

Abstract
Campylobacter jejuni, a leading cause of gastroenteritis world wide, has a unique O-methyl phosphoramidate (MeOPN) moiety attached to its capsular polysaccharide. Investigations into the biological role of MeOPN have revealed that it contributes to the pathogenicity of C. jejuni, and this modification is important for the colonization of C. jejuni. Previously, the reactions catalyzed by four enzymes (Cj1418-Cj1415) from C. jejuni that are required for the biosynthesis of the phosphoramidate modification have been elucidated. Cj1418 (L-glutamine kinase) catalyzes the formation of the initial phosphoramidate bond with the ATP-dependent phosphorylation of the amide nitrogen of L-glutamine. Here we show that Cj1418 catalyzes the phosphorylation of L-glutamine through a three-step reaction mechanism via the formation of covalent pyrophosphorylated (Enz-X-Pβ-Pγ) and phosphorylated (Enz-X-Pβ) intermediates. In the absence of L-glutamine, the enzyme was shown to catalyze a positional isotope exchange (PIX) reaction within β-[18O4]-ATP in support of the formation of the Enz-X-Pβ-Pγ intermediate. In the absence of ATP, the enzyme was shown to catalyze a molecular isotope exchange (MIX) reaction between L-glutamine phosphate and [15N-amide]-L-glutamine in direct support of the Enz-X-Pβ intermediate. The active site nucleophile has been identified as His-737 based on the lack of activity of the H737N mutant and amino acid sequence comparisons. The enzyme was shown to also catalyze the phosphorylation of D-glutamine, γ-L-glutamyl hydroxamate, γ-L-glutamyl hydrazide and β-L-aspartyl hydroxamate, in addition to L-glutamine.
Graphical Abstract
INTRODUCTION
Campylobacter jejuni, a Gram-negative pathogenic bacterium, is a leading cause of gastroenteritis in the US.1 While C. jejuni is pathogenic towards humans, this organism is commensal towards chickens, and as a result contaminated poultry is a common source of human infection. Like many other Gram-negative bacteria, C. jejuni produces a capsular polysaccharide (CPS), which is composed of unusually modified sugars that envelops the organism. The capsular polysaccharides help protect the organism from the environment and are important for the invasion and colonization of the host organisms.2 Numerous strains of C. jejuni have been identified and it is currently believed that each strain produces a unique capsular polysaccharide.3,4 An O-methyl phosphoramidate (MeOPN) modification was discovered in the CPS of the NCTC11168 strain of C. jejuni.4,5 This modification to the CPS is most unusual since phosphorus-nitrogen bonds are quite rare in nature.5
The gene cluster for the biosynthesis of the CPS for the NCTC11168 strain of C. jejuni capsule has been identified, and eight of the 35 genes in this cluster appear to be linked with the presence of the MeOPN modification in the capsular polysaccharide.5,6 Of the eight enzymes that appear to be required for the biosynthesis of the MeOPN modification, two are predicted to be phosphoramidate transferases (Cj1421 and Cj1422) and two are annotated as SAM-dependent methyltransferases (Cj1419 and Cj1420). The other four enzymes (Cj1415, Cj1416, Cj1417, and Cj1418) are directly responsible for the formation of the phosphoramidate moiety. Previously, we have characterized the four enzymes responsible for the formation of the phosphoramidate.7–9 The first enzyme, Cj1418, is a novel glutamine kinase that catalyzes the ATP-dependent phosphorylation of L-glutamine (1) to form L-glutamine phosphate (2).8 L-glutamine phosphate is subsequently used by Cj1416, a CTP:phosphoglutamine cytidylyltransferase, to displace pyrophosphate from CTP to generate CDP-L-glutamine (3).7 Cj1417, a γ-glutamyl-CDP-amidate hydrolase, hydrolyzes CDP-L-glutamine to form L-glutamate and cytidine diphosphoramidate (4).7 Cj1415, a cytidine diphosphoramidate kinase, catalyzes the phosphorylation of the 3’ hydroxyl group of cytidine diphosphoramidate.9 The phosphoramidate moiety of the 3’-phospho 5’-cytidine diphosphoramidate cofactor (5) is likely transferred to the capsule and methylated or, alternatively, methylated and then transferred to the capsule. The reactions are summarized in Scheme 1.
Scheme 1:
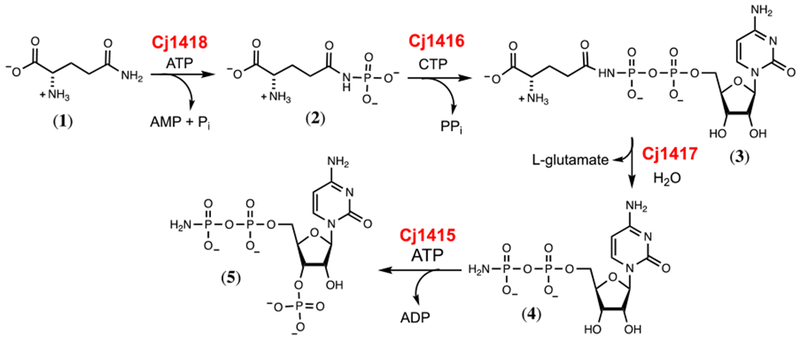
Biosynthesis of 3’-phospho-5’-cytidine diphosphoramidate (5) in C. jejuni NCTC11168.
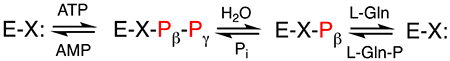
Of these four enzymes, glutamine kinase (Cj1418), is perhaps the most interesting mechanistically, as it is responsible for synthesis of the phosphoramidate bond. Very few enzymes have been shown previously to catalyze the formation of P-N bonds, however two strategies exist. Creatine kinase and arginine kinase catalyze the ATP dependent phosphorylation of their respective substrates and generate ADP as a product.10, 11 Alternatively, enzymes that are related to tRNA synthetases can also be used to generate phosphoramidate bonds through a mechanism similar to adenylylation.12 Cj1418 belongs to a family of kinases that employ an unusual reaction mechanism. Phospho(enol)pyruvate synthase catalyzes the ATP-dependent phosphorylation of pyruvate, generating PEP, AMP and phosphate.13 Pyruvate phosphate dikinase catalyzes the ATP-dependent phosphorylation of phosphate and pyruvate to generate AMP, pyrophosphate and PEP.14 Finally, rifampin phosphotransferase catalyzes the phosphorylation of rifampin with formation of Pi and AMP.15,16 All three enzymes share a similar protein architecture, and contain an ATP-grasp domain, a phospho-histidine domain, and a specialized substrate-binding domain.16–18 These three domains can be identified in Cj1418, where residues 1–219 comprise the predicted ATP-grasp domain, residues 220-693 is likely the specialized substrate-binding domain for L-glutamine, and residues 694-767 form the predicted phospho-histidine domain.17
This enzyme family catalyzes the phosphorylation of substrates via a mechanism that requires three distinct chemical steps. In the first step, the enzyme utilizes a histidine residue to attack ATP at the β-phosphoryl group to liberate AMP and a pyrophosphorylated enzyme intermediate. In the next step the pyrophosphorylated enzyme is hydrolyzed to form inorganic phosphate and a phosphorylated enzyme intermediate. In the final step the phosphorylated enzyme intermediate transfers the phosphate to the acceptor substrate. These transformations are summarized in Scheme 2 for the reaction catalyzed by Cj1418.
Scheme 2:
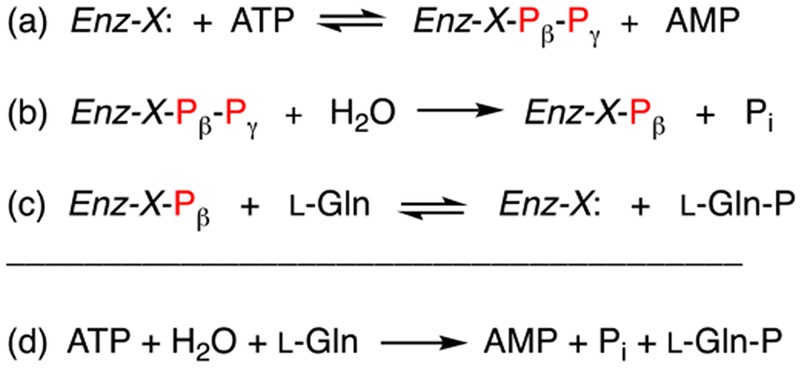
Proposed reaction mechanism for formation of L-glutamine phosphate by Cj1418.
In this investigation we have addressed the reaction mechanism and substrate profile for the reaction catalyzed by glutamine kinase. The formation of the pyrophosphorylated and phosphorylated covalent reaction intermediates have been interrogated by utilization of positional isotope exchange (PIX) and molecular isotope exchange (MIX) techniques.19, 20 The active site histidine residue has been identified by mutagenesis and the enzyme has been shown to catalyze the phosphorylation of the hydroxamate and hydrazide derivatives of glutamate and aspartate.
MATERIALS and METHODS
Materials.
All buffers and salts were purchased from Sigma-Aldrich, unless otherwise specified. L-glutamine (1), γ-L-glutamyl hydroxamate (7), β-L-aspartyl hydroxamate (9), AMP-morpholidate (10), and phosphorus pentachloride were purchased from Sigma Aldrich. γ-L-glutamyl hydrazide (8) was obtained from Santa Cruz Biotechnology. 18O-labeled water (97 atom %) was obtained from Cambridge Isotope Laboratories. [15N-amide]-L-glutamine was obtained from Merck, Sharp and Dohme, Canada. D-Glutamine (98% pure) was purchased from Acros Organics. The restriction enzyme DpnI was obtained from New England Biolabs. Primestar HS Polymerase was purchased from Takara Industries. The plasmid used for the expression of Cj1418 from C. jejuni NCTC 11168 was obtained from Professor Christine Szymanski of the University of Georgia.
Purification of Glutamine Kinase.
The plasmid used for the expression of Cj1418 (UniProt: Q0P8J6) with an N-terminal poly-histidine purification tag was used to transform Rosetta (DE3) Escherichia coli cells by electroporation. Five-mL cultures of LB medium supplemented with 50 µg/mL kanamycin and 25 µg/mL chloramphenicol were inoculated with a single colony and grown overnight at 30 oC. These cultures were used to inoculate 1 L of LB medium (50 µg/mL kanamycin and 25 µg/mL chloramphenicol) and then incubated at 30 oC until an OD600 of ~0.6–0.8 was reached. The cells were induced with 1.0 mM isopropyl β-thiogalactoside (IPTG), grown for 16 h at 16 oC and then harvested by centrifugation at 6000 rpm at 4 oC. The resulting cell pellet was re-suspended into loading buffer (50 mM HEPES/KOH, 300 mM KCl, 20 mM imidazole, pH 8.0) and lysed by sonication. The total cell lysate was passed through a 0.45 µm filter before being loaded onto a prepacked 5-mL HisTrap HP (GE Healthcare) nickel affinity column. Protein was eluted with 50 mM HEPES/KOH, pH 8.0, 300 mM KCl, and 400 mM imidazole over a gradient of 30 column volumes. Excess imidazole was removed by exchanging the buffer against 50 mM HEPES/KOH, pH 8.0, 100 mM KCl, using a 20 mL (10 kDa molecular weight cutoff) concentrator (GE Healthcare). The protein was concentrated to a volume of 3 mL and loaded onto a GE Healthcare HiLoad 16/600 Sephadex 120 mL size exclusion column and eluted with 10 mM HEPES/KOH (pH 8.0) and 100 mM KCl. Fractions were assayed for catalytic activity, pooled, concentrated, and stored at −80 °C until needed.
Construction of H737N Mutant of Glutamine Kinase.
His-787 from Cj1418 was mutated to asparagine. The plasmid used for the expression of wild-type Cj1418 with an N-terminal hexa-histidine tag was used as a template for the construction of the plasmid for the expression of the H737N mutant. The forward (5’-GGGGGTGCTAATTCAAATATGGCCATTCGTGC-3’) and reverse primers (5’-GCACGAATGGCCATATTTGAATTAGCACCCCC-3’) were used to convert the histidine codon (CAT) at residue 737 to asparagine (AAT). Primestar HS polymerase was used to amplify the gene. The reaction used a three-step thermal cycle (98 oC for 10 s, 55 oC for 40 s, and 72 oC for 10 min) and continued through thirty cycles. After amplification, DpnI was used to digest the template DNA for 2 h at 37 oC. Following DpnI digestion, PCR cleanup (Qiagen) was performed and the plasmid transformed into E. coli BL21 (DE3) cells. Single colonies were selected and the DNA sequence of the mutant gene was confirmed.
Determination of Kinetic Constants.
The kinetic constants for Cj1418 were determined using a pyruvate kinase/lactate dehydrogenase/myokinase coupled enzyme assay by following the oxidation of NADH at 340 nm at 25 oC with a SpectraMax340 UV-visible spectrophotometer. Assays were performed in 100 mM HEPES/KOH, pH 8.0, and 100 mM KCl. The coupling system contained 2.0 mM PEP, 0.3 mM NADH, 14 mM MgCl2, 10 mM ATP, 8 units/mL lactate dehydrogenase, 8 units/mL adenylate kinase and 8 units/mL pyruvate kinase (PK). The kinetic constants for D-glutamine (2.0 – 20 mM), γ-L-glutamyl hydroxamate (0.4 – 4.0 mM), γ-L-glutamyl hydrazide (2.0 – 20 mM), β-L-aspartyl hydroxamate (5.0 – 40 mM) were all assayed using 250 nM Cj1418. The kinetic constants were determined by fitting the initial rates to Eqn. 1 using GraFit 5, where υ is the initial velocity of the reaction, Et is the enzyme concentration, kcat is the turnover number, [A] is the substrate concentration, and Km is the Michaelis constant.
| (1) |
Synthesis of [18O4]-Phosphate.
The synthesis of [18O4]-Pi (11) was carried out as reported previously.21 The reaction was conducted in a 10-mL round bottom flask that was placed in an ice bath. The reaction utilized 1.0 g of water containing 97% 18O, and 2.5 g of PCl5. PCl5 was slowly added to the water and then heated to 100 °C and allowed to stir for an additional 2 h. After heating, the reaction was cooled to room temperature, the pH adjusted to 7.0, and aliquots were flash frozen and stored at −80 °C. The 31P NMR spectrum of the product indicated that 93% of the phosphate contained four 18O-atoms and 7% contained three 18O-atoms, for an overall 18O-content of 98%.
Synthesis of β-[18O4]-ATP.
The procedure for the synthesis of 18O-labeled ATP where the β-phosphoryl group contains four 18O-atoms was modified from a previously reported method.22,23 AMP morpholidate (10, 1.0 g) and the tributylammonium salt of [18O4]-phosphate were stirred in dry DMSO (20 mL) at room temperature for three days. The reaction was diluted with water (100 mL) and then loaded onto a column of DEAE Sephadex. The β-[18O4]-ADP (12) was eluted from the column using a linear gradient of triethylammonium bicarbonate (300 mL from 0.01 to 1.0 M) and then concentrated by rotary evaporation. The labeled ADP (20 mM) was incubated with pyruvate kinase (3 units/mL) and 25 mM phospho(enol)pyruvate (PEP) in 50 mM HEPES/KOH, pH 8.0 for 16 h in a volume of 50 mL. The reaction mixture was loaded onto a column of DEAE Sephadex and eluted with triethylammonium bicarbonate (600 mL from 0.01 to 1 M). The fractions containing β-[18O4]-ATP (13) were identified by the absorbance at 260 nm and 31P NMR spectroscopy. Excess triethylamine removed by evaporation and the product stored at −80 °C. The reactions are summarized in Scheme 3.
Scheme 3:
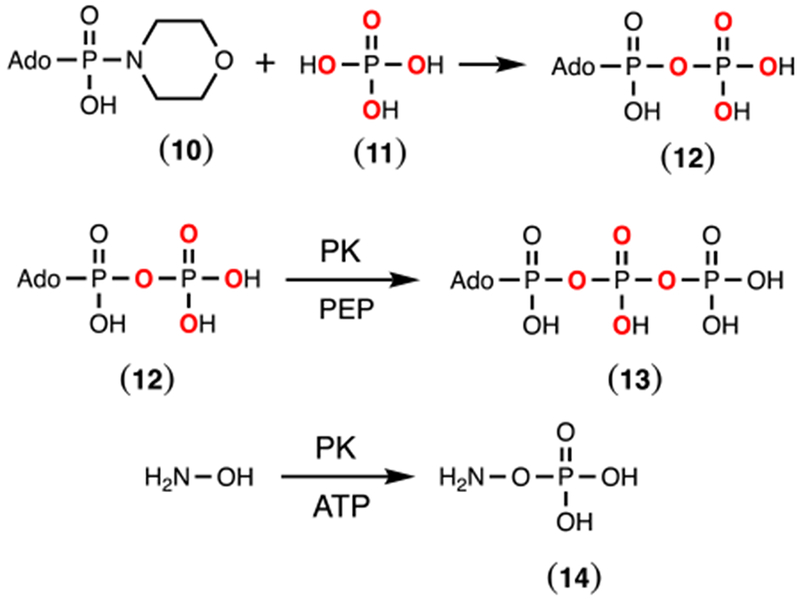
Chemical synthesis of β-[18O4]-ADP (12) and enzymatic syntheses of β-[18O4]-ATP (13) and hydroxylamine O-phosphate ester (14).
Synthesis of Hydroxylamine O-Phosphate Ester.
O-phosphorylated hydroxylamine (14) was synthesized using pyruvate kinase.24 Pyruvate kinase (4 units/mL) was incubated with 20 mM ATP, 25 mM hydroxylamine, 30 mM zinc acetate and 100 mM ammonium bicarbonate (pH 8.0). The reaction was monitored by 31P NMR spectroscopy until all of the ATP was converted to ADP. Once complete, the reaction mixture was diluted with water and loaded onto a DEAE Sephadex anion exchange resin and eluted with a linear gradient of triethylammonium bicarbonate (0.01 to 1.0 M). Fractions containing the O-phosphorylated hydroxylamine were identified by 31P NMR spectroscopy, concentrated, flash frozen and then stored at −20 °C.
Positional Isotope Exchange (PIX).
A reaction mixture containing 5 mM β-[18O4]-ATP (13), 7 mM MgCl2, and 10 μM Cj1418 in 100 mM KCl, 100 mM HEPES/KOH, 50 mM CHES/KOH, pH 8.0, was incubated at 25 °C. At various times, 500 μL of the reaction mixture were removed and quenched with 15 mM EDTA and the pH adjusted to 9.5 before 31P NMR analysis. Samples that were not analyzed immediately were flash frozen and stored at −20 °C. Aliquots were collected at 0, 1, 4, 8, 12, and 15 h and the reaction velocities were determined using Eqn. 2, where X is the fraction of change of the original ATP pool, and F is the fraction of the equilibrium value for positional isotope exchange in the ATP pool at time t.25
| (2) |
Molecular Isotope Exchange (MIX).
The reaction mixture containing 10 mM L-glutamine phosphate and 10 mM [15N-amide]-L-glutamine was incubated with 5 mM MgCl2, and 20 μM Cj1418 for 12 h in 100 mM HEPES/KOH, 100 mM KCl (pH 8.0) at 25 °C. The reaction products were analyzed by 15N and 31P NMR spectroscopy. [15N-amide]-L-glutamine phosphate was made enzymatically as previously described.8
RESULTS
Substrate Specificity of Cj1418.
Cj1418 has previously been identified as a glutamine kinase that catalyzes the ATP-dependent phosphorylation of L-glutamine.8 However, the substrate profile for this enzyme has not been adequately determined. In addition to L-glutamine, 15 structural analogs of L-glutamine were tested as potential substrates for Cj1418 (Scheme S1). Of the compounds tested, only L-glutamine (1), D-glutamine (6), γ-L-glutamyl hydroxamate (7), γ-L-glutamyl hydrazide (8), and β-L-aspartyl hydroxamate (9) exhibited substrate activity based on the formation of AMP and Pi (Scheme 4). The kinetic constants for the phosphorylation of these compounds by Cj1418 are summarized in Table 1.
Scheme 4:
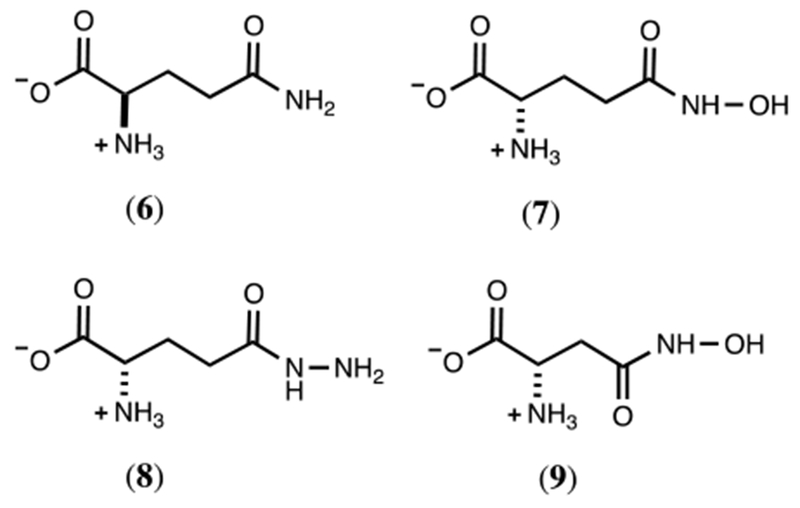
Alternate substrates for Cj1418.
Table 1:
Kinetic constants for Cj1418 at 25 °C, pH 8.0, and 10 mM ATP.
| substrate | kcat (s-1) | Km (mM) | kcat/Km (M-1 s-1) |
|---|---|---|---|
| L-glutamine (1) | 2.5 ± 0.3 | 0.64 ± 0.06 | 3900 ± 800 |
| D-glutamine (6) | 1.5 ± 0.1 | 10.5 ± 1.8 | 140 ± 30 |
| γ-L-glutamyl hydroxamate (7) | 2.1 ± 0.2 | 1.5 ± 0.4 | 1400 ± 360 |
| γ-L-glutamyl hydrazide (8) | 0.41 ± 0.05 | 17.2 ± 3.8 | 24 ± 6 |
| β-L-aspartyl hydroxamate (9) | nda | nda | 1.2 ± 0.2b |
nd, not determined.
A plot of reaction rate versus substrate concentration was linear up to a concentration of 40 mM
Characterization of Reaction Products.
The reaction products for the phosphorylation of the alternate substrates 6-9 were analyzed by 31P NMR spectroscopy (Figures S1 through S4). The product for the reaction with D-glutamine (6) exhibited two new resonances (−3.65 and −4.28 ppm) with the same chemical shifts as reported previously for the reaction of Cj1418 with L-glutamine and formation of L-glutamine phosphate (2).8 These two resonances represent the syn- and anti-conformers of D-glutamine phosphate (Figure S1). With the hydroxamate derivative of L-glutamate (7), two new resonances were observed at 5.91 ppm (major) and 6.33 ppm (minor) ppm after the addition of ATP and Cj1418 (in addition to those for AMP and Pi). However, this product was unstable and a new resonance was observed at 7.92 ppm after ~24 h. The degradation product was shown to be hydroxylamine-O-phosphate (14) by direct comparison of authentic 14 by 31P NMR (Figure S2). This result is consistent with the enzymatic phosphorylation of 7 on the hydroxyl group and the initial formation of compound 15 (Scheme 5). Formation of 14 is proposed to occur via the intramolecular attack of the α-amino group of 15 with the C5 carbonyl group and generation of pyroglutamic acid (16) as shown in Scheme 5. The 13C NMR spectrum of the reaction mixture confirmed the formation of pyroglutaminic acid (Figure S5a).
Scheme 5:
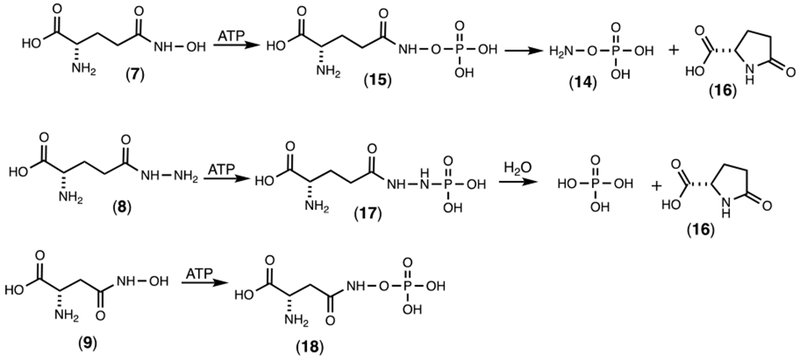
Reaction products for the phosphorylation of 7, 8, and 9 by Cj1418.
With the hydrazide derivative of L-glutamine (8) two new 31P NMR resonances were observed with chemical shifts of 5.66 ppm (major) and 5.47 ppm (minor) (Figure S3a). This product was unstable (17) and subsequently converted to phosphate (Figure S3b). The 13C NMR spectrum is consistent with the formation of pyroglutamate (Figure S5b). The fate of the hydrazine is unknown. The reaction product with the hydroxamate of aspartate (9) exhibited two new 31P NMR resonances with chemical shifts of 6.07 ppm (major) and 6.44 ppm (minor) (Figure S4a). However, unlike the phosphorylated hydroxamate from L-glutamate (15) the phosphorylated hydroxamate product (18) from L-aspartate is stable (Figure S4b). Presumably, this is a direct consequence of the fact that nucleophilic attack by the α-amino group of 16 is hindered by the steric strain imposed by the formation of a β-lactam.
Characterization of Reaction Products with L-Glutamine.
It was previously demonstrated that Cj1418 catalyzes the formation of AMP, Pi, and L-glutamine phosphate from ATP, water, and L-glutamine.8 The proposed mechanism of action (Scheme 2) assumes that the β-phosphoryl group of ATP is ultimately found as L-glutamine phosphate, whereas the γ-phosphoryl group is found as Pi. However, the previous characterization of the reaction products did not differentiate between the β- and γ–phosphoryl groups in the two reaction products.8 To unambiguously determine the origin of the two products from ATP, the reaction was first conducted in 100% [16O]-H2O and then in 50% [18O]-H2O. For the product Pi, there is a single phosphorus resonance when the reaction was conducted in unlabeled water (Figure 1a) but two phosphorus resonances, separated by ~0.023 ppm from one another, when the reaction was conducted in a mixture of 16O- and 18O-H2O (Figure 1b). The up-field resonance is for the phosphate with a single 18O-label and the down-field resonance is for phosphate without an 18O-label.26 The 31P NMR spectrum of the reaction products clearly indicates that only the Pi product was labeled with 18O.
Figure 1.
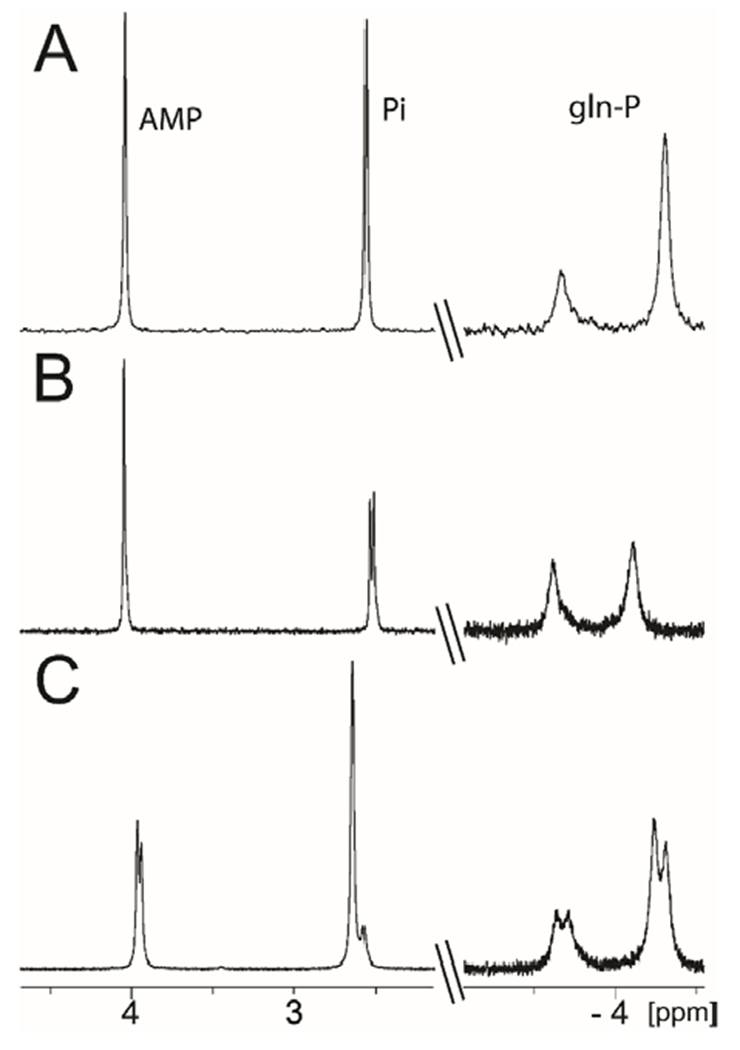
31P NMR spectra of the products in the reaction catalyzed by Cj1418. (A) 31P NMR spectrum after incubation of Cj1418 (5 µM) with L-glutamine (5 mM) and MgATP (5 mM) at pH 8.0. Resonances at −3.66 and −4.31 ppm are from L-glutamine phosphate, 2.43 ppm is from phosphate and 3.91 ppm is from AMP. (B) Same reaction conditions as for spectrum A, but the reaction was conducted in 50% [18O]-H2O. The 31P NMR resonance for phosphate is shifted up-field by 0.023 ppm due to the incorporation of a single atom of 18O in the phosphate product. (C) Same reaction conditions as for spectrum A, except that unlabeled ATP was mixed with 50% of β-[18O4]-ATP (13). The 31P NMR spectrum for L-glutamine phosphate exhibits four resonances. There are two resonances each for the syn- and anti-conformers that are separated by 0.072 ppm due to the incorporation of 3 atoms of 18O. In the spectra, the 31P NMR resonance for AMP appears at 3.91 ppm. In spectrum C the 31P NMR resonance for AMP exhibits two resonance separated by 0.024 ppm due to the presence of a single atom of 18O from the enzymatic cleavage of the bond between the β-P and α/β-bridging oxygen in the labeled ATP used in this reaction.
In a second experiment, the Cj1418-catalyzed reaction was conducted with L-glutamine and a 50/50 mixture of β-[18O4]-ATP (13) and unlabeled ATP. In the 31P NMR spectrum of the reaction products, a single resonance is observed for phosphate, and four resonances are observed for the L-glutamine phosphate product (Figure 1c). One pair of resonances is for the unlabeled product (syn- and anti-conformations) and the other for the product labeled with 18O. The separation in the chemical shifts for the two pairs of resonances is 0.072 ppm, consistent with the incorporation of three 18O-atoms, as expected from the labeled ATP used in this experiment.26 These two experiments clearly indicate that the β-phosphoryl group is ultimately found in L-glutamine phosphate and that the γ-phosphoryl group is ultimately found as phosphate. This confirms part of the mechanism proposed in Scheme 2.
Importance of His-737 as Enzyme Nucleophile.
The closest homologues of Cj1418 include PEP synthase, pyruvate phosphate dikinase, and rifampin phosphotransferase.13,15,16,18 Each of these enzymes has been structurally characterized and shown to be pyrophosphorylated on a conserved histidine residue that originates from either a central or C-terminal domain of the protein.16,18 The multi-protein sequence alignment of Cj1418 (residues 694–767) with the phospho-histidine domains of these proteins (Figure S6) identifies His-737 as the residue that is likely to be pyrophosphorylated in the reaction mechanism of Cj1418 (Scheme 2). This residue was mutated to asparagine (H737N) and the mutant assayed for its ability to phosphorylate glutamine, however, no activity was detected (kcat ≤ .001 s−1). This result is consistent with His-737 as being the nucleophile that attacks the β-phosphoryl group of ATP with formation of a pyrophosphorylated enzyme intermediate.
Detection of Pyrophosphorylated Intermediate.
In the proposed reaction mechanism for Cj1418, the first step is initiated by the nucleophilic attack of the enzyme (likely His-737) on the β-phosphoryl group of ATP to generate a covalent pyrophosphorylated intermediate and AMP (Scheme 2a). The significance of this first step was addressed by measurement of the positional isotope exchange (PIX) reaction using β-[18O4]-ATP (13) in the absence of added L-glutamine.19,20 To observe a PIX reaction with Cj1418, the formation of the Enz-X-PPi intermediate must be reversible and the α-phosphoryl group of AMP bound in the active site must be free to rotate. The positional exchange of the 18O label is outlined in Scheme 6. As the reaction proceeds, the 18O that originally occupies the α/β-bridge position in the labeled ATP (13) will equilibrate with the 16O in the two α-nonbridge positions to form 19 and 20. At equilibrium, 1/3 of the 18O label will remain in the α/β-bridge position and 2/3 will be in the two α-nonbridge positions.
Scheme 6:
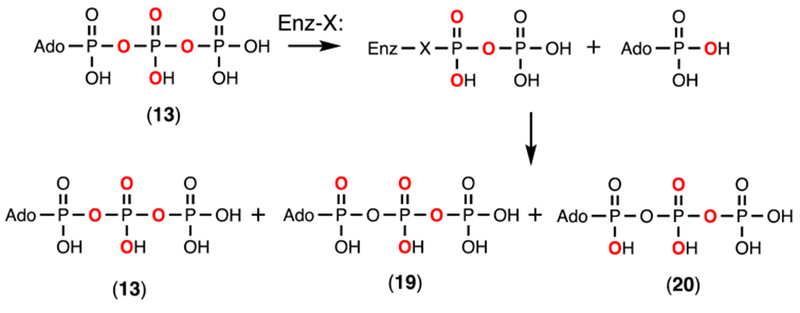
Positional isotope exchange reaction with Cj1418 and β-[18O4]-ATP.
The PIX reaction was followed by 31P NMR spectroscopy since the incorporation of 18O into ATP results in a perturbation in the chemical shift for phosphorus and the magnitude of the shift is dependent on whether the 18-oxygen occupies a bridge or non-bridge position within the polyphosphate moiety of ATP.26 It has been previously shown that 18O in the α/β-bridge position of ATP will cause a +0.017 ppm up-field chemical shift perturbation for the α- and β-phosphoryl groups.26 In the α-nonbridge position there will be a +0.028 ppm up-field shift perturbation for a single 18-oxygen.26 Thus, as the α/β-bridge oxygen in the β-[18O4]-ATP scrambles, there will be a change in the chemical shift of the β-phosphate of approximately −0.017 ppm and an increase in the chemical shift for the α-phosphate by ~0.011 ppm (0.028–0.017 ppm).
The PIX reaction was initiated by incubation of 5 mM β-[18O4]-ATP (13) and 10 μM Cj1418. Aliquots were removed, the reaction quenched with EDTA, and the products analyzed by 31P NMR spectroscopy. The 31P NMR spectra of the α- and β-phosphoryl groups of the labeled ATP, before the addition of the Cj1418 and after 8 h, are presented in Figure 2. For the β-phosphoryl group, the 31P NMR spectrum (Figure 2c) indicates that in the original β-[18O4]-ATP, 86% is labeled with 2 nonbridge and 2 bridge 18O atoms (denoted as 2,2), 7% contain 2 nonbridge and 1 bridge 18O atoms (2,1) and 7% contain 1 nonbridge and 2 nonbridge (1,2) 18O atoms. For the α-phosphoryl group, the 31P NMR spectrum (Figure 2a) indicates that in the original β-[18O4]-ATP, 96% contains 1 bridge 18O atoms (0,1) and 4% contain no label (0,0). The average 18O content of the 4 oxygen atoms attached to the β-phosphoryl group is 96%. After 8 h, the fraction of the ATP with 2 nonbridge and 2 bridge 18O atoms at the β-phosphoryl group (2,2), relative to that fraction of ATP with 2 nonbridge and 1 bridge 18O (2,1), has been reduced from 92% to 58% (Figure 2d). For the α-phosphoryl group, the fraction of ATP with 1 bridge 18O atom (0,1), relative to that with 1 nonbridge 18O atom has decreased from 100% to 38% (Figure 2b). The rate of the PIX reaction (vPIX) was calculated using Eqn. 2 and the data for additional time points are collected in Table 2. The average ratio of the PIX rate to the rate for the loss of ATP (vPIX/vchem) is 1.6 ± 0.3. In the presence of 5 mM AMP, there was no measureable increase in the rate of the PIX reaction under these conditions. The observation of the positional isotope exchange reaction is consistent with the formation of a pyrophosphorylated enzyme in the absence of added L-glutamine.
Figure 2.
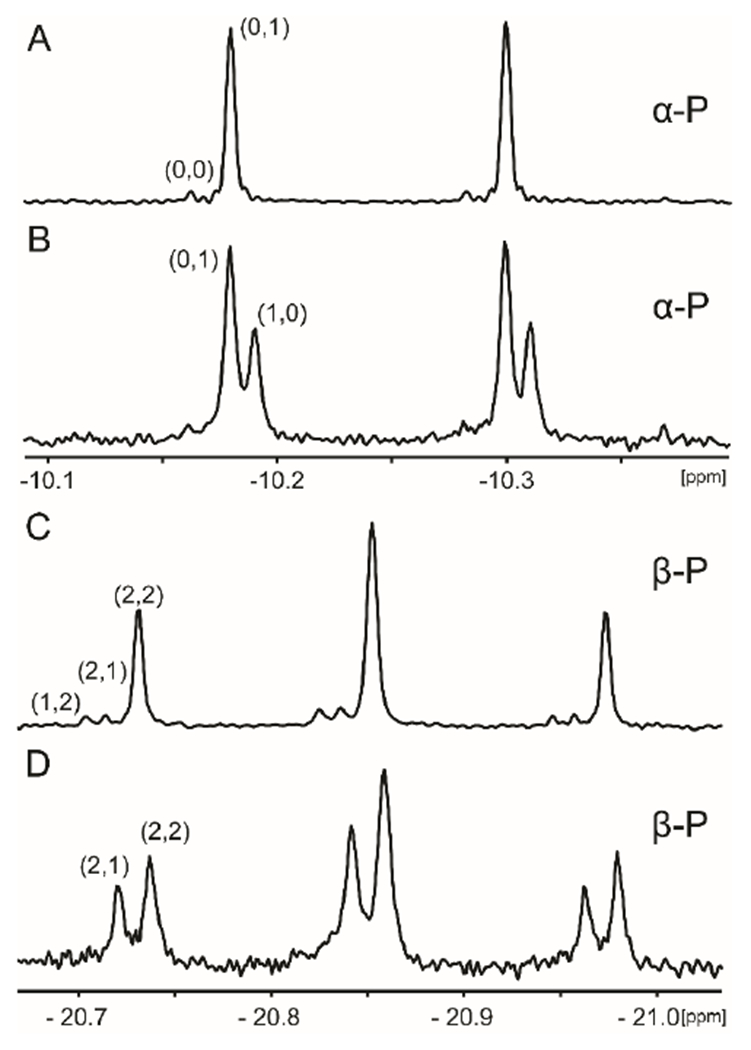
31P NMR spectra of 18O-labeled ATP before and after the addition of Cj1418. (A) Spectrum of the α-phosphoryl group of [18O4]-ATP (13) before the addition of Cj1418. (B) Spectrum of the α-phosphoryl group of the ATP after 8 h of incubation with Cj1418. (C) Spectrum of the β-phosphoryl group of [18O4]-ATP (13) before the addition of Cj1418. D. β-phosphoryl group of the ATP after incubation with Cj1418 for 8 h. Additional details are provided in the text. The first number in parentheses indicates the number of non-bridging 18O atoms attached to respective phosphoryl group in ATP and the second indicates the number of bridging 18O atoms.
Table 2:
Positional isotope exchange for the α- and β-phosphoryl groups with β-[18O4]-ATP.
| Time (h) |
% α-P (0,1) |
% α-P (1,0) | % β-P (2,2) |
% β-P (2,1) |
F | ATP (mM) |
X | ʋex (mM hr−1) |
ʋchem (mM hr−1) |
ʋex/ʋChem |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 100 | 0 | 92 | 8 | 5.00 | |||||
| 1 | 86 | 14 | 82 | 18 | 0.19 | 4.41 | 0.11 | 0.99 | 0.59 | 1.68 |
| 4 | 75 | 25 | 68 | 32 | 0.39 | 3.91 | 0.21 | 0.55 | 0.27 | 2.04 |
| 8 | 62 | 38 | 58 | 42 | 0.57 | 3.03 | 0.39 | 0.41 | 0.25 | 1.64 |
| 12 | 51 | 49 | 53 | 47 | 0.69 | 2.21 | 0.55 | 0.33 | 0.23 | 1.43 |
| 15 | 48 | 52 | 48 | 52 | 0.76 | 1.66 | 0.66 | 0.29 | 0.22 | 1.32 |
Detection of Phosphorylated Intermediate.
In Scheme 2 we have proposed that the pyrophosphorylated enzyme (Enz-X-Pβ-Pγ) is hydrolyzed to generate a phosphorylated enzyme intermediate (Enz-X-Pβ) and in the last step this intermediate is utilized to phosphorylate L-glutamine. If this last step is reversible then a molecular isotope exchange reaction (MIX) can be used to provide experimental support for the formation of Enz-X-Pβ. This exchange reaction is illustrated in Scheme 7. In the MIX experiment, Cj1418 was incubated with 10 mM [15N-amide]-L-glutamine (21) and 10 mM of unlabeled L-glutamine phosphate (2) in the absence of added ATP. The products of the reaction mixture were examined by both 31P and 15N NMR spectroscopy after a period of 12 h. In Figure 3a is the 31P NMR spectrum of the L-glutamine-P (2) prior to the addition of Cj1418 and the [15N-amide]-L-glutamine (21). Two resonances are observed at −3.56 and −3.91 ppm, representing the syn and anti-conformations of the product. After the addition of Cj1418 and [15N-amide]-L-glutamine, four new resonances are observed, representing L-glutamine-P where the two phosphorus resonances are now coupled to 15N (compound 22). In the corresponding 15N NMR spectra (Figure 3c), a single 15N-resonance is observed at 111.92 ppm for the amide nitrogen of the labeled L-glutamine (21). After incubation with L-glutamine-P and Cj1418, four new resonances are observed from 146.06 and 142.18 ppm. These resonances represent the syn and anti-conformations of the L-glutamine-P products and coupling with 31P of 19 Hz.
Scheme 7:
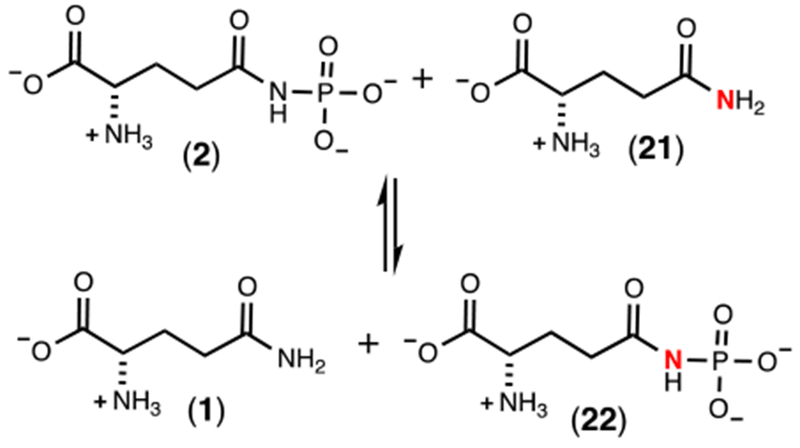
Molecular isotope exchange reaction for Cj1418.
Figure 3.
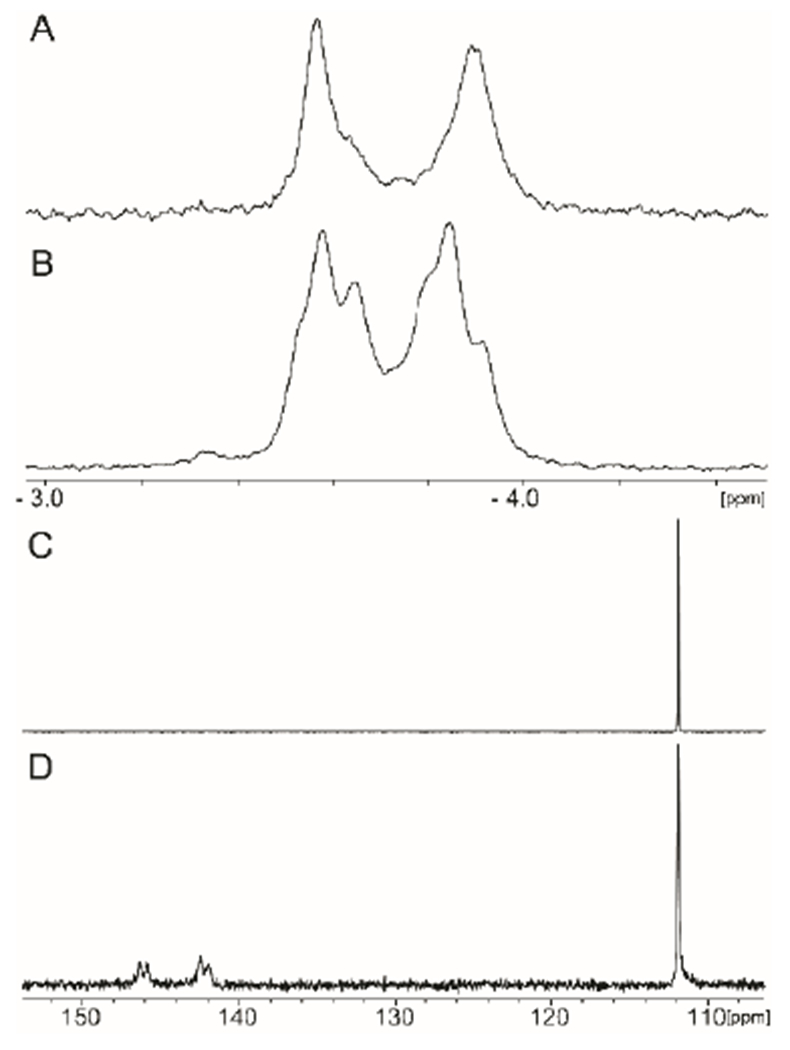
31P and 15N NMR spectra for the MIX reaction with Cj1418. (A) 31P NMR spectrum of L-glutamine phosphate. (B) 31P NMR spectrum after 10 μM Cj1418 was incubated with 10 mM L-glutamine phosphate (2) and 10 mM [15N-amide]-L-glutamine (21) for 12 h. (C) 15N NMR spectrum of [15N-amide]-L-glutamine (21). (D) 15N NMR spectrum after 10 µM Cj1418 was incubated with 20 mM [15N-amide]-L-glutamine and 20 mM L-glutamine phosphate (2) for 12 h.
DISCUSSION
Mechanism of Action.
The proposed mechanism of action for L-glutamine kinase is presented in Scheme 2. In this mechanism a nucleophile in the active site first attacks the β-phosphoryl group of ATP to form a pyrophosphorylated enzyme intermediate (Enz-X-Pβ-Pγ) and AMP. The pyrophosphorylated intermediate is subsequently hydrolyzed to generate a phosphorylated intermediate (Enz-X-Pβ) and Pi. In the last step the phosphorylated intermediate is used to phosphorylate L-glutamine on the amide nitrogen. Utilization of amino acid sequence alignments with the closest functional homologues to Cj1418 and site directed mutagenesis experiments, we have demonstrated that His-737 is the most likely residue in the active site of this enzyme to function as the primary nucleophile.
In the proposed mechanism, the γ-phosphoryl group of ATP is converted to phosphate and the β-phosphoryl group is ultimately used to phosphorylate L-glutamine. An alternative mechanism could have been written with the opposite outcome for these two phosphoryl groups of ATP. However, we have now demonstrated using β-[18O4]-ATP that the β-phosphoryl group is exclusively utilized to phosphorylate L-glutamine. This result was confirmed by conducting the Cj1418-catalyzed reaction in [18O]-H2O, where the only labeled product was inorganic phosphate.
Positional isotope exchange (PIX) and molecular isotope exchange (MIX) experiments were used to further support the formation of the pyrophosphorylated (Enz-X-Pβ-Pγ) and phosphorylated enzyme intermediates (Enz-X-Pβ) in the overall reaction mechanism. In the PIX experiment, β-[18O4]-ATP was incubated with the enzyme in the absence of L-glutamine in an attempt to observe the migration of the α/β-bridge oxygen to the α-nonbridge positions in ATP (Scheme 6). As conducted, the PIX reaction can only occur if the enzyme is pyrophosphorylated by ATP in the absence of L-glutamine and this reaction must be reversible. In essence, the experiment measures the partitioning of the [Enz-X-Pβ-Pγ • AMP] complex. When this complex reverts back to Enz-X and ATP, a PIX reaction will be observed if the α-phosphoryl group of AMP is able to rotate in the active site. The PIX rate will then be governed by the rate at which the ATP can be reformed and dissociate from the active site into solution.
The partitioning forward is potentially governed by two related events. The AMP may dissociate from the active site or the pyrophosphorylated enzyme may be hydrolyzed to the phosphorylated enzyme. The dissociation of AMP is essentially irreversible since the initial concentration of AMP is equivalent to the enzyme concentration used in this experiment (10 μM). We attempted to enhance the PIX rate by adding unlabeled AMP to the reaction mixture.20 If the partitioning forward was governed by AMP dissociation, then increasing the concentration of AMP would enhance the steady-state concentration of the [Enz-X-Pβ-Pα • AMP] complex and the overall PIX rate would increase. However, the addition of AMP had no effect on the PIX rate and no ATP was directly formed from the added unlabeled AMP. This result indicates that the partitioning forward is actually governed by the rate at which the [Enz-X-Pβ-Pγ • AMP] complex is hydrolyzed to [Enz-X-Pβ • Pγ]. Thus, the experimentally determined partitioning ratio (vPIX/vchem) of 1.6 for the [Enz-X-Pβ-Pα • AMP] complex indicates that ATP is reformed and dissociates from the active site 1.6 times faster than this complex is hydrolyzed to irreversibly form [Enz-X-PβPi • AMP].
It should also be noted that the average rate constant for the PIX reaction (obtained from the average value for vex in Table 2) is relatively small at ~54 h−1 (0.54 mM h−1/0.01 mM enzyme). Since the dissociation of AMP and the hydrolysis of the pyrophosphorylated enzyme intermediate are essentially irreversible, the enzyme must somehow be recycled back to free enzyme. This can only occur via the hydrolysis of the [Enz-X-Pβ] complex to [Enz-X]. The rate constant for the net loss of ATP in the absence of L-glutamine (obtained from the average value of vchem in Table 2) is 31 h−1 (0.31 mM h−1/0.01 mM enzyme). The kcat for the overall reaction (2.5 s−1) in the presence of L-glutamine is 2.9 × 104 fold faster than this value. This clearly indicates that L-glutamine is much better than water in reacting with the [Enz-X-Pβ] intermediate. PIX experiments have previously been used to characterize the pyrophosphorylated enzyme intermediate formed in the reaction catalyzed by pyruvate phosphate dikinase.20
Experimental support for the [Enz-X-Pβ] intermediate was obtained with the MIX experiment where free enzyme (Enz-X) was phosphorylated with L-glutamine phosphate in the absence of any added nucleotide (Scheme 7). The phosphorylated enzyme intermediate was subsequently captured by the 15N-labeled L-glutamine to generate the 15N-labeled L-glutamine phosphate (22). This exchange reaction can only occur with the formation of the [Enz-X-P] intermediate.
Substrate Specificity.
We have identified four additional substrates for Cj1418 in addition to L-glutamine. These new substrates include the hydroxamate derivatives of L-glutamate (7) and L-aspartate (9), the hydrazide of L-glutamate (8), and D-glutamine (6). Other compounds were tested as alternative substrates such as L-glutamate and L-asparagine, however neither of these compounds exhibited any detectable activity. L-Glutamine derivatives with either the α-amino or α-carboxy groups removed were not substrates, however, D-glutamine was shown to be a substrate and thus the stereochemistry at the α-carbon is not vital.
The second best substrate for Cj1418 is γ-glutamyl hydroxamate (7) and formation of an unstable O-phosphorylated derivative (15). Previously, compound 15 has been shown to be an inhibitor of γ-L-glutamyl-L-cysteine synthetase where a highly reactive isocyanate intermediate was proposed via a Lossen rearrangement.27 With Cj1418 we did not obtain any evidence for the formation of an isocyanate derivative from 7. However, the 13C and 31P NMR spectra clearly indicate the formation of pyroglutamic acid (16) and the hydroxyl amine O-phosphate ester (14). In contrast, the phosphorylated derivative of β-aspartyl hydroxamate (18) appears to be relatively stable under the reaction conditions. The hydrazide derivative of L-glutamate (9) is also a substrate that produces a relatively unstable phosphorylated product (18). The final products were determined to be phosphate and L-pyroglutamate (16). The formation of phosphate, presumably arises from the nonenzymatic hydrolysis of the hydrazide derivative of phosphate. However, this intermediate was not observed directly in the 31P NMR spectrum of the reaction mixture.
Conclusions.
Here we have determined the substrate profile for L-glutamine kinase, an enzyme that functions in the formation of the O-methyl phosphoramidate modification in the capsular polysaccharide in C. jejuni. We have proposed a chemical reaction mechanism for the phosphorylation of L-glutamine and have provided kinetic evidence for the formation of pyrophosphorylated and phosphorylated enzyme intermediates in this transformation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by grants from the National Institutes of Health (GM 122825).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: doi.org/10.1021/acs.biochem.8b00811
The authors declare no competing financial interest.
REFERENCES
- [1].Young KT, Davis LM, and Dirita VJ (2007) Campylobacter jejuni: molecular biology and pathogenesis, Nat. Rev. Microbiol. 5, 665–679. [DOI] [PubMed] [Google Scholar]
- [2].Roberts IS (1996) The biochemistry and genetics of capsular polysaccharide production in bacteria, Annu. Rev. Microbiol. 50, 285–315. [DOI] [PubMed] [Google Scholar]
- [3].Karlyshev AV, Champion OL, Churcher C, Brisson J-R, Jarrell HC, Gilbert M, Brochu D, St Michael F, Li J, Wakarchuk WW, Goodhead I, Sanders M, Stevens K, White B, Parkhill J, Wren BW, and Szymanski CM (2005) Analysis of Campylobacter jejuni capsular loci reveals multiple mechanisms for the generation of structural diversity and the ability to form complex heptoses, Mol. Microbiol. 55, 90–103. [DOI] [PubMed] [Google Scholar]
- [4].St. MF, S. C. M, Jianjun L, H. CK, Huan KN, Suzon L, W. WW, Jean-Robert B, and A. MM (2002) The structures of the lipooligosaccharide and capsule polysaccharide of Campylobacter jejuni genome sequenced strain NCTC 11168, Eur. J. Biochem. 269, 5119–5136. [DOI] [PubMed] [Google Scholar]
- [5].McNally DJ, Lamoureux MP, Karlyshev AV, Fiori LM, Li J, Thacker G, Coleman RA, Khieu NH, Wren BW, Brisson J-R, Jarrell HC, and Szymanski CM (2007) Commonality and Biosynthesis of the O-Methyl Phosphoramidate Capsule Modification in Campylobacter jejuni, J. Biol. Chem. 282, 28566–28576. [DOI] [PubMed] [Google Scholar]
- [6].Karlyshev AV, Linton D, Gregson NA, Lastovica AJ, and Wren BW (2000) Genetic and biochemical evidence of a Campylobacter jejuni capsular polysaccharide that accounts for Penner serotype specificity, Mol. Microbiol. 35, 529–541. [DOI] [PubMed] [Google Scholar]
- [7].Taylor ZW, Brown HA, Holden HM, and Raushel FM (2017) Biosynthesis of Nucleoside Diphosphoramidates in Campylobacter jejuni, Biochemistry 56, 6079–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Taylor ZW, Brown HA, Narindoshvili T, Wenzel CQ, Szymanski CM, Holden HM, and Raushel FM (2017) Discovery of a Glutamine Kinase Required for the Biosynthesis of the O-Methyl Phosphoramidate Modifications Found in the Capsular Polysaccharides of Campylobacter jejuni, J. Am. Chem. Soc. 139, 9463–9466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Taylor ZW, and Raushel FM (2018) Cytidine Diphosphoramidate Kinase: An Enzyme Required for the Biosynthesis of the O-Methyl Phosphoramidate Modification in the Capsular Polysaccharides of Campylobacter jejuni, Biochemistry 57, 2238–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Morrison JF, Griffiths DE, and Ennor AH (1957) The purification and properties of arginine phosphokinase, Biochem. J. 65, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ennor AH, Rosenberg H, and Armstrong MD (1955) Specificity of Creatine Phosphokinase, Nature 175, 120. [DOI] [PubMed] [Google Scholar]
- [12].Roush RF, Nolan EM, Löhr F, and Walsh CT (2008) Maturation of an Escherichia coli Ribosomal Peptide Antibiotic by ATP-Consuming N−P Bond Formation in Microcin C7, J. Am. Chem. Soc. 130, 3603–3609. [DOI] [PubMed] [Google Scholar]
- [13].Cooper RA, and Kornberg HL (1967) The mechanism of the phosphoenolpyruvate synthase reaction, Biochimica et Biophysica Acta (BBA) - General Subjects 141, 211–213. [DOI] [PubMed] [Google Scholar]
- [14].Evans HJ, and Wood HG (1968) The mechanism of the pyruvate, phosphate dikinase reaction, Proceedings of the National Academy of Sciences 61, 1448–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stogios PJ, Cox G, Spanogiannopoulos P, Pillon MC, Waglechner N, Skarina T, Koteva K, Guarné A, Savchenko A, and Wright GD (2016) Rifampin phosphotransferase is an unusual antibiotic resistance kinase, Nat. Commun. 7, 11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qi X, Lin W, Ma M, Wang C, He Y, He N, Gao J, Zhou H, Xiao Y, Wang Y, and Zhang P (2016) Structural basis of rifampin inactivation by rifampin phosphotransferase, Proceedings of the National Academy of Sciences 113, 3803–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang H-Y, Dosztányi Z, El-Gebali S, Fraser M, Gough J, Haft D, Holliday GL, Huang H, Huang X, Letunic I, Lopez R, Lu S, Marchler-Bauer A, Mi H, Mistry J, Natale DA, Necci M, Nuka G, Orengo CA, Park Y, Pesseat S, Piovesan D, Potter SC, Rawlings ND, Redaschi N, Richardson L, Rivoire C, Sangrador-Vegas A, Sigrist C, Sillitoe I, Smithers B, Squizzato S, Sutton G, Thanki N, Thomas PD, Tosatto Silvio C. E., Wu CH, Xenarios I, Yeh L-S, Young S-Y, and Mitchell AL (2017) InterPro in 2017—beyond protein family and domain annotations, Nucleic Acids Res. 45, D190–D199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lim K, Read RJ, Chen CCH, Tempczyk A, Wei M, Ye D, Wu C, Dunaway-Mariano D, and Herzberg O (2007) Swiveling Domain Mechanism in Pyruvate Phosphate Dikinase, Biochemistry 46, 14845–14853. [DOI] [PubMed] [Google Scholar]
- [19].Rose IA (1979) Positional Isotope Exchange Studies of Enzyme Mechanisms, In Adv. Enzymol. 50, 361. [DOI] [PubMed] [Google Scholar]
- [20].Wang HC, Ciskanik L, Dunaway-Mariano D, Von der Saal W, and Villafranca JJ (1988) Investigations of the partial reactions catalyzed by pyruvate phosphate dikinase, Biochemistry 27, 625–633. [DOI] [PubMed] [Google Scholar]
- [21].Melby ES, Soldat DJ, and Barak P (2011) Synthesis and Detection of Oxygen-18 Labeled Phosphate, PLoS One 6, e18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wehrli W, Verheyden D, and Moffatt J (1965) Dismutation Reactions of Nucleoside Polyphosphates. II. Specific Chemical Syntheses of α-, β-, and γ-P32-Nucleoside 5’-Triphosphates1, J. Am. Chem. Soc. 87, 2265–2277. [DOI] [PubMed] [Google Scholar]
- [23].Midelfort CF, and Rose IA (1976) A stereochemical method for detection of ATP terminal phosphate transfer in enzymatic reactions. Glutamine synthetase, J. Biol. Chem. 251, 5881–5887. [PubMed] [Google Scholar]
- [24].Cottam GL, Kupiecki FP, and Coon MJ (1968) A Study of the Mechanism of O-Phosphorylhydroxylamine Synthesis Catalyzed by Pyruvate Kinase, J. Biol. Chem. 243, 1630–1637. [PubMed] [Google Scholar]
- [25].Litwin S, and Wimmer MJ (1979) Correction of scrambling rate calculation for loss of substrate, J. Biol. Chem. 254, 1859. [PubMed] [Google Scholar]
- [26].Cohn M, and Hu A (1978) Isotopic ((18)O) shift in (31)P nuclear magnetic resonance applied to a study of enzyme-catalyzed phosphate—phosphate exchange and phosphate (oxygen)—water exchange reactions, Proc. Natl. Acad. Sci. U. S. A. 75, 200–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Katoh M, Hiratake J, and Oda J. i. (1998) ATP-Dependent Inactivation of Escherichia coli γ-Glutamylcysteine Synthetase by L-Glutamic Acid γ-Monohydroxamate, Biosci., Biotechnol., Biochem. 62, 1455–1457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.