

Abstract
Non-coding RNAs engage in numerous biological activities including gene regulation. To fully understand RNA function it is necessary to probe biologically relevant conformations in living cells. To address this challenge, we coupled RNA-mediated regulation of the GFPuv-reporter gene to icSHAPE (in cell Selective 2´-Hydroxyl Acylation analyzed by Primer Extension). Our transcript-specific approach provides sensitive, fluorescence-based readout of the regulatory-RNA status as a means to coordinate chemical modification experiments. We chose a plasmid-based reporter compatible with E. coli to allow use of knockout strains that eliminate endogenous effector biosynthesis. The approach was piloted using the Lactobacillus rhamnosus (Lrh) preQ1-II riboswitch, which senses the pyrrolopyrimidine metabolite preQ1. Using an E. coli ΔqueF strain incapable of preQ1 anabolism, the Lrh riboswitch yielded nearly one log unit of GFPuv-gene repression resulting from exogenously added preQ1. We then subjected cells in gene “on” and “off” states to icSHAPE. The resulting differential analysis indicated reduction in Lrh riboswitch flexibility in the P3 helix of the pseudoknot, which comprises the ribosome-binding site (RBS) paired with the anti-RBS. Such expression platform modulation was not observed by in vitro chemical probing, and demonstrates that the crowded cellular environment does not preclude detection of compact and loose RNA-regulatory conformations. Here we describe the design, methods, interpretation and caveats of Reporter Coupled (ReCo) icSHAPE. We also describe mapping of the differential ReCo-icSHAPE results onto the Lrh riboswitch-preQ1 co-crystal structure. The approach should be readily applicable to functional RNAs triggered by effectors or environmental variations.
Introduction
Relatively little is known about the conformations adopted by functional RNAs inside of cells and how such changes modulate chemical interaction networks to impart biological activity.1, 2 Riboswitches are model systems to explore this question because they adopt mutually exclusive folds in response to a cognate effector to exert precise conformational control over gene expression (Fig. 1a).3–5 To this end, we coupled GFPuv-reporter-gene expression by a bacterial riboswitch to icSHAPE (in cell Selective 2´-Hydroxyl Acylation analyzed by Primer Extension),6 which employs an electrophilic acylation reagent to modify individual 2´-hydroxyl groups of RNA (Fig. 1b). Recovery and primer-extension-based detection of the acylated riboswitch RNA — isolated from cells grown under different regulatory conditions — allows us to pinpoint changes that arise from discrete gene ‘on’ and ‘off’ conformational states. Differences in acylation can then be mapped onto a known crystal structure to better understand how form leads to in vivo function. In the latter respect, we chose to investigate the Lactobacillus rhamnosus (Lrh) preQ1-II riboswitch, which has several desirable properties. First, the riboswitch senses a defined small-molecule ligand, preQ17, 8 — the last free intermediate on the queuosine (Q) biosynthesis pathway (Fig. 2a). Although Q is important for translational fidelity, it is not essential for bacterial growth9–11. This allowed genetic ablation of its biosynthesis to gain experimental control over preQ1 levels in a reporter assay. PreQ1 is also highly soluble and is capable of being synthesized in large quantities.12 The preQ1-II riboswitch class also exhibits well-defined aptamer and expression platform domains that fold into a compact pseudoknot in the effector-bound state (Fig. 2b,c).13–15 Effector binding occurs within a three-helix junction that contributes multiple interactions to preQ1 recognition (Fig. 2d), which account for its tight binding (KD,App of 2.76 ± 0.14 nM) (Fig. 2e). PreQ1 stacks directly on A71 — the first base of the highly sequestered ribosome-binding site (RBS) — providing a tangible link between effector binding and the expression platform. Collectively, these structural and functional observations provide the groundwork to establish a robust reporter in which functional readout of GFPuv expression can be coupled with chemical modification of the riboswitch in the context of live bacteria.
Figure 1. Drawing of riboswitch-mediated gene regulation and overview of a riboswitch-mediated GFPuv reporter coupled (ReCo) to in cell (ic)SHAPE.
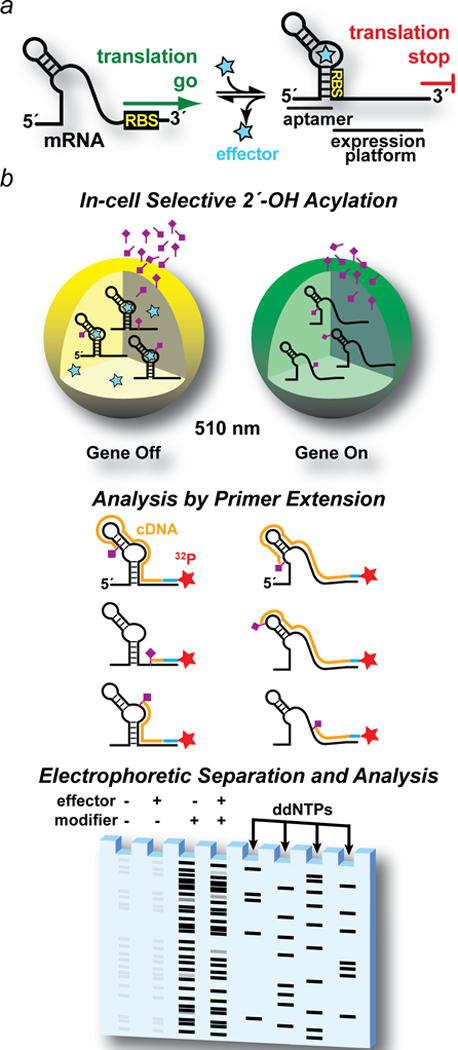
(a) Ideal two-state control of mRNA translation by a riboswitch. When cellular effector levels are low, the ribosome-binding site (RBS) in the 5′-leader is exposed to interact with the ribosome, leading to protein translation (i.e., a gene “on” conformation). When effector levels rise (star), the aptamer domain senses the cognate effector, shifting the conformational equilibrium to a compact state that reduces SDS accessibility in the expression platform, thus attenuating translation (i.e., a gene “off” conformation). (b) Major steps in riboswitch-mediated gene regulation coupled to icSHAPE. Step 1. Cells are grown ± effector to elicit gene “off” and “on” conformational states of the target regulatory RNA. Gene regulation is monitored by GFPuv fluorescence emission at 510 nm recorded using live cells. The RNA modification reagent (tailed rhombus) is incubated with each cell population. Step 2. Cells are lysed to recover RNA. A 32P-labeled primer complementary to the GFPuv gene allows reverse transcription to produce cDNA (orange lines). Primer extension stops at each site of 2´-acylation that occurred in live cells. Step 3. cDNA is separated by size using denaturing polyacrylamide gel electrophoresis. Controls are included for untreated cells (i.e., no chemical modifier ± effector) to correct for factors such as endogenous primer pausing. As a sequence reference, primer extensions are run on unmodified RNA in the presence of ddNTPs to produce a sequence ladder.
Figure 2. Queuosine (Q) biosynthesis, structural depictions of the Lactobacillus rhamnosus preQ1-II riboswitch and RNA quality control.
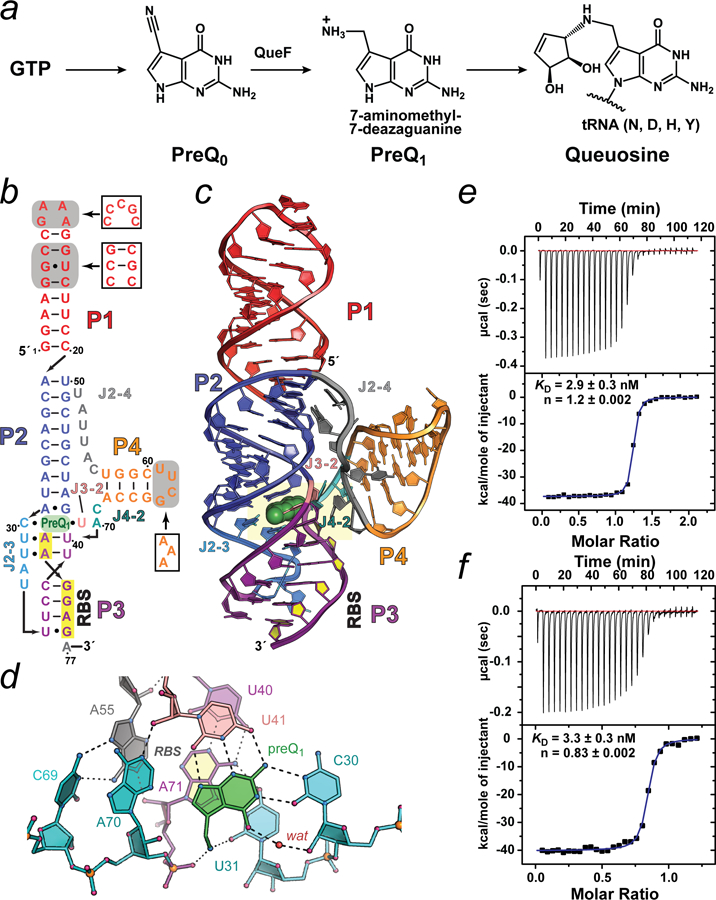
(a) Q is a widespread hypermodified base synthesized in many types of bacteria. Anabolism starts from GTP and proceeds via multiple steps (broken arrow) to the pyrrolopyrimidine intermediate preQ0, which is converted directly to preQ1 by a nitrile reductase encoded by the queF gene. PreQ1 is the last free intermediate prior to insertion into specific tRNAs. Additional in situ modifications (broken arrow) yield Q.45 (b) Secondary-structure diagram of the Lactobacillus rhamnosus (Lrh) preQ1-II riboswitch based on the known structure and computational analysis.13, 44 Pairing (P) and (J) junction regions are shown in distinct colors. Gray boxes represent sequence differences between the construct used for crystallization relative to the wild-type (white boxes). (c) Ribbon diagram depicting the global HLout pseudoknot fold based on the 2.3 Å resolution co-crystal structure (PDB entry 4jf2). The riboswitch comprises three co-axially stacked helices (P1, P2 & P3) with a fourth helix (P4) flanking the P2-P3 interface. The three-helix junction binds preQ1 to complete P2-P3 coaxial helical stacking. The co-crystal structure reveals that the pseudoknot buries the entire RBS. This observation supports the hypothesis that preQ1 binding promotes a cellular conformation of the riboswitch that obstructs RBS hybridization with the 16S rRNA of the ribosome, thus attenuating translation initiation. The RBS nucleotides are filled (yellow) and preQ1 is drawn as a CPK model (green). (d) Close-up of the preQ1-binding pocket (yellow highlighting in c. PreQ1 recognition occurs by hydrogen bonds between C30 and U41 to the effector edge. The methylamine of preQ1 hydrogen bonds to the keto group of U31 and a non-bridging oxygen of A71 — the first nucleotide of the RBS. (e) Isothermal titration calorimetry (ITC) thermogram and curve fit for a 76-mer Lrh preQ1-II riboswitch purified by denaturing methods. The sequence is that of the crystallization construct in b ending at G76. Here and below, errors of the fit are reported. (f) ITC thermogram and curve fit for a 76-mer Lrh preQ1-II riboswitch purified under conditions that preserved native folding. The sequence is that of the crystallization construct in b ending at A76. All structure-related cartoons were produced in PYMOL (Schrödinger, LLC).
A major consideration for the success of chemical modification is the method and choice of reagent (Fig. 1b). We selected SHAPE because it simultaneously interrogates all four nucleotides at single-residue resolution and possesses picomolar sensitivity, making it the gold standard to probe RNA secondary structure.16–20 Acylation occurs with a reactivity that depends on local backbone flexibility. Unconstrained groups sample more conformations that transiently enhance nucleophilicity to produce more modifications.16, 19 To maximize our success, we leveraged recent advances in icSHAPE reagents developed for use in live cells. The electrophile NAI (2-methylnicotinic acid imidazolide) has low toxicity, high solubility in aqueous solution, a relatively long half-life in water (t1/2 ~33 min), and selectivity for RNA 2´-OH groups over other cellular nucleophiles.6 Here we describe the rationale, design, methods, caveats and interpretation of a GFPuv-reporter coupled (ReCo) with icSHAPE (i.e, ReCo-icSHAPE). Although we developed this simple approach to investigate preQ1-II-riboswitch-mediated control of gene expression, this potent and economical tool is adaptable to other riboswitches4, as well as functional RNAs that change conformation in response to environmental stimuli or effectors. Examples include RNA thermometers21, miRNA-mRNA interactions22, viral frameshifting sequences23, or protein-directed RNA switches.24 In cases where high-resolution experimental models are available, differential ReCo-icSHAPE changes can be mapped onto RNA coordinates to rationalize biological function when viewed through the lens of structure.
MATERIALS AND METHODS
RNA Preparation and Purification.
A 76-mer Lrh preQ1-II riboswitch (Fig. 2a) was synthesized by T7 polymerase in vitro transcription and purified by denaturing polyacrylamide gel electrophoresis (PAGE).25 This material was lyophilized and stored at −80 °C prior to folding. Methods to generate the Lrh preQ1-II riboswitch by co-transcriptional folding and native purification were also conducted essentially as described.26 For the latter work, a synthetic DNA of the Lrh preQ1-II riboswitch (GenScript Inc.) was sub-cloned into the pRAV23 vector (AddGene) using EcoRI and KpnI NEB restriction sites. This yielded plasmid pLrhXtal76_RAV23 wherein the Lrh preQ1-II riboswitch is appended to a 3´-glmS ribozyme fused to tandem, downstream MS2 hairpins. T7 polymerase was used for co-transcriptional folding of the encoded riboswitch that was subsequently captured by 6His-MBP-MS2 coat protein (AddGene) immobilized on NiNTA resin (Thermo Scientific). The major steps of this method are depicted in Figure S1a and are described in detail elsewhere.26 After washing, glucosamine-6-phosphate (Sigma-Aldrich) was added to stimulate glmS ribozyme cleavage, yielding the free Lrh preQ1-II riboswitch with a uniform 3´-end at A76. A representative gel depicting key purification steps is provided (Figure S1b). The natively folded riboswitch was eluted from the column by washing and used immediately for ITC (Fig. 2f). Here and elsewhere DNA constructs were confirmed by sequencing (GeneWiz).
Isothermal Titration Calorimetry.
Lyophilized RNA purified by denaturation was suspended in RNA folding buffer (RFB) comprising: 0.010 M Na-HEPES, pH 7.0 and 0.10 M NaCl. The RNA was heated to 65 °C for 5 min in a heat block. MgCl2 was added with vortexing to a concentration of 6.0 mM. The sample was slow cooled to 24 °C. Refolded RNA samples were dialyzed at 4 °C overnight using a 3.5K MWCO Slide-A-Lyzer (Thermo) against 4 L of ITC buffer (ITCB) comprising: 0.05 M Na-HEPES pH 7.0, 0.1 M NaCl and 0.006 M MgCl2. Natively folded RNA was concentrated using a centrifugal concentrator (3K MWCO, Pierce) prior to dialysis. ITCB was used to dilute RNA samples to exactly 3.3 μM for ITC. PreQ1 (LeadGen Labs, LLC) powder was resuspended in used ITCB to a concentration 10-fold higher than the RNA. Measurements were conducted at 20 °C using a VP-ITC (MicroCal Inc) essentially as described.27 PreQ1 in the syringe was titrated into the riboswitch located in the sample cell (cell volume is ~1.7 mL) using 29 injections of 10 μL each, except for the first injection of 3 μL, with 240 s injection intervals. Thermograms were analyzed with Origin 7.0 (MicroCal) using a 1:1 binding model. Experiments were performed in duplicate for the denatured and refolded RNA. A single measurement was made for the natively transcribed and folded riboswitch. Curve fits are provided (Figs. 2e,f).
Construction of the Lrh PreQ1-II Riboswitch GFPuv Reporter.
A synthetic DNA insert comprising the desired wild-type Lrh riboswitch sequence (Figs. 3a,b) was prepared in pUC57 (GenScript Inc) and sub-cloned into the pEnv8(GAAA) vector28 (AddGene) using NsiI and HindIII (NEB) restriction sites. The resulting plasmid, pBR327-Lrh(WT)-GFPuv, was amplified in E. coli DH5α. A positive control was also prepared in which the GFPuv gene is expressed constitutively. This plasmid lacks the Lrh preQ1-II riboswitch (i.e., powder blue sequence; Fig. 3a). This “gene on” control sequence is: 5´-AACTATTAACTGAATCAAGGAGAAACAATATG-3´, which follows the Pribnow box. The underlined sequence is the Lrh RBS and the bold sequence is the GFPuv start codon. A negative control vector was prepared by replacing the RBS of the positive control with its reverse complement: 5´-CUCCUU-3´.
Figure 3. Regulatory sequence of the pBR327-Lrh(WT)GFPuv vector and outcome of GFPuv reporter assay.
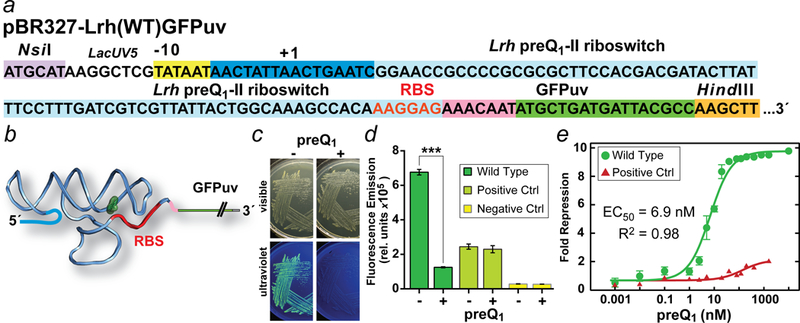
(a) DNA sequence of the pBR327 lacUV5 promoter showing the Pribnow box (yellow) and approximate transcription start site at +1. The wild-type Lrh sequence corresponding to the COG4708 gene13 extends from the −10 box to the GFPuv start codon (green) and comprises multiple regions: the 5´-leader (dark blue box); the preQ1-II riboswitch (powder blue box); the RBS (red letters); and a short linker (pink box) that separates the RBS from GFPuv (green box). The GFPuv gene starts at ATG, which replaces the start site of the naturally occurring Lrh queT gene. Unique restriction sites for sub-cloning the Lrh riboswitch sequence are shown. (b) Schematic diagram for the gene “off” Lrh riboswitch transcript from a depicting sequestration of the RBS. (c) E. coli strain JW2765 ΔqueF harboring the pBR327-Lrh(WT)GFPuv vector plated on CSB agar in the presence (2 μM) and absence of preQ1. Under white light cells grow similarly at 37 °C but show fluorescence differences attributable to GFPuv expression under a handheld UV lamp at 365 nm. (d) Reporter assay analysis of preQ1-mediated repression of GFPuv protein expression. Cells were grown in CSB media with 1 μM preQ1 or no preQ1 to assess difference in GFPuv fluorescence emission from the wild-type reporter and control constructs. Each pair was analyzed statistically. The mean and standard deviation of three independent readings are reported. The wild-type reporter probability (P) equals 0.0002 based on an unpaired, two-tailed Welch’s t-test (t = 51; df = 2.16). ***P < 0.001 is statistically highly significant. Controls ± preQ1 but lacking the riboswitch were not statistically different (P >0.05). (e) The fold repression for the on and off states of the wild-type reporter was calculated and dose-response curve was fit to give an approximate EC50. Measurements were conducted as in d. Similar analysis was conducted on the positive control, which showed <2-fold repression.
GFPuv Fluorescence Measurements.
The sequence-verified reporter plasmid pBR327-Lrh(WT)-GFPuv was transformed into competent E. coli strain JW2765 ΔqueF (Coli Genetic Stock Center, Yale University).29 For agar plates and liquid cultures we employed a chemically defined CSB media30 comprising: 0.033 M NaH2PO4, 0.086 M K2HPO4, 0.015 M (NH4)2SO4, 0.005 M sodium citrate pH 6.0, 0.492 mM MgSO4, 0.109 mM FeCl3, 0.0283 mM ZnCl2, 0.0248 mM CoCl2, 0.0138 mM Na2MoO4, 0.0201 mM CaCl2, 0.0219 mM CuCl2, 0.0234 mM MnCl2, 0.0239 mM H3BO3, and 6% (w/v) D-glucose. Transformed cells were plated on CSB-amp and grown overnight at 37 °C in the presence or absence of preQ1 (Fig. 3c). Fresh single colonies were selected and grown at 37 °C overnight in 3 mL liquid CSB-amp cultures. These growths were then used to inoculate fresh CSB-amp media with preQ1 at the following nM concentrations: 0, 0.001, 0.01, 0.1, 1, 2.5, 5, 10, 20, 40, 80, 120, 200, 800, 400, 1600 and 10000. From a starting optical density (OD600) of 0.05, the cells were grown until mid-log phase, which took 5 hr using our specific cell strain and growth-media combination. GFPuv fluorescence measurements on live bacteria were conducted by 395 nm excitation and 510 nm emission recorded by an EnSpire Plate Reader (PerkinElmer). Fluorescence was recorded on 3–5 growth replicates per preQ1 concentration. Readings were normalized by OD600 nm cell densities. Fold-repression and EC50 (Fig. 3d,e) were calculated as described28, with the following changes that altered the presentation style of repression. The fold-repression fit, y, is represented by applying the formula y(x) = [(1 – (preQ1(x) / preQ1(0)] *10 to the OD600-normalized and background-subtracted fluorescent readings, x.
Acylation of RNA in bacterial cells.
E. coli strain JW2765 ΔqueF cells were transformed with pBR327-Lrh(WT)-GFPuv were grown at 37 °C in two fresh 50 mL cultures of CSB-amp media. At an optical density (OD600) of 0.3, preQ1 was added to one flask to a final concentration of 2 µM. We chose 0.3 based on empirical measurements, which showed that cells were entering log-phase growth. A value of 2 μM preQ1 was chosen because this value is 10-fold higher than the concentration of effector required to reach a plateau in our repression analysis, suggesting saturation (Fig. 3e). To assure that preQ1 was affecting GFPuv reporter regulation, both cultures were grown for an additional 60 min. The rationale for choosing this time is provided below; see Step 1: Acylation of RNA in Bacterial Cells. Subsequently, cells were harvested by centrifugation at 4000 x g at 4 °C. Cell pellets were washed with 10 mL ice-cold Phosphate Buffered Saline (PBS), spun-down, decanted, and resuspended in 810 µL PBS. Cells were split into three tubes. One tube was treated with 30 µL DMSO + 0.6 mM NAI. A second tube was treated with 30 µL DMSO as a control. The third tube was left untreated for dideoxy-sequencing reactions. After a 20 min incubation with NAI/DMSO at 24 °C, the cells were harvested again by centrifugation, decanted, flash-frozen in liquid nitrogen, and stored at −80 °C until further processing. The RNA was extracted from thawed cells using a RiboPure™-Bacteria kit (Invitrogen) as described by the manufacturer. From ~109 cells, 16 – 25 µg of RNA was extracted. Although this approach proved reliable as a means to isolate RNA, we developed an alternative solution in which the MS2 hairpin was substituted in place of helix P1 (Fig. 2b). By applying the isolated whole-cell RNA onto NiNTA charged with 6His-MBP-MS2 coat protein (Figure S1a), we were able to enrich the desired Lrh preQ1-II riboswitch transcript prior to reverse transcription. This approach has the potential to facilitate loading normalization for icSHAPE experiments. In practice it was not used here due to our observation of extensive premature pausing at the P2-P1 junction during reverse transcription. As such, we recommend application of this approach on a case-by-case basis.
Reverse transcription of RNA.
A DNA primer complementary to the 5´-GFPuv gene was used for reverse-transcription primer-extension analysis of the recovered Lrh preQ1-II riboswitch RNA. The DNA-primer (5´-GGATCCTCTAGAGTCGACCTGCAG-3´) was 5´-radiolabelled with ATP-[γ−32P] 6000 Ci mmoL−1 10 mCi mL−1 EasyTide (PerkinElmer Life & Analytical Sciences/NEN) and purified as described.20 A volume of 4 µL of 32P-end–labeled DNA primer was annealed to 3.2 μg of RNA by incubating at 95 °C for 2 min followed by cooling (2 °C per sec) to 4 °C. Primer extension and RNA sequencing were performed as described.20 cDNA fragments were resolved on an 8% denaturing (7 M urea) polyacrylamide gel. The gel was fixed in a solution comprising 10% (v/v) methanol and 10% (v/v) acetic acid for 30 min, dried, and exposed to a phosphor-screen for 16 h. The gel image was visualized using a Typhoon 9410 scanner (GE Lifesciences).
Differential icSHAPE reactivity analysis.
To normalize icSHAPE band intensities and ascertain a linear analysis of response, the intensity of the full-length bands for the four RNA samples (i.e, DMSO control ± preQ1 and NAI treated ± preQ1, Figure S2) were measured using ImageQuant (GE Lifesciences). Multiple sequencing-gels were run with adjusted loading volumes to obtain equal-intensity of full-length primer extension products. This loading-normalized gel was analyzed using Semi-Automated Footprinting Analysis (SAFA) software.31, 32 The band identities were assigned using the sequencing ladder obtained from the ddTTP and ddATP treated lanes. Each band was then quantified by SAFA. To control for non-specific preQ1-dependent modulation, we used band intensities inside and outside of the riboswitch as a basis for normalization in SAFA. Specifically, nucleotides 24, 26, 28 and 45 (of stem P2), and 82, 83 and 86 (located in the spacer between the RBS and GFPuv ORF) were used as invariant bands (Fig. 4a and Figure S2). Furthermore, the data were also corrected in SAFA for background against DMSO-treated control lanes. The normalized band-intensities of the NAI-treated ± preQ1 bands were then used to obtain the differential-reactivity of each nucleotide using the following formula: differential reactivity = [Intensity]+preQ1 - [Intensity]−preQ1. For display, the resulting differential-reactivity values were normalized so that the highest value corresponds to 1 and lowest to −1 (Fig. 4b). Differential icSHAPE reactivity was rendered by Pymol (Schrödinger, LLC) as a heat map on the Lrh preQ1-II riboswitch co-crystal structure (Fig. 4c,d) by reassignment of the PDB temperature factors and use of the ‘chainbows’ color option. Differences (Fig. 4b) were placed on a positive scale by adding the most negative value to each nucleotide value.
Figure 4. Representative ReCo-icSHAPE analysis showing effector-dependent chemical modification of the wild-type Lrh preQ1-II riboswitch.
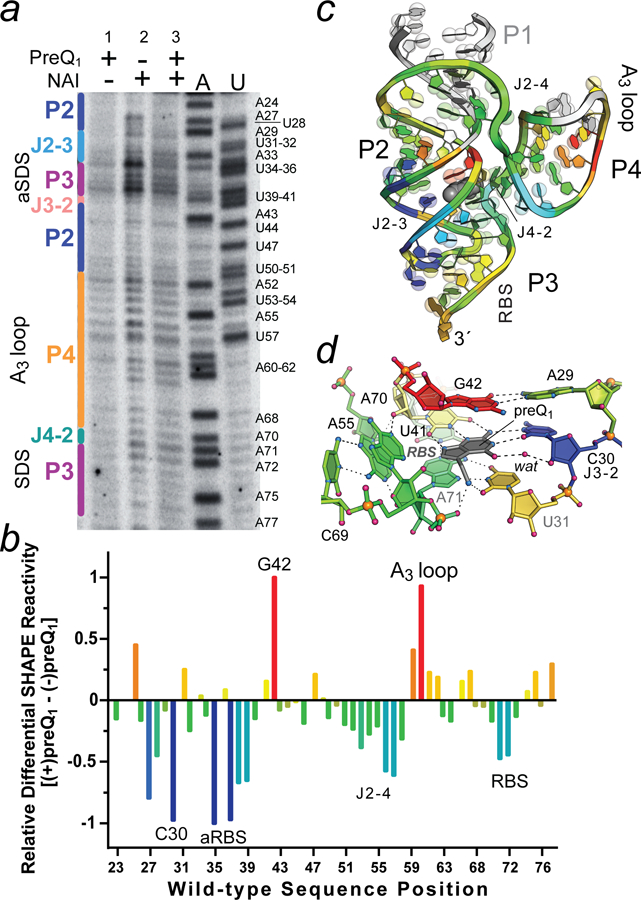
(a) Electrophoretic separation of 32P-labeled cDNAs generated from reporter coupled (ReCo) in cell (ic)SHAPE analysis conducted on bacteria exhibiting GFPuv gene “on” (no preQ1) and “off” (2 μM preQ1) states based on fluorescence emission (Fig. 3d,e and Figure S3). Lane 1: cells grown with preQ1 but no NAI added; lane 2: cells grown with no preQ1 but NAI added (gene on state); and lane 3: cells grown with preQ1 and NAI added (gene off state). A and U indicate reference nucleotide sequences with labels shown for each type. Secondary structure is labeled (left) based on the color scheme in Fig. 2b. (b) Heat map of the ReCo-icSHAPE analysis showing differences in acylation for the gene on and off states. Positive and negative values were normalized to the largest positive (red) and negative (blue) changes, which were set to 1. A24 was excluded from the analysis; aRBS is the anti-RBS sequence. The results are representative of duplicate trials for the conditions shown. (c) Ribbon diagram of the Lrh preQ1-II riboswitch showing the spatial distribution of differential ReCo-icSHAPE reactivity as a heat map from b. White regions in stem P1 (1–22) were not analyzed, and the wild-type P4 AAA (A3) loop was replaced by a UUCG tetraloop in the crystal structure, resulting in a mapping gap. PreQ1 is depicted as a gray CPK model. Bases are shown on semi-transparent spheres to emphasize purines (two spheres) and pyrimidines (one sphere). (d) Close-up of the preQ1-binding pocket rotated +90° relative to Fig. 2d. Heat mapping is derived from panel b.
RESULTS
Design Considerations for the GFPuv Reporter Assay
Background and Rationale.
Prior chemical modification efforts established the feasibility of targeted SHAPE approaches to probe an adenine riboswitch in the context of bacterial cells.33 Although groundbreaking, this approach had shortcomings. The riboswitch aptamer was fused within the tRNA anticodon stem, precluding its ability to function as a gene regulatory element.33 Another complicating factor was the difficulty associated with controlling the synthesis or salvage of the adenine effector. This necessitated use of a non-natural effector in a background of adenine metabolite. Such limitations prompted us to devise a more robust system capable of genuine effector-mediated reporter-gene regulation, leading to a visible signal that clearly differentiates between riboswitch gene on and off states for icSHAPE probing (i.e., Fig. 1). At the time of our assay development, no member of the preQ1-II riboswitch class had been validated as a bona fide gene regulator.7, 8 This validation obstacle is faced by many putative riboswitches.4, 34 Although a prior reporter-gene approach was described for the preQ1-I riboswitch using β-galactosidase,35 this method was limited by the need for integration into the B. subtilis chromosome.36, 37 Recently, preQ1 riboswitch reporter assays demonstrated preQ1-dependent eGFP gene regulation.38 However, this system was restricted to mycobacteria, which apparently lacks the Q biosynthesis pathway. To make our approach broadly applicable, we co-opted a previously described pEnv8(GAAA) vector (Fig. 3a) developed to evaluate cobalamin riboswitch control.28 This plasmid-based system uses the low-copy-number pBR327 vector, which is ideal to probe prokaryotic translation and transcription.39 The plasmid is compatible with E. coli and took advantage of the Keio collection of single-gene knockout mutants.29 In this manner, we were able to conduct our reporter assay in the context of a ΔqueF E. coli strain in which the nitrile reductase (Fig. 1a) required for preQ1 biosynthesis was ablated, thus eliminating endogenous preQ1 production. CSB media was used to control exogenous preQ1 levels and to reduce background fluorescence.28 This robust system should be compatible with a broad number of gene-regulatory elements.
Overview of the GFPuv Reporter Assay and Observed Outcomes
Step 1: Establishing GFPuv Gene Control.
With our reporter plasmid in hand (Fig. 3a), we next evaluated the Lrh preQ1-II riboswitch for effector dependent gene-regulatory function. In the presence of preQ1, we surmised that the resulting transcript would exhibit a well-structured 5´-leader sequence followed by the GFUuv open reading frame, resulting in a gene “off” mRNA state (Fig. 3b). As an initial test of switching function, we transformed the pBR327-Lrh(WT)-GFPuv plasmid into E. coli JW2765 ΔqueF cells and plated them in the presence and absence of preQ1. Both cell populations grew well when visualized with white light (Fig. 3c, top). Using a handheld UV-lamp, it was obvious that the reporter system was functional, although appropriate controls should be implemented (see next step). Cells grown in absence of preQ1 fluoresced, while those grown in the presence of 2 µM preQ1 did not exhibit appreciable fluorescence (Fig. 3c, bottom).
Step 2: Statistical Analysis of Riboswitch-Mediated Gene Regulation and Controls.
Having observed the gene-regulatory competency of the reporter plasmid, we then sought to quantify its level of repression based on comparisons to positive and negative control vectors. Control plasmids were constructed such that the riboswitch sequence was removed while retaining the RBS (positive) and by altering the positive control such that the RBS was defective (negative). In liquid cultures, cells harboring the pBR327-Lrh(WT)-GFPuv plasmid showed statistically significant differences in fluorescence emission that depended upon the presence of preQ1 in the CSB media (Fig. 3d). As expected, the positive control leads to constitutive fluorescence emission independent of preQ1. In contrast, the negative control produced comparatively little fluorescence emission in the presence or absence of preQ1; the negative control was used henceforth to correct for background fluorescence levels. The positive and negative controls showed no statistically significant difference in fluorescence emission as a result of preQ1 addition to the growth media.
Step 3: Assessing Riboswitch-Mediated Repression and Effector EC50 Values.
To measure GFPuv repression as a function of preQ1 concentration, cells were grown in CSB media containing varying amounts of preQ1. The cells containing the riboswitch showed a sigmoidal dose-response corresponding to an ~10-fold overall level of repression with an approximate EC50 value of ~7 nM. In contrast, the positive control cells showed virtually no response to added preQ1 (Fig. 3e).
Outcomes and Perspective.
Notably, overall repression mediated by the Lrh preQ1-II riboswitch is comparable to the ~8.5-fold repression attained for the wild-type env8 HyCbl riboswitch obtained when comparing cells grown in the presence of 5 μM HyCbl versus no effector.28 Our results independently confirm the modularity of this pEnv8(GAAA) reporter, which proved useful not only to probe the env8 HyCBL riboswitch cobalamin-dependent regulatory response, but also to validate other interactions that regulate translation.28, 30, 40 We also note that the concentration of effector required for 50% repression of fluorescence emission was ~7 nM. This value appears tuned to the wild-type KD,App of 17.9 ± 0.6 nM measured by ITC.13 As a caveat, we do not know the actual concentration of preQ1 inside the cells and recognize that the EC50 cannot provide a direct readout of riboswitch-mediated regulation inside the bacteria.
Considerations for Each Step of Reporter Coupled In-Cell SHAPE and Outcomes
Step 1: Acylation of RNA in Bacterial Cells.
The central hypothesis to test is that the Lrh preQ1-II riboswitch exhibits different levels of expression platform flexibility that depend on its gene regulatory states. This supposition is supported by the co-crystal structure, which shows RBS sequestration with bound preQ1 (Fig. 3b), and results of the GFPuv reporter assay that provide strong evidence of gene on and off conformational states that depend on levels of preQ1 and the presence of the riboswitch (Fig. 3c–e). To probe riboswitch regulatory conformations in bacteria, we conducted icSHAPE, which proved effective previously to interrogate 5S rRNA flexibility in muitple cell types.6 We based our reaction conditions on those established for bacteria in the prior study, which sought to introduce a single acylation event per RNA molecule (Fig. 1b). As a caveat, too many modified nucleotides will yield fewer full-length transcripts. Incubating the cells with NAI for 20 min was sufficient to allow reagent penetration of cells leading to adequate RNA modification.6
As an empirical consideration for the duration of preQ1 incubation with cells prior to NAI modification, we explored our observation that E. coli JW2765 ΔqueF cells grow more slowly in CSB in the absence of preQ1. Conversely, even though cells grow faster with preQ1, successful RNA recovery diminished with growth time. Therefore, to optimze the timing of NAI addition to cells, we measured GFPuv fluorescence for cells grown in the presence and absence of preQ1 as a function of time (Figure S3). In as little as 30 min, a signifcant difference in GFPuv fluorescence emission was observed when comparing cells grown in 2 μM to those with no preQ1. By ≥ 60 min, marked differences were apparent in fluorescence emission levels. Since uptake of preQ1 is likely rapid for JW2765 ΔqueF cells during log-phase, we rationalized that the majority of preQ1-II riboswitch transcripts were likely bound to preQ1 shortly after its addition to the media, although steady-state levels of GFPuv protein did not necessarily reflect this. As such, we waited for 60 min after the addition of preQ1 before conducting icSHAPE to assure riboswitch binding to preQ1 and to recover sufficient cellular RNA for primer-extension analysis. Overall, the effector-incubation time will likely vary depending on cell strain, growth-media, riboswitch and the effector itself. These factors will need to be investigated on a case-by-case basis. Finally, despite initiating primer extension reactions with equal amounts of RNA, loading and software normalizations were still required. RNA was recovered as described (see MATERIALS AND METHODS).
Step 2: Processing Modification Data.
The loading-volume-adjusted gel image (Fig. 4a) was processed using SAFA31, 32 (see MATERIALS AND METHODS). Beyond integration of individual nucleotide band intensities and automatic background-subtraction, SAFA enabled normalization of intensities based on invariant residues to attain further parity. SAFA was also used to correct the intensities of NAI-treated lanes to account for background levels of innate primer pausing observed in control lanes. The final intensities were used to calculate the normalized differential reactivity of each base (Fig. 4b).
Outcome:
Significant modulation of the flexibility was observed in the differential reactivity analysis. Negative values were interpreted as less flexible nucleotides (blue) whereas more flexible regions resulting from preQ1 addition were positive (red) (Fig. 4b). These values ostensibly arose due to conformational differences arising from the gene off and on states. Prior analysis of the Lrh preQ1-II riboswitch by in vitro in-line probing did not reveal preQ1-dependent modulation of the RBS, although J2–3 and a portion of the anti-Ribosome Binding Site (aRBS) showed decreased backbone cleavage with added preQ1.13 Similarly, in vitro SHAPE analysis of the related Streptococcus pneumoniae preQ1-II riboswitch did not reveal appreciable preQ1-dependent modulation of the aRBS-RBS P3 helix.15 In contrast, we observed strong modulation for both the RBS and aRBS sequences (Fig. 4b). Unlike prior studies33, our results demonstrate that the crowded cellular environment does not preclude detection of both loose and compact RNA-regulatory conformations. Indeed, variations in P3 acylation (i.e., the expression platform) show changes in flexibility in accord with preQ1-dependent switching that gives rise to downstream gene regulation.
Mapping Differential icSHAPE Results to Structures
Step 1: Quality Control Considerations.
An optional but powerful aspect of our analysis was to consider the differential ReCo-icSHAPE analysis in light of the existing Lrh preQ1-II riboswitch co-crystal structure.13 As a point of quality control, we decided to test directly whether any appreciable differences arose in preQ1 binding as a result of preparing the riboswitch via denaturing purification (i.e., used for crystallization) versus native purification. Although RNA purification by denaturing polyacrylamide gel electrophoresis (PAGE) is common practice, not all RNAs refold into active conformations. This limits their reliability in subsequent functional analysis.26, 41, 42 To proceed, we first prepared an Lrh preQ1-II riboswitch 76-mer comprising the crystallization sequence (Fig. 2b) by denaturing PAGE with heat annealing and Mg2+ addition for refolding13, 25. We then prepared the 76-mer in a manner that preserves the co-transcriptional folding attained by T7 polymerase synthesis (Figure S1).26 ITC analysis of each sample revealed no discernable differences. The denatured and refolded transcript had an average KD,App of 2.76 ± 0.14 nM with an average stoichiometry (n) of 1.16 ± 0.07 (Fig. 2e). The natively purified transcript exhibited a KD,App of 3.3 ± 0.3 nM with n = 0.83 ± 0.002. The close KD values – within error – and thermodynamic parameters for binding provide confidence that the denaturing method used to purify and refold the Lrh preQ1-II riboswitch for crystallization yielded a structure that concurs with the native fold. As such, the crystal structure provides a reliable scaffold to map our differential icSHAPE results to relate atomic-level features to cellular function.
Step 2: Accounting for Sequence Differences.
A second caveat of structural mapping is that the free-state riboswitch structure is unknown. This presents challenges in the analysis, although computational approaches can be helpful to rationalize the results on a molecular level (see below). A minor consideration is that the wild-type Lrh preQ1-II riboswitch sequence is not completely identical to that used for X-ray crystallography in P1 and the P4 loop (Fig. 2b). As such, we excluded P1 from the analysis and considered the absence of the UUCG tetraloop in the mapping process (Fig. 4c, gray regions). Mapping of the differential icSHAPE results extended from base 23 through 77, providing a three-dimensional overview of nucleotide flexibility changes as a result of effector binding.
Step 3: Results and Mapping Interpretation.
Although it is tempting to consider small changes in acylation, a more circumspect approach is to consider the least flexible (blue) and most flexible (red) regions. In these respects, the 2´-OH group of C30 of J2–3 appears much less susceptible to acylation in the preQ1 bound state and appears to be one of the least flexible nucleotides in the differential analysis (Fig. 4b). The crystal structure reveals a site-bound water at this position that forms a bridging contact to the keto group of preQ1. Previously this interaction was attributed to the riboswitch’s promiscuous ability to recognize the N6 group of 2,6-diaminopurine in place of the O6 keto moiety of preQ1.13 Although a water molecule is likely to exchange quickly at this position, this interaction could be a factor in the reduction of local flexibility in the preQ1-bound state. Perhaps a more dominant consideration is that Watson-Crick face of C30 is essential to recognize preQ1 (Fig. 4c,d).7, 13 This interaction likely stabilizes the C30 conformation and reduces acylation susceptibility at the 2´-OH, consistent with the bound-state NMR ensemble of the Streptococcus pneumoniae preQ1-II riboswitch.14 In contrast, one of the most flexible positions resulting from preQ1 binding is nearby G42, which forms the ceiling of the binding pocket (Fig. 4c,d). G42 engages in a cis Watson-Crick interaction with A29 in the bound state. The homologous interaction in the Streptococcus pneumoniae preQ1-II riboswitch structure is a canonical G7-C20 pair that is relatively stable in the 18 lowest energy structures.14 However, noncanonical pairs, such as A29-G42 of the Lrh riboswitch, can increase backbone flexibility43 and the 2´-OH of G42 is solvent exposed in the corresponding bound-state crystal structure. Although the exact basis for the increase in G42 2´-OH flexibility is uncertain, it could arise if the riboswitch were to adopt an alternate, stable, conformation in the apo state. Although there is no apo-state structure of a preQ1-II riboswitch, prior molecular dynamics simulations revealed formation of a Watson-Crick C30-G42 pair in some trajectories of the preQ1-free state on a microsecond timescale. This interaction could account for the reduced acylation of G42 in the apo state, wherein the C30-G42 pair is sandwiched between novel triplex (A29•U28-A43) and U31•U41 interactions that appear when preQ1 is absent.44
Summary.
A major bottleneck in our understanding of RNA is the ability to relate fold to function in live cells. Here we used sensitive fluorescence-based readout of GFPuv in bacteria to validate and report upon the gene regulatory properties and status of a preQ1-II riboswitch. This knowledge was coupled to chemical modification experiments designed to interrogate relevant conformations for this functional RNA, which complements existing in vitro techniques. Our approach can be applied to myriad other riboswitches as well as functional RNAs that have already been identified. Complementary high-resolution structural coordinates will heighten the interpretative power of such results but are not essential to gain useful information.
Supplementary Material
Figure S1:
Native purification schematic diagram and representative results for the Lrh preQ1-II riboswitch Supplemental
Figure S2:
Whole-gel PAGE analysis of icSHAPE cDNA obtained from primer extension Supplemental
Figure S3:
Timecourse of GFPuv fluorescence resulting from gene regulation by the Lrh preQ1-II riboswitch
Acknowledgment.
We thank members of the Wedekind lab, Drs. J.L. Jenkins, C.L. Kielkopf, and D.H. Mathews for helpful discussions. We thank Dr. R.C. Spitale for the generous gift of NAI for icSHAPE experiments and for suggestions regarding RNA recovery and purification.
Funding
This work was supported by National Institutes of Health Grant R01 GM063162 (J.E.W).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
References
- (1).Kubota M, Tran C, and Spitale RC (2015) Progress and challenges for chemical probing of RNA structure inside living cells, Nat Chem Biol 11, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spitale RC, Flynn RA, Torre EA, Kool ET, and Chang HY (2014) RNA structural analysis by evolving SHAPE chemistry, Wiley Interdiscip Rev RNA 5, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Garst AD, Edwards AL, and Batey RT (2011) Riboswitches: structures and mechanisms, Cold Spring Harb Perspect Biol 3, a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).McCown PJ, Corbino KA, Stav S, Sherlock ME, and Breaker RR (2017) Riboswitch diversity and distribution, RNA 23, 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sherwood AV, and Henkin TM (2016) Riboswitch-Mediated Gene Regulation: Novel RNA Architectures Dictate Gene Expression Responses, Annu Rev Microbiol 70, 361–374. [DOI] [PubMed] [Google Scholar]
- (6).Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, and Chang HY (2013) RNA SHAPE analysis in living cells, Nat Chem Biol 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Meyer MM, Roth A, Chervin SM, Garcia GA, and Breaker RR (2008) Confirmation of a second natural preQ1 aptamer class in Streptococcaceae bacteria, RNA 14, 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McCown PJ, Liang JJ, Weinberg Z, and Breaker RR (2014) Structural, functional, and taxonomic diversity of three preQ1 riboswitch classes, Chem Biol 21, 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bienz M, Kubli E, Kohli J, deHenau S, Huez G, Marbaix G, and Grosjean H (1981) Usage of the three termination codons in a single eukaryotic cell, the Xenopus laevis oocyte, Nucleic Acids Res 9, 3835–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Meier F, Suter B, Grosjean H, Keith G, and Kubli E (1985) Queuosine modification of the wobble base in tRNAHis influences ‘in vivo’ decoding properties, EMBO J 4, 823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Noguchi S, Nishimura Y, Hirota Y, and Nishimura S (1982) Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. Function and biosynthesis of queuosine in tRNA, J Biol Chem 257, 6544–6550. [PubMed] [Google Scholar]
- (12).Akimoto H, Imamiya E, Hitaka T, Nomura H, and Nishimura S (1988) Synthesis of Queuine, the Base of Naturally-Occurring Hypermodified Nucleoside (Queuosine), and Its Analogs, Journal of the Chemical Society-Perkin Transactions 1, 1637–1644. [Google Scholar]
- (13).Liberman JA, Salim M, Krucinska J, and Wedekind JE (2013) Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold, Nat Chem Biol 9, 353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kang M, Eichhorn CD, and Feigon J (2014) Structural determinants for ligand capture by a class II preQ1 riboswitch, Proc Natl Acad Sci U S A 111, E663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Souliere MF, Altman RB, Schwarz V, Haller A, Blanchard SC, and Micura R (2013) Tuning a riboswitch response through structural extension of a pseudoknot, Proc Natl Acad Sci U S A 110, E3256–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wilkinson KA, Merino EJ, and Weeks KM (2005) RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts, J Am Chem Soc 127, 4659–4667. [DOI] [PubMed] [Google Scholar]
- (17).Weeks KM, and Mauger DM (2011) Exploring RNA structural codes with SHAPE chemistry, Acc Chem Res 44, 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gherghe CM, Shajani Z, Wilkinson KA, Varani G, and Weeks KM (2008) Strong correlation between SHAPE chemistry and the generalized NMR order parameter (S2) in RNA, J Am Chem Soc 130, 12244–12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Merino EJ, Wilkinson KA, Coughlan JL, and Weeks KM (2005) RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE), J Am Chem Soc 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- (20).Wilkinson KA, Merino EJ, and Weeks KM (2006) Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution, Nat Protoc 1, 1610–1616. [DOI] [PubMed] [Google Scholar]
- (21).Kortmann J, and Narberhaus F (2012) Bacterial RNA thermometers: molecular zippers and switches, Nat Rev Microbiol 10, 255–265. [DOI] [PubMed] [Google Scholar]
- (22).Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, Kasprzak WK, Shapiro BA, and Dinman JD (2014) Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway, Nature 512, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ofori LO, Hilimire TA, Bennett RP, Brown NW Jr., Smith HC, and Miller BL (2014) High-affinity recognition of HIV-1 frameshift-stimulating RNA alters frameshifting in vitro and interferes with HIV-1 infectivity, J Med Chem 57, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yao P, Potdar AA, Ray PS, Eswarappa SM, Flagg AC, Willard B, and Fox PL (2013) The HILDA complex coordinates a conditional switch in the 3’-untranslated region of the VEGFA mRNA, PLoS Biol 11, e1001635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lippa GM, Liberman JA, Jenkins JL, Krucinska J, Salim M, and Wedekind JE (2012) Crystallographic analysis of small ribozymes and riboswitches, Methods Mol Biol 848, 159–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Batey RT, and Kieft JS (2007) Improved native affinity purification of RNA, RNA 13, 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Liberman JA, Bogue JT, Jenkins JL, Salim M, and Wedekind JE (2014) ITC analysis of ligand binding to preQ(1) riboswitches, Methods Enzymol 549, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Johnson JE Jr., Reyes FE, Polaski JT, and Batey RT (2012) B12 cofactors directly stabilize an mRNA regulatory switch, Nature 492, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, and Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection, Mol Syst Biol 2, 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Holmstrom ED, Polaski JT, Batey RT, and Nesbitt DJ (2014) Single-molecule conformational dynamics of a biologically functional hydroxocobalamin riboswitch, J Am Chem Soc 136, 16832–16843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Das R, Laederach A, Pearlman SM, Herschlag D, and Altman RB (2005) SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments, RNA 11, 344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Laederach A, Das R, Vicens Q, Pearlman SM, Brenowitz M, Herschlag D, and Altman RB (2008) Semiautomated and rapid quantification of nucleic acid footprinting and structure mapping experiments, Nat Protoc 3, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tyrrell J, McGinnis JL, Weeks KM, and Pielak GJ (2013) The cellular environment stabilizes adenine riboswitch RNA structure, Biochemistry 52, 8777–8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wedekind JE, Dutta D, Belashov IA, and Jenkins JL (2017) Metalloriboswitches: RNA-based inorganic ion sensors that regulate genes, J Biol Chem 292, 9441–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Roth A, Winkler WC, Regulski EE, Lee BW, Lim J, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, Iwata-Reuyl D, and Breaker RR (2007) A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain, Nat Struct Mol Biol 14, 308–317. [DOI] [PubMed] [Google Scholar]
- (36).Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, and Breaker RR (2003) An mRNA structure in bacteria that controls gene expression by binding lysine, Genes Dev 17, 2688–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Winkler WC, Nahvi A, Sudarsan N, Barrick JE, and Breaker RR (2003) An mRNA structure that controls gene expression by binding S-adenosylmethionine, Nat Struct Biol 10, 701–707. [DOI] [PubMed] [Google Scholar]
- (38).Van Vlack ER, Topp S, and Seeliger JC (2017) Characterization of Engineered PreQ1 Riboswitches for Inducible Gene Regulation in Mycobacteria, J Bacteriol 199, e00656–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).http://www.discoverbiotech.com/wiki/-/wiki/Main/Cloning+Vectors.
- (40).Polaski JT, Holmstrom ED, Nesbitt DJ, and Batey RT (2016) Mechanistic Insights into Cofactor-Dependent Coupling of RNA Folding and mRNA Transcription/Translation by a Cobalamin Riboswitch, Cell Rep 15, 1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kieft JS, and Batey RT (2004) A general method for rapid and nondenaturing purification of RNAs, RNA 10, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Keel AY, Easton LE, Lukavsky PJ, and Kieft JS (2009) Large-scale native preparation of in vitro transcribed RNA, Methods Enzymol 469, 3–25. [DOI] [PubMed] [Google Scholar]
- (43).Hermann T, and Westhof E (1999) Non-Watson-Crick base pairs in RNA-protein recognition, Chem Biol 6, R335–343. [DOI] [PubMed] [Google Scholar]
- (44).Aytenfisu AH, Liberman JA, Wedekind JE, and Mathews DH (2015) Molecular mechanism for preQ1-II riboswitch function revealed by molecular dynamics, RNA 21, 1898–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McCarty RM, and Bandarian V (2008) Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin, Chem Biol 15, 790–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1:
Native purification schematic diagram and representative results for the Lrh preQ1-II riboswitch Supplemental
Figure S2:
Whole-gel PAGE analysis of icSHAPE cDNA obtained from primer extension Supplemental
Figure S3:
Timecourse of GFPuv fluorescence resulting from gene regulation by the Lrh preQ1-II riboswitch