

Abstract
Neuropathy target esterase is a transmembrane protein of unknown function whose specific chemical modification by certain organophosphorus compounds leads to distal axonopathy. Therefore, solving the 3D structure of NTE would advance the understanding of its pathogenic and physiologic roles. In this study, the tertiary structures of the patatin (catalytic) domain and the N-terminal transmembrane domain of NTE were modeled using the crystal structures of patatin (PDB ID 1oxw) and moricin (PDB ID 1kv4) as templates. Sequence alignments and secondary structure predictions were obtained from the INUB server (Buffalo, NY). O and PyMol were used to build the PNTE and NTE TMD chains from these sequence alignments. The PNTE model was refined in the presence of water using the crystallography and NMR system, while the NTE TMD model was refined in vacuo using the GROMOS implementation in the Swiss PDB viewer. The modeled active site of NTE was found to consist of a Ser966-Asp1086 catalytic dyad, which is characteristic of phospholipase A2 enzymes. The Ser966 Oγ was located 2.93 Å from the Oδ2 of Asp1086. In addition, our NTE model was found to consist of a single N-terminal transmembrane domain. This modeling effort provided structural and mechanistic predictions about the catalytic domain of NTE that are being verified via experimental techniques.
Keywords: Delayed neurotoxicity, organophosphorus (OP) compounds, neuropathy target esterase (NTE), patatin, protein modeling
Introduction
Neuropathy target esterase (NTE) is an integral membrane protein of unknown physiological function whose chemical modification by certain organophosphorus (OP) compounds triggers a delayed distal axonopathy with concomitant paralysis and sensory loss (Richardson, 2005). While the primary pathology associated with organophosphorylation of NTE is seen in neurons, this protein is also expressed in non-neuronal cells, including lymphocytes and Leydig cells (Schwab and Richardson, 1986; Winrow et al., 2003). Although the biological role of NTE remains unidentified, recent evidence indicates that it is vital for normal development, because NTE knockout mice are not viable beyond embryonic day 11 (Winrow et al., 2003).
Determining the tertiary structure of NTE would be expected to yield insight into its function. However, it has thus far eluded crystallization, owing in part to its size (1327 amino acid residues) and membrane anchorage to the endoplasmic reticulum (ER) (Li et al., 2003; Richardson, 2005). Therefore, alternative strategies to structural elucidation were considered, starting with computational modeling of discrete domains within the protein.
Previous studies have postulated that NTE is a serine esterase composed of four TMD (TMD1 11–32, TMD2 734–753, TMD3 925–943, and TMD4 956–976) (Lush et al., 1998) and an active site containing an unprecedented catalytic triad (Ser966- Asp960-Asp1086) (Atkins and Glynn, 2000). Atkins and Glynn (2000) also proposed that the Ser 966 nucleophile was located in the center of TMD4. Preliminary bioinformatics analysis of the primary sequence of NTE has shown that its catalytic center resides within a domain that has homology to the plant protein, patatin (Winrow et al., 2003), a 40 kDa protein that constitutes as much as 40% of the soluble protein in potato tubers (Pots et al., 1999; Racusen and Foote, 1980). Like NTE (Glynn, 2005), patatin is known to have acyl hydrolase activity toward certain lipids (Andrews et al., 1988 and van Tienhoven et al., 2002). The crystal structure of the patatin isoform Pat17 (PDB ID 1oxw; isolated from Solanum cardiophyllum) has been solved to a resolution of 2.2 Å and has shown this protein to have a catalytic center composed of a Ser77-Asp215 dyad whose residues (Ser77 Oγ and Asp215 Oδ2) are located 3.8 Å apart (Rydel et al., 2003). This catalytic machinery is similar to that observed in mammalian cytosolic phospholipase A2 (cPLA2; an enzyme that cleaves glycerophospholipids in cell membranes at the sn-2 position). The cPLA2 active site was originally thought to consist of a novel ser-asp-arg triad, but it was revealed to consist of a ser-asp dyad, whose residues are located 2.9 Å apart (Dessen et al., 1999; Rydel et al., 2003). Such catalytic dyads are distinct from the classic ser-his-asp/glu catalytic triads observed in serine esterases and serine proteases (Zhang et al., 2002; Zhu et al., 2005). In addition to patatin having 42 % sequence homology to cPLA2, the crystal structure of Pat17 revealed it to have structural homology to cPLA2, as well, in that both Pat17 and cPLA2 consist of a modified α/β hydrolase fold (Ollis et al., 1992; Rydel et al., 2003).
Evidence for NTE being a serine esterase/lipase is known operationally from the fact that it catalyzes the hydrolysis of esters and that its esterase activity is inhibited by preincubation with OP compounds (Kropp et al., 2004). NTE also contains the two consensus sequences for acyl hydrolases, the active site G-X-S-X-G containing the nucleophilic Ser966 (Schrag and Cygler, 1997) and the G-G-X-R motif, which forms an oxyanion hole in Pat17 and cPLA2 that stabilizes the oxyanion produced during catalysis. In addition, recent evidence has shown that NTE is able to hydrolyze 1-palmitoyl-lysophosphatidylcholine and deacylate phosphatidylcholine to glycerophosphocholine, thereby pointing to a potential role for NTE in the turnover of membrane lipids (van Tienhoven et al., 2002).
The crystal structures of both patatin and cPLA2 have revealed that these proteins contain active sites composed of ser-asp dyads. While the tertiary structure of NTE has yet to be elucidated, the sequence similarity between patatin and the catalytic domain of NTE prompted the hypothesis that, like patatin and cPLA2, the active site of NTE may consist of a ser-asp catalytic dyad. To test this hypothesis, a computationally derived model for the tertiary structure of the patatin (catalytic) domain of NTE (PNTE) has been constructed using the Pat17 x-ray crystal structure as the template. The model serves to suggest hypotheses and provide insights about the catalytic machinery of NTE and provides a preliminary step toward solving the X-ray crystal or NMR structure of the catalytic domain of NTE.
Materials and methods
Bioinformatics
The NTE sequence (accession number NP_006693) was obtained from the NCBI Entrez Protein database (http://www.ncbi.nlm.nih.gov/entrez/). The protein family database (PFAM; Bateman et al., 2004), an online database containing collections of protein domains and families, was used to identify domains in NTE from its primary sequence. Secondary structural predictions and analyses of the patatin and transmembrane domains were done using PredictProtein (Rost et al., 2003) and TMHMM (Krogh et al., 2001), which uses a hidden Markov model to identify membrane-spanning regions based on primary sequence. Hydropathy analyses of the PNTE and NTE TMD primary sequences were undertaken using the GREASE service on the University of Virginia FASTA server (http://fasta.bioch.virginia.edu/fasta_www/grease.htm). BoxShade 3.21 (http://www.ch.embnet.org/software/BOX_form.html), an on-line service for formatting multiple-sequence alignments, was employed to shade identical and homologous amino acid residues in the sequence alignment between PNTE and Pat17.
NTE patatin domain model building
Secondary structure predictions for PNTE along with sequence alignments of PNTE against Pat17 (PDB ID 1oxw) were obtained from the INUB server (Buffalo, NY; Fischer, 2003). Pymol (DeLano Scientific LLC, San Carlos, CA) and O (Jones et al., 1991) were utilized to build the PNTE structure based on the sequence alignments via virtual mutagenesis of the corresponding residues on the template Pat17 structure. Small chain breaks in the model were repaired using the lego-loop procedure in O. The resulting PNTE model was placed in a cubic shell of water molecules 2 Å from the surface of the protein (depth of the solvent shell was 4 Å). Energy minimization was undertaken using the Crystallography and NMR System (Brunger et al., 1998) via simulated annealing to 500 K followed by cooling (step time of 0.5 fs per step) to room temperature (300 K) carried out in 25 K increments. Packing of residues in the active site of the resulting PNTE structure was evaluated visually. The refined model was analyzed in Pymol and Chem3D (CambridgeSoft, Cambridge, MA). Final images were rendered in Pymol.
NTE transmembrane domain model building
In order to verify the structure of the putative N-terminal NTE TMD predicted by PredictProtein and TMHMM, a computationally derived model was developed. Sequence alignments and secondary structure predictions of the NTE TMD against moricin (PDB ID 1kv4; a bacterial pore-forming protein) were obtained from the INUB server (Buffalo, NY; Fischer, 2003). Pymol and O were utilized to build the NTE TMD model via virtual mutagenesis of the corresponding residues in the moricin structure. Due to the inherent hydrophobicity of the NTE TMD, the resulting structure was minimized in vacuo using the GROMOS implementation in Swiss PDB viewer (Guex and Peitsch, 1997). The refined model was analyzed in Pymol and MolMol (Koradi et al., 1996).
Results and Discussion
The NTE active site consists of a Ser966-Asp1086 catalytic dyad
Pfam analysis of the NTE primary sequence confirmed that the NTE active site resides within a patatin homology domain (residues 933–1099) (Figure 1). Alignment of PNTE and Pat17 indicated 30% homology and 18% identity between the 2 sequences (Figure 2A). Analysis of the derived PNTE spatial model indicated that the overall structure of PNTE contains 7 α-helices (named α1- α7) and 6 β-strands (named B1-B6) (Figure 2B). The β-strands are arranged in three β-sheets: one anti-parallel β-sheet and two parallel β-sheets.
Figure 1:
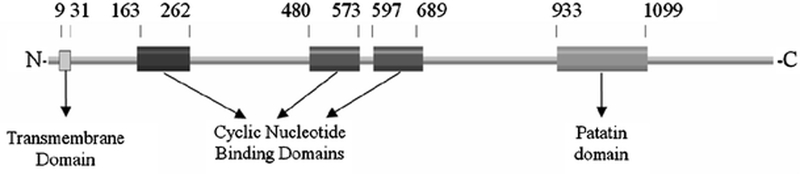
Results of the protein family (Pfam) analysis showing the domains in NTE. Numbering corresponds to amino acid residues at the N- and C- termini of each domain. As shown, NTE consists of a single N-terminal transmembrane domain (TMD; residues 9–31), three cyclic nucleotide domains (CNP domains; residues 163–262, 480–573 and 597–689), and a patatin homology domain (PNTE, residues 933–1099) containing its active site.
Figure 2:
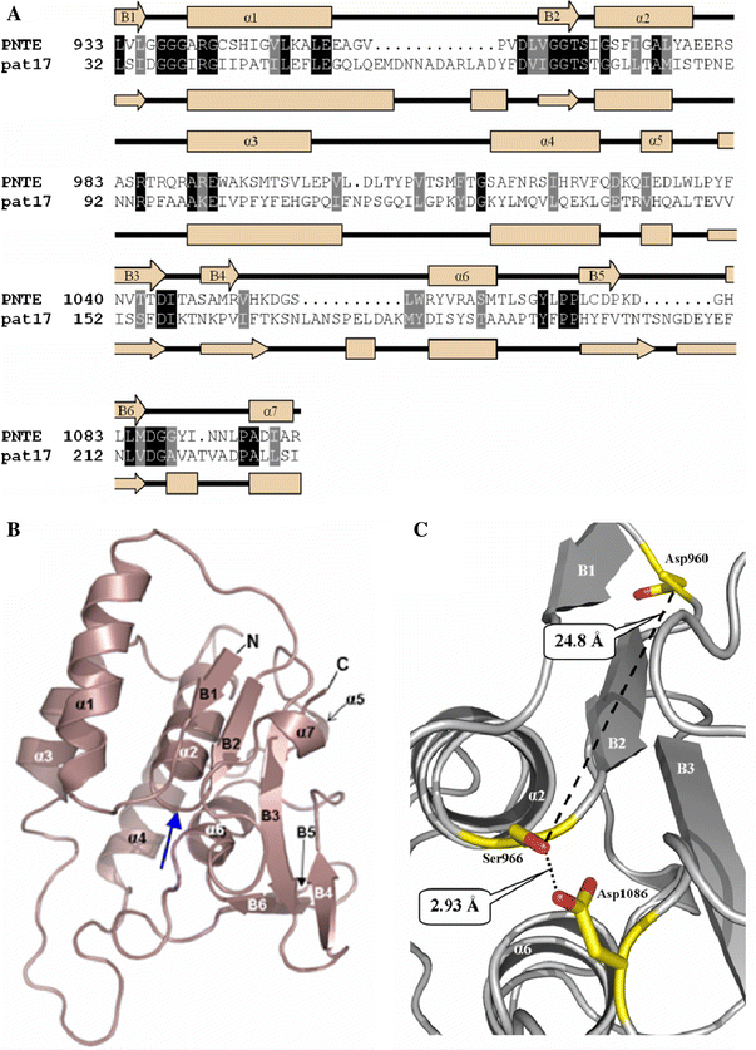
A.) Sequence homologies and identities between PNTE and pat17 sequence alignment of PNTE and pat17. Identical residues are boxed in black, while homologous residues are in grey. Secondary structures of PNTE and pat17 are shown along with their respective primary structures as cream arrows (representing β-strands) and orange rectangles (representing α helices). α-helices and β-strands in the PNTE sequence are numbered α1-α7 and B1-B6, respectively. Shading for homologous and identical residues in the aligned sequences was performed using BoxShade 3.21 (http://www.ch.embnet.org/software/BOX_form.html). B.) A ribbon diagram of the putative tertiary structure for the NTE patatin domain. Blue arrow denotes the location of nucleophilic elbow. C.) Organization of the predicted active site of NTE. Ser966, Asp960, and Asp1086 are depicted as balls-n-sticks. Distances between Ser966 Oγ and Asp960 Oδ2 and Ser966 Oγ and Asp 1086 Oδ2 are shown as dotted lines. Images b. and c. were produced in PyMol.
Analysis of the PNTE model shown in Figure 2C showed that the NTE active site contains a catalytic dyad composed of Ser966 and Asp1086. In our model, Ser966 and Asp1086 are located on conserved loops, B2-α2 and B3-B4, respectively, with Ser966 Oγ located only 2.93 Å from the Asp1086 Oδ2. This is similar to distances observed between ser-asp dyads in other serine esterases/lipases, such as the 3.8 Å span between the Ser77 Oγ and Asp215 Oδ2 in the crystal structure of the patatin active site (Rydel et al., 2003) and the 2.9 Å length between the Ser228 Oγ and Asp549 Oδ2 in the crystal structure of the mammalian cPLA2 active site (Dessen et al., 1999). Moreover, the B2-α2 loop containing the active site Ser966 residue of PNTE has a similar conformation to the ‘nucleophilic elbow’ found in both Pat17 (Rydel et al., 2003) and cPLA2 (Dessen et al., 1999) crystal structures.
In marked contrast to previous results by Atkins and Glynn (2000), our model of PNTE showed that the Oδ2 of Asp960 is located 24.8 Å away from the Ser966 Oγ, which is too far for Asp960 to play a direct role in catalysis (Figure 2C). A possible explanation for discrepancies between the present model and the earlier site-directed mutagenesis results could be that mutation of Asp960 causes local misfolding that could affect the structure of the NTE active site thereby reducing the catalytic activity of the enzyme. Given that our model indicates that Asp960 is exposed to solvent, mutating this residue may affect interactions between PNTE and other residues or domains in full-length NTE, which may in turn, limit catalytic activity. Further biochemical analysis will be required to assess the effects of this mutation. In further support of NTE utilizing a catalytic dyad instead of the traditional ser-his-asp catalytic triad, Atkins and Glynn (2000) showed that mutating all the histidine residues in the catalytic domain to ala residues had no adverse effect on enzyme activity.
In addition to identification of a probable catalytic dyad, it was observed that a conserved Gly938-Gly939-Ala940-Arg941 sequence, corresponding to the consensus sequence for an oxyanion hole (G-G-X-R) was present in close proximity to the NTE active site. Therefore, by analogy to Pat17 and cPLA2, it is proposed that Gly938 and Gly939 define the oxyanion hole of NTE, with the backbone nitrogens of these residues located 4.8 Å and 5.7 Å, respectively, from the Ser966 Oγ. These dimensions from our model are somewhat larger than the corresponding 4.2 Å and 4.8 Å distances found between Ser288 Oγ and the backbone nitrogens of Gly197 and Gly198 in cPLA2, and the 3.4 Å and 3.8 Å distances observed between Ser77 Oγ and the backbone nitrogens of Gly37 and Gly38 in Pat17. However, despite the differences in these distances, the conservation of the G-G-X-R motif in NTE is indicative of a putative oxyanion hole, and the actual measurements between its residues and the active site Ser966 need to be determined experimentally.
Proposed catalytic mechanism
Based on the predictions arising from the present model, it is proposed that the catalytic dyad in NTE functions in a manner similar to catalytic dyads found in other serine hydrolases. The model suggests that Asp1086 will act as both a general acid and general base, which will activate the Ser966 Oγ and turn it into a potent nucleophile (Figure 3A). In this proposed catalytic mechanism, the negatively charged trigonal bipyramidal transition states that result will be stabilized by the oxyanion hole formed by Gly938, Gly939, Ala940, and Arg941.
Figure 3:
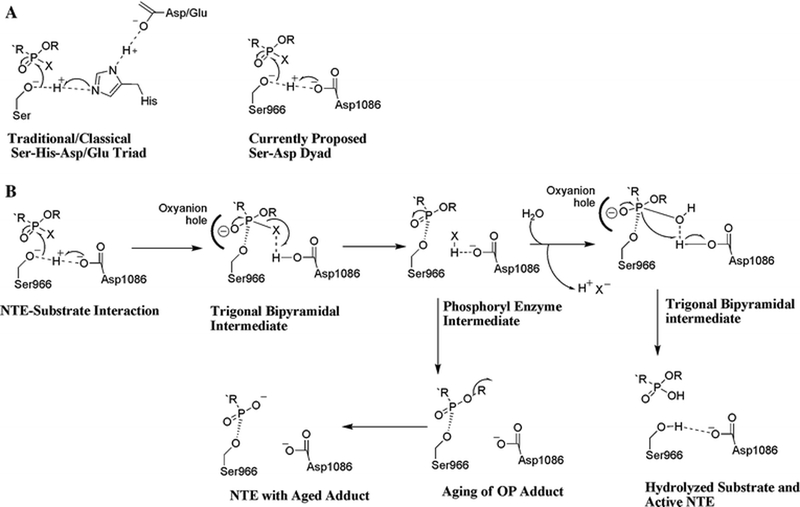
A.) Comparison of mechanisms of catalysis in a traditional Ser-His-Asp/Glu triad and our proposed Ser-Asp dyad for NTE. B.) Proposed mechanism for interactions of an OP ester with the NTE catalytic dyad. OP compounds are substrates that become mechanism-based inhibitors, because hydrolysis of the acyl-enzyme intermediate is usually slow. Moreover, this intermediate can undergo aging via net loss of an alkyl group to yield a negatively charged OP adduct on the active site Ser966 residue. It is currently postulated that aging of an OP adduct on NTE is the initiating step in the pathogenesis of OP compound-induced axonopathy.
In the case of an OP ester (Figure 3B), Asp1086 will act as a general base by abstracting a proton from Ser966 Oγ. Ser966 Oγ will then perform a nucleophilic attack on the phosphorus atom of the substrate producing a trigonal bipyramidal intermediate, which will collapse into a phospho-enzyme intermediate as Asp1086 acts as a general acid donating a proton to the acyl leaving group. Once the acyl chain is released, a water molecule can enter the active site and be activated by Asp1086. This subsequent replacement of the leaving group with a molecule of water would form a second trigonal bipyramidal transition state resulting in the subsequent hydrolysis of the phospho-enzyme intermediate (Figure 3B) (Dessen et al., 1999).
Because hydrolysis of the phosphorylated enzyme is expected to be slow in the case of OP esters, the phosphorylated intermediate would be relatively stable, resulting in inhibition of the enzyme (Kropp et al., 2004). Moreover, the organophosphorylated intermediate has an additional potential pathway involving either SN1- or SN2-mediated displacement of a side chain, leaving a negatively charged phosphoryl moiety covalently attached to the Ser966, which could potentially be stabilized by the Nδ2 of Asn1092, located 5.8 Å away from the Oγ of Ser966. This displacement would be analogous to the ‘aging’ reaction of organophosphorylated AChE and NTE (Doorn et al., 2001; Kropp et al., 2004). Thus, the structure of the active site of the model suggests mechanisms of catalysis, inhibition, and aging reactions that need to be tested experimentally.
PNTE has a hydrophobic face and a hydrophilic face
Hydropathy analysis of the PNTE sequence indicated that approximately 50% of surface residues in our PNTE model are hydrophilic. Interestingly, mapping electrostatic potentials to the surface of the refined PNTE model revealed that one ‘face’ of PNTE is predominantly hydrophobic, while the other ‘face’ of PNTE is chiefly hydrophilic (Figure 4). It was also found that the PNTE active site pocket is located in the hydrophobic face of PNTE. Therefore, while it is possible that PNTE is not a membrane-spanning domain, it might have points of membrane attachment or association, because NTE is known to hydrolyze lipids, and it has been shown to have lysophospholipase activity (Quistad et al., 2003). However, it is also important to consider the possibility that the hydrophobic face of PNTE may represent a surface involved in interactions with other residues and domains in the structure of full-length NTE or other proteins that interact with NTE.
Figure 4:
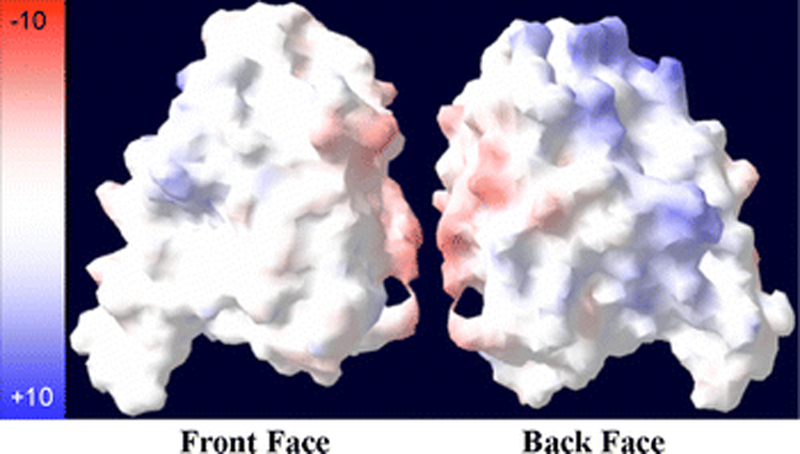
Surface maps of PNTE. Colors represent the degree of charge with red = negatively charged residues, white = uncharged residues, and blue = positively charged residues. Image was produced using the Swiss PDB Viewer (Guex and Peitsch, 1997).
NTE has a single N-terminal transmembrane domain
TMHMM and PFAM analysis of the NTE primary sequence indicated that NTE contains a single N-terminal TMD (residues 9–31) (Figure 1). Moreover, based on the ‘positive inside’ rule (Andersson and von Heijne, 1994), TMHMM predicted a 95.7% probability that the N-terminus of the TMD is located in the lumen of the ER, thereby confirming previous observations (Li et al., 2003) suggesting that the catalytic domain of NTE is located in the cytosol.
Analysis of the NTE TMD and moricin primary sequences revealed 39% sequence homology and 22% sequence identity. Modeling the TMD structure using the moricin (PDB ID 1kv4) structure as a template indicated that the NTE TMD consists of a single right-handed α-helix. This is consistent with the expected structure for mammalian integral membrane protein domains (Ott and Lingappa, 2002). The α-helical nature of the NTE TMD (residues 9–31) was confirmed by examination of the Ramachandran plot for the NTE TMD model, which revealed that all residues in the NTE TMD were in the conformation expected of residues in a right-handed α-helix. In addition, hydropathy analysis of the NTE TMD primary sequence indicated that 100% of its residues are hydrophobic, thereby supporting the conclusion that NTE contains a single N-terminal TMD, and that the catalytic site of NTE is not contained within a membrane-spanning domain.
Summary
Based on the present modeling results and the literature, it is possible to propose a new representation for the overall structure of NTE, as depicted in Figure 5. It is proposed that NTE contains a single N-terminal integral membrane domain (residues 9–31). It is also proposed that the hydrophobic face of PNTE, which contains the NTE active site, is associated with the membrane (based on evidence that NTE hydrolyzes membrane lipids) (Zaccheo et al., 2004). The proposed model suggests that PNTE is not a true membrane-spanning domain, owing to the fact that the active site residues are seen on loops as opposed to α-helices, which are the expected secondary structures in mammalian transmembrane domains (Ott and Lingappa, 2002). However, the details of the relative spatial arrangement of the TMD, putative cyclic nucleotide binding domains, and the catalytic domains must await experimental determination to confirm our model.
Figure 5:
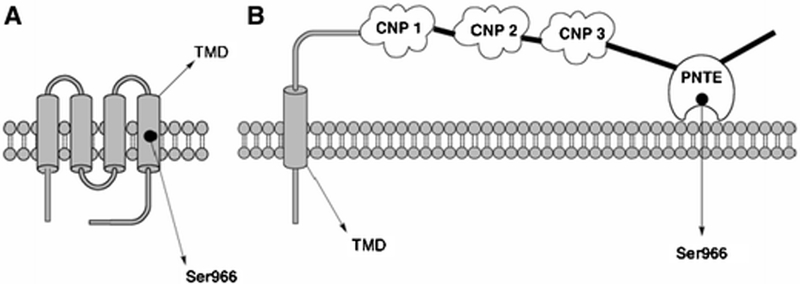
Models for NTE. A.) Model proposed previously by Atkins and Glynn (2000) showing 4 transmembrane domains with the active site Ser966 residue located within the TMD4 (Ser966 is represented by a black dot), and B.) Model proposed based on the present results depicting NTE’s transmembrane domain, the PNTE domain containing the NTE active-site dyad and 3 putative cyclic nucleotide-binding (CNP) binding domains, based on previous results (Glynn, 2000; Nasser et al., 2006). Based on the data presented in this paper, it is proposed that PNTE could be associated with the membrane, but that it cannot be considered an integral membrane domain.
The results obtained in the present study provide models for the putative tertiary structures of the catalytic and transmembrane domains of NTE, thereby affording preliminary insight into its structure. Experimental studies using X-ray crystallography and NMR spectroscopy are currently in progress to confirm the predictions arising from these computational studies.
Acknowledgments
This material is based upon work supported in part by the U.S. Army Research Laboratory (ARL) and the U.S. Army Research Office (ARO) under research grant DAAD19–02-1–0388 (RJR), a research grant from The Dow Chemical Company (RJR), and a grant from the Michigan Economic Development Corporation for the Michigan Life Sciences Corridor Initiative (JAS). SJW was supported by an institutional training grant from the National Institutes of Health (NIH) under Ruth L. Kirschstein National Research Service Award ES07062 (RJR).
Abbreviations:
- CNP
cyclic nucleotide
- cPLA2
cytosolic phospholipase A2
- ER
endoplasmic reticulum
- NTE
neuropathy target esterase
- Pat17
patatin isoform 17
- PNTE
Patatin-homology domain of neuropathy target esterase
- TMD
transmembrane domain
References
- Andersson H, and von Heijne G (1994). EMBO. J. 13, 2267––2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews DL, Beames B, Summers MD, and Park WD (1988). Biochem. J. 252, 199––206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins J, and Glynn P (2000). J. Biol. Chem. 275, 24477––24483. [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, and Eddy SR (2004). Nucleic Acids Res. 32D, 138––141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, and Warren GL (1998). Acta Crystallogr. D Biol. Crystallogr. 54 905––921. [DOI] [PubMed] [Google Scholar]
- DeLano Scientific LLC, San Carlos, CA, USA. http://www.pymol.org.
- Dessen A, Tang J, Schmidt H, Stahl M, Clark JD, Seehra J, and Somers WS (1999). Cell. 97, 349––360. [DOI] [PubMed] [Google Scholar]
- Doorn JA, Schall M, Gage DA, Talley TT, Thompson CM, and Richardson RJ (2001). Toxicol. Appl. Pharmacol. 176, 73––80. [DOI] [PubMed] [Google Scholar]
- Fischer D (2003). Proteins. 51, 434––441. [DOI] [PubMed] [Google Scholar]
- Glynn P (2000). Prog. Neurobiol. 61, 61––74. [DOI] [PubMed] [Google Scholar]
- Glynn P (2005). Biochim. Biophys. Acta. 1736, 87––93. [DOI] [PubMed] [Google Scholar]
- Guex N, and Peitsch MC (1997). Electrophoresis. 18, 2714––2723. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, and Kjeldgaard M (1991). Acta Cryst. A47, 110––119. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, and Wuthrich K (1996). J. Mol. Graphics Modell. 14, 51––55. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, and Sonnhammer EL (2001). J. Mol. Biol. 305, 567––580. [DOI] [PubMed] [Google Scholar]
- Kropp TJ, Glynn P, and Richardson RJ (2004). Biochemistry. 43, 3716––3722. [DOI] [PubMed] [Google Scholar]
- Li Y, Dinsdale D, and Glynn P (2003). J. Biol. Chem. 278, 8820––8825. [DOI] [PubMed] [Google Scholar]
- Lush MJ, Li Y, Read DJ, Willis AC, and Glynn P (1998). Biochem. J. 332, 1––4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser FA, Wijeyesakere SJ, Stuckey JA, and Richardson RJ (2006). The Toxicologist. 90, 299. [Google Scholar]
- Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, and Goldman A (1992). Protein Eng. 5, 197––211. [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, and Liu J, (2003). Nucleic Acids Res. 32, W321––W326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott CM, and Lingappa VR (2002). J. Cell. Sci. 115, 2003––2009. [DOI] [PubMed] [Google Scholar]
- Pots AM, Gruppen H, Hessing M, van Boekel MA, and Voragen AG (1999). J. Agric. Food. Chem. 47 4587––4582. [DOI] [PubMed] [Google Scholar]
- Quistad GB, Barlow C, Winrow CJ, Sparks SE, and Casida JE (2003). Proc. Natl. Acad. Sci. U.S.A. 100, 7983––7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racusen D, and Foote M (1980). J. Food. Biochem. 4, 43––52. [Google Scholar]
- Richardson RJ (2005). In Encyclopedia of Toxicology 2nd ed., Vol. 2 (Wexler P, ed.), Academic Press, New York, pp. 302––306. [Google Scholar]
- Rydel TJ, Williams JM, Krieger E, Moshiri F, Stallings WC, Brown SM, Pershing JC, Purcell JP, and Alibhai MF (2003). Biochemistry. 42, 6696––6708. [DOI] [PubMed] [Google Scholar]
- Schrag JD, and Cygler M (1997). Methods Enzymol. 284, 85––107. [DOI] [PubMed] [Google Scholar]
- Schwab BW, and Richardson RJ (1986). Toxicol. Appl. Pharmacol. 83, 1––9. [DOI] [PubMed] [Google Scholar]
- van Tienhoven M, Atkins J, Li Y, and Glynn P (2002). J. Biol. Chem. 277, 20942––20948. [DOI] [PubMed] [Google Scholar]
- Winrow CJ, Hemming ML, Allen DM, Quistad GB, Casida JE, and Barlow C (2003). Nature Genetics, 33, 477––485. [DOI] [PubMed] [Google Scholar]
- Zaccheo O, Dinsdale D, Meacock PA, and Glynn P (2004). J. Biol. Chem. 279, 24024––24033. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kua J, and McCammon JA (2002). J. Am. Chem. Soc. 124, 10572––10577. [DOI] [PubMed] [Google Scholar]
- Zhu YC, Liu X, Maddur AA, Oppert B, and Chen MS (2005). Insect Biochem. Mol. Biol. 35, 22––32. [DOI] [PubMed] [Google Scholar]