

Abstract
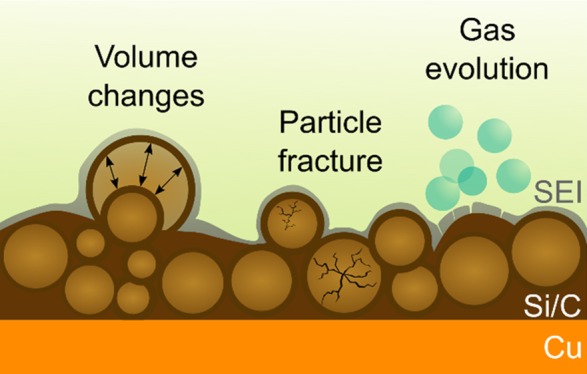
Silicon (Si) and composites thereof, preferably with carbon (C), show favorable lithium (Li) storage properties at low potential, and thus hold promise for application as anode active materials in the energy storage area. However, the high theoretical specific capacity of Si afforded by the alloying reaction with Li involves many challenges. In this article, we report the preparation of small-size Si particles with a turbostratic carbon shell from a polymer precoated powder material. Galvanostatic charge/discharge experiments conducted on electrodes with practical loadings resulted in much improved capacity retention and kinetics for the Si/C composite particles compared to physical mixtures of pristine Si particles and carbon black, emphasizing the positive effect that the core–shell-type morphology has on the cycling performance. Using in situ differential electrochemical mass spectrometry, pressure, and acoustic emission measurements, we gain insights into the gassing behavior, the bulk volume expansion, and the mechanical degradation of the Si/C composite-containing electrodes. Taken together, our research data demonstrate that some of the problems of high-content Si anodes can be mitigated by carbon coating. Nonetheless, continuous electrolyte decomposition, particle fracture, and electrode restructuring due to the large volume changes during battery operation (here, ∼170% in the voltage range of 600–30 mV vs Li+/Li) remain as serious hurdles toward practical implementation.
Introduction
The demands imposed on battery materials with respect to energy storage capacity, durability, cost, and so forth are ever increasing. State-of-the-art high-energy-density Li-ion cells are based on layered lithium transition metal oxides LiMO2 (M = Ni, Co, and Mn or Al) as cathode active materials (≈150–200 mAh g–1 usable discharge capacity)1−4 and graphite, with a theoretical specific capacity of 372 mAh g–1, as an anode active material.4,5 Incorporating some fraction of silicon (Si) (3579 mAh gSi–1 for Li15Si4) into the negative electrode has been shown to be an effective strategy to further improve the specific energy.6−8 For example, Yim et al. prepared and tested anodes with a polyetherimide binder using composites made of graphite and Si powder (5 wt %). The best performing electrodes showed a specific capacity of 514 mAh g–1 after 350 cycles which, according to the authors, was 1.6 times higher than commercial graphite anodes.7 However, achieving stable cycling performance of “high-content” and “high-loading” Si anodes is very challenging.
The most severe drawbacks of Si as the anode active material are (i) the large irreversibilities, especially during the first couple of cycles, because of side reactions with the electrolyte for instance and (ii) the large volume changes upon alloying with Li, leading to particle fracture and anode self-pulverization.9−13 Note that all of these adverse effects not only cause significant impedance build-up during battery operation, but also result in Li consumption from the cathode. Furthermore, it has been shown that the contact loss of Si particles from the conductive carbon black/binder network and/or current collector leads to accelerated capacity degradation.13,14
In recent years, many attempts have been made to overcome the main problems of Si anodes by testing of novel and/or improved electrolytes, additives, and polymer binders.11,13,15−17 In addition, the focus was on the preparation of advanced Si-based redox-active materials. From these studies—only some of which have been considered here—it seems that especially Si and SiOx materials and composites thereof, preferably with nanoscale carbons, hold promise in future energy storage applications.18−21 For example, Breitung et al. produced a composite made of Si particles and carbonaceous nanofibers, showing around two times higher specific capacity than electrodes with pristine Si particles after hundreds of cycles.18 Hu et al. demonstrated improved capacity retention for carbon-coated Si particles, which were obtained by hydrothermal carbonization of glucose in the presence of Si.19 Inspired by these studies, in the present work, we aimed at preparing nanoscale Si/C composites with a core–shell-type structure from polymer precoated Si particles.
The powder samples after carbonization at temperatures ranging from 700 to 900 °C were characterized by a combination of electron microscopy, X-ray diffraction, and Raman spectroscopy, and their Li-storage properties were investigated by galvanostatic cycling experiments in half-cells. In addition, the volume expansion, gassing behavior, and mechanical deformation/degradation of practical electrodes using the Si/C composite particles were analyzed via in situ pressure, differential electrochemical mass spectrometry, and acoustic emission measurements, respectively.
Results and Discussion
Synthesis
After polymerization—details of which are given in the Experimental Section—the precursor material, containing ≈20 wt % Si (see thermogravimetric analysis (TGA) in Figure S1), was heated under an argon atmosphere at temperatures of 700, 800, or 900 °C to convert the organic shell into carbon. However, in this article, we primarily show data for the Si/C composite particles obtained at 700 °C, as there were no significant differences between the materials. The Si content after carbonization was determined by TGA to be ≈60 wt % (Figure S1).
Structure and Composition
The Si/C composite particles after synthesis and subsequent heating at 700 °C were investigated by transmission electron microscopy (TEM). Figure 1a shows a typical bright-field image, indicating that the particles are polydisperse. The average particle size from counting ≈300 particles was (28 ± 9) nm (Figure S2a). High-resolution TEM (HRTEM, Figure 1b) revealed an amorphous-like shell covering the Si particles. Energy-filtered TEM (EFTEM) mapping using the Si K and C K absorption edges (Figure 1c) confirmed the presence of a relatively homogeneous, nanometer thick layer, which is carbonaceous in nature. High-angle annular dark-field (HAADF)-scanning TEM (STEM) coupled with energy-dispersive X-ray (EDX) spectroscopy analysis was also performed, demonstrating that apart from both Si and C, O is also present in the sample (Figure 1d). Such oxygen is likely due to the native oxide layer on the Si particles. The integrated EDX spectrum of the acquired data indicates that the oxygen content is below 3 wt % (Figure S2b).
Figure 1.
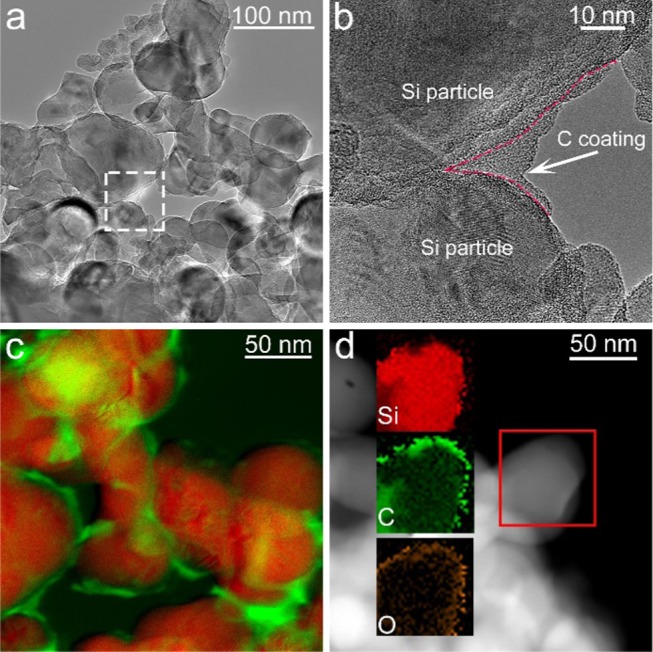
Electron microscopy of Si/C composite particles. (a) Bright-field TEM image. (b) HRTEM image of the area denoted by the dashed white box in (a). (c) EFTEM image indicating the presence of a carbon shell. Si and C are shown in red and green, respectively, in (c). (d) HAADF-STEM image and EDX maps of Si, C, and O for the area denoted by the solid red box.
For further characterization of carbon coating, Raman spectroscopy was used (Figure S3a). Pristine Si particles produced a distinct band at ≈515 cm–1 and a series of weaker multiphonon bands in the range of 200–1000 cm–1. The same Raman bands were also observed for the Si/C composite particles. In addition, there were broad bands at ≈1350 and 1600 cm–1, which can be clearly assigned to the D and G modes of turbostratic carbon from the surface shell. The lateral extent of the graphene layers can be estimated from the ratio of the integrated band intensities.22,23 Consistent with expectations, the degree of carbonization, and thus also of crystallinity, increased with heating temperature (Figure S3b,c).
Furthermore, the pristine Si and Si/C composite particles were analyzed by X-ray diffraction (XRD, Figure S3d). The patterns obtained are basically identical, displaying reflections typical of cubic Si with space group Fd3®m. Overall, this means that coarsening and grain growth effects can be ruled out. Besides, the fact that no carbon features are visible in the pattern of the Si/C composite sample corroborates the disordered (low crystalline) structure of the polymer-derived carbon shell.
Cycling Performance
Electrodes with areal loadings ranging from 1.2 to 2.2 mgSi cm–2 were prepared using both the Si/C composite particles and pristine Si particles for comparison. Because fluoroethylene carbonate (FEC) has been shown in the past to have a positive effect on the cycling performance of especially alloying and conversion-type electrode materials, 1 M LiPF6 in a mixed solvent of FEC and ethyl methyl carbonate (EMC) was used as the electrolyte in the electrochemical testing of Li cells or, in other words, half-cells with a Li counter electrode. After two formation cycles at a rate of C/20 (1C = 4008 mA gSi–1) in the potential range of 1000–10 mV and another three cycles at C/10 between 600 and 30 mV were completed (constant current–constant voltage (CCCV) cycling to electrochemically address the bulk of the electrode and to form a proper solid electrolyte interface (SEI) layer), the cells were cycled at C/2 in the CCCV mode in the latter potential range. A rate performance test was implemented after every 25 cycles (CCCV lithiation at C/2 and constant current (CC) delithiation at different rates of 1C, 3C, and C/10). A detailed description of the cycling protocol used can be found in Table S1.
Figure 2a shows a comparison of the capacity retention between the Si/C composite particles and the pristine Si particles at an average loading of 1.2 mgSi cm–2. During the first lithiation cycle, specific capacities of >4200 mAh gSi–1 were achieved for both the coated and noncoated Si-based electrodes, exceeding the theoretical specific capacity of Li15Si4 (3579 mAh gSi–1) and Li21Si5 (4008 mAh gSi–1). This is probably due to SEI formation and side reactions with the native oxide layer, both of which (irreversibly) consumes a fair amount of active Li, and thus, is a major problem, especially in the full-cell where the cathode is usually the Li source. Over the first 20 cycles, the cells exhibited comparable cycling behavior, but then capacity degradation was observed in the case of pristine Si particles. Though we do not have direct evidence, we suspect that mechanical degradation and detachment of Si particles, carbon black, and polymer binder from the electrode lead to loss of electrochemical activity. The core–shell-type structure of the Si/C composite particles apparently has a stabilizing effect on the cyclability. The corresponding electrode retained a specific (lithiation) capacity of ≈1800 mAh gSi–1 at the 100th cycle, compared to only ≈1250 mAh gSi–1 for the pristine Si particles. As can be also seen from Figure 2a, the cell using the Si/C composite particles showed much improved rate capability from the second rate performance test (after 42 cycles) onwards. This is particularly evident at 1C (see specific delithiation capacities in Figure S4)—the Si/C composite-containing cell delivered specific capacities of about 2 times higher than with the pristine Si particles. By contrast, the overall performance at the 3C rate was similar. The specific capacity dropped immediately to below 200 mAh gSi–1, indicating that the materials employed here are not suited for high-power applications, as somewhat expected.
Figure 2.
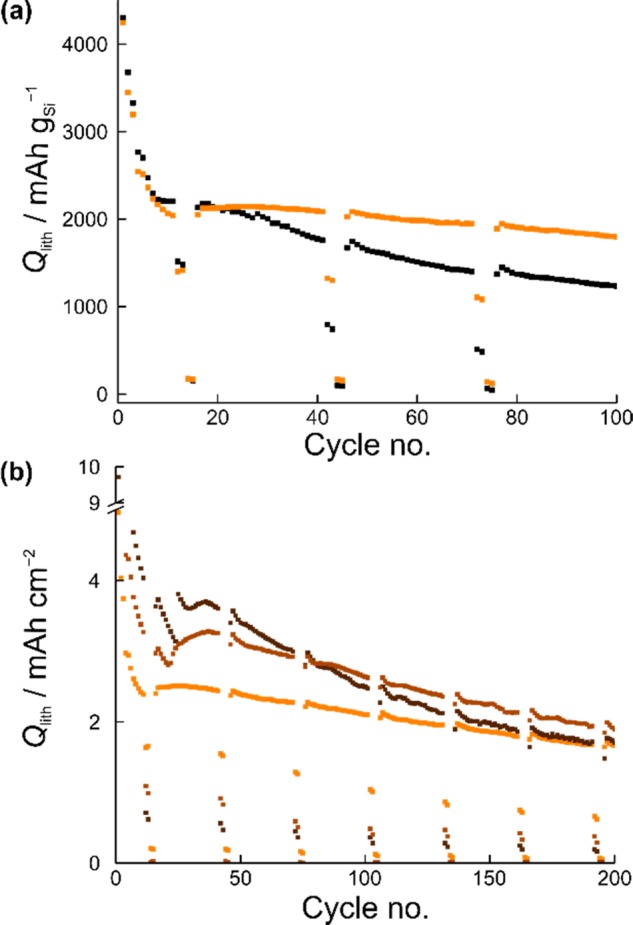
Electrochemical cycling performance of half-cells with the FEC-based electrolyte. (a) Specific lithiation capacity of pristine Si particles (black) and Si/C composite particles (orange). The areal loading was 1.2 mgSi cm–2. (b) Areal capacity achieved with electrodes using Si/C composite particles of different loading: 1.2 (orange), 1.9 (light brown), and 2.2 mgSi cm–2 (dark brown).
Furthermore, electrodes with a higher loading of the Si/C composite particles were tested. The areal capacities achieved over the first 200 cycles are shown in Figure 2b. The electrode with the highest loading exhibited an initial (lithiation) capacity of >9 mAh cm–2. However, in this case, more pronounced fading was observed with prolonged cycling. After 80 cycles, the electrodes with 1.9 and 2.2 mgSi cm–2 revealed similar areal capacities of ≈3 mAh cm–2, and after ≈180 cycles, the low-loading electrode even outperformed the high-loading one. This finding demonstrates that the capacity decay rate is strongly dependent on the active material loading.
The effect of carbonization temperature on the cyclability is shown in Figure S5. Interestingly, the Si/C composite particles after heating at 700, 800, and 900 °C exhibited very similar cycling performance during the first 100 cycles, delivering more or less the same specific capacities. However, slightly better capacity retention and kinetics were observed in the later cycles for the 900 °C sample (e.g., ≈180 mAh gSi–1 difference in specific lithiation capacity between the Si/C composite-containing cells at the 200th cycle), which seems to be due to subtle changes in the properties of the carbon shell, caused by progressive carbonization.
Volume Expansion
One of the key challenges of Si as a negative electrode material is to effectively accommodate the volume changes during battery operation. The large expansion and contraction with alloying and dealloying, respectively, leads to accelerated mechanical degradation (anode self-pulverization) and SEI instability (continuous electrolyte decomposition), and therefore ultimately to cell failure. The volume changes have been studied in the past using various methods such as XRD—by comparing the unit cell volume of cubic Si and Li15Si4—resulting in a total expansion of ≈280%.24,25 XRD, in particular, is well suited to evaluate structural differences between crystalline phases, but it cannot be used to determine volume changes of practical electrodes, containing carbon black additive and polymer binder in addition to the Si anode active material. The volume changes of Si nanopillars with a height of 100 nm and diameters ranging from 100 to 1000 nm were analyzed via atomic force microscopy (AFM).26 For pillars of diameter >200 nm, a total volume expansion of ≈300% was observed, whereas 100 nm diameter pillars showed volume changes below 200%. This study focused on the basic properties of Si upon alloying with Li, but again, it did not consider the effect of other electrode constituents or architectures on the “breathing” behavior. By contrast, Breitung et al. conducted in situ AFM measurements on practical Si electrodes,27 revealing a strong increase in electrode thickness (height) during the initial lithiation cycle. With further cycling, only minor volume changes were observed. In fact, it might well be that the electrode height remains largely unaffected after the first cycle, whereas the Si particles in the bulk of the electrode still undergo volume changes, which cannot be detected by AFM, though. Further results were obtained by Tranchot et al. by measuring thickness variations using dilatometry. They found that electrodes with 85 nm Si particles experience much larger expansion (>300%), with higher irreversibility of the volume changes after the initial cycle, than electrodes with 230 nm Si particles (<150%).28 However, a drawback of the method is that the measurement itself is confined to changes in only one dimension. This is unfavorable, especially when considering that the volume expansion of Si particles is accommodated to some extent by the electrode’s porosity.29
In situ pressure measurements are a viable alternative for monitoring three-dimensional volume changes over many cycles.5,30 Here, a pressure sensor is connected to a custom cell of known headspace volume. Upon cycling, the volume changes of the Si electrode affect the headspace volume, thus leading to periodic pressure changes—after subtraction of irreversible contributions due to gassing. The measured pressure changes are ultimately the sum of the individual contributions of anode and cathode. For this reason, in the present work, (prelithiated) Li4Ti5O12 (LTO), a well-known “zero-strain” material, served as the counter electrode.31 In addition, a lithium reference was used to precisely determine the potential of the Si working electrode. For some unknown reason, the FEC-based electrolyte interfered with the lithium reference, which is why the experiments were performed using the LP57 electrolyte. Figure 3 depicts the volume changes of the Si/C composite-containing electrode (after a couple of cycles in the CC mode when the capacity had leveled off), with lithiation capacities of 1850 mAh gSi–1 on average. As is evident, the total volume expansion amounts to ≈170%. Interestingly, the ΔV versus t curve is rather featureless and has a zigzag shape, with the maxima and minima at the cut-off potentials, consistent with expectations. Taken together, this indicates that the volume expansion/contraction varies relatively linearly with specific capacity or, in other words, Li content. More details and results can be found in Figures S6 and S7 as well as in Appendix S1, including potential profiles and pressure changes over 20 cycles, specific lithiation/delithiation capacities and corresponding Coulombic efficiencies, and calculation of volume changes.
Figure 3.
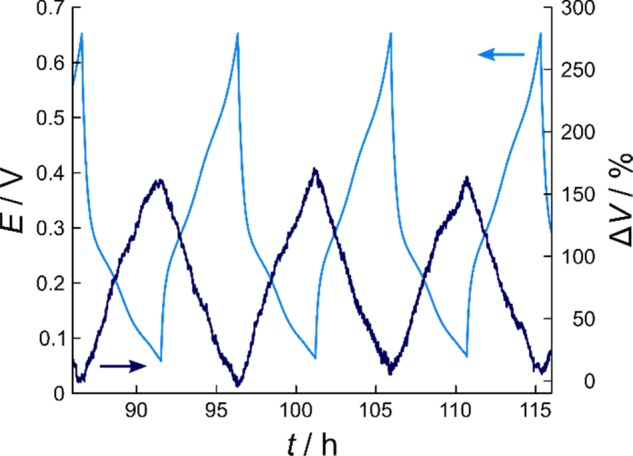
In situ pressure analysis of a cell with three-electrode configuration using prelithiated LTO, Si/C composite particles, and LP57 as the cathode, anode, and electrolyte, respectively. Both the potential profile of the Si working electrode vs Li reference at a rate of C/10 (light blue) and the corresponding volume changes (dark blue) are shown for the 9th–11th cycles.
After cycling the cell for 20 cycles, the Si/C composite-containing electrode was harvested in the delithiated state (≈650 mV) and washed with dimethyl carbonate (DMC) to remove any electrolyte residues. Cross-sectional SEM images obtained on the electrodes before and after cycling are shown in Figure S8. Apparently, the thickness did not change much (≈26 μm, compared to ≈21 μm before cycling), but there are significant differences in morphology and composition (Figure S9). Prior to cycling, the electrode had a rough and grainy appearance, whereas it was relatively smooth with larger pores after cycling. Besides, the electrode constituents were clearly covered by an SEI layer. EDX mapping indicated that, in addition to O and C, which were also present before cycling, the SEI consists of P and F species from reductive decomposition of the supporting salt and carbonate solvents at low potential.
Gas Evolution
Volume change effects during cycling are always accompanied to some extent by the formation of new reactive surfaces, leading to continuous electrolyte decomposition and thickening of the SEI layer. As discussed previously, such processes cause gas evolution, which was monitored in situ by means of differential electrochemical mass spectrometry (DEMS). Figure 4 shows the potential profile of a half-cell with the LP57 electrolyte using the Si/C composite particles and the gas evolution profile of H2, C2H4, and CO2 detected with DEMS during the first five cycles (see also zoomed-in view in Figure S10). The cell was cycled in the CC mode at a rate of C/10. After the initial cycle with upper and lower cut-off potentials of 1.0 V and 10 mV, respectively, was completed, the cell was cycled in the range of 600–30 mV. The main gaseous species evolved were H2 (m/z = 2) and C2H4 (m/z = 26). However, small amounts of CO2 (m/z = 44) were also detected. For clear identification of C2H4, mass signal m/z = 26 was used. The reason is that m/z = 28 may be affected by fragments of CO2, CO, and/or N2. The highest evolution rates of H2 and CO2 were observed in the first 3 h of lithiation, during which the Si electrode potential decreased rapidly from ≈3.0 V to below 100 mV. The main source of H2 formation is probably the reduction of trace H2O, originating from the electrode, electrolyte, separator, and the different cell parts. In later cycles, H2 was always detected during lithiation, with a subtle maximum around 110 mV, whereas virtually no H2 evolution took place during delithiation. C2H4 evolution usually accompanies ethylene carbonate (EC) reduction and is indicative of SEI formation on graphite.32−34 The highest evolution rate of C2H4 was observed in the first lithiation cycle when the major part of the SEI is formed. After the first full cycle, slightly higher rates were detected upon lithiation than delithiation. The increase in the C2H4 evolution rate was nearly linear until a potential of ≈70 mV was reached (close to that of the H2 maximum), and then, it leveled off and finally decreased rapidly with beginning of the delithiation cycle. CO2 evolution was detected as well, with rates an order of magnitude lower compared to C2H4. After the initial cycle, the highest evolution rates were seen at the upper and lower cut-off potentials. Some (minor) CO2 was identified in the past as a side product of the reductive decomposition of EC/EMC- and EC/DMC-based electrolytes on graphite.33,35 We believe that similar, if not the same, processes occur in the case of Si, especially when considering the comparable operating potential range. Note that details about the gassing of FEC-based cells are provided elsewhere.36,37
Figure 4.
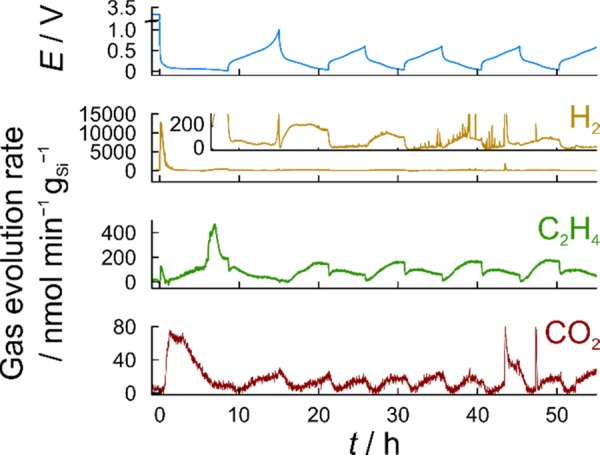
Cell potential at C/10 (light blue) and evolution rates of H2 (yellow), C2H4 (green), and CO2 (red) during DEMS measurement on the Si/C composite-containing electrode. The cell was assembled with the Li metal and LP57 as the counter electrode and electrolyte, respectively.
Particle Fracture/Mechanical Degradation
As mentioned above, a direct result of the large volume changes of Si during Li insertion/extraction with cycling is particle fracture and anode self-pulverization, both of which can be studied by acoustic emission (AE) measurements. To this end, half-cells with the LP57 electrolyte using the Si/C composite-containing electrode were built and then cycled for five cycles. The cycling protocol was similar to that used in the DEMS measurement, except that the initial (formation) cycle was performed at a rate of C/20. As is evident from Figure 5, the rate of events shows strong variations with cell potential. Most hits were detected in the first cycle. Interestingly, there was higher acoustic activity during delithiation than lithiation. This finding is in contrast to studies by Tranchot et al.28,38 and Rhodes et al.,39 where more acoustic events were observed upon lithiation. The high event rate during delithiation may suggest high tensile stress. However, it should be noted that the mechanical properties depend on the composition of the electrode and the morphology and microstructure of the active material.13 With further cycling, a relatively weak peak in the AE hit rate was observed at low potential in the lithiation cycles (starting at ≈60 mV). During delithiation, hits were detected more or less over the entire potential range, with the highest rate close to the upper cut-off potential. Nonetheless, it can also be seen from the data in Figure 5 that the AE activity decreased significantly with cycling, indicating that most of the mechanical degradation (including electrode restructuring) occurs in the first couple of cycles. Unfortunately, data interpretation is nontrivial, as many different processes may contribute to the measured AE such as particle fracture, electrode cracking and delamination, or gas evolution to name a few.
Figure 5.
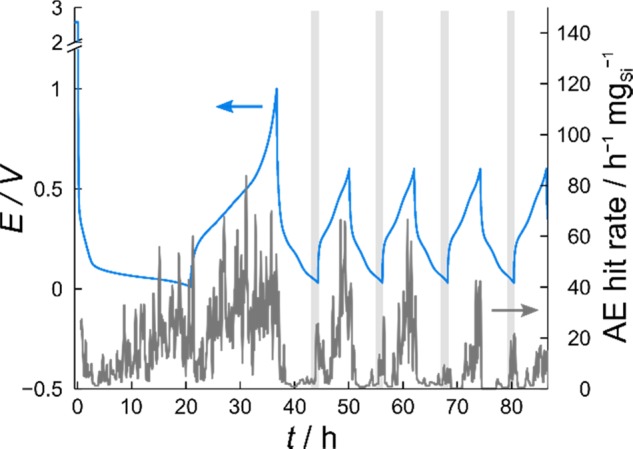
Cell potential (light blue) and AE hit rate (gray) during AE measurement on the Si/C composite-containing electrode. The cell was assembled with the Li metal and LP57 as the counter electrode and electrolyte, respectively. The region close to the lower cut-off potential is highlighted for clarity.
To evaluate the effect of gas evolution on the measured acoustic activity, cells using electrodes made of only Super C65 carbon and polymer binder were studied in the chronoamperometry mode using DEMS and AE measurements. The upper potential was set to 1.5 V and the lower potential was controlled between 500 and 100 mV, while monitoring gas evolution and acoustic activity, respectively (Figure S11). Both H2 and C2H4 evolution was observed at low potential, clearly originating from electrolyte decomposition and trace H2O reduction. However, potential-dependent acoustic activity was not seen in any of the experiments. Thus, we conclude that, in the present case, gassing has no notable effect on the measured AE activity. Additional experiments carried out by cyclic voltammetry in the potential range between 3.0 V and 10 mV corroborate this (Figure S12).
Although AE measurements are very sensitive to mechanical deformation, gassing seems to have negligible influence on the data acquired. This result is in agreement with other studies, indicating that effects related to gas evolution, if detected, exhibit significantly longer rise times (up to 200 μs) than mechanical processes.38,40,41 Finally, we note that to distinguish between different mechanical degradation mechanisms, the waveforms and frequencies of all acoustic hits would need to be analyzed in detail. This, however, is outside the scope of this article.
Conclusions
In summary, we have demonstrated the successful preparation of small-size Si/C composite particles from a novel polymer-coated precursor material. Detailed evaluation through microscopic and spectroscopic analysis showed that the powder sample after carbonization at 700 °C exhibits a kind of core–shell structure, with the surface layer being turbostratic in nature. Despite the relatively low crystallinity of the carbon shell, the Si/C composite outperformed battery cells with pristine Si particles/Super C65 carbon blended electrodes in terms of electrochemical cycling stability. Long-term cycling experiments resulted in an ≈1.5 times higher specific capacity for the Si/C composite particles after 200 cycles. The volume changes of the Si/C composite-containing electrode during alloying and dealloying with Li were studied in situ by pressure measurements. A total volume expansion of ≈170% was observed when cycling in the voltage range between 600 and 30 mV (with Qlith ≈ 1850 mAh gSi−1). Notably, the volume varied virtually linearly with specific capacity. The associated mechanical degradation was analyzed via acoustic emission measurements, indicating a higher rate of events during delithiation than lithiation, with most hits detected in the initial cycle. Furthermore, differential electrochemical mass spectrometry revealed the evolution of H2, C2H4, and CO2 over several cycles, thus providing indirect evidence of solid electrolyte interface instability. The gas evolution rates showed different features, which were thoroughly analyzed. Pronounced surface layer formation as well as changes in the electrode structure and morphology during cell operation were also confirmed by ex situ electron microscopy imaging and elemental mapping analysis. Overall, the data presented in this work demonstrate that core–shell structures of Si or SiOx composites hold promise for future energy storage applications, possibly especially in combination with graphite.
Experimental Section
Materials Synthesis
Si-based precursor material was produced by heating a 1 L glass reactor equipped with a mechanical stirrer, thermometer, and condenser in an oil bath. Monomer and initiator solutions were added via two separate dosing units. Specifically, Si particles (10 g, APS ≤ 50 nm; Alfa Aesar) were dispersed in a 1:1 mixture of hexane and ethyl acetate (227.5 g) under a N2 atmosphere and heated to 65 °C. N-Vinylpyrrolidone (29.9 g), acrylic acid (13.0 g), 1-vinylimidazole (1.5 g), stearyl poly(ethylene glycol) methacrylate 1100 (SPEGMA 1100, 5.5 g), and pentaerythritol triallyl ether (0.21 g) were dissolved in hexane/ethyl acetate (187.5 g) and fed over a period of 3 h. Radical initiators (Wako V65, 0.2 g and tert-butyl peroctoate, 2.3 g) were dissolved in hexane/ethyl acetate (22.5 g) and fed over a period of 5 h, and then, the polymerization was continued for 15 h at 75 °C. The reaction mixture was dispersed in acetone (500 mL), filtered, and the solid product was washed several times with acetone to remove residual hexane/ethyl acetate. Finally, the polymer-coated Si powder sample was heated under an Ar atmosphere at temperatures of 700, 800, or 900 °C in a tube furnace (RT 50-250/11 W/B410; Nabertherm). The temperature profile of the heating process is depicted in Figure S13.
Electrode Preparation
Si electrodes were prepared by water-based slurry coating of 75 wt % Si/C composite particles, 10 wt % carbon black additive (Super C65; Timcal), and 15 wt % poly(vinyl alcohol) binder (Selvol 425; Sekisui) onto 18 μm-thick Cu foil (Gould Electronics). Reference Si electrodes were prepared with 50 wt % pristine Si particles, 35 wt % carbon black, and 15 wt % polymer binder. Super C65 electrodes were prepared with 50 wt % carbon black (≈0.4 mgC cm–2) and 50 wt % polymer binder.
Electrochemical Testing
All cells were assembled in an Ar-filled glovebox ([O2], [H2O] < 1 ppm; MBraun). Electrochemical testing was performed on coin-type cells with the Li counter electrode (Ø13 mm, 600 μm thickness; Albemarle Germany GmbH), glass fiber separator (Ø17 mm, Whatman GF/D; GE Healthcare Life Sciences), Si working electrode (Ø13 mm, 1.2–2.2 mgSi cm–2), and 250 μL of 1 M LiPF6 (Kanto Denka Kogyo Co., Ltd.) in a 1:1 weight mixture of FEC (Solvay) and EMC (BASF SE) as the electrolyte.
DEMS and pressure measurements were performed on custom cells.33,42 For DEMS, cells containing the Li counter electrode (Ø40 mm, 600 μm thickness), glass fiber separator (Ø42 mm, Whatman GF/A), and Si working electrode (Ø40 mm, 0.7 mgSi cm–2) were used. Pressure analysis was performed on cells with a Si anode (Ø40 mm, 0.56 mgSi cm–2), glass fiber separator (Ø40 mm, Whatman GF/A), and prelithiated LTO cathode (Ø40 mm, 14.4 mgLTO cm–2; Sigma-Aldrich). In both cases, 600 μL of LP57 electrolyte (1 M LiPF6 in a 3:7 weight mixture of EC and EMC; BASF SE) were used. Note that both the LTO and Si electrodes had a hole (Ø4 mm) in the middle for gas extraction reason, and a Li reference electrode was used in these experiments.
AE measurements were performed on CR2032 coin cells (Hohsen Corp.) containing the Li counter electrode (Ø14 mm, 600 μm thickness), glass fiber separator (Ø17 mm, Whatman GF/D), Si working electrode (Ø12 mm, 0.7 mgSi cm–2), and 250 μL of LP57 electrolyte.
Prior to use, all electrodes were dried under vacuum at 100 °C for more than 10 h. The water content of LP57 and EMC was determined by Karl Fischer titration to be <10 ppm. According to the supplier (Solvay), the water content of FEC was <10 ppm.
Instrumentation
DEMS and pressure measurements were performed using a BioLogic VSP-300 potentiostat. The open-circuit voltage before cycling was recorded over a time period of 6 h. For DEMS, a constant carrier gas flow (2.5 mLHe min–1, 6.0 purity) was used and the evolved gasses were analyzed by a mass spectrometer (GSD 320, OmniStar Gas Analysis System; Pfeiffer Vacuum GmbH). After each run, a calibration gas of known composition was introduced into the mass spectrometer to convert ionic currents into molar ppm values. More details can be found elsewhere.33,42 For pressure measurements, PAA33X-V-3 pressure sensors (Omega) were used.
Electrochemical cycling was performed using a MACCOR Series 4000 (Tulsa) battery tester. For AE measurements, a BAT-SMALL battery cycler (Astrol Electronic AG) was used. The AE device consisted of a sensor, preamplifier, and data acquisition system (USB-AE Node with AEwin software; MISTRAS Group, Inc.). A differential wideband sensor (MISTRAS), with the operating frequency range between 125 and 1000 kHz and resonant frequency of 125 kHz, was fixed with silicon grease on the current collector of the working electrode. A preamplifier gain of 40 dB, an analog filter of 20–1000 kHz, and a sample rate of 5 MHz were used. AE activity was registered when a hit signal exceeded the threshold of 27 dB. AE signals of less than two counts or with a peak frequency lower than 80 kHz were considered background noise events, and thus, eliminated.
TGA data were collected on a TG 209 F1 Libra (NETZSCH).
Raman spectroscopy was performed at a wavelength of 532 nm using an inVia confocal Raman microscope (Renishaw).
A Mo Kα1,2 diffractometer with a Pilatus 300K-W area detector (DECTRIS) was used for powder XRD.
Specimens for TEM were prepared by powder dispersion in EtOH using an ultrasonic bath. The suspended powder was deposited on a Cu grid with a holey carbon support film. TEM was performed using an FEI Titan 80–300 Cs-corrected microscope operated at 300 keV and equipped with a Gatan 863 Tridiem imaging filter system.
Acknowledgments
This study is part of the projects being funded within the BASF International Network for Batteries and Electrochemistry. This work was partially carried out with the support of the Karlsruhe Nano Micro Facility (KNMF, www.knmf.kit.edu), a Helmholtz research infrastructure at Karlsruhe Institute of Technology (KIT, www.kit.edu). The authors thank Lea de Biasi, Reiner Mönig, and Holger Geßwein as well as Ruby Singh for XRD and Raman spectroscopy measurements, respectively.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02541.
TGA data; particle size distribution; EDX analysis; Raman spectra; XRD patterns; detailed cycling protocol; rate performance analysis; effect of carbonization temperature on specific capacity; pressure measurement on Si/C composite particles; prelithiated LTO/Si cell performance; cross-sectional SEM images and EDX mapping of Si/C composite-containing electrodes; DEMS measurement on Si/C composite particles; chronoamperometric AE and DEMS as well as cyclic voltammetric AE measurements on Super C65 carbon electrodes; carbonization profile; and details about the calculation of volume changes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- de Biasi L.; Kondrakov A. O.; Geßwein H.; Brezesinski T.; Hartmann P.; Janek J. Between Scylla and Charybdis: Balancing Among Structural Stability and Energy Density of Layered NCM Cathode Materials for Advanced Lithium-Ion Batteries. J. Phys. Chem. C 2017, 121, 26163–26171. 10.1021/acs.jpcc.7b06363. [DOI] [Google Scholar]
- Nitta N.; Wu F.; Lee J. T.; Yushin G. Li-Ion Battery Materials: Present and Future. Mater. Today 2015, 18, 252–264. 10.1016/j.mattod.2014.10.040. [DOI] [Google Scholar]
- Myung S. T.; Maglia F.; Park K. J.; Yoon C. S.; Lamp P.; Kim S. J.; Sun Y. K. Nickel-Rich Layered Cathode Materials for Automotive Lithium-Ion Batteries: Achievements and Perspectives. ACS Energy Lett. 2017, 2, 196–223. 10.1021/acsenergylett.6b00594. [DOI] [Google Scholar]
- Scrosati B.; Garche J. Lithium Batteries: Status, Prospects and Future. J. Power Sources 2010, 195, 2419–2430. 10.1016/j.jpowsour.2009.11.048. [DOI] [Google Scholar]
- Schweidler S.; de Biasi L.; Schiele A.; Hartmann P.; Brezesinski T.; Janek J. Volume Changes of Graphite Anodes Revisited: A Combined Operando X-ray Diffraction and In Situ Pressure Analysis Study. J. Phys. Chem. C 2018, 122, 8829–8835. 10.1021/acs.jpcc.8b01873. [DOI] [Google Scholar]
- Dimov N.; Kugino S.; Yoshio M. Mixed Silicon-Graphite Composites as Anode Material for Lithium Ion Batteries - Influence of Preparation Conditions on the Properties of the Material. J. Power Sources 2004, 136, 108–114. 10.1016/j.jpowsour.2004.05.012. [DOI] [Google Scholar]
- Yim C.-H.; Courtel F. M.; Abu-Lebdeh Y. A High Capacity Silicon–Graphite Composite as Anode for Lithium-Ion Batteries Using Low Content Amorphous Silicon and Compatible Binders. J. Mater. Chem. A 2013, 1, 8234–8243. 10.1039/c3ta10883j. [DOI] [Google Scholar]
- Klett M.; Gilbert J. A.; Pupek K. Z.; Trask S. E.; Abraham D. P. Layered Oxide, Graphite and Silicon-Graphite Electrodes for Lithium-Ion Cells: Effect of Electrolyte Composition and Cycling Windows. J. Electrochem. Soc. 2017, 164, A6095–A6102. 10.1149/2.0131701jes. [DOI] [Google Scholar]
- Zuo X.; Zhu J.; Müller-Buschbaum P.; Cheng Y.-J. Silicon Based Lithium-Ion Battery Anodes: A Chronicle Perspective Review. Nano Energy 2017, 31, 113–143. 10.1016/j.nanoen.2016.11.013. [DOI] [Google Scholar]
- Chae S.; Ko M.; Kim K.; Ahn K.; Cho J. Confronting Issues of the Practical Implementation of Si Anode in High-Energy Lithium-Ion Batteries. Joule 2017, 1, 47–60. 10.1016/j.joule.2017.07.006. [DOI] [Google Scholar]
- Szczech J. R.; Jin S. Nanostructured Silicon for High Capacity Lithium Battery Anodes. Energy Environ. Sci. 2011, 4, 56–72. 10.1039/C0EE00281J. [DOI] [Google Scholar]
- Kasavajjula U.; Wang C.; Appleby A. J. Nano- and Bulk-Silicon-Based Insertion Anodes for Lithium-Ion Secondary Cells. J. Power Sources 2007, 163, 1003–1039. 10.1016/j.jpowsour.2006.09.084. [DOI] [Google Scholar]
- Zhang W. J. A Review of the Electrochemical Performance of Alloy Anodes for Lithium-Ion Batteries. J. Power Sources 2011, 196, 13–24. 10.1016/j.jpowsour.2010.07.020. [DOI] [Google Scholar]
- Ryu J. H.; Kim J. W.; Sung Y.-E.; Oh S. M. Failure Modes of Silicon Powder Negative Electrode in Lithium Secondary Batteries. Electrochem. Solid-State Lett. 2004, 7, A306–A309. 10.1149/1.1792242. [DOI] [Google Scholar]
- Erk C.; Brezesinski T.; Sommer H.; Schneider R.; Janek J. Toward Silicon Anodes for Next-Generation Lithium Ion Batteries: A Comparative Performance Study of Various Polymer Binders and Silicon Nanopowders. ACS Appl. Mater. Interfaces 2013, 5, 7299–7307. 10.1021/am401642c. [DOI] [PubMed] [Google Scholar]
- Xu C.; Lindgren F.; Philippe B.; Gorgoi M.; Bjorefors F.; Edstrom K.; Gustafsson T. Improved Performance of the Silicon Anode for Li-Ion Batteries: Understanding the Surface Modification Mechanism of Fluoroethylene Carbonate as an Effective Electrolyte Additive. Chem. Mater. 2015, 27, 2591–2599. 10.1021/acs.chemmater.5b00339. [DOI] [Google Scholar]
- Lee J. H.; Paik U.; Hackley V. A.; Choi Y. M. Effect of Poly(Acrylic Acid) on Adhesion Strength and Electrochemical Performance of Natural Graphite Negative Electrode for Lithium-Ion Batteries. J. Power Sources 2006, 161, 612–616. 10.1016/j.jpowsour.2006.03.087. [DOI] [Google Scholar]
- Breitung B.; Schneider A.; Chakravadhanula V. S. K.; Suchomski C.; Janek J.; Sommer H.; Brezesinski T. Artificial Composite Anode Comprising High-Capacity Silicon and Carbonaceous Nanostructures for Long Cycle Life Lithium-Ion Batteries. Batteries Supercaps 2018, 1, 27–32. 10.1002/batt.201700004. [DOI] [Google Scholar]
- Hu Y. S.; Demir-Cakan R.; Titirici M. M.; Müller J. O.; Schlögl R.; Antonietti M.; Maier J. Superior Storage Performance of a Si@SiOx/C Nanocomposite as Anode Material for Lithium-Ion Batteries. Angew. Chem., Int. Ed. 2008, 47, 1645–1649. 10.1002/anie.200704287. [DOI] [PubMed] [Google Scholar]
- Holzapfel M.; Buqa H.; Scheifele W.; Novák P.; Petrat F.-M. A New Type of Nano-Sized Silicon/Carbon Composite Electrode for Reversible Lithium Insertion. Chem. Commun. 2005, 1566–1568. 10.1039/B417492E. [DOI] [PubMed] [Google Scholar]
- Shen D.; Huang C.; Gan L.; Liu J.; Gong Z.; Long M. Rational Design of Si@SiO2/C Composites Using Sustainable Cellulose as a Carbon Resource for Anodes in Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2018, 10, 7946–7954. 10.1021/acsami.7b16724. [DOI] [PubMed] [Google Scholar]
- Tuinstra F.; Koenig J. L. Raman Spectrum of Graphite. J. Chem. Phys. 1970, 53, 1126–1130. 10.1063/1.1674108. [DOI] [Google Scholar]
- Cançado L. G.; Takai K.; Enoki T.; Endo M.; Kim Y. A.; Mizusaki H.; Jorio A.; Coelho L. N.; Magalhães-Paniago R.; Pimenta M. A. General Equation for the Determination of the Crystallite Size La of Nanographite by Raman Spectroscopy. Appl. Phys. Lett. 2006, 88, 163106 10.1063/1.2196057. [DOI] [Google Scholar]
- Obrovac M. N.; Christensen L. Structural Changes in Silicon Anodes during Lithium Insertion/Extraction. Electrochem. Solid-State Lett. 2004, 7, A93–A96. 10.1149/1.1652421. [DOI] [Google Scholar]
- Obrovac M. N.; Krause L. J. Reversible Cycling of Crystalline Silicon Powder. J. Electrochem. Soc. 2007, 154, A103–A108. 10.1149/1.2402112. [DOI] [Google Scholar]
- Becker C. R.; Strawhecker K. E.; Mcallister Q. P.; Lundgren C. A. In Situ Atomic Force Microscopy of Lithiation and Delithiation of Silicon Nanostructures for Lithium Ion Batteries. ACS Nano 2013, 7, 9173–9182. 10.1021/nn4037909. [DOI] [PubMed] [Google Scholar]
- Breitung B.; Baumann P.; Sommer H.; Janek J.; Brezesinski T. In Situ and Operando Atomic Force Microscopy of High-Capacity Nano-Silicon Based Electrodes for Lithium-Ion Batteries. Nanoscale 2016, 8, 14048–14056. 10.1039/C6NR03575B. [DOI] [PubMed] [Google Scholar]
- Tranchot A.; Idrissi H.; Thivel P. X.; Roué L. Influence of the Si Particle Size on the Mechanical Stability of Si-Based Electrodes Evaluated by In-Operando Dilatometry and Acoustic Emission. J. Power Sources 2016, 330, 253–260. 10.1016/j.jpowsour.2016.09.017. [DOI] [Google Scholar]
- Tranchot A.; Idrissi H.; Thivel P. X.; Roué L. Impact of the Slurry pH on the Expansion/Contraction Behavior of Silicon/Carbon/Carboxymethylcellulose Electrodes for Li-Ion Batteries. J. Electrochem. Soc. 2016, 163, A1020–A1026. 10.1149/2.1071606jes. [DOI] [Google Scholar]
- Schiele A.; Hatsukade T.; Berkes B. B.; Hartmann P.; Brezesinski T.; Janek J. High-Throughput In Situ Pressure Analysis of Lithium-Ion Batteries. Anal. Chem. 2017, 89, 8122–8128. 10.1021/acs.analchem.7b01760. [DOI] [PubMed] [Google Scholar]
- Ohzuku T.; Ueda A.; Yamamoto N. Zero-Strain Insertion Material of Li[Li1/3Ti5/3]O4 for Rechargeable Lithium Cells. J. Electrochem. Soc. 1995, 142, 1431–1435. 10.1149/1.2048592. [DOI] [Google Scholar]
- Novák P.; Joho F.; Imhof R.; Panitz J.-C.; Haas O. In Situ Investigation of the Interaction between Graphite and Electrolyte Solutions. J. Power Sources 1999, 81–82, 212–216. 10.1016/S0378-7753(99)00119-6. [DOI] [Google Scholar]
- Berkes B. B.; Schiele A.; Sommer H.; Brezesinski T.; Janek J. On the Gassing Behavior of Lithium-Ion Batteries with NCM523 Cathodes. J. Solid State Electrochem. 2016, 20, 2961–2967. 10.1007/s10008-016-3362-9. [DOI] [Google Scholar]
- Michalak B.; Berkes B. B.; Sommer H.; Bergfeldt T.; Brezesinski T.; Janek J. Gas Evolution in LiNi0.5Mn1.5O4/Graphite Cells Studied In Operando by a Combination of Differential Electrochemical Mass Spectrometry, Neutron Imaging and Pressure Measurements. Anal. Chem. 2016, 88, 2877–2883. 10.1021/acs.analchem.5b04696. [DOI] [PubMed] [Google Scholar]
- La Mantia F.; Novák P. Online Detection of Reductive CO2 Development at Graphite Electrodes in the 1 M LiPF6, EC:DMC Battery Electrolyte. Electrochem. Solid-State Lett. 2008, 11, A84–A87. 10.1149/1.2890756. [DOI] [Google Scholar]
- Schiele A.; Breitung B.; Hatsukade T.; Berkes B. B.; Hartmann P.; Janek J.; Brezesinski T. The Critical Role of Fluoroethylene Carbonate in the Gassing of Silicon Anodes for Lithium-Ion Batteries. ACS Energy Lett. 2017, 2, 2228–2233. 10.1021/acsenergylett.7b00619. [DOI] [Google Scholar]
- Jung R.; Metzger M.; Haering D.; Solchenbach S.; Marino C.; Tsiouvaras N.; Stinner C.; Gasteiger H. A. Consumption of Fluoroethylene Carbonate (FEC) on Si-C Composite Electrodes for Li-Ion Batteries. J. Electrochem. Soc. 2016, 163, A1705–A1716. 10.1149/2.0951608jes. [DOI] [Google Scholar]
- Tranchot A.; Etiemble A.; Thivel P. X.; Idrissi H.; Roué L. In-Situ Acoustic Emission Study of Si-Based Electrodes for Li-Ion Batteries. J. Power Sources 2015, 279, 259–266. 10.1016/j.jpowsour.2014.12.126. [DOI] [Google Scholar]
- Rhodes K.; Dudney N.; Lara-Curzio E.; Daniel C. Understanding the Degradation of Silicon Electrodes for Lithium-Ion Batteries Using Acoustic Emission. J. Electrochem. Soc. 2010, 157, A1354–A1360. 10.1149/1.3489374. [DOI] [Google Scholar]
- Matsuo T.; Uchida M.; Cho H. Development of Acoustic Emission Clustering Method to Detect Degradation of Lithium Ion Batteries. J. Solid Mech. Mater. Eng. 2011, 5, 678–689. 10.1299/jmmp.5.678. [DOI] [Google Scholar]
- Etiemble A.; Idrissi H.; Roué L. On the Decrepitation Mechanism of MgNi and LaNi5-Based Electrodes Studied by in Situ Acoustic Emission. J. Power Sources 2011, 196, 5168–5173. 10.1016/j.jpowsour.2011.01.098. [DOI] [Google Scholar]
- Berkes B. B.; Jozwiuk A.; Vračar M.; Sommer H.; Brezesinski T.; Janek J. Online Continuous Flow Differential Electrochemical Mass Spectrometry with a Realistic Battery Setup for High-Precision, Long-Term Cycling Tests. Anal. Chem. 2015, 87, 5878–5883. 10.1021/acs.analchem.5b01237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.