

Summary
The incidence of hepatocellular carcinoma (HCC) is rapidly increasing due to the prevalence of obesity and non-alcoholic fatty liver disease, but the molecular triggers that initiate disease development are not fully understood. We demonstrate that mice with targeted loss of function point mutations within the AMP-activated protein kinase (AMPK) phosphorylation sites acetyl-CoA carboxylase 1 (ACC1 Ser79Ala) and ACC2 (ACC2 Ser212Ala) have increased liver de novo lipogenesis (DNL) and liver lesions. The same mutation in ACC1 also increases DNL and proliferation human liver cancer cells. Consistent with these findings, a novel, liver specific ACC inhibitor (ND-654), that mimics the effects of ACC phosphorylation, inhibits hepatic DNL and the development of HCC, improving survival of tumor-bearing rats when used alone and in combination with the multi-kinase inhibitor sorafenib. These studies highlight the importance of DNL and dysregulation of AMPK-mediated ACC phosphorylation in accelerating HCC and the potential of ACC inhibitors for treatment.
Keywords: Non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), fructose, fibrosis, inflammation, malonyl-CoA, cancer metabolism
Graphical Abstract
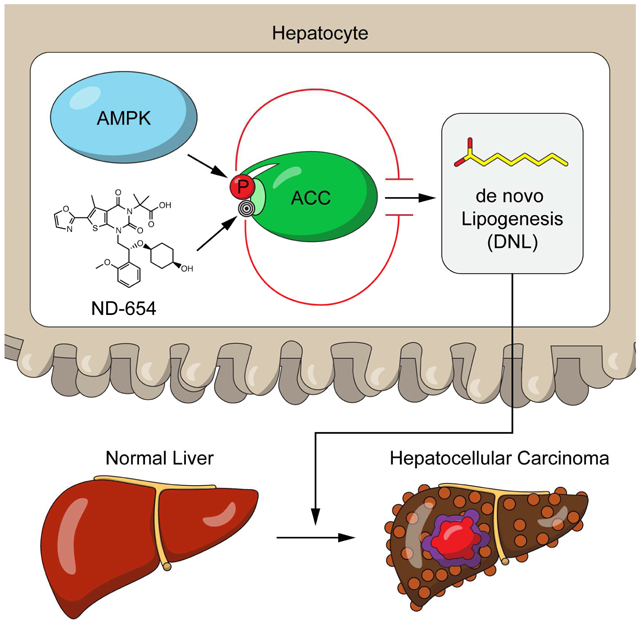
eTOC blurb
Effective therapies are needed for treating hepatocellular carcinoma (HCC). Lally et al. report that fructose consumption and the genetic activation of acetyl-CoA carboxylase (ACC) increase hepatic de novo lipogenesis (DNL) and liver carcinogenesis. The liver-specific ACC inhibitor, ND-654, is found to suppress hepatic DNL, inflammation and HCC development.
Introduction
Liver cancer now accounts for almost 750 000 deaths annually and hepatocellular carcinoma (HCC) is predominant (Torre et al., 2015). Unfortunately, the global incidence of HCC is rapidly increasing, an effect that is attributed in part to the obesity epidemic and subsequent development of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) (Baffy et al., 2012). Despite the growing prevalence of HCC, Sorafenib, a multi-kinase inhibitor which extends life in individuals with advanced HCC by approximately 2-3 months, is currently the only approved first-line therapy (Llovet et al., 2008). Thus, identifying novel and effective therapeutic targets is of critical importance.
Elevated liver de novo lipogenesis (DNL) is a key contributing factor in the development of NAFLD (Donnelly et al., 2005) and many cancers including HCC (Calvisi et al., 2011; Yahagi et al., 2005). Consumption of diets that are high in refined carbohydrate, particularly fructose, increases DNL and, consistent with this finding, also accelerates the development of NAFLD (Rahman et al., 2016) and HCC in mice (Kumamoto et al., 2013) and potentially in humans (Fedirko et al., 2013). One of the central enzymes controlling DNL is acetyl-CoA carboxylase (ACC). ACC facilitates the conversion of acetyl-CoA to the metabolic intermediate malonyl-CoA. Malonyl-CoA is the first committed substrate for DNL and is also an inhibitor of fatty acid oxidation, due to its allosteric inhibition of carnitine palmitoyltransferase-1. Thus, ACC is vital for controlling the flux of carbon intermediates between carbohydrate and fatty acid metabolism (McGarry et al., 1978). Two mammalian isoforms of ACC exist and the enzymatic activity of both human ACC1 and ACC2 can be inhibited by phosphorylation at serine 80 and serine 221 (serine 79 and serine 212 in mice), respectively, by the cellular energy sensor AMP-activated protein kinase (AMPK) (Fullerton et al., 2013). Importantly, in mice with targeted knock-in (KI) mutations in which the serine phosphorylation sites on ACC1 (S79) and ACC2 (S212) are converted to alanine (known herein as ACC KI mice), there is a loss of AMPK-mediated ACC inhibition, elevated hepatic malonyl-CoA, elevated hepatic lipogenesis and early signs of NAFLD and fibrosis development (Fullerton et al., 2013). Studies have shown that inhibition of ACC reduces cell proliferation in some cancers (Wang et al., 2010), but the role of AMPK phosphorylation of ACC in HCC is currently unknown. This is important because AMPK phosphorylation of ACC is reduced with type 2 diabetes and obesity(Ruderman et al., 2013), conditions known to increase NAFLD and HCC risk, and new small molecules that mimic the effects of AMPK on ACC have recently been developed for the treatment of NAFLD/NASH (Harriman et al., 2016) and non-small cell lung cancer (NSCLC) (Svensson et al., 2016).
Results and Discussion
To examine the role of ACC phosphorylation in controlling HCC development, ACC KI and wildtype (WT) mice were maintained on a control chow diet with or without fructose supplemented in the drinking water to enhance rates of DNL. An elevated respiratory quotient (RQ) is indicative of high rates of whole-body DNL (Hellerstein et al., 1996) and under standard chow fed conditions there were no differences in RQ (Figure 1A), activity levels, food and drink consumption, VO2, and VCO2 between WT and ACC KI mice (Supplementary Table 1). As anticipated, fructose treatment increased the RQ of WT mice during the dark (feeding cycle), an effect that was more dramatic in ACC KI mice (Figure 1A) and that was not due to differences in food and water intake or activity levels, which were comparable to WT controls (Supplementary Table 1). Consistent with the increase in RQ, fructose increased the expression of the DNL proteins fatty acid synthase (FASN), ATP citrate lyase (ACLY) and ACC in the livers of WT and ACC KI mice (Figure 1B). Fructose treatment also decreased the activating phosphorylation of AMPK at Thr172 in both WT and ACC KI mice; however, the loss of ACC phosphorylation in ACC KI mice was the only detectable difference between genotypes (Figure 1B & 1C). Fructose fed ACC KI mice also increased liver lipogenesis in vivo (Figure 1D), indicating that AMPK phosphorylation of ACC is vital to suppressing DNL.
Figure 1. AMPK phosphorylation of ACC is vital for limiting hepatocarcinogenesis and cellular proliferation.

(A) The RQ of WT and ACC KI mice during the dark cycle (7 pm- 7 am) after being maintained on either a chow diet alone or chow diet plus fructose for 4 months (n= 4 WT chow, n=5 WT fructose, n=7 ACC KI chow, n=7 ACC KI fructose). (B) and (C) Protein levels of FASN, ACLY, ACC total protein, ACC phospho-Ser79, AMPK α total protein, AMPK α phospho-Thr172 and β-actin in the liver from WT and ACC KI mice (n= 4 WT chow, n=5 WT fructose, n=6 ACC KI chow, n=6 ACC KI fructose). (D) Incorporation of [3H]acetate into total hepatic lipid (n=4). Representative images (E), diameter (F) and number (G) of hepatic lesions in photomicrographs of livers from DEN-treated ACC KI and WT mice maintained on fructose diet for 4 months (Haematoxylin and eosin stain, bar represents 1000 μm, n= 8 WT, n=6 ACC KI).
* significantly different WT fructose vs ACC KI fructose, p < 0.05
** significantly different from WT, p < 0.05
*** main effect of diet, p < 0.05
When mice were injected with the HCC initiator diethylnitrosamine (DEN), which promotes aspects of the human disease (Fuchs et al., 2014), both WT and ACC KI mice had indications of hepatocarcinogenesis, including the presence of altered hepatocyte foci, hyperplastic nodules and hepatocellular adenomas (Figure 1E). Importantly, despite similar sized lesions (Figure 1F). ACC KI mice had twice as many lesions per liver as WT controls (Figure 1G). This increase in the number of lesions was independent of alterations in factors known to accelerate tumorigenesis, including adiposity, liver triglyceride, insulin resistance, inflammatory cytokines, and markers of liver fibrosis, all of which were comparable between genotypes (Supplementary Figure 1A-H). To examine whether the increase in adenomas in ACC KI mice may be due to altered DEN metabolism, DEN-induced 8-hydroxydeoxyguanosine (8-OHdG) DNA adducts levels were assessed in the liver of WT and ACC KI mice 24 hours after intraperitoneal injection and found not to be different between genotypes (Supplementary Figure 1I). These data indicate that AMPK phosphorylation of ACC is vital for restraining the development of hepatocarcinogenesis.
Recently, the discovery of a new class of potent, highly specific, isozyme-nonselective, allosteric, protein-protein interaction ACC inhibitors has been reported.(Harriman et al., 2016) These compounds interact within the phosphopeptide-acceptor and subunit dimerization site of the biotin carboxylase (BC) domain of both ACC1 and ACC2 to prevent dimerization and inhibit enzymatic activity. The first of these drugs, GS-0976, was shown to reduce hepatic steatosis in rats with diet-induced obesity (Harriman et al., 2016) and is now under investigation in clinical trials of NASH (). The second, ND-646, was recently shown to inhibit the growth of NSCLC (Svensson et al., 2016). To further examine the role of ACC in hepatocarcinogenesis, we utilized a third compound in this series, ND-654 (structure shown in Figure 2A inset), for the following studies.
Figure 2. ND-654 selectively targets the liver and inhibits HCC proliferation.

(A inset) The structure of ND-654. (A-B) Rats were treated with a single oral dose of 10 mg/kg ND-654 and the concentration of ND-654 was measured (A) after 1 hour in the liver, muscle and plasma and (B) over 8 hours in the liver and plasma. (C-D) Rats were treated with a single oral dose of different concentrations of ND-654 (0.3, 3, and 30 mg/kg) and the presence of malonyl CoA was determined after 1 hour in (C) the liver and (D) muscle. (E-M) Male Wistar rats were separated into three groups (n = 8 per group). The first group received weekly intraperitoneal (IP) injections of PBS as control for 18 weeks. The second group received weekly IP injections of DEN (50 mg/kg diluted in PBS) for 18 weeks. The third group received weekly IP injections of DEN for 18 weeks as above and were also treated with ND-654 (10 mg/kg) once daily by oral gavage beginning at 15 weeks. In the DEN model, rats develop liver fibrosis after 8 weeks which progresses to cirrhosis at 13 weeks and HCC beginning at 15 weeks. (E) Representative images of gross livers are shown. (F) Tumor nodules ≥5 mm were counted. (G) Liver weight (LW) as a percentage of body weight (BW) was measured at the end of the study. (H) Representative images of H&E, proliferating cell nuclear antigen (PCNA; proliferative marker) and cleaved caspase-3 (apoptosis marker) staining of tumor are shown (100X magnification). The left column shows a representative tumor from the DEN group, the middle column and right columns show representative tumors from the DEN + ND-654 (10 mg/kg) group with reduced proliferation and extensive necrosis (N), respectively. (I) The Histological Activity Index (HAI) and (J) the presence of neutrophils were scored blindly by a GI pathologist. (K) Palmitate levels were measured. (L) Representative images of myeloperoxidase (MPO) staining are shown. (M) Inflammation-related gene expression in liver tissue was quantified.
* significantly different from PBS, p < 0.05
** significantly different from DEN, p < 0.05
ND-654 inhibits human ACC1 with an IC50 of 3 nM, inhibits human ACC2 with an IC50 of 8 nM, inhibits HepG2 cell fatty acid synthesis with an EC50 of 14 nM, and reduces both rat hepatic malonyl-CoA and rat hepatic fatty acid synthesis with ED50 values of 0.34 mg/kg and 0.30 mg/kg at Cmax, respectively using previously described experimental protocols (Harriman et al., 2016). As compared to ND-646, which is broadly distributed (Svensson et al., 2016), ND-654 has been modified to allow for enhanced hepatic uptake. To confirm hepatoselective delivery of ND-654, rats were first treated with a single oral dose of 10 mg/kg ND-654 and the concentration of ND-654 was measured in the liver, muscle and plasma after 1 hour (Figure 2A). Under these conditions, the liver concentration of ND-654 was ~2700 fold higher than that of muscle and ~100 fold higher than that of plasma. Next, rats were treated with a single oral dose of 10 mg/kg ND-654 and the concentration of ND-654 was measured over 8 hours in the liver and plasma (Figure 2B). Concentrations in the plasma were in the nanomolar range and slowly decreased over time, while liver concentrations declined over time but still remained above 1 μM even after 8 hours. To assess the effects of the hepatoselectivity of ND-654 on ACC-mediated activity in hepatic versus extrahepatic tissues, rats were treated with a single oral dose of different concentrations of ND-654 (0.3, 3, and 30 mg/kg), with liver and skeletal (gastrocnemius) muscle malonyl CoA determined 1 hour later as a measure of ACC inhibition (Figure 2C,D). While there was no change in malonyl CoA concentration in the muscle, malonyl CoA levels in the liver were reduced dose-dependently by up to 80% (ED50 = 0.34 mg/kg). These data illustrate that ND-654 is a potent ACC inhibitor which selectively targets the liver.
We next tested whether ND-654 could inhibit hepatocarcinogenesis in a rat model of sequential cirrhosis and HCC that we have previously shown to resemble aspects of the human disease at the biochemical, histological, and molecular level (Fuchs et al., 2014). Oral administration of ND-654 showed dose-dependent increases in plasma and liver pharmacokinetics achieving approximately 40-fold higher liver levels than plasma at the maximal concentration studies (Supplementary Figure 2A). Repeated, weekly intraperitoneal administration of 50 mg/kg DEN in rats causes progressive inflammation and fibrosis followed by development of cirrhosis at 12 weeks and HCC around 15 weeks. In our first study, DEN-injured rats were administered 10 mg/kg ND-654 daily by oral gavage beginning at the start of week 15, when HCCs are first apparent, and were sacrificed at the beginning of week 19. During this short-term administration of ND-654, we observed no significant differences in group mean body weight among DEN-treated animals, although administration of DEN did significantly reduce body weight compared to PBS controls (Supplementary Figure 2B). Likewise, while the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) were elevated in the serum of DEN-injured rats compared to control animals, ND-654 treatment did not cause further elevations (Supplementary Figure 2C,D). Consistent with the inhibition of ACC (Harriman et al., 2016), liver and serum triglyceride levels were reduced with ND-654 (Supplementary Figure 2E,F). Overall, these data demonstrate that ND-654 was effectively delivered to the liver and was well-tolerated in cirrhotic rats.
After 4 weeks of treatment, we counted and measured surface tumors that were ≥ 5 mm, as nodules of this size have previously been shown to be predominantly HCCs in this model (DePeralta et al., 2016; Fuchs et al., 2014; Nakagawa et al., 2016). ND-654 reduced tumor burden in DEN-injured rats by 55%, as the number of tumor nodules decreased from 15.8 ± 1.9 (SEM, n=8) in DEN animals to 7.1 ± 1.3 (SEM, n=8) in DEN animals treated with ND-654 (Figure 2E,F). Liver weight as a percentage of body weight decreased from 6.9 ± 0.5% (SEM, n=8) in DEN animals to 5.1 ± 0.7% (SEM, n=8) in those that received ND-654 (Figure 2G). The reduction in tumor burden was not attributed to decreased DEN metabolism and subsequent DNA damage in response to ND-654 treatment as ND-654 did not inhibit CYP2E1 (the major cytochrome responsible for DEN metabolism (Kang et al., 2007)) and DEN-induced 8-OHdG DNA adduct levels were not significantly different in DEN animals treated with vehicle control or ND654 (Supplementary Figures 2G,H).
As previously shown for ND-630 (Harriman et al., 2016) and ND-646 (Svensson et al., 2016), ND-654 also binds to an arginine residue in the BC domain of the ACC protein (Arg172 in ACC1, Arg 277 in ACC2) to inhibit both dimerization and enzymatic activity. Since this is the same arginine residue in which the AMPK-phosphorylated p-Ser interacts with to inhibit ACC dimerization and enzymatic activity, ND-654 mimics the physiological inhibition of AMPK-induced ACC phosphorylation. Also, as has recently been shown for ND-646 (Svensson et al., 2016), by interacting with this arginine residue, ND-654 promotes constitutive dephosphorylation of this AMPK-phosphorylated serine, and therefore decreased p-ACC levels as a consequence of ND-654 treatment have been proposed as a useful biomarker to assess effective drug targeting (Svensson et al., 2016). In this regard, we observed decreased p-ACC/ACC in both the surrounding cirrhotic liver and tumor nodules from drug-treated animals (Supplementary Figure 2I,J). However, proliferation, as assessed by proliferating cell nuclear antigen (PCNA) protein levels, only decreased in the tumors in the ND-654 group (Supplementary Figure 2I,J). In addition, PCNA staining decreased in tumors in the ND-654 group (Figure 2H, middle row panels) and necrosis was also observed in some tumors from the ND-654 group (Figure 2H, right panels) but there was no evidence of apoptosis as assessed by cleaved caspase-3 staining (Figure 2H lower row panels).
Even though we did not see an increase in inflammatory markers in the ACC KI mice, we did investigate the effects of ND-654 on the chronic inflammation present in the DEN rat model. The extent of inflammation in the DEN-injured rats, as assessed by the Ishak HAI score, was significantly decreased after treatment with ND-654 (Figure 2I). Specifically, the presence of neutrophils in the lobular and portal regions, as distinguished by their segmented nuclei, was significantly diminished after ND-654 treatment (Figure 2J). Palmitic acid is the first fatty acid produced during de novo lipogenesis, and palmitate levels were increased in the livers of DEN-injured rats (Figure 2K). Palmitic acid has been shown to induce pro-inflammatory cytokine production in the liver including the expression of IL-8/Cxcl1 which is known to recruit neutrophils (Joshi-Barve et al., 2007). Treatment with ND-654 reduced palmitate levels (Figure 2K) and decreased expression of pro-inflammatory cytokines including Cxcl1, the rodent equivalent of IL-8 (Figure 2M).
Myeloperoxidase (MPO) has been reported to be a specific marker for neutrophils in the rat liver (Amanzada et al., 2011) although we observed staining of both neutrophils in the sinusoids and some of the resident macrophages in normal rat liver (Figure 2L). After DEN injury, MPO staining of neutrophils dramatically increased in the lobules which would be characteristic of clinical "active hepatitis". By comparison, ND-654 treatment reduced MPO staining of neutrophils in the lobules but increased MPO staining of macrophages in the portal region which is characteristic of "resolving hepatitis". Consistently, we observed decreased expression of CD80, a marker for pro-inflammatory M1 macrophages, and increased expression of CD163, a marker for anti-inflammatory M2 macrophages, after ND-654 treatment (Figure 2M). Thus, ACC inhibition not only directly decreased tumor proliferation, but also reduced neutrophil recruitment, and promoted a switch to restorative M2 macrophages.
Given these promising results, we repeated the study to determine whether ND-654 could increase survival in this lethal model of cirrhosis-driven HCC. For this experiment, DEN-injured rats received vehicle control, 10 mg/kg ND-654, or 30 mg/kg ND-654 by daily oral gavage beginning at the start of week 14. ND-654 markedly and significantly increased the median survival from 114 ± 3.3 (SEM, n=10) days in vehicle controls to 126 ± 4.6 (SEM, n=10) days in the 10 mg/kg group (p = 0.02) and 129 ± 2.7 (SEM, n=10) days in the 30 mg/kg group (p = 0.009) (Figure 3A). Specifically, at day 115, vehicle-treated animals showed 60% mortality whereas ND654-treated animals showed 100% survival and at day 125, vehicle-treated animals showed 90% mortality where ND654-treated animals showed 70% survival. There was no significant difference among the 10 and 30 mg/kg groups and in fact three animals in the 10 mg/kg group lived the longest, suggesting that ND-654 at a dose of 10 mg/kg/day demonstrates maximal survival efficacy in this model (Figure 3A).
Figure 3. ND-654 improves survival and the efficacy of sorafenib in cirrhotic rats with HCC.

(A) Male Wistar rats were separated into three groups (n = 10 per group). The first group received weekly IP injections of DEN (50 mg/kg diluted in PBS), the second and third groups received weekly IP injections of DEN as above and were also treated with either 10 or 30 mg/kg ND-654 once daily by oral gavage beginning at 14 weeks. Survival was examined by a Kaplan-Meier analysis. For combination studies, DEN-injured rats received vehicle control (VEH), 10 mg/kg ND-654 (ND-654), 10 mg/kg sorafenib (SOR), or the combination of 10 mg/kg ND-654 and 10 mg/kg sorafenib (COMBO) by oral gavage for 5 weeks. (B) Representative images of whole livers are shown. (C) Representative images of H&E stainings of tumor illustrating increased necrosis (N) in treated tumors (100X magnification). (D) Tumor nodules ≥5 mm were counted. (E and F) Levels of phosphorylated ACC (p-ACC), total ACC, proliferating cell nuclear antigen (PCNA), and actin were measured by western blot analysis in (E) liver tissue and (F) tumors (representative blot from 3 independent experiments).
* significantly different from PBS, p < 0.05
** significantly different from DEN, p < 0.05
**** significantly different from DEN, DEN + ND-654, and DEN + SOR, p < 0.05
Patients assigned to a control arm of HCC clinical trials receive standard-of-care therapy, which is currently sorafenib for patients with advanced-stage disease. We therefore examined whether ND-654 could increase the efficacy of sorafenib in the DEN rat model. For this experiment, DEN-injured rats received vehicle control, 10 mg/kg ND-654, 10 mg/kg sorafenib, or the combination of 10 mg/kg ND-654 and 10 mg/kg sorafenib by oral gavage daily beginning at the start of week 13. ND-654 and sorafenib had similar efficacy in reducing HCC incidence (the number of tumor nodules was 16.4 ± 2.5 (SEM, n=10) in the control group compared to 9.7 ± 1.5 (SEM, n=10) in the ND-654 group (a 41% decrease) and 7.0 ± 1.5 (SEM, n=10) in the sorafenib group (a 57% decrease)) (Figure 3B-D). While both compounds caused tumor necrosis (Figure 3C), the combination of sorafenib and ND-654 was especially effective at reducing HCC incidence by 81% (the number of tumor nodules was 3.1 ± 0.8 (SEM, n=10) in the ND-654 + sorafenib group) (Figure 3 B-D). Sorafenib led to a reduction in liver weight as a percent of body weight, which was associated with a marked ductular reaction, an effect that was maintained when combined with ND-654 (COMBO) despite modest reduction in body mass in this group (Supplementary Figure 3A-C). While sorafenib increased serum triglycerides (Supplementary Figure 3D), the addition of ND-654 reduced their levels (Supplementary Figure 3D) and also decreased p-ACC levels in both liver and tumor tissue as observed in the first study (Figure 3E-F). Finally, the combination of sorafenib and ND-654 also effectively decreased tumor proliferation as assessed by PCNA protein levels (Figure 3F).
To establish whether AMPK phosphorylation of ACC was also important for modulating the proliferative capacity of cultured human liver cancer cells, the ACC KI mutation was generated in HepG2 cells using CRISPR-Cas9 (Supplementary Figure 4A). Sequencing analysis indicated successful insertion of the ACC1 S80A KI mutation (equivalent to S79A in mice) into the ACC KI HepG2 cells but not control cells. When the HepG2 cells were stimulated with the potent AMPK activator phenformin, AMPKα Thr172 and ACC phosphorylation increased as expected in WT cells, but no ACC phosphorylation was detectable in ACC KI cells (Figure 4A), indicating the ACC KI mutation was successfully inserted and ACC2 phosphorylation was negligible in this cell line. The specificity of the mutation in ACC KI cells was evident as the total protein expression of AMPK and ACC was unaltered as was the phosphorylation of AMPK Thr172 and another AMPK substrate, unc-51 like autophagy activating kinase 1 (ULK1)(Egan et al., 2011) (Figure 4A), indicating that AMPK activity and the ability to phosphorylate other substrates was maintained. Consistent with the elevated DNL and increased hepatocarcinogenesis in ACC KI mice in vivo, HepG2 cells expressing the ACC KI mutation had a 4-fold increase in lipogenesis and 2-fold increase in proliferation (Figure 4B,C). This increase in proliferation did not appear to be the result of alterations in the phosphorylation of other growth signaling pathways, since phosphorylation of representative members of these pathways were comparable between WT and ACC KI cell lines (Supplementary Figure 4B). We also examined the effects of ND-654 on HepG2 cells. As expected, treatment with ND-654 eliminated p-ACC levels (Figure 4D) and reduced the proliferation of HepG2 cells over time (Figure 4E). Also, similar to the above-mentioned observations in the ACC KI cells, this ND-654 mediated reduction in proliferation was not associated with alterations in protein phosphorylation of representative members of other growth signaling pathways (Supplementary Figure 4C). These data indicate that inhibition of ACC activity through phosphorylation at Ser80 is vital for inhibiting HCC proliferation in a cell autonomous manner independently of alterations in other growth signaling pathways.
Figure 4. AMPK phosphorylation of ACC limits HepG2 proliferation.

(A) AMPK Thr172, ACC Ser80, and ULK1 (Ser555) phosphorylation in WT and ACC KI HepG2 cells after treatment with phenformin (2 mM) for 1 hour (representative blot from 3 independent experiments). (B) De novo lipogenesis measured by incorporation of 3[H]acetate into lipid over 1 hour (means of two independent experiments performed in triplicate) and (C) Proliferation in WT and ACC KI HepG2 cells over 72 hours (n=7). (D) HepG2 cells were treated with or without ND-654 (10 μM) for 72 hours and levels of p-ACC, total ACC, and actin were measured by western blot analysis (representative blot from 3 independent experiments). (E) Proliferation in HepG2 cells treated with or without ND-654 (10 μM) was measured at 1 day, 2 days, 4 days, and 6 days post treatment by an MTT assay (representative results from 3 independent experiments). (F) Genomic DNA structural alterations and expression of ACC genes in 374 human HCC tissues are shown according to the transcriptomic HCC subtypes (S1, S2, and S3 subtypes).
* significantly different from WT cells, p < 0.05
Since the approval of sorafenib, HCC clinical trials have largely been unsuccessful and the lack of patient selection in trial design is at least partly responsible for this failure (Goossens et al., 2015). Over the last decade, attempts have been made to define molecular subtypes of HCC and several reports have now shown that these subtypes do correlate with treatment response using in vitro and in vivo experimental systems (Finn et al., 2013; Fuchs et al., 2008; Schmidt et al., 2016; Zhao et al., 2012). We recently described a transcriptomic HCC subtyping system that is highly reproducible between global patient populations and divides HCC into three major subtypes termed S1, S2 and S3 (Hoshida et al., 2009; Tan et al., 2016). Both S1 and S2 tumors are associated with more aggressive biological properties and worse prognosis compared to S3 tumors. To explore ACC pathway-related molecular aberrations in human HCC, we analyzed somatic genomic DNA copy number alterations, mutations, and the transcriptomic subtypes in 374 human HCC tissues. Somatic DNA amplification and mutations in the ACC1 (ACACA) and ACC2 (ACACB) genes were only observed in 17 and 12 tumors (4.5% and 3.2%), respectively, and did not associate with their gene expression nor any of the HCC subtypes, indicating irrelevance of genomic DNA structural alterations during pathogenic ACC pathway activation (Figure 4F). In contrast, S2 tumors, which express several traditional HCC biomarkers like AFP and GPC-3,(Hoshida et al., 2009; Tan et al., 2016) had high ACC1 expression, suggesting that these tumors are readily identifiable in the clinic and might be an attractive target for ACC inhibition.
In conclusion, our studies show that genetic alterations resulting in reduced phosphorylation of ACC augment HCC development in mice and proliferation of human liver cancer cells. Importantly, we also establish that pharmacological targeting of this pathway using ND-654, a novel liver directed small molecule ACC allosteric inhibitor, is effective in reducing HCC and improving survival in rats and that this molecule has additive effects to the current standard of care, sorafenib. While mutations in AMPK or ACC in HCC have not been described, it is well documented that both obesity and type 2 diabetes, two primary risk factors for developing HCC, reduce AMPK activity and the phosphorylation of ACC (Ruderman et al., 2013) as well as increase rates of liver DNL (Donnelly et al., 2005). These data suggest that therapies aimed at mimicking the effects of AMPK phosphorylation on ACC using small molecules such as ND-654 may be valuable for treating HCC particularly in patients in the S2 subclass.
Limitations of Study
One potential limitation of this study is the use of DEN as the sole mechanism for HCC initiation in both the mouse and rat models. In the mouse study, DEN was given as a single injection to juvenile mice where it is metabolized by the proliferating hepatocytes in the post-natal liver into alkylating agents that cause oncogenic DNA adducts resulting in dysplastic lesions that can progress to HCC. Under these commonly-employed conditions, mice develop HCCs in the setting of a relatively normal liver with little inflammation or fibrosis similar to many transgenic models. By comparison, most human HCCs arise in the setting of cirrhosis (Llovet et al., 2016). Since mice do not tolerate repeated injections of DEN, we also used repeated, low-dose DEN injections to rats to generate a disease that more closely resembles the human pathology, with development of fibrosis, and then cirrhosis, and then HCC, to examine the effects of ACC inhibitors on HCC. Histologically and biochemically, this model more closely resembles human disease (Fuchs et al., 2014). In addition, compared to several other models, this model is most reflective of chronic human liver disease at the global transcriptomic level (Nakagawa et al., 2016). While no model can fully recapitulate all aspects of the human disease, the ongoing inflammation and fibrogenesis in the rat DEN model, accompanied with their growth-promoting and immunosuppressive microenvironment, are critical components of the human disease that are absent from many HCC models. We believe that examination of putative therapeutics, like ND-654, under these conditions may ultimately lead to better clinical translation.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bryan C. Fuchs (bfuchs@mgh.harvard.edu).
EXPERIMENTAL MODELS
Mice
ACC KI mice were generated by intercrossing ACC1-S79A KI and ACC2-S212A KI mice, made by OzGene Pty Ltd, (Perth, Australia) using standard methods, on a C57Bl6 background (for detailed recombination strategy see (Fullerton et al., 2013)). All experiments were approved by the Animal Research Ethics Board at McMaster University. Mice were housed in specific pathogen free micro isolator cages maintained under a 12 hour light/dark cycle at a constant temperature of 23°C. All animals were maintained on a control chow diet (Teklad 22/5 rodent diet, Envigo, Mississauga, ON). Mice were allowed access to food and water ad libitum.
HCC Mouse Model
Male mice were chosen for these studies as females are relatively resistant to HCC development. To induce HCC in mice, in separate experiments animals were injected intraperitoneally with the chemical carcinogen diethylnitrosamine (DEN, Sigma, St. Louis, MO) at a concentration of 25 mg/kg body weight at 2 weeks of age (Park et al., 2010). Fructose treated animals were given water supplemented with fructose (30%) as has been performed by others to accelerate HCC development (Ishimoto et al., 2012) starting at 8 weeks continuously for 16 weeks. After 6 months mice were anesthetized using ketamine/xylazine, tissues were removed and a portion was fixed in formalin or frozen in liquid nitrogen for histological and molecular/biochemical analysis.
Rats
5-6 week old male Wistar rats (Charles River Laboratories, Wilmington, MA) were housed in accordance with the guidelines of the Massachusetts General Hospital Institutional Animal Care and Use committee and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” of the National Academy of Sciences. Rats were group-housed in specific pathogen free cages maintained under a 12 hour light/dark cycle at a constant temperature of 23°C. All animals were maintained on a normal chow diet (Isopro 3000) and were allowed access to food and water ad libitum.
HCC Rat Model
Male rats were chosen for these studies as females are relatively resistant to HCC development. Rats were acclimated for 1 week before being subjected to either control PBS or low-dose 50 mg/kg DEN injected IP once per week over the course of 18 weeks. In the first study, DEN-injured rats were randomly chosen to begin treatment with either vehicle control or ND-654 10 mg/kg (n = 8 per group) by oral gavage at 15 weeks and were sacrificed at the end of 18 weeks. In the second study, DEN-injured rats were randomly chosen to begin treatment with either vehicle control, ND-654 10 mg/kg, or ND-654 30 mg/kg (n = 10 per group) by oral gavage at 14 weeks and continued treatment until death. In the third study, DEN-injured rats were randomly chosen to begin treatment with either vehicle control, ND-654 10 mg/kg, sorafenib 10 mg/kg, or ND-654 10 mg/kg + sorafenib 10 mg/kg (n = 10 per group) by oral gavage at 13 weeks and were sacrificed at the end of 18 weeks. At the time of sacrifice, rats were anesthetized using ketamine/xylazine and a terminal blood collection was performed by cardiac puncture. Livers were removed for measurement of weight, snap frozen for further analysis, or fixed in formalin for histology.
Cell lines
HepG2 (ATCC® HB-8065™) human liver cancer cells were authenticated by DNA fingerprinting with small tandem repeat (STR) profiling. HepG2 cells were originally derived from the liver tissue of a 15-year old male Caucasian with a well-differentiated hepatocellular carcinoma. HepG2 cells were propagated in DMEM (4.5 mg/mL glucose, 2 mmol/L l-glutamine) with 10% fetal bovine serum, supplemented with 100 units/mL penicillin and 100 mg/mL streptomycin (all from Life Technologies, Carlsbad, CA). Cells were maintained at 37°C in a humidified incubator with 5% CO2 in air.
The ACC1 KI mutation was introduced into HepG2 cells via transfection with gRNA/Cas9 all in one vector (custom made from GenScript, Piscataway, NJ) with (KI) and without (WT, empty vector) donor ACC1 (Acetyl-CoA carboxylase 1) Ser80Ala knock in sequence. Puromycin selection medium was added to transfected cells for 48 hrs. Pooled cells were propagated and analyzed by PCR and SURVEYOR mutation detection analysis. Clones that harboured the mutation were further selected, analyzed and purified by single cell cloning and the knock-in mutation was confirmed by sequencing. For experimental procedures, cells were maintained in MEM supplemented with 10% fetal bovine serum and 1% Antibiotic-Antimycotic (Gibco, Life Technologies).
METHOD DETAILS
Histology
For mouse studies, the major lobes of the liver were removed and fixed in 10% neutral formalin, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E). The number of altered foci and tumor nodules per liver cross section were quantified from scanned images by researchers who were blinded to the treatment groups using NIS-Elements software (Nikon Canada, Mississauga, ON). With the exception of a single WT animal all mice developed signs of hepatocarcinogenesis.
For rat studies, formalin-fixed samples were embedded in paraffin, cut into 5 µm-thick sections and stained with H&E according to standard procedures. Additional sections were stained with antibodies specific for proliferating cell nuclear antigen (PCNA, #13110), cleaved caspase-3 (#9664, both from Cell Signaling Technologies, Beverly, MA), and myeloperoxidase (MPO, #A039829-2, Agilent Technologies, Santa Clara,CA). The Ishak Histological Activity Index (HAI) was scored on H&E stained sections. In addition, neutrophils in the lobular (0: absent; 1: 0-1 foci per 10X field; 2: 2-4 foci per 10X field; 3: 5-10 foci per 10X field; 4: >10 foci per 10X field) and portal areas (0: none; 1: mild, some or all portal areas; 2: moderate, some or all portal areas; 3: moderate, all portal areas; 4: marked, all portal areas) were scored and combined to create a neutrophil score. All slides were reviewed blindly by the same liver pathologist.
Indirect Calorimetry
Physical activity levels, food and water consumption, and VO2 and VCO2 were monitored in mice using the Oxymax Comprehensive Lab Animal Monitoring System (Columbus Instruments, Columbus, OH). Mice were acclimated to the apparatus for 16 hours before data collection commenced and allowed access to control chow diet and water (control) or water containing 30 % fructose ad libitum. Respiratory quotient was calculated using RQ = VCO2/VO2. Body fat percentage was determined using a time-domain NMR whole-body composition analyzer (minispec LF90II, Bruker Biospin, Milton, ON) and normalized to body weight.
Glucose and Insulin Tolerance Tests
Mice were fasted for 6 hours starting at the beginning of the 12 hour light cycle and basal blood glucose was measured on whole blood obtained by making a small incision at the end of the tail using a handheld glucometer. Mice were given an intraperitoneal bolus of glucose (glucose tolerance test) or insulin (insulin tolerance test) at a concentration of 2 mg/kg or 0.8 IU/kg body weight respectively and blood glucose was measured at 20, 40, 60, 90 and 120 minutes thereafter.
Hepatic lipogenesis
In vivo hepatic lipogenesis was measured in mice by assessing 3[H] acetate incorporation into the total hepatic lipid fraction over the first 4 hours of the dark cycle (1800– 2200 hours) corresponding to the peak period of de novo lipogenesis as determined by indirect calorimetry experiments. Animals were fasted for the last 4 hours of the light cycle at which point 3[H] acetate in saline was injected intraperitoneally at a concentration of 0.67 μCi/g body weight. Mice were allowed to feed ad libitum in the dark for 4 hours, at which point they were anesthetized using ketamine/xylazine, a portion of liver was removed and flash frozen in liquid nitrogen for subsequent analysis. The lipid fraction was isolated using a modified Folch extraction and 3[H] incorporation was quantified. De novo lipogenesis in HepG2 WT and ACC KI cells was measured in cells that were grown to 80-90% confluence in MEM supplemented with 10% fetal bovine serum, 0.5 mM acetate and 3[H] acetate (1 μCi/ml) for 1 hour. Media was removed and cells were washed three times with ice-cold PBS, scraped and pelleted. The lipid fraction was extracted using a modified Folch method and 3[H] incorporation into the lipid fraction was determined.
Liver triglyceride measurements
Liver triglyceride levels in mice were determined using Triglyceride Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI). Liver triglyderide levels in rats were measured by Metabolon (Durham, NC).
DNA adduct measurements
8-OHdG DNA adduct levels were measured in mouse and rat liver tissue by ELISA according to the manufacturer instructions (Cell Biolabs, San Diego, CA).
Serum Collection
Blood was allowed to clot for 1 hour at room temperature before centrifugation at 2,000 rpm for 10 min. at 4°C. Serum was isolated and stored at −80°C prior to use. Serum levels of several biochemical markers including alanine transaminase (ALT), aspartate transaminase (AST), albumin (Alb), glucose (Glu), and triglycerides (TAGs) were measured as previously described (DRI-CHEM 4000 Analyzer, Heska, Switzerland) (DePeralta et al., 2016; Fuchs et al., 2014).
Cytochrome 2E1 activity
ND-654 (up to 50 μM) was incubated with human liver microsomes (0.10 mg/ml) and 1 mM NADPH in the presence of Chlorzoxazone as substrate. Sodium diethyldithiocarbamate trihydrate (DDC) was used as the positive control inhibitor and was run in parallel. The assay was designed so that conditions were linear with respect to time and protein concentration. Substrate was present at concentrations slightly lower than the Km values. The formation of 6-hydroxychlorzoxazone from Chlorzoxazone (substrate concentration = 60 μM) was followed over 20 min at 37°C. The reactions were terminated by the addition of acetonitrile containing Labetalol as an internal standard. The samples were then centrifuged, and the supernatants were analyzed by LC-MS/MS. Formic acid (final concentration = 0.1% v/v) in deionized water was added to the final sample prior to analysis. LC-MS/MS assays were performed at Gilead Sciences. The 6-hydroxychlorzoxazone metabolite plus the internal standard Labetalol were analyzed by LC-MS/MS using electrospray ionization. The Waters Acquity UPLC BEH C18 (1.7 μm, 2.1 × 50 mm) column was held at 40°C and the injection volume was 10 μL. The metabolite/internal standard PAR (Peak Area Ratio) was measured on a Waters Micromass Xevo-TQS triple quadrupole mass spectrometer coupled to a Waters Acquity binary solvent manager and a Waters/CTC PAL 2777 ultrapressure autosampler. The initial mobile phase consisted of water with 0.1% (v/v) formic acid, or acetonitrile with 0.1% (v/v) formic acid pumped at 0.6 mL/min. Elution was achieved by a series of gradients of increasing proportions of acetonitrile from 5% to 95%, with a cycle time of 4.0 minutes. Peak area ratios (PAR) for 6-hydroxychlorzoxazone and Labetalol were used to quantify the metabolites. The PAR values in the presence of inhibitors were compared to those of vehicle controls and activities expressed as the percentage of control activity remaining. Reaction velocities were calculated from the rates of formation of the metabolites and were compared to those seen with the vehicle control (100% activity). IC50 values were then calculated, using GraphPad Prism 7.03, by non-linear regression with a log-response No constraint model.
Real-time PCR
Total RNA was extracted from cells and tissue using TRIzol (Life Technologies) according to the manufacturer's instructions and subsequently treated with DNase I (Promega). Total RNA (1 μg) from each sample was used to create cDNA by single strand reverse transcription (Superscript III First-Strand Synthesis SuperMix; Life Technologies). Gene expression was analyzed by quantitative real-time PCR using TaqMan gene expression assays (Life Technologies) on an Applied Bioscience 7900HT Fast Real Time PCR System using 96 well plates or a Rotogene 6000 Real Time PCR Machine with a reaction volume of 20 μL according to the manufacturer’s instructions. mRNA relative quantification was calculated using the 2−ΔCT method normalizing to 18S or TATA binding protein. TaqMan Assay IDs are as follows: for rat studies: 18S: Hs03003631_g1; Cxcl1: Rn00578225_m1; Il-1b: Rn00580432_m1; Il-6: Rn01410330_m1; Tnfa: Rn99999017_m1; Cd80: Rn00709368; and Cd163: Rn01492519_m1, and for mouse studies: Tbp: Mm01277042_m1; Il-6 Mm00446190_m1; Tnfa Mm00443258_m1; Timpl: Mm01341361_m1; Col1a1: Mm00801666_g1; Co14a1: Mm01210125_m1; Acata2: Mm00725412_s1. All reactions were done in duplicate and the experiment was repeated to ensure reproducible results.
Transcriptomic Analysis
Somatic genomic DNA copy number alterations, mutations, and genome-wide transcriptome profiles of 374 human HCC tissues were obtained from The Cancer Genome Atlas data portal (https://gdc.cancer.gov). Transcriptomic Molecular HCC subtypes determined by our previous transcriptome meta-analysis (Hoshida et al., 2009) were determined by using Nearest Template Prediction (NTP) algorithm (Hoshida, 2010).
Western blots
To test AMPK activation and signalling, WT and KI cells were seeded in 6 wells plates, grown to 70% confluence and incubated with or without 2 mM Phenformin (Sigma) for 1 hour. Media was removed and 100 μl of lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 100 mM NaF, 10 Na-pyrophosphate, 5 EDTA mM, 250 mM sucrose, 1 mM DTT, and 1 mM Na-orthovanadate, 1% Triton X, and Complete protease inhibitor cocktail (Roche Diagnostics, Laval, QC)) was added to each well. Cells were scraped and immediately flash frozen in liquid nitrogen and stored at −80 °C for Western Blot analysis. The protein concentration of the cell lysate was determined by Pierce BCA assay (Life Technologies), 4X SDS sample buffer (40% glycerol, 240 mM, Tris-HCl pH 6.8, 8% SDS, 0.04% bromophenol blue and 20 mM DTT) was added and 10 μg of protein lysate was used for SDS-PAGE.
To assess the effects of ND-654 on phospho-ACC, HepG2 cells were plated at a density of 5 × 104/mL in a 10 cm dish. After 24 h, cells were incubated in fresh media for 72 hours with vehicle control or ND-654 (10 μM), after which the cells were washed in ice-cold PBS and harvested in 500 μL of radioimmunoprecipitation assay buffer (RIPA, Boston BioProducts) containing protease and phosphatase inhibitors (Sigma). Frozen portions of tumors were thawed, minced and sonicated in RIPA buffer containing protease and phosphatase inhibitors before proceeding with western blotting. Protein concentration of lysates was analyzed by the bicinchoninic acid (BCA) method (Pierce Chemical Co.). Cellular lysates (40 μg) were prepared in Laemmli's reducing sample buffer (Boston BioProducts), separated by electrophoresis on 6% to 10% polyacrylamide gradient gels, and transferred electrophoretically onto a polyvinylidene difluoride membrane (Millipore). Nonspecific binding on the membrane was blocked with TBS/0.1% Tween 20 (TBS/T) containing 5% powdered cow’s milk or in 5% Bovine Serum Albumin. Membranes were incubated with primary antibodies overnight at 4°C and with appropriate secondary antibodies conjugated to horseradish peroxidase (HRP; GE Healthcare) for 1.5 hrs at room temperature. After each incubation period, membranes were washed thrice with TBS/T. Immunoreactive bands were visualized on a LI-COR with a chemiluminescent HRP substrate (GE Healthcare). Relative levels of total and phosphorylated proteins were determined by western blot analysis with the following commercially available antibodies: ACC (#3676), ACC pSer79/221 (#3661), AMPK pan (#2532), AMPK pThr172 (#2535), FASN (#3189), ACLY (#4332), β-Actin (#4967), GAPDH (#5174), ULK1 (#8054) ULK1 Ser555 (#5869) all from Cell Signaling Technologies. Each western blot was repeated to ensure reproducible results. Western blots were quantified with image analysis software (Image J).
Kinase arrays
Phospho-kinase array data in HepG2 WT and ACC KI cells was performed using a Human Phospho-Kinase Array Kit from R&D Systems Inc. and was analyzed as recommended by the manufacturer. To assess the effects of ND-654 on phospho-kinase signaling, HepG2 cells were treated with vehicle control or ND-654 (10 μM) for 6 days. The experiment was repeated to ensure reproducible results.
Cell Proliferation
In order to compare proliferation rates, HepG2 WT and ACC KI cells were seeded in a 96-well plate at a density of 1500 cells per well and incubated for 72 hours. The media was removed and cells were fixed using formalin. Cells were stained using 0.5% Crystal Violet, rinsed with distilled water and allowed to dry. The stain was solubilized by the addition of NaH2PO4 and the absorbance was measured at 570 nm.
To assess the effects of ND-654 on HepG2 proliferation, cells were plated in triplicate at a density of 5 × 104/mL in a 24-well plate. After 24 h, medium containing vehicle control or ND-654 (10 μM) was added. After 1, 2, 4, or 6 days of drug exposure, cell number was estimated by colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT; Sigma). The absorbance at 562 nm was measured with a spectrophotometric plate reader (Emax, Molecular Devices) and the experiment was repeated twice to ensure reproducibility.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are shown as means with error bars representing the SEM. Significant differences between means were determined using Student’s t-test for single treatment comparisons or 2-way ANOVA for multiple treatment comparisons with Bonferroni’s post hoc test where appropriate using either Microsoft Excel or GraphPad Prism. Ap value of ≤ 0.05 was considered significant. Statistics are included in the Figure Legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal (D3H8P) Proliferating cell nuclear antigen (PCNA) | Cell Signaling Technologies | 13110 |
| Rabbit monoclonal (Asp175) Cleaved-caspase-3 | Cell Signaling Technologies | 9664 |
| Rabbit polyclonal Myeloperoxidase (MPO) | Agilent Technologies | A039829-2 |
| Rabbit monoclonal (C83B10) Acetyl CoA Carboxylase (ACC) | Cell Signaling Technology | 3676 |
| Rabbit polyclonal phospho-Acetyl CoA Carboxylase (Ser79/221) | Cell Signaling Technology | 3661 |
| Rabbit polyclonal AMP-activated protein kinase (AMPK) | Cell Signaling Technologies | 2532 |
| Rabbit monoclonal (40H9) phospho-AMPK (Thr172) | Cell Signaling Technologies | 2535 |
| Rabbit polyclonal Fatty acid synthase (FASN) | Cell Signaling Technologies | 3189 |
| Rabbit polyclonal ATP-citrate lyase (ACLY) | Cell Signaling Technologies | 4332 |
| Rabbit polyclonal beta-Actin | Cell Signaling Technologies | 4967 |
| Rabbit monoclonal (D16H11) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Cell Signaling Technologies | 5174 |
| Rabbit monoclonal (D8H5) UNC-51-like kinase 1 (ULK1) | Cell Signaling Technologies | 8054 |
| Rabbit monoclonal (D1H4) phospho-ULK1 (Ser555) | Cell Signaling Technologies | 5869 |
| Bacterial and Virus Strains | ||
| None | ||
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Diethylnitrosamine | Sigma | N-0756 |
| ND-654 | Nimbus/Gilead | |
| Sorafenib | Selleck | S1040 |
| Phenformin | Sigma | P7045 |
| Critical Commercial Assays | ||
| Proteome Profiler™ Array, Human Phospho-Kinase Array Kit | R&D Systems | ARY003B |
| OxiSelect™ Oxidative DNA Damage ELISA Kit (8-OHdG Quantitation) | Cell Biolabs | STA-320 |
| Deposited Data | ||
| None | ||
| Experimental Models: Cell Lines | ||
| HepG2 | ATCC | HB-8065 |
| Experimental Models: Organisms/Strains | ||
| Wistar rats | Charles River Laboratories | Strain 003 |
| Acc1–S79A KI and Acc2–S212A KI mice | OzGene Pty Ltd | |
| Oligonucleotides | ||
| 18S | Life Technologies | Hs03003631_g1 |
| Cxcl1 | Life Technologies | Rn00578225_m1 |
| Il-1b | Life Technologies | Rn00580432_m1 |
| Il-6 | Life Technologies | Rn01410330_m1 |
| Tnfa | Life Technologies | Rn99999017_m1 |
| Cd80 | Life Technologies | Rn00709368_m1 |
| Cd163 | Life Technologies | Rn01492519_m1 |
| Tbp | Life Technologies | Mm01277042_m1 |
| Il-6 | Life Technologies | Mm00446190_m1 |
| Tnfa | Life Technologies | Mm00443258_m1 |
| Timp1 | Life Technologies | Mm01341361_m1 |
| Col1a1 | Life Technologies | Mm00801666_g1 |
| Col4a1 | Life Technologies | Mm01210125_m1 |
| Acta2 | Life Technologies | Mm00725412_s1 |
| Recombinant DNA | ||
| pGS –gRNA-Cas9-PuroGeneCRISPR | GenScript | |
| Software and Algorithms | ||
| Excel | Microsoft | |
| Prism Version 5.02 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Nearest Template Prediction | ||
| NIS-Elements Software | Nikon | |
| Other | ||
Highlights.
Fructose and ACC activating mutations increase hepatic carcinogenesis
A liver specific ACC inhibitor (ND-654) suppresses DNL and HCC proliferation
ND-654 reduces neutrophil recruitment and improves HCC survival
Humans with fatal HCC subtypes have increases in ACC expression
Acknowledgements
This work was supported by grants from the National Cancer Institute (T32 CA071345 to D.K.D., O.M., and K.K.T. and K01 CA140861 to B.C.F.), the Uehara Memorial Foundation (N.F.), the National Institute of Diabetes and Digestive and Kidney Diseases (DK099558 to Y.H.), the European Union (ERC-2014-AdG-671231 HEPCIR to Y.H.), the Irma T. Hirschl Trust (Y.H.), the U.S. Department of Defense (W81XWH-16-1-0363 to Y.H.), the Canadian Cancer Society Research and Innovation Fund (G.R.S.), the Canadian Institutes of Health Research (G.R.S. 125980-1, 144625-1), and Nimbus Therapeutics (to B.C.F.). A.E.G received a scholarship from the Canadian Liver Foundation. G.R.S. is supported by a Canada Research Chair and the J. Bruce Duncan Chair in Metabolic Diseases.
Footnotes
Declaration of Interests
Jamie Bates, Hailing Sun, Ting Wang, Henry Liu, and Adrian S. Ray are employees of Gilead Sciences. Jeremy Greenwood and Sathesh Bhat are employees of Schrodinger. Geraldine Harriman, Wenyan Miao, Jennifer L. Rocnik, William F. Westlin, H. James Harwood, Jr., and Rosana Kapeller are employees of Nimbus Therapeutics. Bryan C. Fuchs received research support from Nimbus Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amanzada A, Malik IA, Nischwitz M, Sultan S, Naz N, and Ramadori G (2011). Myeloperoxidase and elastase are only expressed by neutrophils in normal and in inflamed liver. Histochem Cell Biol 135, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baffy G, Brunt EM, and Caldwell SH (2012). Hepatocellular carcinoma in nonalcoholic fatty liver disease: an emerging menace. J Hepatol 56, 1384–1391. [DOI] [PubMed] [Google Scholar]
- Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, et al. (2011). Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 140, 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePeralta DK, Wei L, Ghoshal S, Schmidt B, Lauwers GY, Lanuti M, Chung RT, Tanabe KK, and Fuchs BC (2016). Metformin prevents hepatocellular carcinoma development by suppressing hepatic progenitor cell activation in a rat model of cirrhosis. Cancer 122, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedirko V, Lukanova A, Bamia C, Trichopolou A, Trepo E, Nothlings U, Schlesinger S, Aleksandrova K, Boffetta P, Tjonneland A, et al. (2013). Glycemic index, glycemic load, dietary carbohydrate, and dietary fiber intake and risk of liver and biliary tract cancers in Western Europeans. Ann Oncol 24, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Aleshin A, Dering J, Yang P, Ginther C, Desai A, Zhao D, von Euw E, Busuttil RW, and Slamon DJ (2013). Molecular subtype and response to dasatinib, an Src/Abl small molecule kinase inhibitor, in hepatocellular carcinoma cell lines in vitro. Hepatology 57, 1838–1846. [DOI] [PubMed] [Google Scholar]
- Fuchs BC, Fujii T, Dorfman JD, Goodwin JM, Zhu AX, Lanuti M, and Tanabe KK (2008). Epithelial-to-mesenchymal transition and integrin-linked kinase mediate sensitivity to epidermal growth factor receptor inhibition in human hepatoma cells. Cancer Res 68, 2391–2399. [DOI] [PubMed] [Google Scholar]
- Fuchs BC, Hoshida Y, Fujii T, Wei L, Yamada S, Lauwers GY, McGinn CM, DePeralta DK, Chen X, Kuroda T, et al. (2014). Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 59, 1577–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O'Neill HM, Ford RJ, Palanivel R, O'Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens N, Sun X, and Hoshida Y (2015). Molecular classification of hepatocellular carcinoma: potential therapeutic implications. Hepat Oncol 2, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, Tong L, Saha AK, Westlin WF, Kapeller R, et al. (2016). Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci U S A 113, E1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerstein MK, Schwarz JM, and Neese RA (1996). Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr 16, 523–557. [DOI] [PubMed] [Google Scholar]
- Hoshida Y (2010). Nearest Template Prediction: A Single-Sample-Based Flexible Class Prediction with Confidence Assessment. PLoS One 5, e15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K, Hashimoto M, et al. (2009). Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 69, 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, Jackman MR, Asipu A, Roncal-Jimenez CA, Kosugi T, et al. (2012). Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A 109, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, and McClain CJ (2007). Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 46, 823–830. [DOI] [PubMed] [Google Scholar]
- Kang JS, Wanibuchi H, Morimura K, Gonzalez FJ, and Fukushima S (2007). Role of CYP2E1 in diethylnitrosamine-induced hepatocarcinogenesis in vivo. Cancer Res 67, 11141–11146. [DOI] [PubMed] [Google Scholar]
- Kumamoto R, Uto H, Oda K, Ibusuki R, Tanoue S, Arima S, Mawatari S, Kumagai K, Numata M, Tamai T, et al. (2013). Dietary fructose enhances the incidence of precancerous hepatocytes induced by administration of diethylnitrosamine in rat. Eur J Med Res 18, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. (2008). Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359, 378–390. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, and Gores G (2016). Hepatocellular carcinoma. Nat Rev Dis Primers 2, 16018. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Leatherman GF, and Foster DW (1978). Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J Biol Chem 253, 4128–4136. [PubMed] [Google Scholar]
- Nakagawa S, Wei L, Song WM, Higashi T, Ghoshal S, Kim RS, Bian CB, Yamada S, Sun X, Venkatesh A, et al. (2016). Molecular Liver Cancer Prevention in Cirrhosis by Organ Transcriptome Analysis and Lysophosphatidic Acid Pathway Inhibition. Cancer Cell 30, 879–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, and Karin M (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, Smith T, Neish AS, Li H, Tan S, et al. (2016). Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderman NB, Carling D, Prentki M, and Cacicedo JM (2013). AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 123, 2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Wei L, DePeralta DK, Hoshida Y, Tan PS, Sun X, Sventek JP, Lanuti M, Tanabe KK, and Fuchs BC (2016). Molecular subclasses of hepatocellular carcinoma predict sensitivity to fibroblast growth factor receptor inhibition. Int J Cancer 138, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. (2016). Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 22, 1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PS, Nakagawa S, Goossens N, Venkatesh A, Huang T, Ward SC, Sun X, Song WM, Koh A, Canasto-Chibuque C, et al. (2016). Clinicopathological indices to predict hepatocellular carcinoma molecular classification. Liver Int 36, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, and Jemal A (2015). Global cancer statistics, 2012. CA Cancer J Clin 65, 87–108. [DOI] [PubMed] [Google Scholar]
- Wang C, Rajput S, Watabe K, Liao DF, and Cao D (2010). Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2, 515–526 [DOI] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, et al. (2005). Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer 41, 1316–1322. [DOI] [PubMed] [Google Scholar]
- Zhao H, Desai V, Wang J, Epstein DM, Miglarese M, and Buck E (2012). Epithelial-mesenchymal transition predicts sensitivity to the dual IGF-1R/IR inhibitor OSI-906 in hepatocellular carcinoma cell lines. Mol Cancer Ther 11, 503–513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.