

Abstract

Remediation process produces high-value functional material from low-cost or valueless waste feedstock. Current research demonstrates an innovative solvothermal approach to effectively react sulfuric acid on polyethylene (PE) chains, modifying the PE at a moderate temperature. In this process, the polymer undergoes a cross-linking step above 120 °C, whereas above 500 °C, it transforms into turbostratic carbon structures. Scanning electron micrographs confirmed the free-standing carbon sheet architecture. Raman spectroscopy and X-ray diffraction verified the amorphous/disordered sp2/sp3 hybrid carbon structure in the produced carbons. A high Brunauer–Emmett–Teller surface area of 752.3 and 673.5 m2/g for low-density PE-derived carbon (LDPE-C) and high-density PE-derived carbon (HDPE-C), respectively, was recorded. Thermogravimetric analysis analysis established a total mass retention of 50 and 46% for LDPE and HDPE, respectively, from sulfonated materials. Li-ion battery composite anode comprising LDPE-C and HDPE-C, with a binder and a carbon additive (vs lithium), produced 230 and 350 mA h/g specific capacities for LDPE-C and HDPE-C, respectively, when cycled at room temperature at C/5 rate. Elevated temperature (50 °C) battery cycling produced 290 and 440 mA h/g specific capacities for LDPE-C and HDPE-C, respectively, at C/5 rate. On the basis of the literature survey, this is the first report, which demonstrates that a solvothermal sulfonation process followed by thermal treatment successfully converts waste LDPE and HDPE plastic bags to functional energy-storing carbons.
Introduction
Globally, around 300 million tons of plastic were produced in 2013 to fulfill the growing demand.1 Soon after, most of the used plastic (>85%) makes its way to landfill and oceans, where it takes hundreds of years to decompose into small fragments.2 As plastic slowly degrades with sunlight, it becomes smaller and starts releasing toxic chemicals into the environment.3 This approach disturbs our ecosystem. In 2014, Eriksen et al. estimated that at least 5.25 trillion plastic particles weighing around 269 000 tons4 are submerged or floating in the sea. Currently, there is an unsustainable trend, where waste plastics are shipped for recycling to countries with lower environmental standards.1 However, this approach is not economical, appealing, or benign for our environment and the overall ecosystem.
Typically, closed-loop recycling approach is used in most thermoplastics because energy recovery from plastics is very poor, air emission controls are needed, and byproduct ash could be hazardous. Moreover, polyethylene (PE) with colorants, dyes, labels, and sticky additives5 increases the difficulty in recycling. PE has been proposed for more than four decades as a low cost (1 $/kg) carbon precursor with a high (86%) carbon content. In 1972, Saglio et al. pyrolyzed saturated chlorinated6 PE to obtain carbon. Zhang and Sun produced carbon fibers7 by reacting ultrahigh molecular weight PE with H2SO4 acid at 180 °C followed by carbonization in the He atmosphere at 1100 °C, yielding 55 wt % carbon fibers. Recently, Hameed et al. prepared microporous8 carbon with high-density PE (HDPE) using H2SO4 at 120 °C and carbonization at 1150 °C under a N2 atmosphere, obtaining a 51.9 wt % yield. However, with the former methods, carbon was produced in limited quantities without potential applications.
Hydrothermal carbonization is known to produce carbonaceous materials from biomass precursors using water.9−12 Because of self-generated pressures at low temperatures in a hydrothermal system, it resembles the natural process of coal formation.9 Previously, dry autoclaving of PE (thermolysis under self-generated pressure at an elevated temperature in a closed system) produced solid, dense, low surface area functional carbon spheres. The as-obtained carbon spheres were studied as lithium-ion battery anodes,13 sodium-ion battery anodes,14 and lubrication additives.15 PE is also remediated into carbon nanotubes (CNTs)16 via dry autoclaving. Typically, carbonaceous materials are synthesized from hydrocarbons through expensive and complex multistep processes, with the special requirement of plasma and reducing gases becoming complicated to scale-up. Our innovative solvothermal upcycling approach generates high pressure at a low temperature, which improves PE sulfonation reproducibly with a high carbon yield. Our solvothermal upcycling approach totally gets rid of plastic waste, converting them into functional carbonaceous materials for multiple applications including battery anodes.
Experimental Section
Low-density PE (LDPE) and HDPE bags were obtained from Ziploc and HILEX, respectively. After their general household usage, they are used as feedstock in our solvothermal process. Concentrated H2SO4 (98%) was obtained from BDH and used as received.
Synthesis of Sulfonated PEs
Typically, 1 g of LDPE or HDPE was immersed in 20 mL of sulfuric acid (H2SO4) in a 120 mL capacity Teflon vessel with a Teflon cap. Because of the dominant sulfuric acid component, we define our process as solvothermal and not hydrothermal. Sulfuric acid is known to be a powerful protonating agent, oxidizing agent, and dehydrating agent (remove or replace molecules from organic compounds). The PE/acid-loaded Teflon vessel was inserted into a solvothermal Parr reactor and heated to 110 °C for 12 h, followed by natural cooling to room temperature. Typically, waste solid PE thermally melts at 115–135 °C, above which, it transforms into hazardous gases (CO2, CO, H2O, and CxHy) without converting into a valuable carbon product. To achieve carbonaceous materials, it is essential to transform some H atoms of PE chains to SO3H groups. After opening the reactor, the obtained black sheets and flakes were washed with deionized water several times to remove the unreacted acid. The obtained sulfonated flakes were dried overnight at 80 °C, which are named as LDPE-S and HDPE-S, respectively. After solvothermal synthesis, both sulfonated materials (LDPE-S and HDPE-S) increase their masses by 28 ± 6 and 23 ± 9%, respectively.
Carbonization of Sulfonated PEs
In a tubular split furnace, LDPE-S or HDPE-S was separately carbonized for 2 h in an alumina crucible inside a quartz tube under Ar flow (200 mL min–1) at 900 °C. The heating and cooling rates were 10 °Cmin-1. The exit of the tube furnace was connected to a water tap, which collected the formed byproducts during the carbonization process of sulfonated flakes. Sample yields of carbon products were weighted after carbonization and named as LDPE-C and HDPE-C. These carbonaceous materials were then ground in an agate mortar to form a free-flowing carbon black powder. Carbonization shows a total mass yield (PE to carbon conversion) of 63 ± 5% for LDPE and 54 ± 3% for HDPE.
Characterization Techniques
Scanning electron microscopy (SEM) and elemental mapping were performed by using a JEOL scanning electron microscope instrument (JCM-6000PLUS, JED-2300 AnalysisStation). Raman spectra were collected using a Thermo Scientific DXR Raman microscope with a 633 nm laser; a low laser power of 2 mW was used to avoid sample burning/modification. X-ray diffraction (XRD) patterns were collected using a Riguka SmartLab X-ray diffractometer operated at 40 kV and 40 mA with a Cu Kα radiation source (λ = 0.154 nm); the samples were measured from 10° to 80° with a scan rate of 2.5° per minute. Surface area analysis of black carbon powders/sheets was performed in a Quantachrome Instruments analyzer after outgassing the materials at 300 °C for 24 h. Thermogravimetric analysis (TGA) was performed using a thermal analyzer system (i1000, IS Inc.) with flowing argon gas (99.997%) at a heating rate of 10 °C min–1 from room temperature to 1000 °C.
Electrochemical Tests
Lithium-ion battery anode electrodes were prepared with 80% of waste PE-derived carbons, 10% of polyvinylidene difluoride (KYNAR HSV900, Arkema Inc.) binder, and 10% of carbon black additive (TIMCAL Super P Li). The composite slurry was homogenized for 20 min by employing a Thinky planetary mixture in the presence of seven to eight drops of N-methylpyrrolidone at a constant speed of around 8000 rpm. Using the Dr. Blade technique,17 the slurries were deposited on a battery-grade copper foil using an MTI laminate coater with approximately 40 μm thickness, and the obtained foils were dried overnight at 80 °C. Electrodes of 12 mm diameter were punched out, and CR-2032-type half-coin cells (MTI Corp) were assembled in a high-purity argon glovebox (NEXUS II Vacuum Atmospheres Co.) with <1 ppm of O2 and moisture. Celgard 2500 polypropylene was used as the separator, a lithium metal foil (MTI Corp) was used as the counter electrode, and 1 M LiPF6 with EC:DMC (1:1) was used as the organic electrolyte. Cyclic voltammetric analysis was carried out for the fabricated lithium cells comprising either LDPE-C or HDPE-C derived carbon between 0.005 and 2.0 V at the scan rate of 0.2 mV s–1 by employing a Gamry 600 potentiostat/galvanostat/ZRA. Galvanostatic charge–discharge cycling was performed using an Arbin cycler between 0.005 and 2.0 V with a theoretical capacity of 372 mA h/g, which was used to calculate the corresponding C rates (1 C = 372 mA/g). For kinetic and cell degradation studies, an elevated temperature charge–discharge cycling was conducted at 50 °C in an ESPEC oven.
Results and Discussion
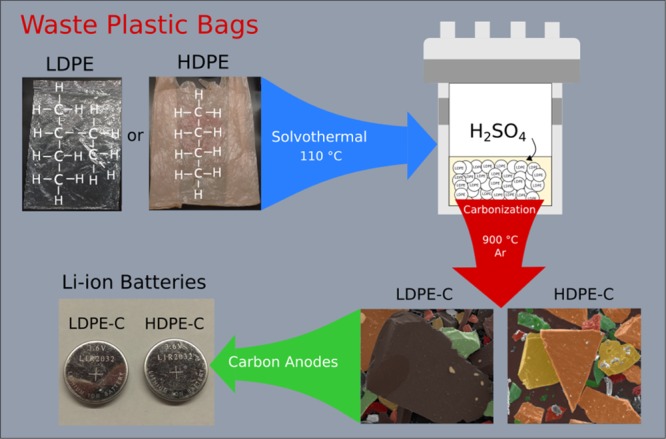
Scheme 1 demonstrates the novel solvothermal approach for the fabrication of carbon chips from the used plastic bags and its effective use in lithium-ion battery anodes. The chemical formula of PE, the solvothermal reactor assembly comprising acid, inert gas treatment, chiplike morphology of carbon (artistically decorated), and the prepared coin cells are presented. Typically, PE thermally melts at 115–135 °C, above which it transforms into hazardous gases (CO2, CO, H2O, and CxHy) without converting into a valuable carbon product. To methodically achieve carbonaceous materials, an oxidative dehydrogenation accompanied by SO3H functionalization is carried out in a solvothermal Teflon inner lining reactor. Hydrothermal reaction facilitates SO3H functionalization. During the high-temperature treatment, the removal of sulfonic acid groups takes place followed by cross-linking18 above 500 °C to remove all of the remaining −H2, −CO, −CO2, −H2O, and SO2 to yield pure carbon. Finally, lithium-ion coin or button cells were made, and Li-ion storage performance was studied.
Scheme 1. Solvothermal Process for the Manufacture of Carbon Chips from Used Low- and High-Density Plastic Bags, Their Chiplike Morphology, and Effective Use in Lithium-Ion Battery Anodes.
To comprehend the morphology of the obtained carbonaceous materials, SEM images and elemental mapping analysis were taken for the LDPE-C and HDPE-C carbon materials, as described in Figures 1 and 2. The larger plastic pieces (both LDPE and HDPE) are converted to carbon pieces, which got fragmented into free-standing carbon sheet/flake architectures as shown in Figures 1 and 2. The particle width is dependent on the grinding process. Gentle milling of the obtained LDPE-C and HDPE-C in an agate mortar with pestle leads to carbon chiplike particles with several microns width. The thickness of the carbon chips is 25.28 ± 4 and 11.86 ± 2.4 μm for LDPE-C and HDPE-C, respectively. The thickness is dependent on the initial feedstock precursor (LDPE and HDPE bags) thickness. The observed shrinkage in diameter from the feedstock waste plastics to the carbon product is approximately 25%. This shrinkage could be explained by the release of molecules (−H2, −CO, −CO2, −H2O, or SO2) during the elevated temperature treatment.18 Carbon chips prepared from the HDPE yielded more porous carbon structures compared to LDPE, possibly due to the thermochemical reaction with inorganic colorants during the elevated temperature heat treatment. Furthermore, the presence of carbon and the trace amount of sulfur are measured in the LDPE-C carbon material, as described in Figure 1c,d. In HDPE, trace amount of colorants is present that leads to additional impurities, revealed in the XRD (Figure 4b) and elemental mapping (Figure 2d–h) for the obtained carbon product. As seen in Figure 2c, the carbon obtained from HDPE possesses more than 90% carbon (Figure 2d) and remaining S (Figure 2e), Mg (Figure 2f), Al (Figure 2g), and O (Figure 2h). The presence of colorant impurities are consistent with the XRD HDPE-C (Figure 4b).
Figure 1.

Morphological investigation and elemental mapping analysis of LDPE-C: (a,b) SEM images indicating the morphology of free-standing carbon sheet/flake architectures and (c,d) corresponding SEM image elemental mapping analysis for the presence of (c) carbon and (d) trace amount of sulfur in the LDPE-C carbon material.
Figure 2.

Morphological investigation and elemental mapping analysis of HDPE-C: (a) SEM and (b) high-resolution SEM images indicating the morphology of free-standing carbon sheet/flake architectures with porosity; elemental mapping analysis for the SEM image (c), indicating the presence of (d) carbon and remaining (e) sulfur, (f) magnesium, (g) aluminum, and (h) oxygen in the HDPE-C carbon material.
Figure 4.
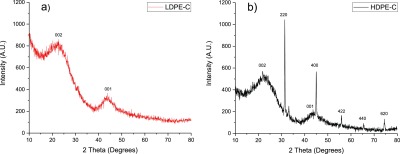
XRD patterns of carbon chips derived from plastic feedstock: LDPE-C (a) and HDPE-C (b).
Amorphous/turbostratic nature of LDPE-C and HDPE-C was analyzed by Raman spectroscopy and XRD patterns. Figure 3a,b depicts the Raman spectra of LDPE and HDPE and its derivative materials. LDPE bags show Raman bands (red curve) at 1067, 1132, 1299, 1441, 1455, and 1465 cm–1 corresponding to different behaviors, such as bending, twisting, and stretching of CH2. HDPE shows different Raman bands (red) because of additional components/impurities such as pigments of different colors. A strong peak at 1580 cm–1 (blue) of LDPE-S and HDPE-S is indicative of the cross-linking and aromatization during the solvothermal synthesis.18 LDPE-C and HDPE-C confirm the disordered carbon nature (yellow curves) from PE plastic bags, showing peaks at 1350 and 1580 cm–1 that relate to the D and G bands.18 Raman spectroscopy studies confirm the transformation of plastic bags into carbonaceous materials during solvothermal processing followed by pyrolysis.
Figure 3.
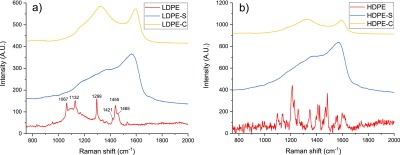
Raman spectra of (a) as-received LDPE after the hydrothermal synthesis (LDPE-S) and carbonization (LDPE-C) process and (b) as-received HDPE after the solvothermal synthesis (HDPE-S) and carbonization (HDPE-C) process.
The XRD pattern depicted peaks corresponding to (002) and (001) Miller turbostratic or amorphous planes of carbon in LDPE-C (Figure 4a) and HDPE-C (Figure 4b). These broad diffraction peaks indicate the amorphous carbon structure.19 However, HDPE-C shows strong peaks corresponding to (220), (400), (422), (440), and (620) Miller planes. These diffraction peaks are analogous to the MgAl2O4 crystal structure. It is known that HDPE could have pigments/colorants that contains MgO and alumina. With the thermal treatment, these impurities in the raw material are converted into the MgAl2O4 structure. So, the derived carbon materials of HDPE-C contain around 10% MgAl2O4. These colorant impurities are observed in SEM–energy-dispersive X-ray spectroscopy (Figure 2c–h) as well as TGA measurements (Figure 6).
Figure 6.
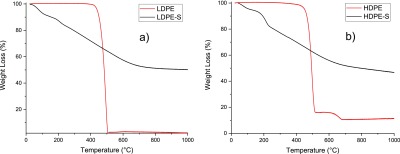
TGA of LDPE and LDPE-S (a) and HDPE and HDPE-S (b).
Additional detailed characterization of carbon chips was performed through N2 adsorption and desorption isotherms of LDPE-C (Figure 5a) and HDPE-C (Figure 5b). These curves reveal the behavior of H3 isotherm (IUPAC classification) for both the samples;20 HDPE-C compared with LDPE-C shows a high hysteresis suggesting the existence of macropores.21 SEM images in HDPE-C show macropores, possibly generated because of the thermochemical reaction of colorant impurities with HDPE during pyrolysis treatment. The decrease in the surface area is also a result of the contaminant present in HDPE, as shown in Table 1. Density functional theory (DFT) pore size distribution is shown for LDPE-C (Figure 5c) and HDPE (Figure 5d). In addition to the pores at around 1.5 nm, in HDPE-C, 4 nm pores are also observed possibly due to the contribution from the inorganic contaminants.
Figure 5.
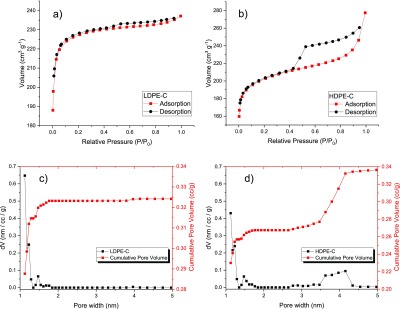
N2 adsorption–desorption isotherm of (a) LDPE-C and (b) HDPE-C. The DFT pore size distribution and pore volume for (c) LDPE-C and (d) HDPE-C.
Table 1. BET Surface Area and DFT Pore Volume of the Carbons Derived from LDPE-C and HDPE-C.
| sample | BET surface area (m2/g) | DFT pore volume (cm3/g) | mean pore width (nm) |
|---|---|---|---|
| LDPE-C | 752.3 | 0.33 | 1.13 |
| HDPE-C | 673.5 | 0.37 | 1.13 |
TGA of feedstock LDPE plastics (red) and solvothermally treated plastics (black) were performed under an argon atmosphere to determine the remaining mass yields. Figure 6a demonstrates that LDPE starts degrading above 400 °C and completely becomes gaseous phase, with no solid mass remaining above 500 °C. This confirms that the thermal decomposition of LDPE in an inert atmosphere does not lead to fabricated carbon-based materials. Alternatively, the sulfonated PE starts showing mass loss from the beginning until it reaches 600 °C. This is a result of conversion of cross-linked PE to a carbon-based material. Around 50% yield remains after the TGA characterization of LDPE. Figure 6b presents the TGA of feedstock-trashed HDPE (red) and solvothermally sulfonated HDPE (black). Analogous to LDPE, the thermal decomposition of HDPE is observed till 500 °C. However, between 500 and 700 °C, additional kink is measured because of inorganic colorant/pigment transformation. Remaining 10 wt % content is the pigments that were detected as MgAl2O4 in the presented XRD pattern (Figure 4b). However, sulfonated HDPE has analogous behavior to LDPE with additional kink between 100 and 200 °C, as a result of sulfur melting and/or its interactions with inorganic pigments. Continuous loss of sulfur due to its weak bonding with polymers and analogous behavior has been reported in sulfur/CNT22 and carbon compartment/sulfur23 materials.
To investigate the electrochemical characteristics, the cyclic voltammetric analysis was carried out for the assembled lithium cells comprising LDPE-C and HDPE-C carbon materials between 0.005 and 2.0 V versus Li+/Li at the scan rate of 0.2 mV s–1 for three cycles, as shown in Figure 7a (LDPE-C) and Figure 7b (HDPE-C). In the first cycle of cyclic voltammograms, significant irreversible redox processes appear, which are associated with the solid electrolyte interface (SEI) formation on the carbon surface,24,25 intercalation–deintercalation process, and followed by the capacitive ionization/deionization26,27 and adsorption/desorption28,29 processes in line with the electromotive force. The obtained strong reduction peak is at 0.8 V related to SEI formation24,25 during the first lithiation on the carbon surface of LDPE-C and HDPE-C carbon materials. Obviously, it can be seen that the Li+ ion storage occurs by the capacitive ionization/deionization26,27 and adsorption/desorption28,29 processes on the carbon surface that leads to the cyclic voltammograms with a wide hysteresis.24,25 In addition to this phenomena, the Li-ion intercalation–deintercalation process takes place during cycling in the short-range order amorphous carbon.30
Figure 7.

Cyclic voltammetric analysis of fabricated lithium cells using (a) LDPE-C and (b) HDPE-C carbon materials between 0.005 and 2.0 V vs Li+/Li at 0.2 mV s–1 for the first three cycles.
Figure 8a,b shows the first and second discharge (lithiation) and charge (delithiation) curves for LDPE-C and HDPE-C samples, respectively. First cycle capacity losses of 40 and 43% (25 and 50 °C, respectively) for the LDPE-C sample with higher lithiation/delithiation potential is evident. Analogous 43 and 45% (25 and 50 °C, respectively) capacity losses in HDPE-C are also noticed, reflecting significant SEIformation. This loss is associated with electrolyte breakdown, forming SEI on larger surface area carbon chips with irreversible adsorption of Li at surface defects and surface-attached O or S atoms.31 The decrease in the charge–discharge potentials during the second cycle is an outcome of stable SEI formation and its stabilization on later cycles.32 The recorded specific capacities are slightly enhanced than the theoretical (372 mA h/g) capacity of graphite in the LDPE-derived carbon sample. HDPE-derived carbon yields improved capacities compared to LDPE-derived carbons because of additional inorganic impurities which participated in the electrochemical redox or alloying reactions.33
Figure 8.
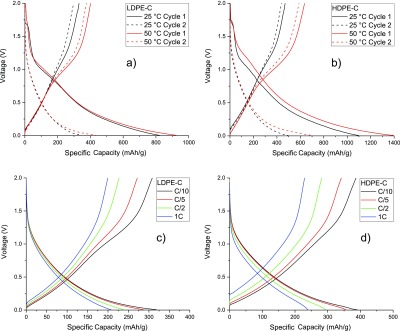
Room (25 °C) and elevated temperature (50 °C) voltage profiles of (a) LDPE-derived C and (b) HDPE-derived C obtained at C/10 rate (1 C = 372 mA/g current density). Room-temperature voltage profiles using various current densities for LDPE-C (c) and HDPE-C (d) samples.
The room-temperature voltage profiles for LDPE-C and HDPE-C electrodes cycled at various currents are shown in Figure 8c,d, respectively. Both LDPE-C and HDPE-C showed similar behavior in the first two cycles. Even at 1 C high current density, the charge and discharge capacities are greater than 200 mA h/g because of the shorter diffusion distance in carbon chips. For standard graphitic materials, typically, the anode particle size is 10–20 μm requiring additional time for charging or discharging.30
Figure 9 summarizes the galvanostatic cycling results of LDPE-C and HDPE-C anodes. Figure 9a,b shows the rate studies performed utilizing various current densities, yielding reversible capacities of 230 and 290 mA h/g (25 and 50 °C, respectively, for LDPE-C). A slight increase in capacity to 350 and 440 mA h/g (25 and 50 °C, respectively for HDPE-C) is recorded at C/5 rate. At 1 C rate (i.e., 1 h charging and 1 h discharging), the capacity drops to 160 and 210 mA h/g (25 and 50 °C, respectively) for LDPE-C and 240 and 270 mA h/g (25 and 50 °C, respectively) for HDPE-C. Long cycle stability of the carbon sample was studied through a cycling rate of C/5 (Figure 9c). After initial 20 cycles, a stable reversible capacity of 200 mA h/g is achieved for both LDPE-C (red) and HDPE-C (black).
Figure 9.
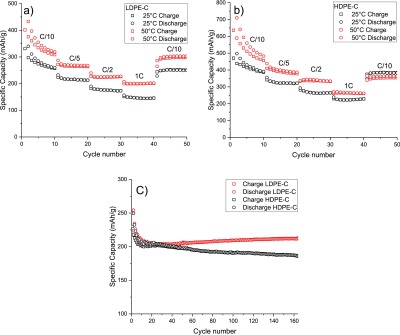
Rate performance at four different currents for LDPE-C (a) and HDPE-C (b) at 25 and 50 °C, respectively. (c) Long-term cycling using C/5 rate for LDPE-C (red) and HDPE-C (black) at 25 °C.
Conclusions
The described novel solvothermal upcycling approach totally gets rid of low- and high-density waste plastic bags by converting them into functional carbonaceous materials. The presented solvothermal upcycling approach generates high pressure at a low temperature, improving PE sulfonation reproducibly with a high carbon yield and forming mesmerizing carbon chips. Raman spectroscopy and XRD verified the turbostratic/amorphous carbon structure for both LDPE-C and HDPE-C. However, XRD identified MgAl2O4 impurities in HDPE-C, aroused from the pigment-derived impurities. Solvothermal upcycling process obtained a moderate Brunauer–Emmett–Teller (BET) surface area of 752.3 and 673.5 m2/g for LDPE-C, and HDPE-C, respectively. TGA analysis of low- and high-density plastics and their sulfonated derivatives provided the scientific reaction mechanism with the understanding of the cross-linking phenomena. Li-ion batteries prepared from upcycled LDPE-C and HDPE-C anode materials demonstrated their fascinating function in Li-ion storage at elevated and room temperatures.
Acknowledgments
The authors would like to acknowledge the Davidson School of Chemical Engineering, Purdue University for the financial support for performing “Plastic Upcycling” work. S.V.-S. and S.F.A.G. would like to express their gratitude to CONCYTEQ and UTEQ for the financial support and paperwork necessary to make this project successful. S.V.-S. would like to thank Purdue University graduate students (Arthur Dysart, Jialiang Tang, and Ryan Adams) for their help in performing XRD and SEM measurements.
The authors declare no competing financial interest.
References
- Global Plastic Production Rises, Recycling Lags. http://vitalsigns.worldwatch.org/sites/default/files/vital_signs_trend_plastic_full_pdf (accessed Dec 15, 2017).
- Pol V. G. Upcycling: Converting Waste Plastics into Paramagnetic, Conducting, Solid, Pure Carbon Microspheres. Environ. Sci. Technol. 2010, 44, 4753–4759. 10.1021/es100243u. [DOI] [PubMed] [Google Scholar]
- Jahnke A.; Arp H. P. H.; Escher B. I.; Gewert B.; Gorokhova E.; Kühnel D.; Ogonowski M.; Potthoff A.; Rummel C.; Schmitt-Jansen M.; Toorman E.; MacLeod M. Uncertainty and Confronting Ignorance about the Possible Impacts of Weathering Plastic in the Marine. Environ. Sci. Technol. Lett. 2017, 4, 85–90. 10.1021/acs.estlett.7b00008. [DOI] [Google Scholar]
- Eriksen M.; Lebreton L. C. M.; Carson H. S.; Thiel M.; Moore C. J.; Borerro J. C.; Galgani F.; Ryan P. G.; Reisser J. Plastic Pollution in the World’s Oceans: More than 5 Trillion Plastic Pieces Weighing over 250,000 Tons Afloat at Sea. PloS One 2014, 9, e111913 10.1371/journal.pone.0111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopewell J.; Dvorak R.; Kosior E. Plastics Recycling: Challenges and Opportunities. Philos. Trans. R. Soc., B 2009, 364, 2115–2126. 10.1098/rstb.2008.0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglio N.; Berticat P.; Vallet G. Study of the Pyrolysis of Saturated Chlorinated Polyethylene Fibers. J. Appl. Polym. Sci. 1972, 16, 2991–3002. 10.1002/app.1972.070161123. [DOI] [Google Scholar]
- Zhang D.; Sun Q. Structure and Properties Development During the Conversion of Polyethylene Precursors to Carbon Fibers. J. Appl. Polym. Sci. 1996, 62, 367–373. . [DOI] [Google Scholar]
- Li C.; Zhu H.; Salim N. V.; Fox B. L.; Hameed N. Preparation of Microporous Carbon Materials via In-depth Sulfonation and Stabilization of Polyethylene. Polym. Degrad. Stab. 2016, 134, 272–283. 10.1016/j.polymdegradstab.2016.10.019. [DOI] [Google Scholar]
- Marinovic A.; Pileidis F. D.; Titirici M.-M.. Hydrothermal Carbonisation (HTC): History, State-of-the-Art and Chemistry. Porous Carbon Materials from Sustainable Precursors; Royal Society of Chemistry, 2015, pp 129–155. [Google Scholar]
- Titirici M.-M.; Antonietti M. Chemistry and Materials Options of Sustainable Carbon Materials made by Hydrothermal Carbonization. Chem. Soc. Rev. 2010, 39, 103–116. 10.1039/b819318p. [DOI] [PubMed] [Google Scholar]
- Falco C.; Marco-Lozar J. P.; Salinas-Torres D.; Morallón E.; Cazorla-Amorós D.; Titirici M. M.; Lozano-Castelló D. Tailoring the Porosity of Chemically Activated Hydrothermal Carbons: Influence of the Precursor and Hydrothermal Carbonization Temperature. Carbon 2013, 62, 346–355. 10.1016/j.carbon.2013.06.017. [DOI] [Google Scholar]
- Salinas-Torres D.; Lozano-Castelló D.; Titirici M. M.; Zhao L.; Yu L.; Morallón E.; Cazorla-Amoros D. Electrochemical Behaviour of Activated Carbons Obtained via Hydrothermal Carbonization. J. Mater. Chem. A 2015, 3, 15558–15567. 10.1039/c5ta03574k. [DOI] [Google Scholar]
- Pol V. G.; Thackeray M. M. Spherical carbon particles and carbon nanotubes prepared by autogenic reactions: Evaluation as anodes in lithium electrochemical cells. Energy Environ. Sci. 2011, 4, 1904–1912. 10.1039/c0ee00256a. [DOI] [Google Scholar]
- Pol V. G.; Lee E.; Zhou D.; Dogan F.; Calderon-Moreno J. M.; Johnson C. S. Spherical Carbon as a New High-Rate Anode for Sodium-ion Batteries. Electrochim. Acta 2014, 127, 61–67. 10.1016/j.electacta.2014.01.132. [DOI] [Google Scholar]
- Alazemi A. A.; Etacheri V.; Dysart A. D.; Stacke L.-E.; Pol V. G.; Sadeghi F. Ultrasmooth Submicrometer Carbon Spheres as Lubricant Additives for Friction and Wear Reduction. ACS Appl. Mater. Interfaces 2015, 7, 5514–5521. 10.1021/acsami.5b00099. [DOI] [PubMed] [Google Scholar]
- Pol V. G.; Thiyagarajan P. Remediating Plastic Waste into Carbon Nanotubes. J. Environ. Monit. 2010, 12, 455–459. 10.1039/b914648b. [DOI] [PubMed] [Google Scholar]
- Berni A.; Mennig M.; Schmidt H.. Doctor Blade. Sol–Gel Technologies for Glass Producers and Users; Springer: Boston, MA, 2004; pp 89–92. [Google Scholar]
- Barton B. E.; Patton J.; Hukkanen E.; Behr M.; Lin J.-C.; Beyer S.; Zhang Y.; Brehm L.; Haskins B.; Bell B.; Gerhart B.; Leugers A.; Bernius M. Chemical Transformation of Hydrocarbons to Carbon using SO3 Sources. Carbon 2015, 94, 465–471. 10.1016/j.carbon.2015.07.029. [DOI] [Google Scholar]
- Tripathy S.; Bhattacharya D. Rapid Synthesis and Characterization of Mesoporous Nanocrystalline MgAl2O4 via Flash Pyrolysis Route. J. Asian Ceram. Soc. 2018, 1, 328–332. 10.1016/j.jascer.2013.08.006. [DOI] [Google Scholar]
- Thommes M.; Kaneko K.; Neimark A. V.; Olivier J. P.; Rodriguez-Reinoso F.; Rouquerol J.; Sing K. S. W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. 10.1515/pac-2014-1117. [DOI] [Google Scholar]
- Pol V. G.; Shrestha L. K.; Ariga K. Tunable, Functional Carbon Spheres Derived from Rapid Synthesis of Resorcinol-formaldehyde Resins. ACS Appl. Mater. Interfaces 2014, 6, 10649–10655. 10.1021/am502324m. [DOI] [PubMed] [Google Scholar]
- Guo J.; Xu Y.; Wang C. Sulfur-Impregnated Disordered Carbon Nanotubes Cathode for Lithium-Sulfur Batteries. Nano Lett. 2011, 11, 4288–4294. 10.1021/nl202297p. [DOI] [PubMed] [Google Scholar]
- Dysart A. D.; Burgos J. C.; Mistry A.; Chen C.-F.; Liu Z.; Hong C. N.; Balbuena P. B.; Mukherjee P. P.; Pol V. G. Towards Next Generation Lithium-Sulfur Batteries: Non-Conventional Carbon Compartments/Sulfur Electrodes and Multi-Scale Analysis. J. Electrochem. Soc. 2016, 163, A730–A741. 10.1149/2.0481605jes. [DOI] [Google Scholar]
- Choi C.; Seo S.-D.; Kim B.-K.; Kim D.-W. Enhanced Lithium Storage in Hierarchically Porous Carbon Derived from Waste Tea Leaves. Sci. Rep. 2016, 6, 39099. 10.1038/srep39099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; et al. Interconnected highly graphitic carbon nanosheets derived from wheat stalk as high performance anode materials for lithium ion batteries. Green Chem. 2016, 18, 2078–2088. 10.1039/c5gc02122g. [DOI] [Google Scholar]
- Ryoo M.-W.; Seo G. Improvement in capacitive deionization function of activated carbon cloth by titania modification. Water Res. 2003, 37, 1527–1534. 10.1016/s0043-1354(02)00531-6. [DOI] [PubMed] [Google Scholar]
- Oren Y. Capacitive deionization (CDI) for desalination and water treatment — past, present and future (a review). Desalination 2008, 228, 10–29. 10.1016/j.desal.2007.08.005. [DOI] [Google Scholar]
- Wang H.; Yoshio M. Carbon-coated natural graphite prepared by thermal vapor decomposition process, a candidate anode material for lithium-ion battery. J. Power Sources 2001, 93, 123–129. 10.1016/s0378-7753(00)00552-8. [DOI] [Google Scholar]
- Lee J.-H.; Bae W.-S.; Choi J.-H. Electrode reactions and adsorption/desorption performance related to the applied potential in a capacitive deionization process. Desalination 2010, 258, 159–163. 10.1016/j.desal.2010.03.020. [DOI] [Google Scholar]
- Etacheri V.; Hong C. N.; Pol V. G. Upcycling of Packing-Peanuts into Carbon Microsheet Anodes for Lithium-Ion Batteries. Environ. Sci. Technol. 2015, 49, 11191–11198. 10.1021/acs.est.5b01896. [DOI] [PubMed] [Google Scholar]
- Lotfabad E. M.; Kalisvaart P.; Kohandehghan A.; Karpuzov D.; Mitlin D. Origin of non-SEI Related Coulombic Efficiency Loss in Carbons Tested Against Na and Li. J. Mater. Chem. A 2014, 2, 19685–19695. 10.1039/c4ta04995k. [DOI] [Google Scholar]
- Etacheri V.; Wang C.; O’Connell M. J.; Chan C. K.; Pol V. G. Porous carbon sphere anodes for enhanced lithium-ion storage. J. Mater. Chem. A 2015, 3, 9861–9868. 10.1039/c5ta01360g. [DOI] [Google Scholar]
- Etacheri V.; Seisenbaeva G. A.; Caruthers J.; Daniel G.; Nedelec J.-M.; Kessler V. G.; Pol V. G. Ordered network of interconnected SnO2 nanoparticles for excellent lithium-ion storage. Adv. Energy Mater. 2014, 5, 1401289. 10.1002/aenm.201401289. [DOI] [Google Scholar]