of the genes for protein, glycerolipid, and fatty acid degradation. This is consistent with the proposed symbiotic life of these archaea (31). However, the Woesearchaeota MAG in this study contained genes encoding archaeal type III (RuBisCO) (data not shown). It is likely that the species of Woesearchaeota in B3 could provide its symbiont with a carbon source.
Possible functional interconnectivities in the B3 community.
Functional predictions of the metabolic pathways in these MAGs from the SCS provide our first glimpses into roles of these distinct microbes in carbon cycling (Fig. 7). Overall, the ability to use inorganic carbon is common among these lineages. H2 utilization in the W-L pathway is assisted by hydrogenases (43). Except for LCP-89, all the microorganisms that contained the W-L pathway in this study also harbor NiFe hydrogenases. The TA06 and B38 bacteria, which are probably not able to conduct autotrophic CO2 fixation, might serve as H2 providers.
FIG 7.
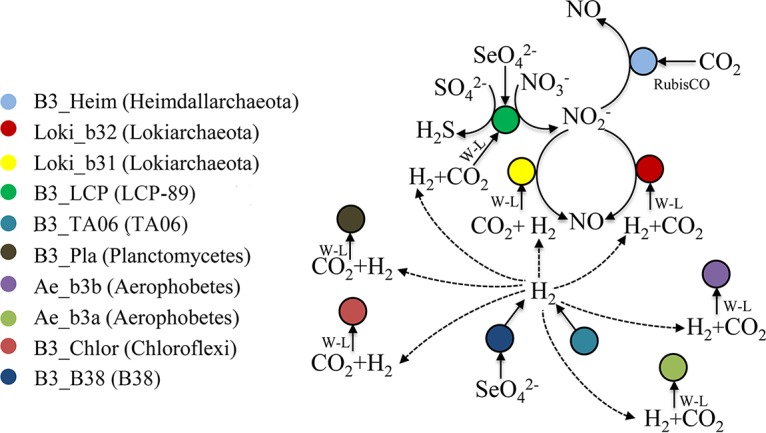
Hypothetical metabolic interactions between B3 microorganisms.
The microorganisms in the B3 sediments were predicted to be capable of the anaerobic respiration of nitrate, selenite, and sulfate. Electronic acceptors, such as nitrate and sulfate, possibly came from the downward circulation of benthic water (1). The reduction of nitrate to nitric oxide might be accomplished by the cooperation among species of LCP-89, Heimdallarchaeota, and Lokiarchaeota (Fig. 7). The reduction of sulfate to H2S was probably solely carried out by the LCP-89. Considering the net product of the predicted metabolic network, the microbes in B3 were probably converting sulfur and nitrogen oxides to the reductive compounds (sulfide and nitric oxide).
Transcriptional activity of lineages in the SCS sediments.
Sequencing of the B3-1 and B3-2 metatranscriptomes generated ∼5.2 Gbp and ∼4.0 Gbp raw data, respectively. After removal of rRNA transcripts, repeats, low-quality reads, and contaminants, there remained ∼0.21-Gbp and ∼0.66-Gbp transcripts for B3-1 and B3-2, respectively. To deduce the transcriptional profile of the 11 MAGs, the transcriptomic reads of B3-1 and B3-2 were independently mapped to the coding sequences (CDSs) of the 11 MAGs. The transcriptomic reads from B3-1 were primarily recruited to B3_Chlor (Chloroflexi) (49.5%) and Ae_b3a (Aerophobetes) (37.6%). A total of 55.1% of the transcripts from B3-2 were assigned to B3_Chlor and 25.8% to Ae_b3a (see Fig. S13A). This suggests that Chloroflexi and Aerophobetes genomes were the most active among B3 MAGs. Fragments per kilobase of transcript per million fragments mapped (FPKM; an estimate of transcriptional level) were calculated for all the genes from these 11 organisms in both B3 layers (see Fig. S14 and Table S4).
The CAZy utilization pathways were detectable in all the bacterial transcriptomes (Fig. 6). A total of 92% of the B3_Chlor (Chloroflexi) CAZy genes were identified in the B3-1 transcriptome, and 46% of the Ae_b3a (Aerophobetes) CAZy genes were identified; fewer genes from archaeal MAGs were detected. In the B3-2 transcriptome, the number of these CAZy genes decreased, and for archaeal MAGs, only one gene belonging to AA4 family from Loki_b31 was found (Fig. 6). Most CAZy genes of Ae_b3a (Aerophobetes) and B3_Chlor (Chloroflexi) were probably expressed in both layers (see Table S5), as suggested by the presence of their transcripts. These genes were more transcriptionally active in Ae_b3a than B3_Chlor in the B3-1 layer based on RNA FPKM/DNA FPKM (Table S5). The transcripts for alpha-mannosidase (GH38), beta-galactosidase (GH2), and alpha-rhamnosidase (CBM67) belonging to Ae_b3a (Aerophobetes) were abundant in B3-1 (Table S5), indicating these carbohydrate-degrading activities were much stronger in B3-1 than in B3-2.
Genes encoding Acs subunit alpha/beta and formate-tetrahydrofolate ligase were also detected in B3_Chlor (Table S4), suggesting that some of the Chloroflexi bacteria may be also fixing CO2. Additionally, the presence of F420-nonreducing hydrogenase transcripts in the B3-1 transcriptome is supportive of H2 metabolism in the Chloroflexi bacteria. Transcriptomics data also indicated the expression of several subunits of the Rnf complex and the NADH:ubiquinone oxidoreductase (complex I) in Aerophobetes MAGs (Ae_b3a and Ae_b3b) (Table S4), indicating that Aerophobetes may rely on these processes for energy conservation. The W-L pathway (Acs subunits, group 3b bifunctional Ni/Fe hydrogenase alpha subunit, and formate-tetrahydrofolate ligase) was also transcriptionally active in Ae_b3a, while only the Acs delta subunit and Ni/Fe hydrogenase alpha subunit transcripts were identified in Ae_b3b (Table S4). Also, transcripts for MAG Ae_b3a ABC transporter genes predicted to be involved in the transport of sugars, peptides, branched-chain amino acids, and iron were abundant (Fig. S10B). The Rnf complex, heterotrophic potential, and W-L pathway were simultaneously active in Aerophobetes detected here. As described above, the integration of the three modules would provide more ATPs for this Rnf-dependent acetogen (27). This may explain why the Aerophobetes bacteria (harboring Ae_b3a) were more abundant than other microbes in B3.
For the candidate phyla B38, TA06, LCP-89, Lokiarchaeota, and Heimdallarchaeota, a small number of genes was detected in the metatranscriptomes, which is comparable to their low abundance in the metagenome (Fig. S13B). Transcripts of the NiFe hydrogenase gene were found in B3-1 transcriptomes and were assigned to Lokiarchaeota and TA06 (Table S4). This indicates that these organisms were probably actively metabolizing H2.
Conclusions.
Although the genomes of several uncultured prokaryotic lineages have been obtained from shallow sediments in recent years (11, 25, 51), we know little concerning the organisms that inhabit the deep ocean sediments. In this study, we obtained MAGs and transcriptomes of 11 distinct deep-sea microbes belonging to nine distinct phyla. Among the lineages, there are metabolic reconstructions of representatives of the candidate phyla LCP-89, TA06, and B38. LCP-89 appears to be involved in the reduction of selenium, nitrogen, and sulfur oxides. LCP-89 and Heimdallarchaeota may degrade detrital proteins by extracellular cysteine proteinase and aminopeptidase. B38 and TA06 are predicted to produce H2, with the identification of hydrogenase in the MAGs. Simultaneously, these organisms are versatile, utilizing a variety of organic carbon compounds in deep-sea sediments. Surprisingly, nearly all the organisms appear to have the capacity to assimilate inorganic carbon for the self-sustainability of the ecological niche, highlighting the critical role of H2-producing microbes in the nutrient-deprived deep-sea sediments. These microorganisms play different roles in utilizing inorganic carbon and scavenging diverse organic compounds involved in the deep-sea carbon cycle. Overall, the MAGs recovered here contributed undescribed species to the tree of life and laid the foundation for future study on these novel phyla persisting in marginal sediments of the SCS.
MATERIALS AND METHODS
Sample collection and measurement of mineral contents.
During a cruise on board the R/V SHIYAN 3, a gravity core (43 cm) was collected on 15 July 2015 from north of the SCS (22°10.18′N, 118°24.00′E), 1,100 m below sea level. Sediment samples were obtained from two layers of the core. The upper layer, B3-1, was estimated to be 30 cm below the sea floor, and the lower layer, B3-2, was ∼10 cm deeper than B3-1. All the samples were collected rapidly from the innermost of the core with sterile instruments, placed in sterile tubes, and then kept at −80°C until use. Approximately 50 g of the sediment samples was mixed with RNAlater (Ambion, Carlsbad, CA, USA), stored at 4°C overnight, and then frozen at −80°C for further processing.
Mineral contents of the samples were analyzed by X-ray diffraction (XRD) conducted on a diffractometer system, XPERT-PRO (Cu Kα radiation at 45 kV and 40 mA; goniometer was PW3050/60 [theta/theta], scanning from 3° to 85° with 0.03 step size° [°2Th]). Diffraction angles were unique to each mineral with different atomic structures. The constitution of minerals was converted to weight percent.
Nucleic acid extraction.
Genomic DNA was extracted from 10 g (wet weight) of each sample using the PowerMax Soil DNA Isolation kit (Mo Bio, Carlsbad, CA, USA), according to the manufacturer’s standard protocol. RNA was extracted from the samples preserved in RNAlater using a PowerSoil Total RNA Isolation kit (Mo Bio, Carlsbad, CA, USA). RNase- and DNase-free sterile tubes and tips were used in the operation to avoid contaminants from the environment. DNA in the RNA extractions was removed by a Turbo DNA-free kit (Ambion, Carlsbad, CA, USA) and was checked by PCR using universal primer 341F (5′-CCTAYGGGRBGCASCAG-3′) and reverse fusion primer 802R (5′-TACNVGGGTATCTAATCC-3′) targeting V3-V4 region of 16S rRNA genes (52). The primers were examined in Silva (https://www.arb-silva.de/) for primer coverage of Asgard archaea and novel bacterial phyla. Subsequently, cDNA was obtained by Ovation RNA-Seq System V2 kit (NuGEN, San Carlos, CA, USA), and purified using a MinElute Reaction Cleanup kit (Qiagen, Hildon, Germany). A blank control (sterile water) was included in this step. The quality and quantity of genomic DNA and cDNA were checked by gel electrophoresis and a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA, USA).
16S rRNA gene amplicon sequencing and bioinformatics analyses.
Using the genomic DNA as the template, the universal primers 341F and 802R with a 6-nucleotide barcode were used to amplify partial 16S rRNA genes. The PCR was executed as follows: denaturation at 98°C for 10 s, 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 15 s, and extension at 72°C for 30 s, and with a final extension at 72°C for 5 min. Illumina libraries for the 16S rRNA gene amplicons were prepared with a TruSeq Nano DNA LT kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina MiSeq platform (2 × 300 bp). Raw reads were separated for different samples based on the barcode information and filtered by NGS QC Toolkit (v2.3.3) (53) with default parameters (base quality <20 for >70% of a given read), followed by merging paired reads with PEAR (v0.9.5) (54). The resulted fragments were processed with the QIIME pipeline (55). First, fragments with similarity >97% were clustered into OTUs by UCLUST (56). The longest fragment in each OTU was picked as the representative read and used for alignment with the references in the Silva database (version 128) by PyNAST (57). Chimeric reads were discarded based on the detection result of ChimeraSlayer (58). Taxa assigned to chloroplast, mitochondria, and eukaryotes were ignored. Taxonomy assignment of representative reads was finally performed by online SILVAngs (https://www.arb-silva.de/ngs) with default settings.
Metagenomic sequencing and de novo assembly.
The genomic DNA was sheared randomly to ∼500 bp by ultrasonication. DNA libraries were prepared with a TruSeq Nano DNA LT kit (Illumina, San Diego, CA, USA) and sequenced on an Illumina MiSeq platform (2 × 300 bp), yielding 27-Gbp raw paired-end reads. Quality control of raw reads was initiated by removing reads that contained adaptors or were associated with a low average quality (<20 over 70% of the reads) using NGS QC Toolkit (v2.3.3). The bases in low quality (<20) at both ends were trimmed. The resulted reads shorter than 50 bp were ignored. The high-quality paired reads of samples B3-1 and B3-2 were coassembled into scaffolds by SPAdes (v3.6.2) (59) with parameters “–careful -k 21, 33, 55, 77, 99, 127.”
Genome binning.
The coverage of scaffolds was calculated by mapping qualified reads to the assembled scaffolds using Bowtie 2 (v2.2.4) with default settings (60). Genome binning was performed according to the protocol described in reference 61. A two-dimensional plotting could split the scaffolds with different coverage levels in B3-1 and B3-2, and then correspondence analysis (CA) might exclude potential contaminants by checking the consistency of tetranucleotide frequencies (TNF) among the scaffolds. Conserved single-copy genes (CSCGs) were identified using hmmsearch (v3.0) with default cutoffs (62). The CSCG data set for bacteria was provided by Albertsen et al. (61), while for archaea, the data set was described by Wang et al. (63). A final assessment of the completeness and contamination was conducted using CheckM (v1.0.5) (64).
Annotation of MAGs.
In addition, CDS and proteins in scaffolds were predicted by Prodigal (v2.6.2) with metagenome mode (65). Functional predictions of proteins were accomplished by comparisons using BLASTP search against protein databases, including NCBI-nr, CAZy (66), KEGG (67), and MEROPS (merops.sanger.ac.uk) with an E value cutoff of 1e−05. tRNA genes were predicted by tRNAscan-SE (v1.3.1) (68) with different models for bacteria and archaea.
Metatranscriptome sequencing and analysis.
The cDNA libraries for B3-1, B3-2, and the blank control were prepared with an Ovation Ultralow System V2 kit (NuGEN, San Carlos, CA, USA) and were then sequenced on an Illumina MiSeq platform. Quality control of raw reads included the following steps: filtration of reads containing adaptors, exclusion of reads with ambiguous bases, removal of reads with average quality lower than 20 over 70% of the length, and deletion of reads with poly(T), poly(A), and poly(AT) content more than 50% of the length and those less than 50 bp by in-house scripts. Then, the reads for the blank control were assembled by Trinity (v2.3.2) (69). The potential contaminant reads in B3-1 and B3-2 were searched by aligning them on the assembled fragments of the blank control using Bowtie 2 (v2.2.4) (with parameters -N 1, –local). The contaminants were further removed by searching against a database containing genomes for the species potentially detected in reagents (70). rRNA genes were identified by SortMeRNA (v2.1) (71) and then excluded. Transcriptome reads were sorted for the CDSs belonging to the different genome bins by mapping the quality-filtered reads on the CDSs using Bowtie 2 (v2.2.4) with default settings. Expression level of the genes was reported in fragments per kilobase of transcript per million fragments mapped (FPKM) (72) performed by RSEM (v1.2.9) (73) (with parameters –fragment-length-mean 160 –fragment-length-sd 110).
Phylogenetic analysis of 16S rRNA and conserved genes.
The 16S rRNA genes were identified in the genomes using rRNA_HMM (74). An initial placement of the 16S rRNA genes in phylogenetic tree was performed by combining the sequences with all the references from the Silva database (version 125) for inference of phylogenetic relationships (75). We collected a total of 984 rRNA sequences representing 89 phyla (73 for Bacteria and 16 for Archaea) in the Silva database. The alignment of the sequences was performed using MAFFT L-INS-i (v7.294b) (76). Poorly aligned regions were trimmed by trimAl (v1.4) (with parameter -automated1) (77). A phylogenetic tree was constructed by maximum likelihood (ML) algorithm using raxmlGUI (v1.5) (78) with GTRGAMMA model for 1,000 replicates. The reference rRNAs with phylogenetic affinity to our 16S rRNA genes were selected for the construction of a new ML tree. Together with the sequences from the MAGs, a total of 157 16S rRNA genes that represented 41 different prokaryotic phyla were pooled to build the tree with the same method. Reference microorganisms and accession numbers used for 16S rRNA gene phylogenetic analysis are shown in Table S6 in the supplemental material. Genomes of the selected phyla were obtained to build a protein-based phylogenetic tree. The genomes used for the phylogenetic analysis can be downloaded from NCBI (Table S2). A total of 24 ribosomal proteins (see Table S2) shared by Bacteria and Archaea were identified in the MAGs and the reference genomes. The ribosomal proteins were first aligned with MAFFT L-INS-i (v7.294b) and then concatenated, which was followed by treatment with trimAl (v1.4). A phylogenetic ML tree was built using raxmlGUI (v1.5) with PROTCATLG model for 1,000 replicates.
Data availability.
Data supporting the results of this article have been deposited in NCBI under BioProject number PRJNA383916. The SRA accession for sequence reads for 16S rRNA amplicons is SRR5685235, and genome accessions are NJBQ00000000 (Ae_b3a), NJBP00000000 (Ae_b3b), NJBO00000000 (B3_TA06), NJBN00000000 (B3_LCP), NJBM00000000 (B3_Pla), NJBL00000000 (B3_B38), NJBK00000000 (B3_Chlor), NJBI00000000 (Loki_b31), NJBH00000000 (Loki_b32), NJBG00000000 (B3_Woes), and NJBF00000000 (B3_Heim).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Z. M. Gao for efforts during the sampling cruise and L. S. He for experimental assistance.
This study was supported by National Science Foundation of China (NSFC; numbers 41476104 and 31460001), The National Key Research and Development Program of China (2016YFC0302500), and the Strategic Priority Research Program B of Chinese Academy of Sciences (numbers XDB06010201 and XDB06010103). The sampling cruise was supported by NSFC Open Research Cruise (cruise number NORC2015-06) funded by Shiptime Sharing Project of NSFC.
Y.W. conceived of and designed experiments. J.-M.H., B.J.B., and J.-T.L. performed the experiments and analyzed the data. J.-M.H., B.J.B., and Y.W. wrote the manuscript. Y.W. directed and supervised all the research.
We declare no competing interests in this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00523-19.
REFERENCES
- 1.D'Hondt S, Bo BJ, Miller DJ, Batzke A, Blake R, Cragg BA, Cypionka H, Dickens GR, Ferdelman T, Hinrichs KU. 2004. Distributions of microbial activities in deep subseafloor sediments. Science 306:2216–2221. doi: 10.1126/science.1101155. [DOI] [PubMed] [Google Scholar]
- 2.Kallmeyer J, Pockalny R, Adhikari RR, Smith DC, D'Hondt S. 2012. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc Natl Acad Sci U S A 109:16213. doi: 10.1073/pnas.1203849109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrenk MO, Huber JA, Edwards KJ. 2010. Microbial provinces in the subseafloor. Annu Rev Mar Sci 2:279–304. doi: 10.1146/annurev-marine-120308-081000. [DOI] [PubMed] [Google Scholar]
- 4.Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH. 2008. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc Natl Acad Sci U S A 105:10583–10588. doi: 10.1073/pnas.0709942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orsi WD, Edgcomb VP, Christman GD, Biddle JF. 2013. Gene expression in the deep biosphere. Nature 499:205. doi: 10.1038/nature12230. [DOI] [PubMed] [Google Scholar]
- 6.Beman JM, Francis CA. 2006. Diversity of ammonia-oxidizing archaea and bacteria in the sediments of a hypernutrified subtropical estuary: Bahia del Tobari, Mexico. Appl Environ Microbiol 72:7767–7777. doi: 10.1128/AEM.00946-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huber JA, Welch DBM, Morrison HG, Huse SM, Neal PR, Butterfield DA, Sogin ML. 2007. Microbial population structures in the deep marine biosphere. Science 318:97–100. doi: 10.1126/science.1146689. [DOI] [PubMed] [Google Scholar]
- 8.Santelli C, Orcutt B, Banning E, Bach W, Moyer C, Sogin M, Staudigel H, Edwards K. 2008. Abundance and diversity of microbial life in ocean crust. Nature 453:653. doi: 10.1038/nature06899. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Su X, Chen F, Wang Y, Jiao L, Dong H, Huang Y, Jiang H. 2012. Microbial diversity in cold seep sediments from the northern South China Sea. Geosci Front 3:301–316. doi: 10.1016/j.gsf.2011.11.014. [DOI] [Google Scholar]
- 10.Cui HP, Su X, Chen F, Wei SP, Chen SH, Wang JL. 2016. Vertical distribution of archaeal communities in cold seep sediments from the Jiulong methane reef area in the South China Sea. Biosci J 32:1059–1068. doi: 10.14393/BJ-v32n4a2016-33994. [DOI] [Google Scholar]
- 11.Baker BJ, Lazar CS, Teske AP, Dick GJ. 2015. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:14. doi: 10.1186/s40168-015-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, Tringe SG, Chain PSG, Scholz MB, Lo C-C, Raymond J, Quake SR, Hedlund BP. 2013. Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4:1854. doi: 10.1038/ncomms2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor B, Hayes DE. 2013. Origin and history of the South China Sea basin, ocean margin systems, p 457–477. Springer, Berlin, Germany. [Google Scholar]
- 14.Hayes DE, Nissen SS. 2005. The South China Sea margins: implications for rifting contrasts. Earth Planet Sci Lett 237:601–616. doi: 10.1016/j.epsl.2005.06.017. [DOI] [Google Scholar]
- 15.Barckhausen U, Engels M, Franke D, Ladage S, Pubellier M. 2014. Evolution of the South China Sea: revised ages for breakup and seafloor spreading. Mar Pet Geol 58:599–611. doi: 10.1016/j.marpetgeo.2014.02.022. [DOI] [Google Scholar]
- 16.Hui GG, Li S, Li X, Guo LL, Suo YH, Somerville ID, Zhao S, Hu M, Lan H, Zhang J. 2016. Temporal and spatial distribution of Cenozoic igneous rocks in the South China Sea and its adjacent regions: implications for tectono-magmatic evolution. Geol J 51:429–447. doi: 10.1002/gj.2801. [DOI] [Google Scholar]
- 17.Franke D. 2013. Rifting, lithosphere breakup and volcanism: comparison of magma-poor and volcanic rifted margins. Mar Pet Geol 43:63–87. doi: 10.1016/j.marpetgeo.2012.11.003. [DOI] [Google Scholar]
- 18.Liang Z, Luan X. 2013. Stability of submarine slopes in the northern South China Sea: a numerical approach. Chin. Chin J Ocean Limnol 31:146–158. doi: 10.1007/s00343-013-2060-z. [DOI] [Google Scholar]
- 19.Suess E. 2005. RV SONNE Cruise Report SO177, Sino-German cooperative project, South China Sea continental margin: geological methane budget and environmental effects of methane emissions and gashydrates. IFM-GEOMAR, Kiel, Germany. [Google Scholar]
- 20.Han X, Suess E, Huang Y, Wu N, Eisenhauer A, Bohrmann G, Su X, Abegg F, Tao J, Fang Y. 2005. Jiulong methane reef: first direct evidence of methane seepage in the South China Sea. Geophys Res Abstr 7:4055. [Google Scholar]
- 21.Wu Y, Qiu J-W, Qian P-Y, Wang Y. 2018. The vertical distribution of prokaryotes in the surface sediment of Jiaolong cold seep at the northern South China Sea. Extremophiles 22:499–510. doi: 10.1007/s00792-018-1012-0. [DOI] [PubMed] [Google Scholar]
- 22.Han X, Suess E, Huang Y, Wu N, Bohrmann G, Su X, Eisenhauer A, Rehder G, Fang Y. 2008. Jiulong methane reef: microbial mediation of seep carbonates in the South China Sea. Mar Geol 249:243–256. doi: 10.1016/j.margeo.2007.11.012. [DOI] [Google Scholar]
- 23.Spang A, Saw JH, Jorgensen SL, Zaremba-Niedzwiedzka K, Martijn J, Lind AE, van Eijk R, Schleper C, Guy L, Ettema TJ. 2015. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521:173–179. doi: 10.1038/nature14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seitz KW, Lazar CS, Hinrichs KU, Teske AP, Baker BJ. 2016. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J 10:1696–1705. doi: 10.1038/ismej.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Bäckström D, Juzokaite L, Vancaester E, Seitz KW, Anantharaman K, Starnawski P, Kjeldsen KU, Stott MB, Nunoura T, Banfield JF, Schramm A, Baker BJ, Spang A, Ettema T. 2017. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 541:353–358. doi: 10.1038/nature21031. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Gao ZM, Li JT, Bougouffa S, Tian RM, Bajic VB, Qian PY. 2016. Draft genome of an Aerophobetes bacterium reveals a facultative lifestyle in deep-sea anaerobic sediments. Sci Bull 61:1176–1186. doi: 10.1007/s11434-016-1135-6. [DOI] [Google Scholar]
- 27.Schuchmann K, Muller V. 2016. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microbiol 82:4056–4069. doi: 10.1128/AEM.00882-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thomas PK, Thomas T, Thomas J, Pandian MS, Banerjee R, Babu PVR, Gupta S, Vimal R. 2014. Role of hydrothermal activity in uranium mineralisation in Palnad Sub-basin, Cuddapah Basin, India. J Asian Earth Sci 91:280–288. doi: 10.1016/j.jseaes.2014.02.013. [DOI] [Google Scholar]
- 29.Parry WT. 1982. Geochemistry of hydrothermal chlorite replacing igneous biotite. Clays Clay Miner 30:81–90. doi: 10.1346/CCMN.1982.0300201. [DOI] [Google Scholar]
- 30.Yarza P, Yilmaz P, Pruesse E, Glöckner F, Ludwig W, Schleifer K, Whitman W, Euzéby J, Amann R, Rosselló-Móra R. 2014. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 31.Castelle CJ, Wrighton KC, Thomas BC, Hug LA, Brown CT, Wilkins MJ, Frischkorn KR, Tringe SG, Singh A, Markillie LM, Taylor RC, Williams KH, Banfield JF. 2015. Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25:690–701. doi: 10.1016/j.cub.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 32.de Souza MP, Lytle CM, Mulholland MM, Otte ML, Terry N. 2000. Selenium assimilation and volatilization from dimethylselenoniopropionate by Indian mustard. Plant Physiol 122:1281–1288. doi: 10.1104/pp.122.4.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walczak R, Westhof E, Carbon P, Krol A. 1996. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. RNA 2:367. [PMC free article] [PubMed] [Google Scholar]
- 34.Mariotti M, Santesmasses D, Capella-Gutierrez S, Mateo A, Arnan C, Johnson R, D'Aniello S, Yim SH, Gladyshev VN, Serras F, Corominas M, Gabaldón T, Guigó R. 2015. Evolution of selenophosphate synthetases: emergence and relocation of function through independent duplications and recurrent subfunctionalization. Genome Res 25:1256–1267. doi: 10.1101/gr.190538.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kryukov GV, Gladyshev VN. 2004. The prokaryotic selenoproteome. EMBO Rep 5:538–543. doi: 10.1038/sj.embor.7400126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mariotti M, Lobanov AV, Manta B, Santesmasses D, Bofill A, Guigó R, Gabaldón T, Gladyshev VN. 2016. Lokiarchaeota marks the transition between the archaeal and eukaryotic selenocysteine encoding systems. Mol Biol Evol 33:2441–2453. doi: 10.1093/molbev/msw122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim Biophys Acta 1827:94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Kim JN, Ahn S-J, Burne RA. 2015. Genetics and physiology of acetate metabolism by the Pta-Ack pathway of Streptococcus mutans. Appl Environ Microbiol 81:5015–5025. doi: 10.1128/AEM.01160-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy MJ, Siegel LM, Tove SR, Kamin H. 1974. Siroheme: a new prosthetic group participating in six-electron reduction reactions catalyzed by both sulfite and nitrite reductases. Proc Natl Acad Sci U S A 71:612–616. doi: 10.1073/pnas.71.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crane BR, Siegel LM, Getzoff ED. 1997. Structures of the siroheme- and Fe4S4-containing active center of sulfite reductase in different states of oxidation: heme activation via reduction-gated exogenous ligand exchange. Biochemistry 36:12101–12119. doi: 10.1021/bi971065q. [DOI] [PubMed] [Google Scholar]
- 41.Ito M, Guffanti AA, Oudega B, Krulwich TA. 1999. mrp, a multigene, multifunctional locus in Bacillus subtilis with roles in resistance to cholate and to Na+ and in pH homeostasis. J Bacteriol 181:2394–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu CH, McTernan PM, Walter ME, Adams MW. 2015. Production and application of a soluble hydrogenase from Pyrococcus furiosus. Archaea 2015:912582. doi: 10.1155/2015/912582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakagawa S, Takai K. 2008. Deep-sea vent chemoautotrophs: diversity, biochemistry and ecological significance. FEMS Microbiol Ecol 65:1–14. doi: 10.1111/j.1574-6941.2008.00502.x. [DOI] [PubMed] [Google Scholar]
- 44.Sato T, Atomi H, Imanaka T. 2007. Archaeal type III RuBisCOs function in a pathway for AMP metabolism. Science 315:1003–1006. doi: 10.1126/science.1135999. [DOI] [PubMed] [Google Scholar]
- 45.Lloyd KG, Schreiber L, Petersen DG, Kjeldsen KU, Lever MA, Steen AD, Stepanauskas R, Richter M, Kleindienst S, Lenk S, Schramm A, Jorgensen BB. 2013. Predominant archaea in marine sediments degrade detrital proteins. Nature 496:215–218. doi: 10.1038/nature12033. [DOI] [PubMed] [Google Scholar]
- 46.Matschiavelli N, Oelgeschlager E, Cocchiararo B, Finke J, Rother M. 2012. Function and regulation of isoforms of carbon monoxide dehydrogenase/acetyl coenzyme A synthase in Methanosarcina acetivorans. J Bacteriol 194:5377–5387. doi: 10.1128/JB.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Zhou Z, Pan J, Baker BJ, Gu JD, Li M. 2018. Comparative genomic inference suggests mixotrophic lifestyle for Thorarchaeota. ISME J 12:1021–1031. doi: 10.1038/s41396-018-0060-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider E. 2001. ABC transporter catalyzing carbohydrate uptake. Res Microbiol 152:303–310. doi: 10.1016/S0923-2508(01)01201-3. [DOI] [PubMed] [Google Scholar]
- 49.Doeven MK, Van DBG, Krasnikov V, Poolman B. 2008. Probing receptor-translocator interactions in the oligopeptide ABC transporter by fluorescence correlation spectroscopy. Biophys J 94:3956–3965. doi: 10.1529/biophysj.107.120964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuchmann K, Muller V. 2012. A bacterial electron-bifurcating hydrogenase. J Biol Chem 287:31165–31171. doi: 10.1074/jbc.M112.395038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lazar CS, Baker BJ, Seitz KW, Teske AP. 2017. Genomic reconstruction of multiple lineages of uncultured benthic archaea suggests distinct biogeochemical roles and ecological niches. ISME J 11:1118–1129. doi: 10.1038/ismej.2016.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao Z-M, Zhou G-W, Huang H, Wang Y. 2017. The cyanobacteria-dominated sponge Dactylospongia elegans in the South China Sea: prokaryotic community and metagenomic insights. Front Microbiol 8:1387. doi: 10.3389/fmicb.2017.01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel RK, Jain M. 2012. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina paired-end read merge R. Bioinformatics 30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 57.Caporaso JG, Bittinger K, Bushman FD, Desantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, Petrosino JF, Knight R, Birren BW. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH. 2013. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31:533–538. doi: 10.1038/nbt.2579. [DOI] [PubMed] [Google Scholar]
- 62.Krogh A, Brown M, Mian IS, Sjölander K, Haussler D. 1994. Hidden Markov models in computational biology. J Mol Biol 235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y, Huang JM, Cui GJ, Nunoura T, Takaki Y, Li WL, Gao ZM, Takai K, Zhang AQ, Stepanauskas R. 2019. Genomics insights into ecotype formation of ammonia-oxidizing archaea in the deep ocean. Environ Microbiol 21:716–729. doi: 10.1111/1462-2920.14518. [DOI] [PubMed] [Google Scholar]
- 64.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hyatt D, Locascio PF, Hauser LJ, Uberbacher EC. 2012. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 28:2223–2230. doi: 10.1093/bioinformatics/bts429. [DOI] [PubMed] [Google Scholar]
- 66.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kanehisa M, Goto S. 2000. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salter S, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman N, Walker AW. 2014. Reagent contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kopylova E, Noé L, Touzet H. 2012. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28:3211–3217. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 72.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang Y, Gilna P, Li W. 2009. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 25:1338–1340. doi: 10.1093/bioinformatics/btp161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2012. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katoh K, Toh H. 2010. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 26:1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Capellagutiérrez S, Sillamartínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12:335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the results of this article have been deposited in NCBI under BioProject number PRJNA383916. The SRA accession for sequence reads for 16S rRNA amplicons is SRR5685235, and genome accessions are NJBQ00000000 (Ae_b3a), NJBP00000000 (Ae_b3b), NJBO00000000 (B3_TA06), NJBN00000000 (B3_LCP), NJBM00000000 (B3_Pla), NJBL00000000 (B3_B38), NJBK00000000 (B3_Chlor), NJBI00000000 (Loki_b31), NJBH00000000 (Loki_b32), NJBG00000000 (B3_Woes), and NJBF00000000 (B3_Heim).