

Abstract
Currently, platinum group metals play a central role in the electrocatalysis of the oxygen reduction reaction (ORR). Successful design and synthesis of new highly active materials for this process mainly rely on understanding of the so-called electrified electrode/electrolyte interface. It is widely accepted that the catalytic properties of this interface are only dependent on the electrode surface composition and structure. Therefore, there are limited studies about the effects of the electrolyte components on electrocatalytic activity. By now, however, several key points related to the electrolyte composition have become important for many electrocatalytic reactions, including the ORR. It is essential to understand how certain “spectator ions” (e.g., alkali metal cations) influence the electrocatalytic activity and what is the contribution of the electrode surface structure when, for instance, changing the pH of the electrolyte. In this work, the ORR activity of model stepped Pt [n(111) × (111)] surfaces (where n is equal to either 3 or 4 and denotes the atomic width of the (111) terraces of the Pt electrodes) was explored in various alkali metal (Li+, Na+, K+, Rb+, and Cs+) hydroxide solutions. The activity of these electrodes was unexpectedly strongly dependent not only on the surface structure but also on the type of the alkali metal cation in the solutions with the same pH, being the highest in potassium hydroxide solutions (i.e., K+ ≫ Na+ > Cs+ > Rb+ ≈ Li+). A possible reason for the observed ORR activity of Pt [n(111) × (111)] electrodes is discussed as an interplay between structural effects and noncovalent interactions between alkali metal cations and reaction intermediates adsorbed at active catalytic sites.
1. Introduction
The successful development of electrocatalysts strongly correlates with a detailed understanding of processes happening at the electrode/electrolyte interface.1−5 Platinum and platinum group metals play a crucial role in catalyzing numerous reactions, and one of the electrocatalytic reactions of great importance, particularly for energy conversion and storage systems, is the oxygen reduction reaction (ORR).6,7 Several factors determine the overall activity and selectivity of Pt and platinum group metals toward the ORR: the electrode surface composition,8−10 electrode surface structure,11−16 and the electrolyte composition. Certain approaches and methodologies such as Sabatier-type volcano plots3,17−19 or so-called coordination–activity relations20 can in many cases provide a good explanation of the observed catalytic activity trends for various electrode materials. There have also been studies regarding the influence of electrolyte components,21−29 which is less understood. For example, in sulfuric acid, sulfate anions form strongly adsorbed layers on Pt electrodes, hamper the adsorption of ORR intermediates, and decrease the electrode activity toward oxygen electroreduction in comparison with HClO4 electrolytes.30,31 On the other hand, in HClO4 media, it has been recently shown that perchlorate anions can interact with OH species adsorbed at the platinum surface and likely influence the ORR active sites.32,33 However, there is much less understanding of how certain cation “spectator species” (such as Na+ or K+), which are widely used as components of supporting electrolytes, affect the ORR activity at different pHs and how they interact with the electrocatalytic surface sites.
There are at least two concurrent hypotheses proposed to explain changes in the electrocatalytic activity of metal electrodes, for example, if one switches the electrolyte pH from acidic to alkaline. The first hypothesis is that the activity of an electrocatalyst simply depends on the pH of the electrolyte because of entropy barriers at the electrified interface34 or due to structuring in the electrical double layer35,36 and/or different solvation effects.37 The second hypothesis is based on a big role of noncovalent interactions between the electrocatalyst active sites and spectator species, for instance, the alkali metal cations.25,27 In the case of the ORR activity at Pt(111) electrodes in different alkaline solutions, the highest activity was observed in 0.1 M Cs+-containing solutions, and it was suggested that Cs+ exhibits the weakest stabilization of the ORR intermediate, OH*,25 leading to a near-optimal OH-binding energy for Pt(111) surfaces.
Herein, we demonstrate that the ORR activity of well-defined high-index model surfaces of Pt[n(111) × (111)] [where n = 3 for Pt(331), or n = 4 for Pt(221), and denotes the atomic width of the (111) terraces of the Pt electrodes with (111) step types] is significantly influenced by different alkali metal cations, namely, Li+, Na+, K+, Rb+, and Cs+. The particular choice of these electrodes for this study is due to their exceptionally high ORR activities in HClO4 media.21,38 We compared the results obtained for the above-mentioned stepped single-crystal surfaces with the similar ones for Pt(111) electrodes known from the literature. The highest activity for the Pt(331) and Pt(221) electrodes was observed in K+-containing electrolytes. However, the activities of the high-index Pt surfaces appeared to be systematically lower in alkaline than in acidic media, which is opposite to what is observed for Pt(111) electrodes. The influence of the alkali metal cations on the active sites located at the steps and terraces of Pt(331) and Pt(221) electrodes is discussed.
2. Results and Discussion
2.1. Electrochemical Measurements in Acidic and Various Basic Solutions
Cyclic voltammograms (CVs) of stepped Pt(221) and Pt(331) single crystals were recorded in Ar-saturated acidic and alkaline electrolytes. The resulting CVs in perchloric acid solutions are shown in Figure 1A; CVs of Pt(111) single-crystal electrodes in 0.1 M HClO4 and 0.1 M KOH are also presented as a reference. The voltammograms of stepped surfaces demonstrate distinctive features, which are associated with different surface structures and the distribution of adsorption sites as compared to Pt(111).21−23,43−45
Figure 1.
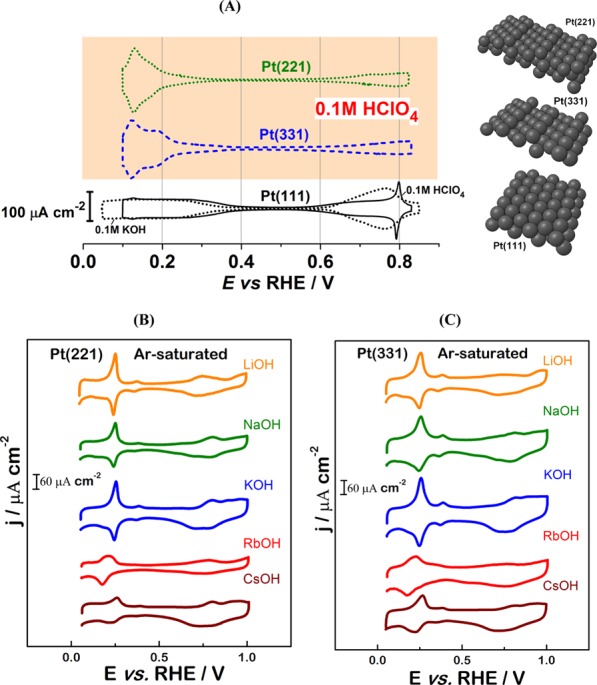
(A) Typical CVs of Pt(221) and Pt(331) electrodes obtained in Ar-saturated 0.1 M HClO4 together with the reference voltammogram for Pt(111) in 0.1 M HClO4 and 0.1 M KOH as indicated in the figure. Representative single-crystal surface structures are also shown for clarity. Typical CVs of (B) Pt(221) and (C) Pt(331) electrodes in Ar-saturated 0.1 M AM–OH (AM = Li+, Na+, K+, Rb+, and Cs+) electrolytes. The scan rate is 50 mV/s.
For all three electrodes, CVs obtained in 0.1 M HClO4 consist of clearly distinguishable regions with pairs of reversible peaks (Figure 1A). In the case of Pt(111), adsorption/desorption of the underpotentially deposited hydrogen species can be revealed between ∼0.1 and ∼0.4 V versus reversible hydrogen electrode (RHE). Within the same potential range, CV features in 0.1 M HClO4 are significantly different for the Pt(221) and Pt(331) electrodes as compared to that for Pt(111). Additional peaks appear at ca. 0.12 V versus RHE, which are attributed to the adsorption of OH species on undercoordinated sites at steps.39,40 Notably, in 0.1 M alkaline electrolytes (Figure 1B,C), the CVs in the potential region between ∼0.1 and ∼0.4 V versus RHE for Pt(221) and Pt(331) are also considerably different: the sharp peaks are shifted toward ∼0.25 V versus RHE. These peaks can in principle be attributed to the adsorption/desorption of the hydroxide species. However, there is no conclusive experimental study confirming the nature of the adsorption species involved in these processes in alkaline media. Most of the relevant studies were carried out under ultrahigh vacuum conditions and did not consider the influence of metal cations and other ions present in actual experimental solutions (see ref (41)). Recently, Koper et al. suggested that coadsorption of cations is the origin of the apparent pH dependence of hydrogen adsorption on stepped Pt single-crystal electrodes.27
At the potential range between ∼0.35 and ∼0.65 V versus RHE, the so-called double-layer region is observed (Figure 1), the width of which is, however, noticeably different for Pt electrodes in acidic and alkaline media. At the potential region more positive than ∼0.65 V versus RHE, even more drastic changes in the voltammetric behavior are observed. Whereas for Pt(111), the reversible peaks related to the OH adsorption at terraces are clearly observed between ∼0.6 and ∼0.9 V versus RHE, the hydroxyl anions adsorb significantly weaker at similar sites of Pt(221) and Pt(331) electrodes. Notably, the positive OH shift is more pronounced in the alkaline electrolyte. Summarizing this part, it is important to admit that changes in the electrolyte pH and composition drastically alter the adsorption properties of Pt electrodes and would also likely influence the electrocatalytic activity of these systems.
The ORR activities of stepped Pt(221) and Pt(331) single crystals were studied under the hanging meniscus rotating-disk electrode (HM-RDE) configuration in O2-saturated 0.1 M HClO4 and 0.1 M AM–OH (AM = Li+, Na+, K+, Rb+, and Cs+) electrolytes. Typical anodic polarization curves for Pt(221) are shown in Figure 2A. In Figure 2B, we show the activities of the Pt(221) electrodes in O2-saturated 0.1 M HClO4 and 0.1 M KOH and for Pt(111) electrodes in O2-saturated 0.1 M HClO4 and 0.1 M CsOH electrolytes taken from the study of Marković et al.25 for benchmarking.
Figure 2.
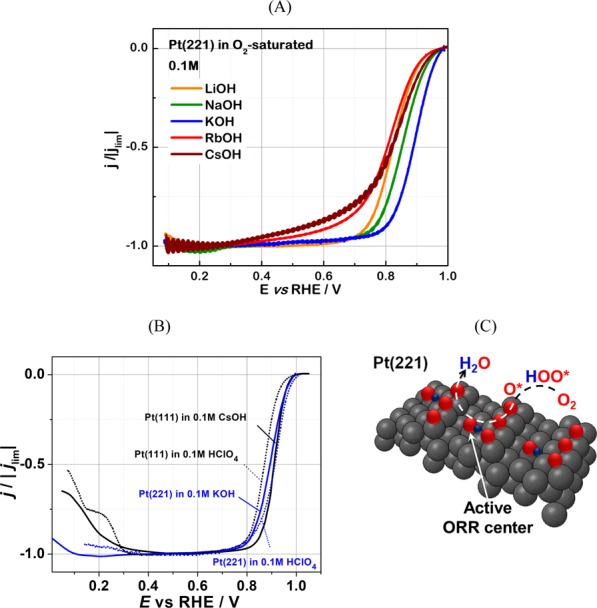
(A) Typical anodic scans of the RDE voltammograms (iR-corrected) of the stepped Pt(221) electrodes in O2-saturated 0.1 M AM–OH (AM = Li+, Na+, K+, Rb+, and Cs+) electrolytes. The scan speed was set to 50 mV/s, and the electrode was rotated at 1600 rpm. (B) iR-corrected RDE voltammograms of the Pt(221) electrodes in O2-saturated 0.1 M HClO4 and 0.1 M KOH and for Pt(111) electrodes in O2-saturated 0.1 M HClO4 and 0.1 M CsOH electrolytes (shown for comparison, adapted from ref (25)). (C) Model illustrating the location of the most active catalytic ORR centers at the surface of Pt(221) in perchloric acid according to our previous work (see ref (38)). These centers have an optimal coordination in terms of so-called generalized coordination numbers20 and are consequently close to optimal binding toward the most important ORR intermediate, OH*. Oxygen atoms, which permanently block the undercoordinated sites at Pt(221), are represented as red spheres. Blue and black spheres represent the hydrogen and platinum atoms, respectively.
It was reported that Pt(221) electrodes demonstrated the highest oxygen reduction activity among Pt single crystals in the 0.1 M HClO4 electrolyte.21,38 In accordance with those results, stepped Pt(221) crystals indeed show higher activity compared to Pt(111) in acid (Figure 2B). The reason for the increased ORR activity of Pt(221) is the maximal density of surface catalytic sites with optimal coordination (in terms of generalized coordination numbers)20,42 and consequently the optimal binding toward the most important ORR intermediate, OH*, as discussed in detail in ref (38). The most active ORR catalytic sites are “on-top” sites located close to the concavities at steps, where the binding to OH intermediates is ca. 0.093 eV weaker than that of the “on-top” sites located at extended Pt(111) surfaces (Figure 2C).
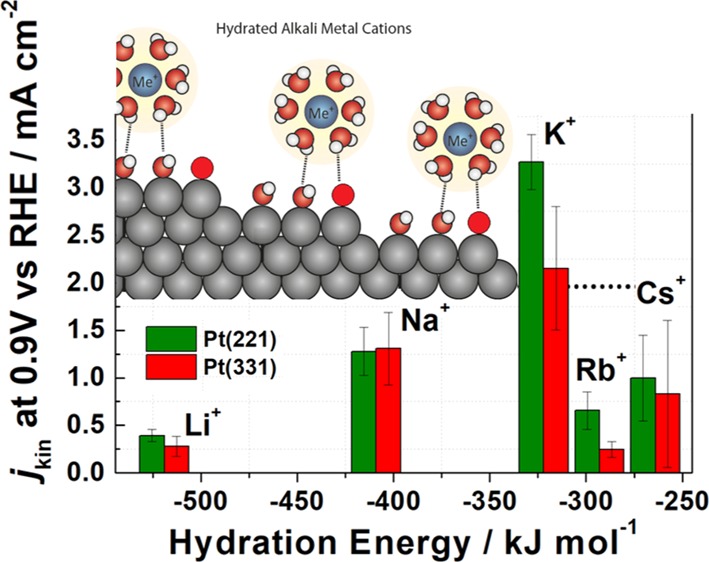
For pristine Pt(111), the ORR activity increases when the electrolyte is switched from the acidic to some alkaline 0.1 M AM–OH (AM = K+ and Cs+) electrolytes. It increases monotonously from Li+- to Cs+-containing solutions, and the activity of Pt(111) electrodes is significantly higher in 0.1 M CsOH than that in 0.1 M HClO4 (Figure 2B).25 Marković et al. suggested that the monotonous increase in the activity from Li+ to Cs+ is a direct consequence of less OH stabilization at (111) terraces of Pt(111) electrodes by “less-solvated” Cs+ (and K+) ions compared to Li+.25 Interestingly, in contrast to Pt(111), the opposite is observed for Pt(221) and Pt(331) electrodes: the ORR activity is systematically higher for the stepped surfaces in the acidic electrolyte compared to the basic ones, as summarized in Figure 3. Our data are also consistent with the study by Rizo et al.23 where the ORR activities of stepped single crystals were lower in 0.1 M NaOH compared to that in the acidic solutions. In general, the Pt(221) electrodes demonstrated higher ORR activities than Pt(331) in all the basic electrolytes. The highest ORR activity for both stepped single-crystal electrodes was observed in KOH in the following order: K+ ≫ Na+ > Cs+ > Rb+ ≈ Li+.
Figure 3.
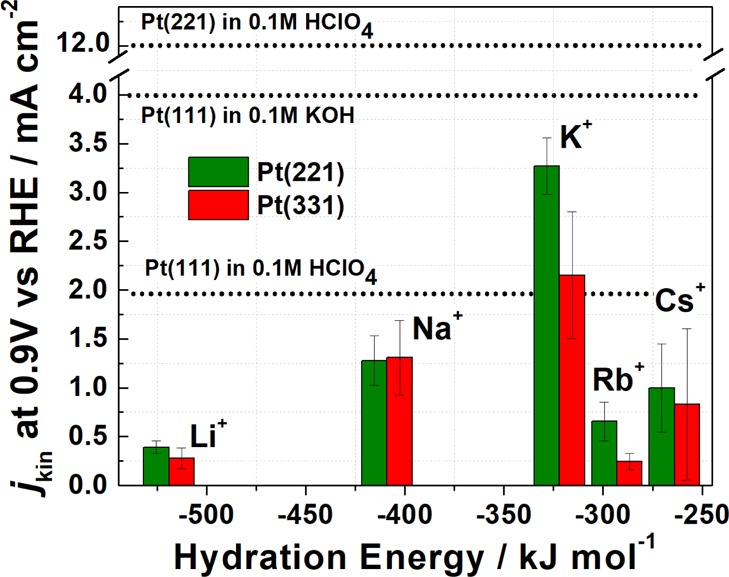
Bar chart showing the ORR kinetic current densities for Pt(331) and Pt(221) electrodes in 0.1 M AM–OH (AM = Li+, Na+, K+, Rb+, and Cs+) electrolytes at the reference electrode potential 0.9 V versus RHE. Dotted lines specify the activity of Pt(111) in 0.1 M HClO4, 0.1 M KOH, and Pt(221) in 0.1 M HClO4 for comparison.
Furthermore, Figure 4A shows the comparison of the CVs of Pt(221) in 0.1 M LiOH and KOH electrolytes. In the K+-containing solution, compared to Li+-containing electrolytes, the adsorption of OH species and oxide formation are observed at more positive potentials, and the shape of the peak observed is slightly different, possibly because of the different coverages of OH/O* species. Similar trends are observed for the Pt(331) electrodes (Figure 4B). Because the OH species are also important ORR intermediates, clearly not only pH but also the nature of the alkali metal cations influence the properties of the adsorption sites (catalytic centers) at the surfaces of high-index Pt single crystals, similar to the case of Pt(111).25 Summarizing the observations from Figures 1–4, one can assume that the observed activity of different Pt electrodes in alkaline media is a result of the interplay between structural effects and noncovalent interactions between alkali metal cations and reaction intermediates adsorbed at active catalytic centers.
Figure 4.

Typical CVs of (A) Pt(221) and (B) Pt(331) electrodes in Ar-saturated 0.1 M LiOH and KOH electrolytes. The adsorption of OH species and surface oxide formation are shifted toward more positive potentials in the K+-containing electrolytes relative to the Li+-containing solution, as indicated by the arrows.
2.2. Influence of Alkali Metal Cations on the ORR Activity of High- and Low-Index Pt Electrodes
Activity measurements for high-index Pt electrodes raise curiosity about the role of alkali metal cations on the electrocatalyst active sites and what is the role of the surface structure in the resulting ORR activity in alkaline media. In other words, why introduction of steps increases the activity in acidic media and decreases it when in basic solutions in the presence of the alkali metal cations? We address this query in this section by considering varying influences of alkali metal cations on different active sites present at the surface of Pt electrodes in question.
There are nonequivalent OH-adsorption sites at the surface of Pt(111) electrodes and the stepped single-crystal Pt surfaces, as schematically shown in Figure 5A,B. The OH-binding energies for these types of adsorption sites for Pt(111) and Pt(221) surfaces are summarized in Table 1. These adsorption sites can also be considered as the most probable catalytic centers for the oxygen electroreduction. Taking a closer look at Figure 5A, one can see that for the Pt(111) surface, only one type of “on-top” sites is possible (designated as type 1 in Figure 5A). For the “on-top” centers at the Pt(111) surfaces in acidic media, the corresponding OH-binding energy is ca. 0.1 eV stronger than the optimal one (ΔEOH – ΔEOH(optimum) value for type 1 in Table 1).
Figure 5.
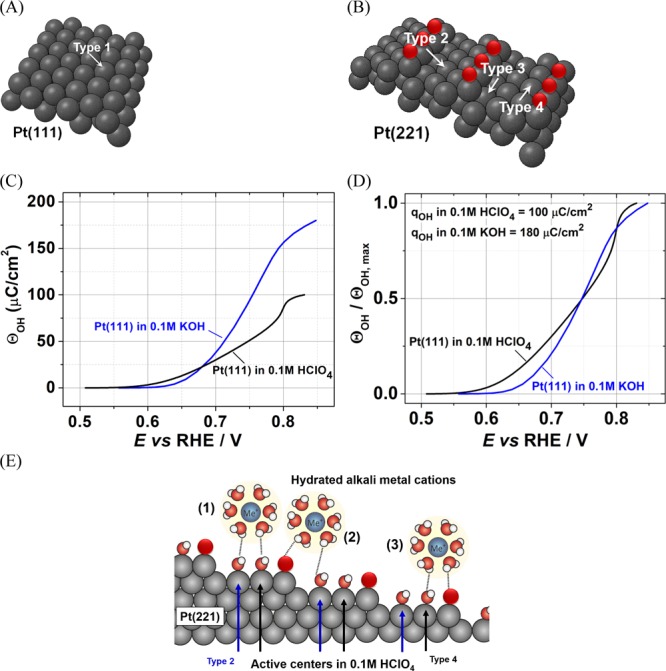
Schematic representation of “on-top” ORR catalytic centers at the surface of (A) Pt(111) and (B) Pt(221) at 0.9 V vs RHE. Oxygen atoms, which are represented by red spheres, permanently block the undercoordinated sites at Pt(221). (C) OH-adsorption isotherm for Pt(111) electrodes in 0.1 M HClO4 and 0.1 M KOH obtained from the anodic parts of the voltammograms shown in Figure 1A. (D) OH-adsorption isotherms in (C) normalized by maximal surface coverages. The maximal OH charges obtained from the CVs are given in the inset. (E) Schematic description of how alkali metal cations interact with the steps and terraces of Pt(221) surfaces. White, red, black, and dark blue spheres represent hydrogen, oxygen, platinum atoms, and the alkali metal cations in the electrolyte, respectively.
Table 1. Parameters Characterizing Adsorption Sites Indicated in Figure 5 in Acidic Media.
| adsorption sites (according to Figure 5) | generalized coordination number (adsorbed O species and Pt are assumed to contribute equally38) | ΔEOH – ΔEOH(Pt(111)) according to refs (20, 38, 42) (eV) | ΔEOH – ΔEOH(optimum) (eV) |
|---|---|---|---|
| type 1 | 7.5 | 0.00 | –0.1 |
| type 2 | 8.0 | 0.0925 | –0.0075 |
| type 3 | 9.83 | 0.43 | 0.33 |
| type 4 | 7.5 | 0.00 | –0.1 |
Alkali metal cations seem to have another peculiar effect on the amount of the OH adsorbed on the Pt(111) surface; the amount of OH adsorbates seems to increase in the presence of alkali metal cations, namely, K+ (Figure 5C,D). OH-adsorption isotherms for Pt(111) electrodes in 0.1 M HClO4 and 0.1 M KOH are shown in Figure 5C. Additionally, the OH-adsorption isotherms normalized by maximal surface coverages are shown in Figure 5D. The surface coverages in respective electrolytes were obtained by calculating the area (charge) under the peaks (anodic parts) at the potentials between ∼0.55 and ∼0.85 V versus RHE from the CVs of Pt(111) in HClO4 and KOH shown in Figure 1A. In KOH solution, the obtained maximum coverage OH charges are ∼2-fold higher than that in HClO4. This observation could be explained by considering repulsive and attractive forces between the same and oppositely charged molecules. Because hydroxide molecules have repulsive forces between each other, they cannot form a densely packed layer on the Pt(111) surface. However, the presence of positively charged metal cations seems to alleviate those repulsive forces. Because only reversible OH adsorption occurs on the Pt(111) surfaces in alkaline media,43 one can assume that all adsorbed OH will participate in the reaction. That is why the increased amount of adsorbed intermediates would directly result in higher ORR activity, as observed for Pt(111) in alkaline solutions.
Moreover, for the Pt(221) electrodes, there are more nonequivalent “on-top” locations as represented in Figure 5B; that is why the same above-mentioned behavior in alkaline solutions is not observed for the high-index stepped Pt surfaces. It seems that the nonuniform surface of Pt(221), Pt(331) (steps and terraces as shown in Figure 5B,E) and the presence of several “on-top” active sites inhibit/hinder the occurrence of the similar phenomena in alkaline solutions. In order to clarify this, we should consider structural and energetic characteristics of stepped Pt(221) and Pt(331) surfaces. For the sake of simplicity, let us consider four types of surface sites on Pt(221) (each corresponding to one of the four atoms on the width of the (111) terraces of the Pt electrodes).
There are two sacrificial surface sites: the first are permanently blocked undercoordinated sites located on the top edges (Figure 5B) and the second are hindered sites labeled as type 3 in Figure 5B. The existence of these sacrificial sites is necessary for creating the highly active sites, shown as type 2 (Figure 5B). In acidic media, the type 2 sites of Pt(221), as mentioned earlier, demonstrate the most optimal OH-binding energy for the intermediates and are responsible for the high ORR activity.
In other words, when there is only relatively moderate influence from the electrolyte components (like in perchloric acid media), the site with an optimum binding energy is type 2 site on the Pt(221) surface, which binds the OH intermediates only ca. 0.0075 eV stronger than the optimum (see Table 1).
In the presence of alkali metal cations (in alkaline solutions), their prominent effects on the type 2 sites divert their OH-binding energy from the near-optimal values. As a result, one always observes lower ORR activity of the stepped Pt electrodes in basic electrolytes compared to that in the acidic ones. Additionally, the reason why Pt(331) shows lower activity compared to Pt(221) in the basic electrolytes could be due to the absence of type 4 sites on Pt(331). The type 4 sites on the Pt(221) surface are similar to the type 1 sites of Pt(111) (see Figure 5A,B), and a comparable behavior (discussed above) in alkaline solutions can be assumed.
There is another peculiarity, which distinguishes the high-index single-crystal Pt surfaces from Pt(111) in alkaline media, apart from the fact that the ORR activity of Pt(221) and Pt(331) is systematically lower than that in HClO4. The ORR activities of Pt(221) and Pt(331) are nonlinear functions of the hydration energy of the alkali metal cations (Figure 3), in contrast to almost a linear relation between these parameters for Pt(111). According to the model proposed in ref (25), the hydrated alkali metal cations should interact with the hydroxide ions adsorbed on the surface via hydrogen bonds. The model assumes that alkali metal cations do not lose their solvation shell and stay intact. Our findings on the influence of different alkali metals on stepped Pt surfaces are mostly consistent with that proposed model25 for Pt(111). A schematic description of how the alkali metal cations can interact with different adsorption sites at the Pt(221) surface is shown in Figure 5E, where possible three interactions of solvated alkali metal cations with the Pt(221) active sites are depicted. Furthermore, in contrast to the planar Pt(111) surface, where the cations can interact with the whole first water layer unhindered, for stepped surfaces the step edges can shield the terrace depending on the size of the solvated metal cations. This shielding occurs because of the steric hindrance resulting from the neighboring Pt atoms at the steps on the platinum surface, which prevents the hydrated alkali cation to noncovalently interact with some of the atoms at the surface. As a result, active sites on the Pt(221) and Pt(331), especially type 2, will be affected differently depending on the type of the metal cations. One can assume the presence of an optimum metal cation hydration energy/shell size, which correlates with the ORR activity of the electrocatalyst. For instance, Li+ and Na+ might have too high hydration energy and small hydration shell radii, whereas Rb+ and Cs+ might have too low hydration energy and large hydration shell radii, which results in lowering of the ORR activity. Therefore, it can be concluded that the reason why the electrolytes containing K+ result in a higher ORR activity compared to other metal cations could be due to the hydration energy/shell size and/or due to the divergent effect of each alkali metal on the OH*-binding energies on the active sites of the high-index Pt surface.
In addition, to demonstrate the possible negative activity role of, namely, alkali metal cations, not only the pH effect the following experiments were performed. It is mostly assumed that perchlorate anions do not adsorb on the electrode surface and do not interact with the reactants/intermediates. Taking this as a working hypothesis, in a separate experiment we explored the effect of K+ concentration on the activity of Pt(221) (Figure 6). The ORR kinetic current density of Pt(221) in 0.1 M KOH and in 0.1 M KOH + 0.1 M KClO4 electrolytes was measured and shown in Figure 6A, and we also show the kinetic current densities at 0.9 V versus RHE as a bar chart in Figure 6B. The activity of the electrocatalyst significantly decreased with the increasing amount of K+ at the same pH further explaining the difference in the activities between alkaline and acidic media shown in Figure 3. However, additional influence of ClO4– anions cannot be excluded.
Figure 6.

(A) Typical kinetic current density and (B) bar chart showing the kinetic current densities at 0.9 V vs RHE for Pt(221) electrodes in 0.1 M KOH and in 0.1 M KOH + 0.1 M KClO4 electrolytes. The latter electrolyte was prepared by mixing HClO4 (where the highest activity in acidic media is observed) and KOH (where the highest activity in alkaline media is observed). The error bars are from five different measurements.
3. Conclusions
In summary, the influence of metal cations on the ORR activities of the Pt(221) and Pt(331) surfaces was investigated in five different AM–OH alkaline electrolytes (AM = Li+, Na+, K+, Rb+, and Cs+). We observed that the influence of the nature of the alkali metal cations on the ORR at Pt(221) and Pt(331) is different when compared to that of Pt(111). For Pt(111), the activity increases when the solution is switched from acidic to basic media. However, for Pt(221) and Pt(331), it is noticeably decreased. The presence of several types of active sites on high-index Pt single crystals interacting with the alkali metal cations seems to have a key role in decreased ORR activity in alkaline solutions. The ORR activity of the stepped Pt electrodes was strongly dependent on the type of the metal cation in alkaline solution and the trend was as follows: K+ ≫ Na+ > Cs+ > Rb+ ≈ Li+. The Pt(221) electrodes showed higher ORR activity than Pt(331) in all alkaline solutions. In combination of the divergent effect of each alkali metal cation on the OH-binding energies on the active sites of the high-index Pt surface and the hydration energy/shell size of solvated metal cations, our results confirmed that not only the pH but alkali metal cations also play a decisive role in changes of the ORR activity of complex Pt surfaces. Further research can be carried out on stepped Pt surfaces with various terrace widths in order to endorse the proposed mechanism of the alkali metal cation influence.
4. Experimental Methods
Single crystals of bead type: Pt(331) (Icryst, Jülich, Germany) and Pt(221) (provided by Prof. Juan Feliu, Alicante, Spain) were used in all of the experiments. For the preparation of the electrode surface crystal structure, the single crystals were flame-annealed with an isobutene gas flame and then cooled down in the mixture of CO (4.7, Air Liquide, Germany) and Ar (5.0, Air Liquide, Germany). The electrode surface structure quality was investigated by taking CVs in Ar-saturated 0.1 M HClO4 solution, which are proven to be extremely sensitive to both miniscule electrolyte contaminations and surface imperfections.44−46 Afterward, CVs of the Pt electrodes were recorded in Ar-saturated 0.1 M solutions of alkali metal hydroxides (Li+, Na+, K+, Rb+, and Cs+). Then, the electrode activities toward ORR were measured under the HM-RDE configuration (see refs (47) and (48)) in O2-saturated (5.0, Air Liquide, Germany) electrolytes. The electrodes were rotated at 1600 rpm for all measurements. The similar experiments were performed in a solution containing 0.1 M KOH and 0.1 M KClO4.
All glassware and experimental cells were cleaned with a 7:3 ratio mixture of H2SO4 and H2O2 (both Suprapur and purchased from Merck, Germany). Afterward, they were cleaned and rinsed several times with boiling ultrapure water (Evoqua, Germany). A VSP-300 potentiostat (Bio-Logic, France) was used for all of the electrochemical measurements. For an initial cycle of each measurement, the working electrodes were introduced into the solution under potential control at 0.05 V versus RHE.
A polycrystalline platinum wire and a mercury–mercury sulfate electrode (SI Analytics, Germany) were used as a counter and a reference electrode, respectively. All of the reported potentials in this study are referred to the RHE scale.
The solutions of perchloric acid were prepared by diluting the 70% HClO4 (Suprapur, Merck, Germany) with ultrapure water (Evoqua, Germany). The alkali metal hydroxide solutions were prepared from LiOH·H2O (99.998%, TraceSELECT, Sigma-Aldrich), NaOH (99.99%, semiconductor grade, Sigma-Aldrich), KOH (99.99%, trace metal basis, Sigma-Aldrich), RbOH (99.9%, 50 wt % solution, Sigma-Aldrich), and CsOH (99.9%, 50 wt % solution, Sigma-Aldrich). For the preparation of the mixture solution of KClO4 + KOH, first, a 0.4 M KOH solution and a 0.2 M HClO4 solution were prepared from previously mentioned respective chemicals and subsequently mixed in a 1:1 ratio.
Acknowledgments
Financial support from the cluster of excellence Nanosystems Initiative Munich (NIM) and DFG project BA 5795/4-1 is gratefully acknowledged. This work was supported by the German Research Foundation (DFG) and the Technical University of Munich within the Open Access Publishing Funding Programme.
Author Contributions
∥ B.G. and S.X. contributed equally.
The authors declare no competing financial interest.
References
- Koper M. T. M. Structure sensitivity and nanoscale effects in electrocatalysis. Nanoscale 2011, 3, 2054–2073. 10.1039/c0nr00857e. [DOI] [PubMed] [Google Scholar]
- Marković N.; Ross P. N. Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep. 2002, 45, 117–229. 10.1016/s0167-5729(01)00022-x. [DOI] [Google Scholar]
- Bandarenka A. S.; Koper M. T. M. Structural and electronic effects in heterogeneous electrocatalysis: Toward a rational design of electrocatalysts. J. Catal. 2013, 308, 11–24. 10.1016/j.jcat.2013.05.006. [DOI] [Google Scholar]
- Gasteiger H. A.; Kocha S. S.; Sompalli B.; Wagner F. T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal., B 2005, 56, 9–35. 10.1016/j.apcatb.2004.06.021. [DOI] [Google Scholar]
- Mahmoud M. A.; Garlyyev B.; El-Sayed M. A. Determining the mechanism of solution metallic nanocatalysis with solid and hollow nanoparticles: homogeneous or heterogeneous. J. Phys. Chem. C 2013, 117, 21886–21893. 10.1021/jp4079234. [DOI] [Google Scholar]
- Calle-Vallejo F.; Koper M. T. M.; Bandarenka A. S. Tailoring the catalytic activity of electrodes with monolayer amounts of foreign metals. Chem. Soc. Rev. 2013, 42, 5210–5230. 10.1039/c3cs60026b. [DOI] [PubMed] [Google Scholar]
- Stamenkovic V. R.; Mun B. S.; Arenz M.; Mayrhofer K. J. J.; Lucas C. A.; Wang G.; Ross P. N.; Markovic N. M. Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat. Mater. 2007, 6, 241–247. 10.1038/nmat1840. [DOI] [PubMed] [Google Scholar]
- Chou S.-W.; Lai Y.-R.; Yang Y. Y.; Tang C.-Y.; Hayashi M.; Chen H.-C.; Chen H.-L.; Chou P.-T. Uniform size and composition tuning of PtNi octahedra for systematic studies of oxygen reduction reactions. J. Catal. 2014, 309, 343–350. 10.1016/j.jcat.2013.09.008. [DOI] [Google Scholar]
- Greeley J.; Stephens I. E. L.; Bondarenko A. S.; Johansson T. P.; Hansen H. A.; Jaramillo T. F.; Rossmeisl J.; Chorkendorff I.; Nørskov J. K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. 10.1038/nchem.367. [DOI] [PubMed] [Google Scholar]
- Coleman E. J.; Co A. C. Galvanic displacement of Pt on nanoporous copper: An alternative synthetic route for obtaining robust and reliable oxygen reduction activity. J. Catal. 2014, 316, 191–200. 10.1016/j.jcat.2014.05.012. [DOI] [Google Scholar]
- Mukerjee S.; Srinivasan S.; Soriaga M. P.; McBreen J. Role of Structural and Electronic Properties of Pt and Pt Alloys on Electrocatalysis of Oxygen Reduction. J. Electrochem. Soc. 1995, 142, 1409–1422. 10.1149/1.2048590. [DOI] [Google Scholar]
- Stamenkovic V.; Mun B. S.; Mayrhofer K. J. J.; Ross P. N.; Markovic N. M.; Rossmeisl J.; Greeley J.; Nørskov J. K. Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure. Angew. Chem. 2006, 118, 2963–2967. 10.1002/ange.200504386. [DOI] [PubMed] [Google Scholar]
- Pfisterer J. H. K.; Liang Y.; Schneider O.; Bandarenka A. S. Direct instrumental identification of catalytically active surface sites. Nature 2017, 549, 74–77. 10.1038/nature23661. [DOI] [PubMed] [Google Scholar]
- Kodama K.; Jinnouchi R.; Takahashi N.; Murata H.; Morimoto Y. Activities and stabilities of Au-modified stepped-Pt single-crystal electrodes as model cathode catalysts in polymer electrolyte fuel cells. J. Am. Chem. Soc. 2016, 138, 4194–4200. 10.1021/jacs.6b00359. [DOI] [PubMed] [Google Scholar]
- Ueno T.; Tanaka H.; Sugawara S.; Shinohara K.; Ohma A.; Hoshi N.; Nakamura M. Infrared spectroscopy of adsorbed OH on n (111)-(100) and n (111)-(111) series of Pt electrode. J. Electroanal. Chem. 2017, 800, 162–166. 10.1016/j.jelechem.2016.11.028. [DOI] [Google Scholar]
- Stoffelsma C.; Rodriguez P.; Garcia G.; Garcia-Araez N.; Strmcnik D.; Marković N. M.; Koper M. T. M. Promotion of the oxidation of carbon monoxide at stepped platinum single-crystal electrodes in alkaline media by lithium and beryllium cations. J. Am. Chem. Soc. 2010, 132, 16127–16133. 10.1021/ja106389k. [DOI] [PubMed] [Google Scholar]
- Stephens I. E. L.; Bondarenko A. S.; Grønbjerg U.; Rossmeisl J.; Chorkendorff I. Understanding the electrocatalysis of oxygen reduction on platinum and its alloys. Energy Environ. Sci. 2012, 5, 6744–6762. 10.1039/c2ee03590a. [DOI] [Google Scholar]
- Čolić V.; Bandarenka A. S. Pt alloy electrocatalysts for the oxygen reduction reaction: from model surfaces to nanostructured systems. ACS Catal. 2016, 6, 5378–5385. 10.1021/acscatal.6b00997. [DOI] [Google Scholar]
- Bandarenka A. S.; Ventosa E.; Maljusch A.; Masa J.; Schuhmann W. Techniques and methodologies in modern electrocatalysis: evaluation of activity, selectivity and stability of catalytic materials. Analyst 2014, 139, 1274–1291. 10.1039/c3an01647a. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Tymoczko J.; Colic V.; Vu Q. H.; Pohl M. D.; Morgenstern K.; Loffreda D.; Sautet P.; Schuhmann W.; Bandarenka A. S. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 2015, 350, 185–189. 10.1126/science.aab3501. [DOI] [PubMed] [Google Scholar]
- Kuzume A.; Herrero E.; Feliu J. M. Oxygen reduction on stepped platinum surfaces in acidic media. J. Electroanal. Chem. 2007, 599, 333–343. 10.1016/j.jelechem.2006.05.006. [DOI] [Google Scholar]
- Maciá M. D.; Campiña J. M.; Herrero E.; Feliu J. M. On the kinetics of oxygen reduction on platinum stepped surfaces in acidic media. J. Electroanal. Chem. 2004, 564, 141–150. 10.1016/j.jelechem.2003.09.035. [DOI] [Google Scholar]
- Rizo R.; Herrero E.; Feliu J. M. Oxygen reduction reaction on stepped platinum surfaces in alkaline media. Phys. Chem. Chem. Phys. 2013, 15, 15416–15425. 10.1039/c3cp51642c. [DOI] [PubMed] [Google Scholar]
- Wang C.; Markovic N. M.; Stamenkovic V. R. Advanced platinum alloy electrocatalysts for the oxygen reduction reaction. ACS Catal. 2012, 2, 891–898. 10.1021/cs3000792. [DOI] [Google Scholar]
- Strmcnik D.; Kodama K.; van der Vliet D.; Greeley J.; Stamenkovic V. R.; Marković N. M. The role of non-covalent interactions in electrocatalytic fuel-cell reactions on platinum. Nat. Chem. 2009, 1, 466–472. 10.1038/nchem.330. [DOI] [PubMed] [Google Scholar]
- Colic V.; Pohl M. D.; Scieszka D.; Bandarenka A. S. Influence of the electrolyte composition on the activity and selectivity of electrocatalytic centers. Catal. Today 2016, 262, 24–35. 10.1016/j.cattod.2015.08.003. [DOI] [Google Scholar]
- Chen X.; McCrum I. T.; Schwarz K. A.; Janik M. J.; Koper M. T. M. Co-adsorption of Cations as the Cause of the Apparent pH Dependence of Hydrogen Adsorption on a Stepped Platinum Single-Crystal Electrode. Angew. Chem., Int. Ed. 2017, 56, 15025–15029. 10.1002/anie.201709455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S.; Watzele S.; Čolić V.; Brandl K.; Garlyyev B.; Bandarenka A. S. Reconsidering Water Electrolysis: Producing Hydrogen at Cathodes Together with Selective Oxidation of n -Butylamine at Anodes. ChemSusChem 2017, 10, 4812–4816. 10.1002/cssc.201701802. [DOI] [PubMed] [Google Scholar]
- Moureaux F.; Stevens P.; Chatenet M. Effect of lithium and potassium cations on the electrocatalytic properties of carbon and manganese oxide electrocatalysts towards the oxygen reduction reaction in concentrated alkaline electrolyte. Electrocatalysis 2013, 4, 123–133. 10.1007/s12678-013-0127-4. [DOI] [Google Scholar]
- Kolics A.; Wieckowski A. Adsorption of Bisulfate and Sulfate Anions on a Pt(111) Electrode. J. Phys. Chem. B 2001, 105, 2588–2595. 10.1021/jp003536f. [DOI] [Google Scholar]
- Gamboa-Aldeco M. E.; Herrero E.; Zelenay P. S.; Wieckowski A. Adsorption of bisulfate anion on a Pt(100) electrode: A comparison with Pt(111) and Pt(poly). J. Electroanal. Chem. 1993, 348, 451–457. 10.1016/0022-0728(93)80151-7. [DOI] [Google Scholar]
- Huang Y.-F.; Kooyman P. J.; Koper M. T. M. Intermediate stages of electrochemical oxidation of single-crystalline platinum revealed by in situ Raman spectroscopy. Nat. Commun. 2016, 7, 12440. 10.1038/ncomms12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G. A.; Brew A.; Hunter K.; Sharman J.; Wright E. Specific adsorption of perchlorate anions on Pt{hkl} single crystal electrodes. Phys. Chem. Chem. Phys. 2014, 16, 13689–13698. 10.1039/c4cp00564c. [DOI] [PubMed] [Google Scholar]
- Rossmeisl J.; Chan K.; Skúlason E.; Björketun M. E.; Tripkovic V. On the pH dependence of electrochemical proton transfer barriers. Catal. Today 2016, 262, 36–40. 10.1016/j.cattod.2015.08.016. [DOI] [Google Scholar]
- Ledezma-Yanez I.; Wallace W. D. Z.; Sebastián-Pascual P.; Climent V.; Feliu J. M.; Koper M. T. M. Interfacial water reorganization as a pH-dependent descriptor of the hydrogen evolution rate on platinum electrodes. Nat. Energy 2017, 2, 17031. 10.1038/nenergy.2017.31. [DOI] [Google Scholar]
- Garlyyev B.; Xue S.; Watzele S.; Scieszka D.; Bandarenka A. S. Influence of the Nature of the Alkali Metal Cations on the Electrical Double-Layer Capacitance of Model Pt(111) and Au(111) Electrodes. J. Phys. Chem. Lett. 2018, 9, 1927–1930. 10.1021/acs.jpclett.8b00610. [DOI] [PubMed] [Google Scholar]
- He Z.-D.; Hanselman S.; Chen Y.-X.; Koper M. T. M.; Calle-Vallejo F. Importance of solvation for the accurate prediction of oxygen reduction activities of Pt-based electrocatalysts. J. Phys. Chem. Lett. 2017, 8, 2243–2246. 10.1021/acs.jpclett.7b01018. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Pohl M. D.; Reinisch D.; Loffreda D.; Sautet P.; Bandarenka A. S. Why conclusions from platinum model surfaces do not necessarily lead to enhanced nanoparticle catalysts for the oxygen reduction reaction. Chem. Sci. 2017, 8, 2283–2289. 10.1039/c6sc04788b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Niet M. J. T. C.; Garcia-Araez N.; Hernández J.; Feliu J. M.; Koper M. T. M. Water dissociation on well-defined platinum surfaces: The electrochemical perspective. Catal. Today 2013, 202, 105–113. 10.1016/j.cattod.2012.04.059. [DOI] [Google Scholar]
- Pohl M. D.; Colic V.; Scieszka D.; Bandarenka A. S. Elucidation of adsorption processes at the surface of Pt(331) model electrocatalysts in acidic aqueous media. Phys. Chem. Chem. Phys. 2016, 18, 10792–10799. 10.1039/c5cp08000b. [DOI] [PubMed] [Google Scholar]
- van der Niet M. J. T. C.; Berg O. T.; Juurlink L. B. F.; Koper M. T. M. The Interaction between H2O and Preadsorbed O on the Stepped Pt(533) Surface. J. Phys. Chem. C 2010, 114, 18953–18960. 10.1021/jp106412e. [DOI] [Google Scholar]
- Calle-Vallejo F.; Martínez J. I.; García-Lastra J. M.; Sautet P.; Loffreda D. Fast prediction of adsorption properties for platinum nanocatalysts with generalized coordination numbers. Angew. Chem., Int. Ed. 2014, 53, 8316–8319. 10.1002/anie.201402958. [DOI] [PubMed] [Google Scholar]
- Marković N. M.; Gasteiger H. A.; Ross P. N. Oxygen reduction on platinum low-index single-crystal surfaces in alkaline solution: rotating ring disk Pt( hkl ) studies. J. Phys. Chem. 1996, 100, 6715–6721. 10.1021/jp9533382. [DOI] [PubMed] [Google Scholar]
- Taguchi S.; Feliu J. M. Electrochemical reduction of nitrate on Pt(S)[n(111)×(111)] electrodes in perchloric acid solution. Electrochim. Acta 2007, 52, 6023–6033. 10.1016/j.electacta.2007.03.057. [DOI] [Google Scholar]
- Climent V.; Gómez R.; Feliu J. M. Effect of increasing amount of steps on the potential of zero total charge of Pt(111) electrodes. Electrochim. Acta 1999, 45, 629–637. 10.1016/s0013-4686(99)00241-8. [DOI] [Google Scholar]
- Scortichini C. L.; Reilley C. N. Surface characterization of Pt electrodes using underpotential deposition of H and Cu. J. Electroanal. Chem. Interfacial Electrochem. 1982, 139, 247–264. 10.1016/0022-0728(82)85124-3. [DOI] [Google Scholar]
- Villullas H. M.; Teijelo M. L. The hanging-meniscus rotating disk (HMRD) Part 1. Dependence of hydrodynamic behavior on experimental variables. J. Electroanal. Chem. 1995, 384, 25–30. 10.1016/0022-0728(94)03716-g. [DOI] [Google Scholar]
- Villullas H. M.; Teijelo M. L. The hanging meniscus rotating disk (HMRD) Part 2. Application to simple charge transfer reaction kinetics. J. Electroanal. Chem. 1995, 385, 39–44. 10.1016/0022-0728(94)03763-s. [DOI] [Google Scholar]