

Abstract
Introduction:
Five new agents have been shown to prolong survival in patients with metastatic castration-resistant prostate cancer, including two targeting androgen receptor signaling (abiraterone acetate plus prednisone; enzalutamide). Recognition that these tumors remain driven by androgen receptor signaling has prompted clinical evaluation of these agents at earlier states in the prostate cancer disease continuum, along with the continued development of new agents targeting this pathway.
Areas covered:
This article focuses on apalutamide, a next-generation nonsteroidal antiandrogen, with current literature queried in PubMed/Medline. A narrative review strategy describes studies from engineering of the compound through to a 5-year outlook.
Expert commentary:
In the phase III SPARTAN study, apalutamide significantly improved metastasis-free survival in patients with nonmetastatic castration-resistant prostate cancer—the first treatment approved by the US Food and Drug Administration for this indication. Phase III studies are underway to determine the clinical benefit of apalutamide in other disease states. Given the multiplicity of prostate cancer treatment options now available, there is a need to maximize individual patient benefit through the development and validation of predictive biomarkers of sensitivity to drugs that can be used in real time to determine the optimal sequence and combinations of treatments for patients in need.
Keywords: androgen receptor inhibitors, apalutamide, efficacy, metastatic, nonmetastatic, pharmacodynamics, pharmacokinetics, prostate cancer, safety, tolerability
1.0. Introduction
1.1. Overview of the market
Prostate cancer is the second most common cancer in men worldwide, accounting for 15% of all malignancies diagnosed in men [1]. Incidence rates vary by more than 25-fold worldwide, with approximately 70% of all cases occurring in developed regions. The highest age-standardized rates (per 100,000 men) are found in Australia/New Zealand (111.6), North America (97.2), Western Europe (85.8) and Northern Europe (85.0), whereas the lowest rates are found in southern and eastern regions of Asia (4.5–10.5) [1]. Other regions with significant prostate cancer rates include the Caribbean (79.8), Southern Africa (61.8), and South America (60.1). In the United States, prostate cancer was diagnosed in an estimated 161,360 men in 2017 and caused 26,730 deaths, accounting for 9.6% and 4.4% of all new cancer cases and deaths, respectively [2]. The prevalence of prostate cancer in the United States is almost 3.1 million [2].
There has been a slight increase in the number of men aged 50–69 years diagnosed with distant stage prostate cancer making the availability of more effective therapies for use in earlier advanced disease states critical to improve outcomes [3].
The need for treatment and choice of treatment is based on a patient’s prognosis and on the presence or absence of disease-related symptoms along the disease continuum [4] (Figure 1). According to the dynamic progression model, prostate cancer can be partitioned into clinical states based on the natural disease and treatment history [5]. The clinical states represent milestones in the disease, which range from newly diagnosed localized, locally advanced, and metastatic disease in the noncastrate state, to biochemical failure after hormonal therapy, and nonmetastatic (nmCRPC) and metastatic castration-resistant prostate cancer (mCRPC) [4,5].
Figure 1. Prostate cancer clinical states model (Prostate Cancer Clinical Trials Working Group 3 framework).
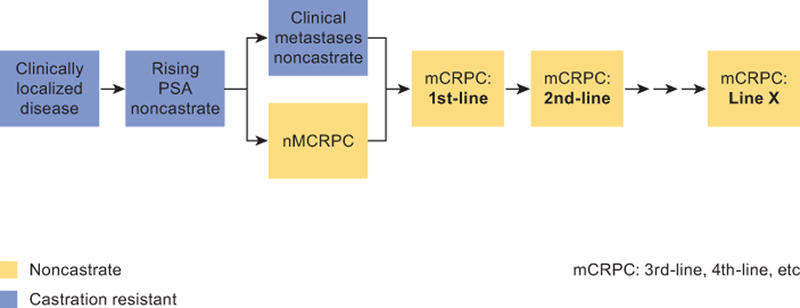
The prostate cancer clinical states model for patient treatment and drug development as developed by Prostate Cancer Clinical Trials Working Group 3.
mCRPC: metastatic castration-resistant prostate cancer; nmCRPC, nonmetastatic castration-resistant prostate cancer; PSA: prostate-specific antigen.
Source: Scher et al., 2016[44]. Reprinted with permission. © (2016) American Society of Clinical Oncology. All rights reserved.
1.2. Unmet needs associated with currently available therapies
In 2004, docetaxel plus prednisone became the first treatment shown to significantly improve overall survival (OS) of men with mCRPC [6]. Subsequently, five new agents, each with a different mechanism of action, were shown to significantly improve OS in patients with mCRPC, including cabazitaxel (a tubulin-binding taxane) [7], sipuleucel-T (an autologous active cellular immunotherapy) [8], abiraterone acetate (a cytochrome P450 c17 inhibitor that blocks androgen biosynthesis) plus prednisone [9–12], enzalutamide (an androgen receptor [AR] signaling inhibitor) [13,14], and radium-223 (an alpha emitter that targets bone metastases) [15]. The optimal sequence for use of these agents remains to be clarified.
The survival benefit seen with enzalutamide and abiraterone acetate plus prednisone in mCRPC, independent of prior exposure to docetaxel, underscores the critical and continued role of the AR in advanced prostate cancer, and has led to their investigation in earlier clinical states. It also supports the continued role of the AR as a therapeutic target for the design of new agents, including next-generation antiandrogens. In the STAMPEDE study, abiraterone acetate plus prednisone added to androgen deprivation therapy (ADT) was associated with significantly higher 3-year OS (83% vs 76%; p < 0.001) and failure-free survival (75% vs 45%; p < 0.001) compared with ADT alone in patients with locally advanced or metastatic prostate cancer not treated previously with hormone therapy [16]. Similarly, in the LATITUDE study, abiraterone acetate plus prednisone added to ADT significantly prolonged 3-year OS compared with ADT alone (66% vs 49%; p < 0.001) in patients with high-risk metastatic hormone-sensitive prostate cancer (mHSPC) [17]. The findings of the latter study led to the recent European Commission and US Food and Drug Administration (FDA) approvals of abiraterone plus prednisone for treatment of high-risk mHSPC [18].
A continued area of interest is whether more complete androgen signaling inhibition, explored first in the 1980’s, would be more beneficial than ADT alone for patients with a more favorable prognosis than those with high-risk mHSPC. This includes patients with high-risk or very high–risk localized or locally advanced disease (i.e., Gleason scores ≥8, clinical stage of cT2c or greater, baseline prostate-specific antigen (PSA) ≥20 ng/ml or regional node involvement), for whom metastatic progression and prostate cancer–related deaths still occur frequently after ADT in combination with radiation therapy, the current standard of care [19]. More effective treatments for these patients represent an unmet need [20]. Support for the evaluation of either enzalutamide or abiraterone acetate plus prednisone in combination with a luteinizing hormone–releasing hormone agonist (LHRHa) in this setting is reinforced by the finding of pathologic complete responses and minimal residual disease in the prostates of men treated in the neoadjuvant setting [21,22]. Predictive biomarkers to identify patients likely to benefit from these drugs are also needed to avoid exposure to ineffective treatment.
Although some patients with mCRPC experience durable benefit from these agents, a proportion have tumors that are resistant de novo, and virtually all tumors acquire resistance over time [23,24]. Whether combinations of these agents, or new agents targeting known mechanisms of resistance such as ARv7 splice variants, AR ligand specific mutations or AR overexpression can delay the emergence of clinical resistance or restore sensitivity once it has developed remains to be elucidated.
1.3. Additional AR-targeted compounds in clinical development
1.3.1. Enzalutamide
Enzalutamide is a selective AR inhibitor that has five- to eight-fold higher binding affinity for the AR compared with the antiandrogen bicalutamide [25,26]. It was identified in a cell-based screen in prostate cancer cell lines with overexpressed AR. Enzalutamide is thought to induce a conformational change of the AR distinct from that caused by bicalutamide, thereby inhibiting AR translocation to the nucleus, recruitment of AR cofactors and AR binding to DNA [26]. It has a half-life of 6 days, and crosses the blood–brain barrier. In the PROSPER study of patients with nmCRPC who continued on ADT, enzalutamide reduced the relative risk of developing mCRPC by 71% compared with ADT alone, prolonging metastasis-free survival (MFS) from 14.7 to 36.6 months (p < 0.0001) [27]. Several randomized controlled phase III trials are ongoing, including EMBARK (), ARCHES (), and PEACE III (). In patients (N = 1860) with nonmetastatic prostate cancer (biochemical failure after local therapy), the primary endpoint of MFS is being examined in groups randomized to enzalutamide, leuprolide or both, with a primary completion date of March 2021 (EMBARK) [28]. In the mHSPC setting, ARCHES is designed to examine the primary endpoint of radiographic progression-free survival (rPFS) in 1150 patients randomized to enzalutamide plus ADT versus ADT alone, with a primary completion date of April 2020 [29]. In patients (N = 560) with asymptomatic or mildly symptomatic mCRPC (with ≥2 bone metastases and no visceral metastases), a primary endpoint of rPFS is being examined in PEACE III in arms randomized to enzalutamide plus radium-223 or enzalutamide alone, with a primary completion date of November 2019 [30]. Finally, in the Alliance trial of patients with mCRPC (N = 1311), complete androgen annihilation is being tested with combination use of enzalutamide and abiraterone acetate plus prednisone versus enzalutamide alone, with data pending for the primary endpoint of overall survival [31]. The results of these phase III trials will help to better define the use of enzalutamide within the prostate cancer disease continuum.
1.3.2. Darolutamide
Darolutamide (ODM-201; BAY1841788) is an AR inhibitor [32,33]. Darolutamide has an eight-fold greater affinity for the AR compared with enzalutamide and, notably, remains antagonistic against mutant AR forms, including the F877L mutation that confers resistance to enzalutamide. The main metabolite (ORM-15341) is pharmacologically active, with a profile similar to that of the parent compound; unlike enzalutamide, neither parent nor metabolite crosses the blood–brain barrier [32,33]. The phase I/II ARADES study provided initial evidence that darolutamide is active in patients with progressive mCRPC despite ongoing ADT: the 12-week PSA response rates (defined by ≥50% reductions in serum PSA from baseline) were 29%, 33% and 33% at daily doses of 200, 400 and 1400 mg, respectively [32]. PSA response rates were highest for patients who were naïve to both chemotherapy and abiraterone acetate plus prednisone. In the study, darolutamide was deemed to have a favorable safety profile; the most common treatment-emergent adverse events (TEAEs) among patients receiving those three dose levels were fatigue or asthenia (12%). Ongoing phase III trials include the ARAMIS study (), which is comparing darolutamide versus placebo in 1488 patients with high-risk nmCRPC, with MFS as the primary endpoint [34], and the ARASENS study (), which is comparing darolutamide versus placebo added to standard ADT and docetaxel therapy in 1300 patients with mHSPC, with OS as the primary endpoint [35]. The primary completion dates for ARAMIS and ARASENS are April 2018 and August 2022, respectively.
2.0. Introduction to apalutamide
2.1. Chemistry
Apalutamide (ARN-509; JNJ-56021927) (Figure 2) is a AR inhibitor that was discovered using a structure–activity relationship-guided medicinal chemistry approach designed to find more potent antiandrogens that retain full antagonist activity with no significant agonistic activity in the setting of increased AR expression [25,36].
Figure 2. Structure of apalutamide.
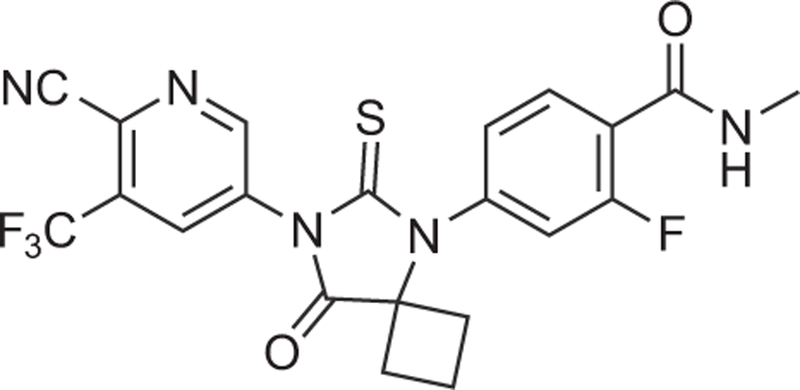
Apalutamide is a synthetic biaryl thiohydantoin compound discovered using a structure/activity relationship-guided medicinal chemistry program to design more potent antiandrogens without significant agonist activity.
2.2. Pharmacodynamics
Apalutamide binds to the ligand-binding domain of the AR with seven- to 10-fold greater affinity compared with bicalutamide as shown in LNCaP cells, which were transfected to overexpress AR (LNCaP/AR cells) in order to mimic the castration-resistant clinical state. Under equilibrium conditions, apalutamide inhibited binding of [18F]fluoro-5α-dihydrotestosterone (FDHT) with a half-maximal inhibitory concentration (IC50) of 16 nM. In comparison, the IC50 values for enzalutamide and bicalutamide were 21.4 and 160 nM, respectively [36]. The binding of apalutamide was selective for AR compared with other nuclear hormone receptors as shown by competitive binding assays in vitro with purified androgen, estrogen, progesterone and glucocorticoid receptors at drug concentrations to 100 µM. Apalutamide exhibited low affinity (IC50 = 3.0 µM) for the neurotransmitter GABAA receptor in radioligand binding assays, which was comparable to the activity shown by enzalutamide. However, steady-state brain tissue levels of apalutamide were four-fold lower than those of enzalutamide, suggesting a lower seizurogenic potential for apalutamide as compared with enzalutamide [36].
Apalutamide screened with enzalutamide both inhibited the synthetic androgen (R1881)-induced expression of 13 target genes, including PSA, in LNCaP/AR cells [36]. Bicalutamide was substantially less effective in blocking R1881-induced gene expression but, in the absence of R1881, bicalutamide altered gene expression consistent with its well-documented agonist activity in the setting of AR overexpression. In the absence of R1881, neither apalutamide nor enzalutamide exhibited any agonist activity at concentrations up to 10 µM. To assess the effects of apalutamide on AR binding to DNA, experiments were conducted in Hep-G2 cells expressing a VP16-AR fusion protein and an antioxidant response element–driven luciferase reporter [36]. Apalutamide and enzalutamide inhibited R1881-induced transcription of VP16-AR with an IC50 of 0.2 µM, whereas bicalutamide exhibited weak partial antagonist activity with an IC50 of 0.35 µM. In the absence of R1881, unlike bicalutamide, apalutamide did not partially activate VP16-AR transcription at concentrations to 10 µM, indicating that apalutamide functions as a full antagonist.
Apalutamide exhibited full antagonist activity in cells lines with AR overexpression and in those with mutations conferring resistance to bicalutamide such as T878A and W741C [37]. To better understand acquired resistance, LNCaP and LNCaP/AR cell lines were exposed chronically in vitro to high concentrations of apalutamide and enzalutamide [37]. Several cell lines were identified with a novel missense mutation in the AR ligand-binding domain (F877L), which conferred partial agonist activity to both apalutamide and enzalutamide at high concentrations. These findings support the hypothesis that nonsteroidal antiandrogens select for compound-specific gain-of-function AR mutations not observed at a high frequency in the untreated population. Transcriptional reporter–based studies comparing cell lines containing the selected AR mutants with lines containing wild-type AR or other mutations commonly found in CRPC patients concluded that the F877L mutation is sufficient to confer agonist activity to apalutamide and enzalutamide [37]. It has been suggested that the F877L mutation may influence ligand-induced conformational changes in helix 12 of the AR-binding domain, thereby affecting the dissociation rate of apalutamide and enzalutamide as well as AR responses to these ligands. In equilibrium AR-binding assays, apalutamide and enzalutamide had 30- and 48-fold higher affinity, respectively, for the F877L mutant compared with wild-type AR, implying potential agonist activity in this setting [37]. Of note, small molecule inhibitors (e.g. TRC253) of multiple AR mutations are currently being evaluated in early-stage clinical trials [38].
The antitumor efficacy of apalutamide was demonstrated in castrate mice bearing LNCaP/AR(cs) tumors [36]. In one series of experiments, apalutamide (10 mg/kg/day for 28 days) produced tumor regression (defined by >50% regression in tumor volume) in eight of 10 animals (two tumors were no longer palpable) compared with one of 10 animals for bicalutamide (10 mg/kg/day). The tumors from apalutamide-treated mice exhibited a 60% decrease in proliferative index and a 10-fold increase in apoptotic rate compared with vehicle as measured by Ki-67 staining and TUNEL, respectively. Similar findings were observed in castrate SCID mice bearing LNCaP/AR-luc xenograft tumors.
Apalutamide was also compared with enzalutamide in subsequent experiments in castrate mice bearing LNCaP/AR(cs) xenografts [36]. At 30 mg/kg/day, apalutamide produced a >50% decrease in tumor volume in 13 of 20 animals compared with three of 19 animals for enzalutamide. At 100 mg/kg/day, apalutamide and enzalutamide produced >50% decreases in a similar number of animals (13 of 19 and 12 of 19, respectively). On the basis of these findings, the optimal biological dose for producing tumor regression in the LNCaP/AR(cs) xenograft model was between 10 and 30 mg/kg/day for apalutamide compared with between 30 and 100 mg/kg/day for enzalutamide.
Steady-state plasma and tumor concentrations of apalutamide and enzalutamide were measured in the castrate mice bearing LNCaP/AR(cs) xenografts [36]. Following 28 days of continuous dosing, steady-state plasma concentrations of apalutamide were lower than those of enzalutamide (3.3 vs 11.0 µg/ml at 10 mg/kg/day), whereas intratumoral drug levels were similar (3.26 vs 3.39 µg/g tissue). As a result, apalutamide exhibited a higher tumor:plasma ratio compared with enzalutamide (107% vs 31%). In addition, apalutamide had a higher steady-state volume of distribution compared with enzalutamide (2.1 vs 0.82 L/kg), in part reflecting an approximately two-fold greater free fraction in plasma.
These studies in the murine CRPC model suggest that apalutamide may be more efficacious per unit dose and per unit steady-state plasma concentration compared with enzalutamide. In turn, the ability to achieve maximal efficacy at a lower dose and lower plasma concentration may be expected to result in a higher therapeutic index. This may potentially be explained by the more extensive distribution of apalutamide compared with enzalutamide, thereby allowing higher drug concentrations to be achieved within the tumor and contributing to greater antitumor effects.
2.3. Pharmacokinetics and metabolism
The first-in-human phase I study (ARN-509–001) was conducted in 30 patients with progressive mCRPC [39]. Patients were assigned sequentially to nine dose levels (30, 60, 90, 120, 180, 240, 300, 390 and 480 mg) using a traditional 3+3 dose escalation design. One of the primary study objectives was to assess the pharmacokinetics of apalutamide. Patients received a single dose of apalutamide and then, 1 week later, started continuous daily dosing. Blood samples for pharmacokinetic analysis were collected at multiple time points following the single dose (up to 168 hours) and on day 22 of cycle 1 (up to 24 hours). In addition, samples were collected weekly during cycle 1 and then before each new 28-day treatment cycle. Peak apalutamide plasma concentrations were achieved at 2–3 hours after dosing, and then drug levels declined slowly, with a mean half-life at steady-state of 3–4 days. Apalutamide exhibited dose-proportional pharmacokinetics for both maximal drug concentration (Cmax) and area under the plasma concentration–time curve (AUC). Trough apalutamide plasma concentrations increased over time, with most patients reaching steady-state after 3 weeks of continuous dosing. Both time to steady state and drug half-life were independent of the apalutamide dose.
Other pharmacokinetic studies have been conducted with apalutamide, and will be reported in separate publications. The effect of multiple doses of the CYP3A4 inhibitor itraconazole or the CYP2C8 inhibitor gemfibrozil on the single-dose pharmacokinetics of apalutamide was assessed in a phase I study () in 45 healthy male subjects [40]. The effect of apalutamide 240 mg once daily on the pharmacokinetics of single doses of probe substrates, including midazolam, warfarin, vitamin K, omeprazole, fexofenadine, pioglitazone and rosuvastatin was assessed in another phase I study () in 25 patients with CRPC [41].
3.0. Clinical efficacy
3.1. Phase I study
The primary objectives of the aforementioned first-in-human phase I study (ARN-509–001) were to assess the safety, tolerability and pharmacokinetics of apalutamide and to identify a recommended phase II dose [39]. The secondary objective was to evaluate antitumor effects based on PSA kinetics, imaging and circulating tumor cell (CTC) number. The study cohort of 30 mCRPC patients had a median age of 68 years (range, 45–81) and median baseline PSA of 42 ng/ml (range, 2.3–327); five (16.7%) had received prior chemotherapy [39]. Patients participated in the study for a median of 9.5 months. The most commonly reported TEAEs were fatigue (47%), back pain (30%), diarrhea (30%), dyspnea (30%) and nausea (30%); all were grade 1 or 2 except for one case of grade 3 nausea. No patients discontinued due to toxicity. One patient (in the 300 mg cohort) with a history of irritable bowel syndrome had a dose-limiting toxicity: grade 3 abdominal pain that resolved with drug interruption and subsequent dose reduction to 240 mg (120 mg twice daily). No seizures were reported, nor were any grade ≥4 adverse events (AEs). The dose-limiting toxicity may have been attributable to the soft gel capsule formulation used at the time. Conversion to tablets for phase II and subsequent studies was associated with a lower incidence of TEAEs, including those in the gastrointestinal system as well as a lower proportion of patients requiring dose reduction or interruption.
FDHT positron-emission tomography (PET)/computed tomography (CT) was used to evaluate AR inhibition in 16 patients [39]. The percent decline in standard uptake value (maximum–average) (SUVmax–avg) after 4 weeks of apalutamide treatment increased in a dose-dependent manner, reaching a plateau of inhibition at ≥120 mg daily (Figure 3). At doses of ≥120 mg, the decline from baseline in SUVmax–avg at 4 weeks was >90%. Although available AR-binding sites appeared to be fully occupied at the 120-mg dose level, the mean apalutamide trough plasma concentration with this dose in men (2.5 µg/ml) was at the lower end of the range associated with tumor regression in the murine LNCaP/AR model. In comparison, the steady-state trough plasma concentration at 240 mg was well within the range associated with tumor regression at the 10-mg/kg/day dose in the murine model. As a result, the 240-mg dose was selected as the recommended phase II dose.
Figure 3. Recommended phase II dose of apalutamide.
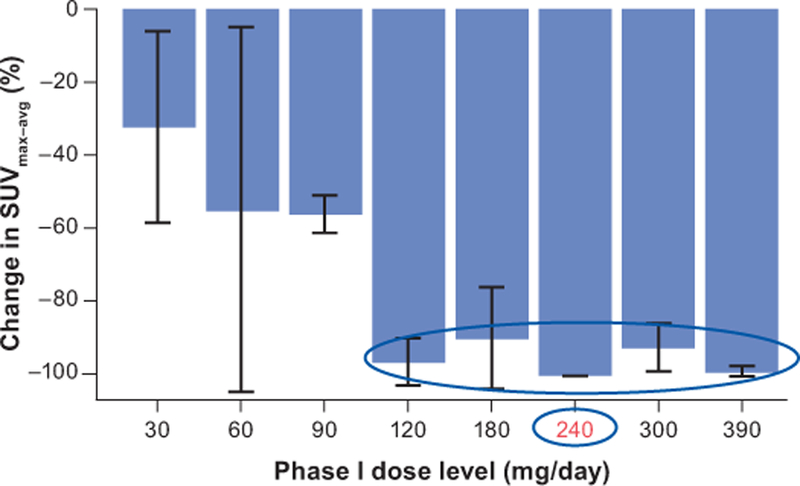
FDHT-PET imaging data from 16 patients in the phase I study support 240 mg as the optimal dose of apalutamide.
FDHT: [18F]fluoro-5α-dihydrotestosterone; PET: positron emission tomography; SUVmax–avg: standard uptake value (maximum–average).
Source: Rathkopf et al., 2013[39] (with permission). Reprinted with permission. © (2013) American Society of Clinical Oncology. All rights reserved.
Apalutamide exhibited antitumor efficacy based on PSA kinetics, radiographic response and CTC enumeration [39]. At 12 weeks, 14 patients (47%) had ≥50% declines from baseline in PSA level. Over the entire study course, 18 patients (60%) had PSA responses, including six (20%) with ≥90% decreases in PSA from baseline. Five of 10 patients (50%) with measurable soft-tissue disease maintained stable disease for >6 months. Finally, four of seven patients with unfavorable CTC levels at baseline (i.e. ≥5 cells/7.5 ml blood) converted to favorable levels. In conclusion, the phase I study showed that apalutamide was safe and well tolerated, and provided initial evidence that it has antitumor activity in men with mCRPC.
3.2. Phase II study
The development program for apalutamide faced several key challenges, including the availability of abiraterone acetate plus prednisone and enzalutamide, and the need to conduct a placebo-controlled trial in the nmCRPC setting while retaining patients on study in the setting of rising PSA levels. The phase II program focused on three distinct patient populations: 1) high-risk nmCRPC, 2) chemotherapy-naïve and abiraterone acetate plus prednisone–naïve mCRPC and 3) progressive mCRPC after abiraterone acetate plus prednisone [42,43]. In order to better reflect a change in clinical status, whenever possible, patients remained on study treatment (apalutamide 240 mg/day) until radiographic or symptomatic progression was documented, or until clinical progression such as skeletal-related events, or until the treating physician decided to initiate a new systemic anticancer therapy. The primary endpoint was PSA percentage change from baseline at 12 weeks in the high-risk nmCRPC cohort and PSA response rate in the mCRPC cohorts determined according to Prostate Cancer Working Group 2 criteria. Secondary endpoints included time to PSA progression in all cohorts, MFS in the high-risk nmCRPC cohort and rPFS and objective response rate in the mCRPC cohorts.
3.2.1. High-risk nmCRPC cohort
Eligible patients had nmCRPC with castrate serum testosterone levels (<50 ng/ml), Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 and a high risk of developing metastases (defined by PSA ≥8 ng/ml within 3 months before enrollment or a PSA doubling time ≤10 months) [43]. Patients with distant metastases, a history of seizures or prior treatment with enzalutamide or abiraterone acetate plus prednisone were excluded.
The high-risk nmCRPC cohort included 51 patients; the median age was 71 years (range, 51–88) and the median baseline PSA was 10.7 ng/ml (range, 0.5–201.7) [43]. Most patients (80%) had been treated previously with one or more first-generation antiandrogens, most commonly bicalutamide. Study results were reported after a median follow-up of 28 months, at which time 18 patients (35%) remained in the study. The main reasons for study discontinuation were disease progression (22%) or AEs (18%).
PSA declined from baseline by a median of 85% at week 12, corresponding to a PSA response rate of 89% [43]. The maximal percentage decline in PSA at any point during the study was 93%, corresponding to a PSA response rate of 94%. During the follow-up period, 25 patients (53%) had PSA progression. The median time to PSA progression was 24 months (95% CI (confidence interval) 16.3–not reached [NR]), whereas median time to metastasis (TTM) was not reached. The most commonly reported treatment-related TEAEs were fatigue (45%), diarrhea (29%) and nausea (25%); the vast majority was grade 1 or 2. Seizures were not reported. Grade ≥3 TEAEs were uncommon; fatigue, hypertension and malignant melanoma were each reported in two patients (4%). Overall, the safety profile was consistent with the prior phase I data. In this high-risk nmCRPC cohort, apalutamide was safe and well tolerated, and exhibited robust activity as evidenced by durable PSA responses and disease control.
3.2.2. mCRPC cohorts
Patients with mCRPC had either not received prior abiraterone acetate plus prednisone (AAP-naïve) or had been treated with abiraterone acetate plus prednisone for ≥6 months (post-AAP) [42]. Patients in both cohorts had castrate serum testosterone levels ≤50 ng/ml and ECOG performance status of 0 or 1; those in the AAP-naïve cohort had progressive disease based on a rising PSA ≥2 ng/ml within 2 weeks of enrollment, evidence of measurable disease on CT/magnetic resonance imaging (MRI) scans or radiographic progression with at least two new bone lesions. Patients previously treated with enzalutamide, ketoconazole or chemotherapy for mCRPC and those with a history of seizures or conditions predisposing to seizures were excluded. The data analysis for these cohorts included swim lane plots, as recommended by the Prostate Cancer Clinical Trials Working Group 3 (PCWG3), to distinguish progression according to standard criteria versus the decision to stop treatment [44].
The AAP-naïve and post-AAP cohorts included 25 and 21 patients, respectively [42]. The median age in these respective cohorts was 68 years (range, 53–91) and 67 years (range, 48–83), and the median time since initial prostate cancer diagnosis was 61 and 107 months, respectively. Median baseline PSA in the AAP-naïve and post-AAP cohorts was 14.7 ng/ml (range, 1.1–2552) and 58.4 ng/ml (range, 1.1–6074), respectively. Study results were reported after a median follow-up of 22.1 months in the AAP-naïve cohort and 5.6 months in the post-AAP cohort. The most common reasons for study discontinuation were disease progression (36% and 38%, respectively) and AEs (12% and 5%). Two patients in the AAP-naïve cohort died from disease progression [42].
The PSA response rates at 12 weeks in the AAP-naïve and post-AAP cohorts were 88% and 22%, respectively, and the maximal PSA response rates were 92% and 28%, respectively [42]. Swim lane plots showed that the proportions of patients remaining on apalutamide for ≥6 months in the AAP-naïve and post-AAP cohorts were 80% and 43%, respectively (Figure 4). For many patients, there was a disconnect between time on treatment and the degree of PSA decline. Use of swim plots demonstrates the patient experience with study treatment, which may be useful for physicians when evaluating patients who are no longer benefiting clinically from treatment.
Figure 4. Patient experience with apalutamide in phase II study of patients with mCRPC. (A) AAP-naïve and (B) post-AAP (B) cohorts.
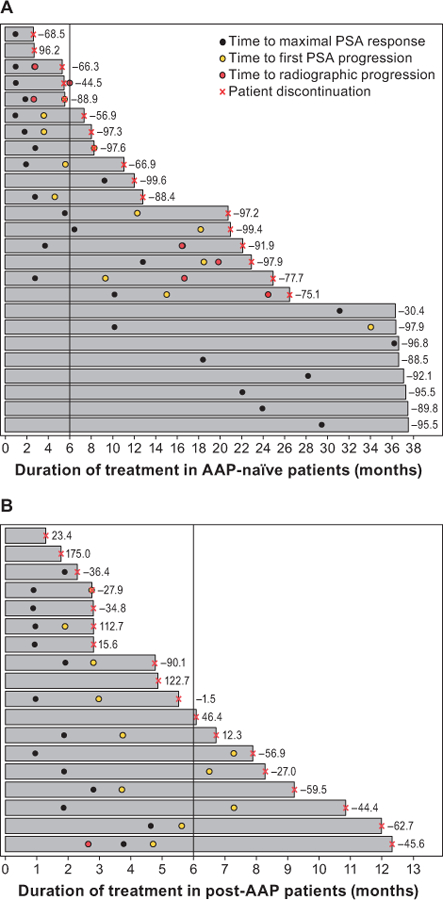
[43] These swim lane plots show the duration of apalutamide treatment from shortest (top lane) to longest (bottom lane) in the AAP-naïve and post-AAP mCRPC cohorts.
AAP: abiraterone acetate plus prednisone; PSA: prostate-specific antigen.
Source: Rathkopf et al., 2017[42]. Reprinted with permission.
Based on Response Evaluation Criteria In Solid Tumors criteria, four of eight AAP-naïve patients with measurable target lesions at baseline achieved partial responses, with two additional patients having stable disease lasting for 14 and 2.8 months [42]. Of the 10 post-AAP mCRPC patients with measurable disease, four had stable disease lasting for 2.5–5.6 months. The median time to PSA progression was 18.2 months (95% CI 8.3–NR) in the AAP-naïve cohort and 3.7 months (95% CI 2.8–5.6) in the post-AAP cohort.
The safety profile was consistent with that observed in the phase I study; the most common drug-related TEAEs in the AAP-naïve and post-AAP mCRPC cohorts were fatigue (48% and 52%, respectively), diarrhea (32% and 19%, respectively) and nausea (32% and 24%, respectively) [42]. Grade 3 TEAEs reported in >1 patient in each cohort were anemia (in two AAP-naïve patients [8%]) and back pain (in two post-AAP patients [10%]). Again, no seizures were reported. In summary, apalutamide was safe and well tolerated in mCRPC patients, and exhibited clinical antitumor activity, particularly in the AAP-naïve cohort.
3.2.3. Mutational analysis in CRPC patients
An exploratory analysis was conducted to identify the type and frequency of known AR ligand-binding domain mutations in CRPC patients enrolled in the ARN-509–001 study [45]. Eleven known mutations affecting six key amino acids in the ligand-binding domain (V716, W742, H875, F877, T878 and M896) were evaluated in CTC DNA using a digital polymerase chain reaction method known as BEAMing (Beads, Emulsification, Amplification, and Magnetics). BEAMing combined emulsion polymerase chain reaction using magnetic beads coated with gene-specific primers, hybridization and flow cytometry to detect and quantify known mutations in circulating tumor DNA.
Of the 97 patients enrolled in the phase II study, 93 (95.9%) were evaluable for the mutational analysis at baseline and 82 (84.5%) were evaluable at disease progression [45]. The overall frequency of detected AR mutations was 7.5% (7/93) at baseline and 7.3% (6/82) at progression. The most common mutations at baseline were T878A (n = 3; associated with abiraterone resistance) and F877L (n = 2; associated with apalutamide resistance), whereas the most common identified at progression was F877L (n = 3). Overall, the low rate of F877L acquisition suggests that it may not be a common contributor to resistance to apalutamide.
3.3. Phase III study
The phase III SPARTAN study was conducted at 332 sites in 26 countries in North America, Europe and the Asia-Pacific region (Figure 5A) [46]. SPARTAN enrolled patients with nmCRPC who were at high risk of developing metastases as defined by a PSA doubling time of ≤10 months. Patients with malignant pelvic lymph nodes below the iliac bifurcation that were smaller than 2 cm in short axis (N1) were eligible. Patients were stratified by PSA doubling time, use of bone-sparing agents and classification of nodal status as N0 or N1, and then randomized (2:1) to receive apalutamide 240 mg or placebo on a continuous daily basis. ADT was continued throughout the study. Patients continued study treatment until progression, unacceptable toxicity or withdrawn consent. After developing metastases, patients were treated at their physicians’ discretion, with an option to receive study-provided abiraterone acetate plus prednisone; the study blind was not broken on progression.
Figure 5. (A) SPARTAN study design. (B) metastasis-free survival in the SPARTAN study.
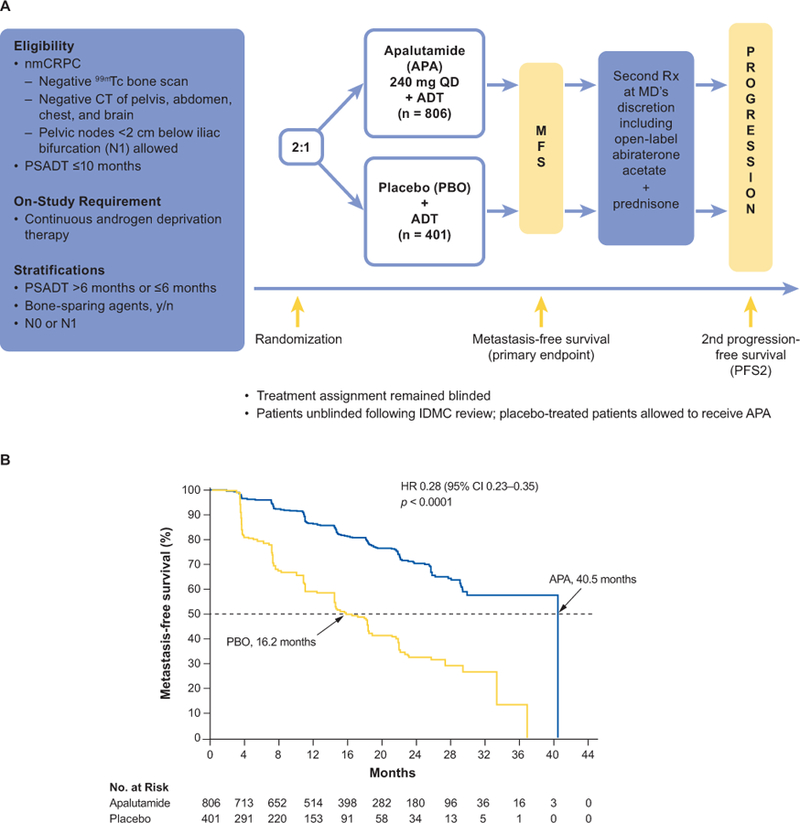
The study design of SPARTAN, a phase III study of apalutamide in patients with nonmetastatic castration-resistant prostate cancer, and the Kaplan–Meier curves for the primary end point of metastasis-free survival showing significant improvement in the apalutamide group.
CT: computed tomography; IDMC: independent data monitoring committee; MFS: metastasis-free survival; nmCRPC, nonmetastatic castration-resistant prostate cancer; PSADT, prostate-specific antigen doubling time; QD: once daily.
Source: Smith et al., 2018[46] and Small et al., 2018 [60](with permission from Small, EJ).
Disease assessments were performed every 16 weeks, and all imaging was assessed prospectively by blinded independent central review [46]. The primary endpoint was MFS, defined as the time from randomization to first evidence of blinded independent central review–confirmed, radiographically detectable distant metastasis (bone or soft tissue) or death. Secondary endpoints included TTM, PFS, time to symptomatic progression and time to initiation of cytotoxic chemotherapy. Second PFS (PFS2)—defined as the time from randomization to investigator-assessed disease progression or death, during the first subsequent treatment for mCRPC—was evaluated as an exploratory endpoint, as were time to PSA progression, PSA decline, and patient-reported outcomes.
SPARTAN was designed to enroll 1200 patients to observe 372 MFS events in order to detect a hazard ratio (HR) of 0.7 for MFS, with 90% power at two-sided significance level of 0.05 [46]. This treatment effect would correspond to an increase in median MFS of 11 months (from 25 to 36 months), which would be considered clinically relevant. A single, final analysis was planned for the primary endpoint of MFS and for TTM and PFS. A hierarchical adaptive group sequential testing of secondary efficacy endpoints according to the prespecified O’Brien-Fleming-type α spending function was used in the following order: TTM, PFS, symptomatic progression, OS and time to initiation of cytotoxic chemotherapy.
A total of 1207 patients were enrolled, including 807 in the apalutamide arm and 401 in the placebo arm [46]. Both treatment groups were well balanced with respect to demographics and baseline characteristics. Overall, the median age was 74 years (range, 48–97); patients were enrolled at a median of 7.9 years after initial prostate cancer diagnosis. Of the stratification factors, 71.3% had a PSA doubling time ≤6 months, 10.0% used a bone-sparing agent and 83.6% had N0 nodal status. The majority (73.1%) had previously used a first-generation antiandrogen. At the time of the clinical cutoff after a median follow-up of 20.3 months, 60.9% of patients in the apalutamide group compared with 29.9% of those in the placebo group were still receiving their assigned study treatment.
Apalutamide significantly improved median MFS compared with placebo (40.5 vs 16.2 months; HR 0.28; 95% CI 0.23–0.35; p < 0.001) (Figure 5B) [46]. The treatment difference represents an increase of >2 years in MFS, and corresponds to a 72% reduction in risk of metastasis or death. These data were obtained after 378 MFS events were observed, including 184 (23%) in the apalutamide group and 194 (48%) in the placebo group. The treatment effect of apalutamide on MFS was consistently favorable across all prespecified subgroups, including PSA level, PSA doubling time and nodal status.
Apalutamide also provided consistent improvement across all secondary endpoints (Table 1) [46]. Because TTM, PFS and time to symptomatic progression were statistically significant, they were considered final analyses based on the protocol-specific statistical analysis. The analysis of OS was immature, as only 104 of the required 427 events had occurred. At this early time point, there was a trend toward improved survival in the apalutamide group, with an HR of 0.70. A preplanned final, event-driven analysis for OS and the next hierarchical secondary endpoint, time to cytotoxic chemotherapy, will be undertaken after sufficient events have occurred.
Table 1.
Summary of clinical efficacy of apalutamide in phase II and phase III studies
| Study | Population | Patients | Primary endpoint | Secondary endpoints |
|---|---|---|---|---|
| ARN-509–001 (phase II) [42] | High-risk nmCRPC | N = 51; median age 71 years (range, 51–88); median PSA 10.7 ng/ml (range, 0.5–201.7) | Median change from baseline in PSA at week 12: –85% (range, –99.9% to 52.2%) corresponded to PSA response rate at 12 weeks of 89% | • Median TTPP: 24 months (95% CI 16.3–NR) • Median MFS: NR (95% CI 33.4 months–NR) |
| ARN-509–001 (phase II) [41] | AAP-naïve mCRPC | N = 25; median age 68 years (range, 53–91); median PSA 14.7 ng/ml (range, 1.1–2552) | PSA response rate at 12 weeks: 88% (95% CI 69–97) | • Median TTPP: 18.2 months (95% CI 8.3–NR) • Median PFS: NR (95% CI 16.7 months–NR) after 22.1 months of follow-up |
| Post-AAP mCRPC | N = 21; median age 67 years (range, 48–83); median PSA 58.4 ng/ml (range, 1.1–6074) | PSA response rate at 12 weeks: 22% (95% CI 6–48) | • Median TTPP: 3.7 months (95% CI 2.8–5.6) • Median PFS: NR (95% CI NR–NR) after 5.6 months of follow-up |
|
| SPARTAN (phase III) [45] | High-risk nmCRPC | N = 1207; median age 74 years (range, 48–97); median PSA 7.8 ng/ml (range not provided) Stratification factors at baseline: PSA doubling time ≤6 months, 71.5%; use of bone-sparing agent, 10.2%; N0 nodal status, 83.5% |
Median MFS for apalutamide vs placebo: 40.5 vs 16.2 months; (HR 0.28; 95% CI 0.23–0.35); p < 0.001 |
• Median TTM: 40.5 vs 16.6 months; HR 0.27 (95% CI 0.22–0.34); p < 0.001 • Median PFS: 40.5 vs 14.7 months; HR 0.29 (95% CI 0.24–0.36); p < 0.001 • Median time to symptomatic progression: NR vs NR; HR 0.45 (95% CI 0.32–0.63); p < 0.001 • Median OS: NR vs 39.0 months*; HR 0.70 (95% CI 0.47–1.04); p = 0.07 • Median time to initiation of cytotoxic chemotherapy: NR vs NR*; HR 0.44 (95% CI 0.29–0.66) |
Results from first interim analysis for OS and time to initiation of cytotoxic chemotherapy.
AAP: abiraterone acetate plus prednisone; CI: confidence interval; HR: hazard ratio; mCRPC: metastatic castration-resistant prostate cancer; MFS: metastasis-free survival; nmCRPC: nonmetastatic castration-resistant prostate cancer; NR: not reached; OS: overall survival; PFS: progression-free survival; PSA: prostate-specific antigen; TTM: time to metastasis; TTPP: time to PSA progression.
At the time of the independent data and safety monitoring committee’s recommendation to unblind the study, 279 placebo patients and 314 apalutamide patients had already discontinued study treatment, mostly as a result of having reached the MFS endpoint [46]. Of these, 77.8% of placebo and 52.5% of apalutamide patients subsequently received FDA-approved therapy for mCRPC, most frequently abiraterone acetate plus prednisone. Use of apalutamide in the nmCRPC setting significantly reduced risk of disease progression during the first subsequent treatment for mCRPC (HR 0.49; 95% CI 0.36–0.66; p < 0.0001). Of the other exploratory endpoints, time to PSA progression was also significantly prolonged by apalutamide (HR 0.06; 95% CI 0.05–0.08; p < 0.0001). PSA response at 12 weeks was observed in a greater proportion of patients in the apalutamide group compared with the placebo group (90% vs 2%).
In conclusion, SPARTAN demonstrates that apalutamide significantly reduces risk of metastasis or death, and prolongs the time to symptomatic progression in men with high-risk nmCRPC. The efficacy of apalutamide was consistent across multiple endpoints, supporting the robustness of the improvement in the primary endpoint of MFS.
4.0. Safety and tolerability
4.1. Phase III SPARTAN study
Most patients in the apalutamide and placebo groups had AEs during the course of the SPARTAN study (96.5% and 93.2%, respectively); the most common TEAEs were fatigue, hypertension, rash and diarrhea, with the majority rated as grade 1 or 2 [46].
However, when adjusted for exposure, the incidence of hypertension was no longer greater in the apalutamide arm compared with the placebo arm. The incidence of grade 3 or 4 AEs was higher in the apalutamide group than in the placebo group (45.1% vs 34.2%); differences between treatments were seen for rash, fractures, falls and weight loss. Discontinuations due to AEs occurred in 10.6% of patients in the apalutamide group and 7.0% in the placebo group. Deaths associated with AEs were reported for 10 patients in the apalutamide group (prostate cancer and sepsis as the cause in two patients each, and acute myocardial infarction, cardiorespiratory arrest, cerebral hemorrhage, myocardial infarction, multiple organ dysfunction, and pneumonia as the cause in one patient each) and in one patient in the placebo group (cardiorespiratory arrest).
Seizure was reported in two patients (0.2%) in the apalutamide group; one was grade 1 and the other was grade 2 [46]. Both were reported as serious AEs, and both patients permanently discontinued study treatment per protocol. The first patient was an 85-year-old man who had sustained multiple falls and was diagnosed with Parkinson’s disease during the study. He had a grade 3 fall, resulting in a grade 3 hematoma and presentation to an emergency department where he was witnessed to have a grade 2 seizure. The seizure resolved on the same day. The second patient had a relevant medical history of febrile seizures during infancy (history was elicited after the seizure). The patient’s spouse reported jerking motions while the patient was sleeping, and upon evaluation a neurologist felt the symptoms described were consistent with a seizure.
Patient-reported assessments with Functional Assessment of Cancer Therapy–Prostate and the European Quality of Life-5 Dimensions questionnaires indicated that patients who received apalutamide in addition to ADT maintained stable overall health-related quality of life over time [46].
4.2. Management strategies for apalutamide-related AEs
Several treatment-related AEs were observed in SPARTAN, including rash, hypothyroidism and fracture, for which specific management strategies are recommended.
Skin rash was reported at a higher rate in the apalutamide group than in the placebo group (23.8% vs 5.5%) [46]. Grade 3 rash covering >30% of body surface area was seen in 5.2% of patients treated with apalutamide compared with 0.3% of those receiving placebo. The skin rash associated with apalutamide was commonly described as macular or maculo-papular, and had an onset after a median of 82 days of treatment. Rash typically resolved after drug interruption and/or dose reduction; most patients with grade 1 or 2 rash were able to continue apalutamide treatment. Rash led to discontinuation in 19 patients (2.4%), dose reduction in 22 patients (2.7%) and dose interruption in 55 patients (6.8%) in the apalutamide group. The corresponding values in the placebo group were 0, 1 (0.3%) and 5 (1.3%), respectively.
For grade 1 rash, apalutamide may be continued at the current dose, and dermatologic treatment considered with a topical steroid cream and/or oral antihistamine. For grade 2 rash, apalutamide treatment may be interrupted and dermatologic treatment initiated with a topical steroid cream and oral antihistamine; short courses of oral corticosteroids may be considered. When the rash improves to grade ≤1, apalutamide can be reinitiated and a reduction of one dose level considered. For grade 3 rash, treatment with apalutamide should be interrupted and topical steroid cream, oral antihistamine, and short courses of oral corticosteroids should be considered; reassessment should be done after 1–2 weeks. Apalutamide may be reinitiated at a one-dose-level reduction if rash had resolved to grade ≤1. If rash remains unchanged or worsened, oral corticosteroids should be initiated if not already done so, and the patient referred to a dermatologist. Oral steroids should be continued for at least 1 week after resumption of apalutamide at the reduced dose. Apalutamide must be discontinued if oral corticosteroids will be required for >28 days. With these treatments, skin rash resolved within a median of 60 days in 80.6% of patients in SPARTAN.
Hypothyroidism was reported in 65 patients (8.1%) in the apalutamide group and eight (2.0%) in the placebo group; all events were grade 1 or 2 [46]. One patient elected to discontinue apalutamide due to hypothyroidism, and a second had a dose reduction. Hypothyroidism did not lead to a dose interruption in any patient. The median time to the first elevated thyroid-stimulating hormone level was 113 days. Apalutamide is thought to cause hypothyroidism by inducing UDP-glucuronosyl-transferase, leading to reduced exposure to levothyroxine. This AE is managed by initiating or increasing the dose of thyroid replacement therapy.
ADT is associated with decreased bone mineral density and increased risk of clinical fracture [47,48]. Apalutamide appears to further increase fracture risk in men receiving long-term ADT [46]. In SPARTAN, fall and fractures occurred in 125 (15.6%) and 94 (11.7%) patients in the apalutamide group, respectively, with many of the fractures preceded by a fall. Patients should be evaluated for fracture risk and treated according to clinical practice guidelines when fracture risk warrants therapy using approved medications, such as denosumab, zoledronic acid or alendronate [19]. In SPARTAN, patients were allowed bone-sparing agents indicated for the treatment of osteoporosis at indicated doses and dosing schedules; however, agents indicated for the prevention of skeletal-related events in patients with solid tumors were prohibited [46]. Physical activity and lifestyle modification should also be included in patient management.
5.0. Regulatory affairs
The US FDA approved apalutamide on February 14, 2018, for the treatment of patients with nmCRPC [49,50]. Apalutamide is the first FDA-approved treatment for nmCRPC, and the first to be based on use of the endpoint of MFS. According to the package insert, patients treated with apalutamide should also receive concurrent ADT or should have had bilateral orchiectomy. An application to market apalutamide in the European Union was submitted to the European Medicines Agency on February 9, 2018.
6.0. Expert commentary
Apalutamide was engineered using structure–activity relationship-guided medicinal chemistry to identify a next-generation clinically beneficial nonsteroidal antiandrogen [36]. It exhibits properties satisfying that objective, acting as a competitive inhibitor of the AR and as a full antagonist to AR overexpression, a common and important feature in CRPC [36,37]. Apalutamide binds AR with greater affinity than the first-generation antiandrogen bicalutamide, and displays a higher steady-state tumor/plasma ratio compared with enzalutamide [36].
The phase II study of apalutamide highlights the concept that treatment should be stopped when a patient is no longer clinically benefiting instead of relying on standard criteria for evidence of disease progression, consistent with PWCG3 recommendations [44]. Using standard criteria may lead to the drug being discontinued prematurely. The phase II study showed that the clinical benefit of apalutamide in mCRPC was greater in AAP-naïve patients than in post-AAP patients; the PSA response rates in these populations were 88% and 22%, respectively [42]. According to PCWG3-recommended swim plots, 80% of the AAP-naïve cohort continued apalutamide treatment for ≥6 months compared with 43% of the post-AAP cohort.
These data are important to highlight for several reasons: 1) the response post AAP is less in frequency and duration relative to primary treatment but 43% of patients still benefitted from sequential therapy for 6+ months, 2) some patients continued to derive clinical benefit from continuing treatment, even with a rising PSA, underscoring the importance of the patient experience in considering treatment decisions and 3) all patients experienced a rise in PSA at the time of clinical or radiographic progression, suggesting that the AR still remains an important target in mCRPC and newer therapies rationally designed to overcome AR resistance will be critical in this setting.
In the phase II study of apalutamide in CRPC, the frequency of AR mutations, including F877L and T878A, was relatively low at the start of treatment and did not increase in frequency at the time of progression, suggesting that AR mutations are unlikely to play a major role in the mechanism of primary or acquired resistance to apalutamide in CRPC [45]. Besides ligand-binding domain mutations, other AR alterations are known to lead to clinical resistance, including AR splice variants with an N-terminal domain that mediates receptor transactivation [51,52]. Other suspected resistance mechanisms include AR bypass signaling (in which other hormone receptors, e.g. the glucocorticoid receptor, co-opt downstream AR signaling by activating AR target genes), increased steroidogenesis and development of androgen-independent tumor cells [53,54]. Additional avenues of exploration include combining AR therapy with alternative signaling pathways known to contribute to resistance such as PI3K signaling for selected patients with phosphatase and tensin homolog (PTEN) loss [55].
Larger prospective studies using assays that can detect AR mutations and other AR alterations are still needed to more completely address de novo and acquired resistance [51]. Clinical studies with novel agents that target suspected mechanisms of resistance are under way, such as AR N-terminal domain inhibitors that will help evaluate the role of AR splice variants in mCRPC [52]. Combining AR signaling inhibitors with agents that target potential resistance mechanisms may offer the potential to prolong therapeutic benefit and improve patient outcome. Another approach that warrants mention is MetaCURE, which has a continuously enrolling multiarm, multistage design, and allows new experimental arms to be added on a continual basis, thereby allowing resistance-targeting approaches to be evaluated in a timely manner [56]. This approach shifts the focus onto early measures of response instead of time-to-event measures that are associated with greater uncertainty and confounding by postprotocol interventions.
For patients without metastatic disease who are progressing on ADT (nmCRPC), apalutamide is the first drug to be approved in the United States. The phase III SPARTAN study demonstrated that apalutamide reduced risk of metastasis or death by 72% compared with placebo, and prolonged median MFS by >2 years in men with high-risk nmCRPC [46]. The MFS benefit was consistent across all subgroups, and was supported by improvements across all evaluable efficacy endpoints. Survival data are still being collected. Patients in SPARTAN maintained their overall health-related quality of life with the addition of apalutamide to ADT. Drug-related AEs included a higher than expected rate of fracture and falls for which appropriate prophylaxis should be considered while on treatment. A higher incidence of falls of any grade was also observed with enzalutamide versus placebo (11% vs 4%) in the PROSPER trial, suggesting a class effect. Overall, grade ≥3 adverse events that were more common with enzalutamide than placebo were fatigue (3% vs 1%) and hypertension (5% vs 2%). Phase 3 safety data with darolutamide are not yet available; falls were not reported in the phase 2 ARADES trial of darolutamide, with most common adverse events of fatigue and asthenia (both 12%). The clinical experience to date with long-term exposure to apalutamide after apalutamide-related skin rash suggests that the drug remains well tolerated in postmarketing safety surveillance.
Overall, apalutamide has demonstrated efficacy in phase II and III trials for patients with nonmetastatic and metastatic CRPC, with a tolerable safety profile, and is approved for men with nmCRPC who are in need of treatment options.
7.0. Five-year view
The role of apalutamide within the prostate cancer disease continuum will be better defined as results from additional phase III trials become available, including ACIS () in patients with chemotherapy-naïve mCRPC [57], TITAN () in patients with mHSPC [58] and ATLAS () in patients with high-risk, localized or locally advanced prostate cancer receiving primary radiation therapy (Table 2) [59].
Table 2.
Ongoing phase III clinical trials of apalutamide
| Study (ClinicalTrials.gov Identifier) | Target population | Comparator arms | Primary endpoint(s) | Secondary endpoints | Primary completion date |
|---|---|---|---|---|---|
| ACIS () [56] | Chemotherapy-naïve mCRPC | Apalutamide and abiraterone acetate plus prednisone vs placebo and abiraterone acetate plus prednisone | rPFS | • OS • Time to chronic opioid use • Time to initiation of cytotoxic chemotherapy • Time to pain progression |
September 2018 |
| TITAN () [57] | mHSPC | Apalutamide plus LHRHa vs placebo plus LHRHa | rPFS and OS | • Time to pain progression • Time to skeletal-related event • Time to chronic opioid use • Time to initiation of cytotoxic chemotherapy |
November 2020 |
| ATLAS () [58] | High-risk localized or locally advanced prostate cancer receiving primary radiation therapy | Apalutamide plus LHRHa vs placebo (bicalutamide for first 4 months) plus LHRHa | MFS | • Time to local-regional recurrence • Time to CRPC • Time to distant metastasis • OS |
December 2022 |
CRPC: castration-resistant prostate cancer; LHRHa: luteinizing hormone–releasing hormone agonist; mCRPC: metastatic castration-resistant prostate cancer; MFS: metastasis-free survival: mHSPC, metastatic hormone-sensitive prostate cancer; OS: overall survival; rPFS: radiographic progression-free survival.
ACIS is designed to examine whether combined AR signaling inhibition with apalutamide and abiraterone acetate plus prednisone provides clinical benefit beyond that observed with androgen synthesis inhibition with abiraterone acetate plus prednisone alone in patients with chemotherapy-naïve mCRPC [57]. A total of 983 patients have been randomized 1:1 to receive apalutamide (240 mg once daily) or placebo; both groups will receive abiraterone acetate (1000 mg once daily) plus prednisone (5 mg twice daily), and also will continue to receive ADT. Treatment will continue in 28-day cycles until disease progression, withdrawal of consent or unacceptable toxicity. The primary endpoint is rPFS, and secondary endpoints include OS, time to chronic opioid use, time to initiation of cytotoxic chemotherapy and time to pain progression. The primary completion date is December 2018.
TITAN is comparing apalutamide plus ADT versus placebo plus ADT in patients with mHSPC [58]. The rationale for clinical benefit is based in part on the recent results of LATITUDE showing the efficacy of abiraterone acetate plus prednisone in this patient population. The ongoing ARCHES study with enzalutamide and ARASENS study with darolutamide are also examining the benefit of antiandrogens in the mHSPC setting. In TITAN, a total of 1052 patients with at least one bone lesion (with or without visceral metastases) were randomized to apalutamide (240 mg) or placebo. The ADT regimen is at the investigator’s discretion. The primary endpoints are rPFS and OS, with secondary endpoints including time to pain progression, time to skeletal-related event, time to chronic opioid use and time to initiation of cytotoxic chemotherapy. The primary completion date is November 2020.
ATLAS will examine whether adding apalutamide to LHRHa will improve outcomes in patients with high-risk localized or locally advanced prostate cancer who are receiving primary radiation therapy [59]. “High risk” is defined by Gleason score ≥8 with clinical stage ≥cT2c, or by Gleason score ≥7 and PSA ≥20 ng/ml with clinical stage ≥cTc2. Eligible patients will be randomized 1:1 to receive apalutamide (240 mg) or placebo; all patients will receive LHRHa and radiation therapy. The hormonal treatments will be continued for 30 months; patients in the placebo group will receive bicalutamide for the first four cycles (placebo will be given in the apalutamide arm to maintain blinding). Planned accrual is 1500 patients. The primary endpoint is MFS; secondary endpoints include time to local-regional recurrence, time to CRPC, time to distant metastasis and OS. The primary completion date is December 2022.
As the therapeutic armamentarium for prostate cancer increases, it will be important to identify biomarkers predictive of therapeutic benefit as well as to determine the optimal sequence and combinations for delivering these medications.
Key Issues.
Apalutamide was engineered as a next-generation AR inhibitor to improve potency and tolerability across the prostate cancer disease continuum.
Apalutamide binds directly to the ligand-binding domain of the AR, and thereby prevents AR nuclear translocation, inhibits DNA binding, and impedes AR-mediated transcription.
Apalutamide selectively blocks androgen signaling to decrease tumor cell proliferation and increase cell death, leading to potent antitumor activity in preclinical prostate cancer models.
Apalutamide was generally safe and well tolerated, and exhibited durable clinical responses in a phase II trial in nmCRPC patients and in mCRPC patients, particularly in those naïve to abiraterone acetate plus prednisone.
In the phase III SPARTAN study, apalutamide prolonged median MFS by >2 years in patients with high-risk nmCRPC and exhibited a manageable safety profile, which led to FDA approval for this indication.
Ongoing phase III studies are evaluating the clinical benefit of apalutamide in additional prostate cancer disease states.
Acknowledgements
Writing assistance was provided by Ira Mills, PhD, of PAREXEL, funded by Janssen Global Services, LLC
Funding
HI Scher reports funding from the National Institute of Health National Cancer Institute Prostate SPORE Grant P50-CA92629, National Institute of Health National Cancer Institute Cancer Center Support Grant P30 CA008748, Department of Defense Congressionally Directed Medical Research Program W81XWH-15–2-0018, Prostate Cancer Foundation, Sidney Kimmel Center for Prostate and Urologic Cancers. Janssen Global Services, LLC, provided funding for writing assistance.
List of Abbreviations:
- AAP
Abiraterone acetate plus prednisone
- AEs
Adverse events
- ADT
Androgen deprivation therapy
- AR
Androgen receptor
- AUC
Area under the plasma concentration–time curve
- BEAMing
Beads, Emulsification, Amplification, and Magnetics
- Cmax
Maximal drug concentration
- CI
Confidence interval
- CT
Computed tomography
- CTC
Circulating tumor cell
- ECOG
Eastern Cooperative Oncology Group
- FDA
US Food and Drug Administration
- FDHT
[18F]fluoro-α-dihydrotestosterone
- HR
Hazard ratio
- IC50
Half maximal inhibitory concentration
- LHRHa
Luteinizing hormone-releasing hormone agonist
- mCRPC
Metastatic castration-resistant prostate cancer
- MFS
Metastasis-free survival
- mHSPC
Metastatic hormone-sensitive prostate cancer
- MRI
Magnetic resonance imaging
- nmCRPC
Nonmetastatic castration-resistant prostate cancer
- NR
Not reached
- OS
Overall survival
- PCWG3
Prostate Cancer Clinical Trials Working Group 3
- PET
Positive-emission tomography
- PFS
Progression-free survival
- PFS2
Second progression-free survival
- PSA
Prostate-specific antigen
- PTEN
Phosphatase and tensin homolog
- rPFS
Radiographic progression-free survival
- SUVmax–avg
Standard uptake value (maximum–average)
- TEAEs
Treatment-emergent adverse events
- TTM
Time to metastasis
Footnotes
Declaration of interest
Dana E. Rathkopf reports research funding from Janssen and uncompensated research funding from Janssen, Astellas/Medivation, Genentech/Roche, Celgene, TAIHO, Tracon, Novartis, Millenium, and Ferring. Howard I. Scher reports non-financial support as an uncompensated consultant from Aragon relevant to this review; and personal fees as a compensated consultant from Astellas and Clovis Oncology; non-financial support as an uncompensated consultant from Ferring Pharmaceuticals and Janssen Research & Development; personal fees as a compensated consultant from Merck, Sanofi Aventis, and WCG Oncology; institutional grants to Memorial Sloan Kettering Cancer Center from Illumina, Inc, Innocrin Pharma, and Janssen; and personal fees as a compensated member of the Board Of Directors from Asterias Biotherapeutics outside the submitted work.
Reviewer disclosures
A reviewer on this manuscript has disclosed that they act as a consultant for Janssen, Astellas, Bayer. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose.
References
[*of interest, ** of considerable interest]
- 1.International Agency for Research on Cancer. Prostate cancer: estimated incidence m, and prevalence worldwide GLOBOCAN Web site. [Internet]. [June 29, 2017]. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx
- 2.SEER cancer stat facts: prostate cancer National Cancer Institute, Bethesda, MD: [Internet]. [July 6, 2017]. Available from: https://seer.cancer.gov/statfacts/html/prost.html [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 4.Scher HI, Solo K, Valant J, et al. Prevalence of prostate cancer clinical states and mortality in the United States: estimates using a dynamic progression model. PLoS One 2015;10(10):e0139440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Heller G. Clinical states in prostate cancer: toward a dynamic model of disease progression. Urology 2000;55(3):323–7. [DOI] [PubMed] [Google Scholar]
- 6.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351(15):1502–12. [DOI] [PubMed] [Google Scholar]
- 7.de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010;376(9747):1147–54. [DOI] [PubMed] [Google Scholar]
- 8.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363(5):411–22. [DOI] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rathkopf DE, Smith MR, de Bono JS, et al. Updated interim efficacy analysis and long-term safety of abiraterone acetate in metastatic castration-resistant prostate cancer patients without prior chemotherapy (COU-AA-302). Eur Urol 2014;66(5):815–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368(2):138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol 2015;16(2):152–60. [DOI] [PubMed] [Google Scholar]
- 13.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371(5):424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012;367(13):1187–97. [DOI] [PubMed] [Google Scholar]
- 15.Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med 2013;369(3):213–23. [DOI] [PubMed] [Google Scholar]
- 16.James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med 2017;377(4):338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med 2017;377(4):352–360. [DOI] [PubMed] [Google Scholar]
- 18.Janssen Biotech I Zytiga (abiraterone acetate) tablets. Prescribing information Horsham, PA: February 2018. [Google Scholar]
- 19.National Comprehensive Cancer Network. Prostate cancer (Version 1.2018) [Internet] 2018. [March 13, 2018]. Available from: https://ww.nccn.org/
- 20.Mottet N, Bellmunt J, Briers E, et al. EAU-ESTRO-SIOG guidelines on prostate cancer [Internet] 2016. [updated March 2016;February 28, 2018]. Available from: https://uroweb.org/wp-content/uploads/EAU-Guidelines-Prostate-Cancer-2016-Pocket.pdf
- 21.McKay RR, Montgomery B, Xie W, et al. Post prostatectomy outcomes of patients with high-risk prostate cancer treated with neoadjuvant androgen blockade. Prostate Cancer Prostatic Dis 2017. [DOI] [PMC free article] [PubMed]
- 22.Taplin ME, Montgomery B, Logothetis CJ, et al. Intense androgen-deprivation therapy with abiraterone acetate plus leuprolide acetate in patients with localized high-risk prostate cancer: results of a randomized phase II neoadjuvant study. J Clin Oncol 2014;32(33):3705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Graham L, Schweizer MT. Targeting persistent androgen receptor signaling in castration-resistant prostate cancer. Med Oncol 2016;33(5):44. [DOI] [PubMed] [Google Scholar]
- 24.Karantanos T, Evans CP, Tombal B, et al. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol 2015;67(3):470–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung ME, Ouk S, Yoo D, et al. Structure-activity relationship for thiohydantoin androgen receptor antagonists for castration-resistant prostate cancer (CRPC). J Med Chem 2010;53(7):2779–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tran C, Ouk S, Clegg NJ, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324(5928):787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hussain M, Fizazi K, Saad F, et al. PROSPER: A phase 3, randomized, double-blind, placebo (PBO)-controlled study of enzalutamide (ENZA) in men with nonmetastatic castration-resistant prostate cancer (M0 CRPC). J Clin Oncol 36, 2018. (suppl 6S; abstr 3). [Google Scholar]
- 28.Safety and efficacy study of enzalutamide plus leuprolide in patients with nonmetastatic prostate cancer (EMBARK) ClinicalTrials.gov website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02319837.
- 29.A study of enzalutamide plus androgen deprivation therapy (ADT) versus placebo plus ADT in patients with metastatic hormone sensitive prostate cancer (mHSPC) (ARCHES) ClinicalTrials.gov website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02677896.
- 30.Phase III radium 223 mCRPC-PEACE III (PEACE III) ClinicalTrials.gov website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02194842.
- 31.Enzalutamide with or without abiraterone and prednisone in treating patients with castration-resistant metastatic prostate cancer [Internet] [June 19, 2018]. Available from: https://clinicaltrials.gov/ct2/show/NCT01949337.
- 32.Fizazi K, Massard C, Bono P, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol 2014;15(9):975–85. [DOI] [PubMed] [Google Scholar]
- 33.Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep 2015;5:12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Efficacy and safety of BAY1841788 (ODM-201) in men with high-risk nonmetastatic castration-resistant prostate cancer (ARAMIS) [Internet] ClinicalTrials.gov. [February 28, 2018]. Available from: https://clinicaltrials.gov/show/NCT02200614.
- 35.ODM-201 in addition to standard ADT and docetaxel in metastatic castration sensitive prostate cancer (ARASENS) [Internet] ClinicalTrials.gov. [February 28, 2018]. Available from: https://clinicaltrials.gov/show/NCT02799602.
- 36.Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 2012;72(6):1494–503.**Provides experimental data on the mechanism of action of apalutamide.
- 37.Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov 2013;3(9):1020–9. [DOI] [PubMed] [Google Scholar]
- 38.Phase 1/2A study of TRC253, an androgen receptor antagonist, in metastatic castration-resistant prostate cancer patients [Internet] ClinicalTrials.gov. [March 19, 2018]. Available from: https://clinicaltrials.gov/show/NCT02987829.
- 39.Rathkopf DE, Morris MJ, Fox JJ, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol 2013;31(28):3525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Study to assess drug-drug interaction between itraconazole or gemfibrozil and JNJ-56021927 [Internet] ClinicalTrials.gov. [March 13, 2018]. Available from: https://clinicaltrials.gov/show/NCT02230033.
- 41.A study to evaluate the effect of multiple doses of JNJ-56021927 on the pharmacokinetics of multiple cytochrome P450 and transporter substrates in participants with castration-resistant prostate cancer [Internet] ClinicalTrials.gov. [February 28, 2018]. Available from: https://clinicaltrials.gov/show/NCT02592317.
- 42.Rathkopf DE, Antonarakis ES, Shore ND, et al. Safety and antitumor activity of apalutamide (ARN-509) in metastatic castration-resistant prostate cancer with and without prior abiraterone acetate and prednisone. Clin Cancer Res 2017;23(14):3544–3551.*Provides evidence of apalutamide activity in the mCRPC setting.
- 43.Smith MR, Antonarakis ES, Ryan CJ, et al. Phase 2 study of the safety and antitumor activity of apalutamide (ARN-509), a potent androgen receptor antagonist, in the high-risk nonmetastatic castration-resistant prostate cancer cohort. Eur Urol 2016;70(6):963–970.*Provides evidence of apalutamide activity in the nmCRPC setting.
- 44.Scher HI, Morris MJ, Stadler WM, et al. Trial Design and Objectives for Castration-Resistant Prostate Cancer: Updated Recommendations From the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol 2016;34(12):1402–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rathkopf DE, Smith MR, Ryan CJ, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol 2017;28(9):2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med 2018; 378(15):1408–1418.**Provides evidence of apalutamide activity in the nmCRPC setting that led to the first FDA-approved treatment for nmCRPC and the first to be based on the endpoint of metastasis-free survival.
- 47.Shahinian VB, Kuo YF, Freeman JL, et al. Risk of fracture after androgen deprivation for prostate cancer. N Engl J Med 2005;352(2):154–64. [DOI] [PubMed] [Google Scholar]
- 48.Smith MR, Lee WC, Brandman J, et al. Gonadotropin-releasing hormone agonists and fracture risk: a claims-based cohort study of men with nonmetastatic prostate cancer. J Clin Oncol 2005;23(31):7897–903. [DOI] [PubMed] [Google Scholar]
- 49.Janssen Products L Erleada (apalutamide) tablets. Prescribing information Horsham, PA: February 2018. [Google Scholar]
- 50.U.S. Food & Drug Administration. FDA approves new treatment for a certain type of prostate cancer using a novel clinical trial endpoint US FDA website. [Internet]. 2018. [February 26, 2018]. Available from: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm596768.htm
- 51.Antonarakis ES, Armstrong AJ, Dehm SM, et al. Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis 2016;19(3):231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antonarakis ES, Chandhasin C, Osbourne E, et al. Targeting the N-Terminal Domain of the Androgen Receptor: A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 2016;21(12):1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galletti G, Leach BI, Lam L, et al. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat Rev 2017;57:16–27. [DOI] [PubMed] [Google Scholar]
- 54.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015;15(12):701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ipatasertib plus abiraterone plus prednisone/prednisolone, relative to placebo plus abiraterone plus prednisone/prednisolone in adult male patients with metastatic castrate-resistant prostate cancer (IPATential150) [Internet] ClinicalTrials.gov. [March 19, 2018]. Available from: https://clinicaltrials.gov/show/NCT03072238.
- 56.Teo MY, O’Shaughnessy MJ, McBride SM, et al. Drug development for noncastrate prostate cancer in a changed therapeutic landscape. Nat Rev Clin Oncol 2018;15(3):168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.An efficacy and safety study of apalutamide (JNJ-56021927) in combination with abiraterone acetate and prednisone versus abiraterone acetate and prednisone in participants with chemotherapy-naive metastatic castration-resistant prostate cancer (mCRPC) ClinicalTrials.gov website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02257736.
- 58.A study of apalutamide (JNJ-56021927, ARN-509) plus androgen deprivation therapy (ADT) versus ADT in participants with mHSPC (TITAN) ClinicalTrials.gov. website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02489318.
- 59.An efficacy and safety study of JNJ-56021927 (Apalutamide) in high-risk prostate cancer subjects receiving primary radiation therapy: ATLAS. ClinicalTrials.gov website [Internet]. [February 26, 2018]. Available from: https://clinicaltrials.gov/show/NCT02531516.
- 60.Small EJ, Saad F, Chowdhury S, et al. SPARTAN, a phase 3 double-blind, randomized study of apalutamide (APA) versus placebo (PBO) in patients (pts) with nonmetastatic castration-resistant prostate cancer (nmCRPC). J Clin Oncol 36, 2018. (suppl 6S; abstr 161). [Google Scholar]