

Abstract

In this paper, the mechanistic studies on the isomerization of hydroxyl and silyl derivatives of bicyclic cyclopropanes under the catalytic action of Zeise’s salt have been reported. The catalytic activity of both the monomeric and the dimeric forms of Zeise’s salt has been studied by applying the high-level quantum mechanical method. Results from this investigation reveal that the reaction goes favorably under the catalysis of the dimeric form of Zeise’s salt. The calculated activation barrier for the catalytic process using Zeise’s dimer reveals that the rearrangement occurs with an activation barrier of 19–25 kcal mol–1. Depending on the nature of substituents present on the substrate, formation of various products has been explained. This study also includes the heteronuclear counter part of Zeise’s dimer where one of the Pt-metals is replaced by palladium (Pd) and nickel (Ni) successively. The calculated activation barrier using these heteronuclear catalysts is found to be close enough to that calculated for the catalytic pathway using Zeise’s dimer.
Introduction
The reactions of cyclopropane
derivatives under the transition-metal
catalytic conditions have got considerable attention in various fields
of chemistry.1 In many organic reactions,
the cyclopropane ring system behaves like an unsaturated olefin moiety
because of the similarities in their electronic structures.2 As a result, the tendency of the cyclopropane
system to form coordinate bonds with other transition-metal ions resembles
with the property of the unsaturated compounds that acts as the π-ligand
in many complex molecules for organic synthesis.3,4
The literature survey reveals that the cyclopropane derivative
when coordinated to a platinum atom, invariably undergoes an oxidative
insertion reaction to produce a platinacyclobutane derivative.5
Under several circumstances, the formation of this four-membered ring was detected by spectroscopic measurement, yet the instability of the ring is quite evident by its reactivity with other groups or reagents present within the substrate or environment that leads to a variety of rearranged products. In the past few decades, several instances were noticed where the substrates are found to be mostly bicyclic in nature.6 In particular, if the substrate contains an oxy substituent, the ring opening and rearrangement reactions are quite facile.7 Thus, reports from Jennings et al. reveal the effective utilization of Zeise’s salt for the ring opening reactions in conversion of several hydroxy- and alkoxy-substituted cyclopropane derivatives to methyl ketones8 (Scheme 1a,b). Another report by Sonoda et al. shows an efficient catalytic isomerization of the cyclopropane derivative to allyl silyl ether at ambient temperature when the siloxy group is present on the ring junction9 (Scheme 1c). Madsen et al. extends such ring-opening reaction in a derivative of the natural product for the synthesis of 2-C-branched carbohydrates. Such reaction can be performed by using a variety of O-nucleophiles including alcohols, phenols, and water.10
Scheme 1. Rearrangement Reactions of Bicyclic Derivatives of Cyclopropane under the Catalysis of Zeise’s Salt.

While proposing the mechanism of these reactions, all authors assumed the involvement of a mononuclear Pt-complex for catalyzing the reaction (Scheme 2). At the very first step of the mechanisms, the Pt-metal gets inserted into the cyclopropane moiety by oxidative addition reaction to generate a four-membered platinacyclobutane ring as an intermediate.
Scheme 2. Reported Mechanism of the Conversion of the Bicyclic Substrate to the Monocyclic One under Metal Complex-Catalyzed Condition.

Depending on the nature of various substituents present on the ring junction, the generated platinacyclobutane intermediate rearranges to different products. In particular, the presence of the hydroxy group at the bridge head of the bicyclic skeleton promotes the formation of keto compounds by cleavage of the Pt–C bond. During such process, the mandatory transfer of the proton from the hydroxy group at the ring junction to cyclopropane carbon was further confirmed by the deuterium labeling experiment and other NMR spectroscopy. When the silyl group is present at the ring junction of the substrate, an exocyclic double bond is formed in the product; such formation could be explained by a [1,2]-hydrogen shift between two adjacent carbon atoms present on the ring. All mechanisms proposed here utilize a single Pt-metal ion for the catalytic process; however, the experimental method reveals clearly the utilization of dimeric Zeise’s salt to carry out the reaction. Our previous density functional theory (DFT) study revealed that the involvement of Zeise’s dimer with the substrate provides a more favorable pathway than that observed using a single platinum atom for the rearrangement of oxaspirohexane to 3-methylenetetrahydrofuran.11 Here, depending on the nature of various substituents in the bicyclic skeleton, we report our investigation on the mechanism of the reactions by using the monomeric and dimeric form of the metal catalyst and show the importance of the dimeric form (Zeise’s salt) in reducing the activation barrier.
Results and Discussion
To perform our mechanistic study, we have selected the bicyclo[3.1.0]hexane derivatives with hydroxy or siloxy substituents at the ring junction (1 and 2 in Scheme 3) that were previously reported in the experiment. The overview of the studied pathways is shown in Scheme 3.
Scheme 3. Overview of Plausible Mechanisms Involving the Monomeric and Dimeric Forms of the Pt-Catalyst Condition.

Though a single Pt atom is shown as a catalyzing element, we have studied the effect of both the monomeric and dimeric form of Zeise’s salt involved in catalyzing the reaction to compare the effectiveness of the catalytic activity of monometallic and dimetallic complexes. However, representing the single and double activation process, we have used the letter “M” and “D” attached to the name of the pathways or stationary points; for example, the single activation process of pathway-1 is represented as pathway-1M, whereas the corresponding double activation process is designated as pathway-1D.
The hydroxyl derivative of the bicyclic compound (R=H in Scheme 3) furnishes 2-methylcyclopentanone (designated as pathway-1), whereas the silyl derivative of the same bicyclic structure results 2-methylene-1-siloxycyclopentane as the final product (designated as pathway-2). The first step involves the coordination of the substrate (1 or 2) to the metal center of the catalyst to form the intermediate complex MI1/DI1. This step is followed by the oxidative addition of a Pt-metal ion into the cyclopropane moiety to generate four-membered platinacyclobutane intermediate MI2/DI2. Opening of the platinacyclobutane ring takes place through the cleavage of the Pt–C bond to produce intermediate MI3/DI3 (R=SiH3) or MI4/DI4 (R =H). For the hydroxyl derivative, protodeplatination takes place in intermediate MI4 or DI4 by transferring a proton from the hydroxyl group to generate methylcyclopentanone as the final product (pathway-1). However, the silyl derivative MI3 or DI3 promotes a 1,2-hydride shift which ultimately undergoes a deplatination process to form the cyclopentane derivative with an exocyclic double bond (pathway-2).
As the starting reactant may exist in several conformational isomers, we have selected the most favorable one by constructing it from cyclopentane geometry. While constructing the bicyclic structure, we have chosen the envelope geometry of the cyclopentane ring and utilized its two adjacent eclipsed carbons to insert the cyclopropane ring for generating the bicyclic structure (Scheme 4).
Scheme 4. Construction of an Endo and Exo Bicyclic Structure from Envelope Conformation of the Cyclopentane Ring.

Out of the two possible orientations of the cyclopropane structure relative to the cyclopentane ring system (exo and endo in Scheme 4), the exo orientation is found to be more stable than the endo isomer by 3.8 kcal mol–1. With this exo isomer, the initial complex is generated by forming a Pt–O coordinate bond between monomeric or dimeric Zeise’s salt and the oxygen atom of the substrate molecule. Our investigations on the energy and topology of molecular orbitals of the substrates have given us a clue about the number and orientation of metal centers that can be employed to bind with the geometry of the substrates (Figure 1).
Figure 1.
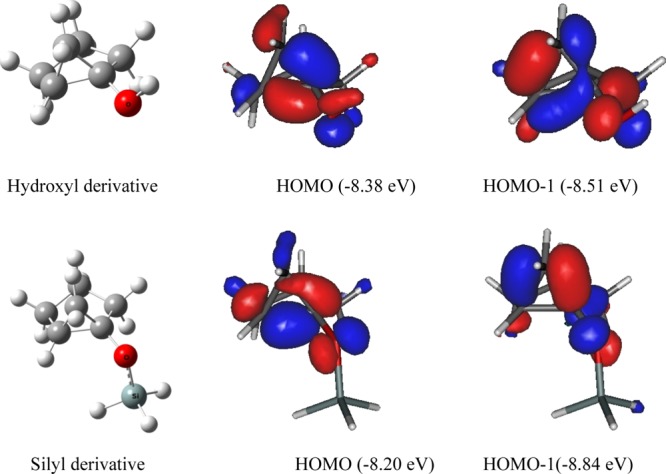
Structure and the outermost molecular orbitals of the bicyclic compounds.
The outermost two filled molecular orbitals [highest occupied molecular orbital (HOMO) and HOMO – 1] have almost the same energies and are suitable for making the coordinate bond with a metal center. The next molecular orbital (HOMO – 2) is situated well below of these two orbitals and may be assumed to be inert for making a coordinate bond with a metal center. Scrutiny of the topology of the outermost two molecular orbitals (Figure 1) clearly reveals that the lobes of the wave functions are largely associated with the edges of the cyclopropane moiety and with the substituent oxygen atom as well. We surmised that the electrons associated to these molecular orbitals and suitable to coordinate with maximum two metal atoms are present at the vicinity of the substrate.
Study on the Mechanism of Rearrangement of 1-Hydroxy-bicyclo[3.1.0]hexane
The potential energy surface indicating the transition states and intermediates involved in all the proposed mechanisms of pathway-1M and pathway-1D (Scheme 3) are shown in Figure 2. Pathway-1Ma (red color) is an alternative pathway diverged from pathway-1M (brown color) that shows a variation of mechanism for the transformation of the bicyclic derivative 1-hydroxy-bicyclo[3.1.0]hexane to 2-methylcyclopentanone under the catalytic action of the monomeric form of Pt-salt. Similarly, pathway-1Da (magenta color) is another alternative pathway diverged from pathway-1D (blue color) that progress under the catalysis of the dimeric form of Zeise’s salt.
Figure 2.

PES along with the thermodynamic parameters and structures of the stationary points of pathway-1M (brown color), pathway-1Ma (red color), pathway-1D (blue color), and pathway-1Da (magenta color) under the catalytic condition of the monomeric and dimeric form of Pt-salt. Energy values that not enclosed in brackets are calculated using M06-2X functional. Values indicated in the round bracket are calculated using M06 functional.
Catalysis by Activation of the Substrate with One Metal Atom
The pathway-1M and pathway-1Ma (Figure 2) in which single Pt-metal is used as a catalyst follow three major steps: the first step is the formation of the four-membered platinacyclobutane intermediate, the second step is the opening of the four-membered platinacyclobutane intermediate, and the third step that is the final step involves with the protodeplatination process. The initial complex (1MI1), in which the Pt-metal is coordinated to the hydroxyl oxygen of the substrate, undergoes an oxidative insertion reaction by crossing an activation barrier of 18.99 kcal mol–1 through the transition-state 1MTS1, and the four-membered platinacyclobutane intermediate (1MI2) is formed as a product. The ball and stick models (Figure 3) of the initial complex 1MI1 reveals that the distance between the hydroxyl oxygen of the substrate and the coordinated Pt-metal is 2.17 c.
Figure 3.
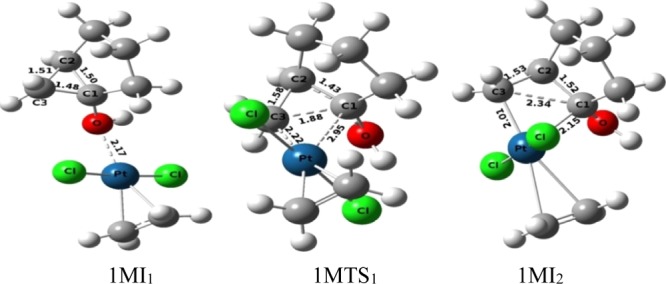
Ball and stick models of the stationary points during the formation of the platinacyclobutane intermediate under the catalytic condition of the monomeric form of Pt-salt.
While forming the transition structure (1MTS1) in the oxidative insertion process, the shorter bond length between C3 and Pt (2.22 Å) in comparison to that of C1–Pt (2.95 Å) clearly indicates that the bond formation between C3 and the Pt-metal atom takes place at the early stage of the reaction. While forming the intermediate from the transition-state 1MTS1, the C1 atom gets the bonding electron mostly from the Pt atom and reduces its positive charge to some extent (+0.526 to +0.423). It has also been observed that C1–C3 distance of the cyclopropane moiety in the initial substrate 1MI1 (1.48 Å) increases to generate the four-membered platinacyclobutane intermediate 1MI2 (2.34 Å) through the distance 1.88 Å in 1MTS1. Intermediate 1MI2 then undergoes a conformational change for the formation of the H bond and intermediate 1MI3 is formed through transition-state 1MTS2 with an activation barrier of 5.30 kcal mol–1.
Opening of the platinacyclobutane ring then takes place by the cleavage of one Pt–C1 bond of intermediate 1MI3 to form intermediate 1MI4 by increasing the C1–Pt distance from 2.19 Å (in 1MI3, Figure 4) to 3.52–352 Å (in 1MI4, Figure 5). This cleavage occurs by crossing a very low activation barrier of 3.10 kcal mol–1 through the transition-state 1MTS3. NBO analysis shows that during this cleavage, the C1 atom acquires more positive charge (+0.459 in 1MI3 to +0.729 in 1MI4 through +0.665 in transition-state 1MTS3), whereas Pt and C3 atoms progressively get negative charge (−0.535 to −0.687 on C3 and +0.365 to +0.270 on Pt). This indicates that the opening of the platinacyclobutane ring by the cleavage of the C1–Pt bond is mostly heterolytic in nature. After the opening of the platinacyclobutane ring, the intermediate 1MI4 undergoes the protodeplatination process. A direct proton transfer from hydroxyl oxygen to C3 carbon atom (through 1MTS5) of pathway-1M results in the detachment of the metal atom to generate the product (1MI6). This step requires 13.62 kcal mol–1 energy to overcome the activation barrier and the relative energy of this transition state is 23.16 kcal mol–1 for the formation of the end product 1MI6 (C2–C1 = 1.51 Å, Figure 5). However, intermediate 1MI5, from which the proton transferring reaction starts, differs from intermediate 1MI4 by a small conformational orientation of the metal atom with respect to the rest of the structure. Several trials to find out the transition structure between these two conformers failed, indicating a very low energy barrier between them. Replacement of the Pt-metal by the migration of protons results in the reduction of electron density on the Pt-metal as apparent from the change of NBO charge (+0.270 to +0.429). It reveals that the delocalization of the bonding electron between the Pt and C3 atom to the C3 and H atom takes place during the protodeplatination process. Global activation barrier of pathway-1M is 26.09 kcal mol–1. In an alternative pathway of protodeplatination (pathway-1Ma), the metal atom is replaced by the proton from the reactant 1MI7a through the SN2 type process (with an activation barrier of 26.41 kcal mol–1 through transition-state 1MTS7a) to form intermediate 1MI8a. To continue such pathway, a conformational change of the intermediate (1MI4) is required through some steps, out of which one has been detected through transition-state 1MTS5a (activation barrier 8.05 kcal mol–1), resulting in intermediate 1MI6a. Global activation barrier of pathway-1Ma is 35.95 kcal mol–1. However, this pathway requires more activation energy with respect to the previous pathway-1M and thus is not favorable. The proton transfer, as shown here, in an intramolecular fashion was already confirmed by the labeling experiment in the previous report.
Figure 4.
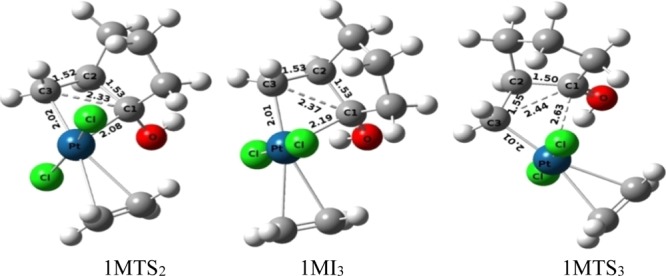
Ball and stick models of the stationary points during the conformational change and cleavage of the platinacyclobutane intermediate under the catalytic condition of the monomeric form of Pt-salt.
Figure 5.

Ball and stick model of the stationary points of protodeplatination in pathway-1M (upper) and 1Ma (lower).
Catalysis by Activation of the Substrate with Two Metal Centers
Rearrangement of 1-hydroxy-bicyclo[3.1.0]hexane has also been studied under the catalytic activity of Zeise’s dimer and is shown in Figure 2 (blue and magenta in color). The blue-colored pathway, pathway-1D, has been diverged to pathway-1Da, whereas deplatination reaction takes place by the migration of a proton. In most of the steps, the rearrangement follows a similar sequence as that of pathway-1M or pathway-1Ma. As evident of the Figure 2, the initial oxidative addition of the Pt metal of the cyclopropane ring to generate platinacyclobutane intermediate 1DI2 (C1–C3 = 2.33 Å in Figure 6) occurs by crossing a very low activation barrier (1.69 kcal mol–1) through transition structure 1DTS1 (C1–C3 = 1.82 Å in Figure 6).
Figure 6.
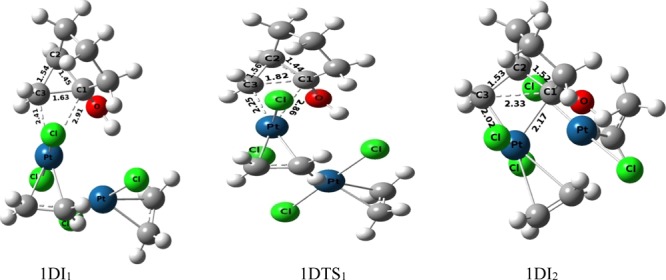
Ball and stick models of the stationary points associated to the pathway of formation of the platinacyclobutane intermediate under the catalysis of the dimeric form of Pt-salt.
Comparison of the geometries of the transition states of oxidative addition under both monomeric and dimeric metal catalytic conditions clearly reveals that the geometry of the monometallic transition state is considerably distorted.
Normally the plane of the square planar geometry of the Pt-complex is nearly perpendicular to the plane of the cyclopropane ring in the transition structure of the oxidative addition of the metal atom that generates platinacyclobutane (Figure 7a). However, a distorted geometry is identified in the transition state of the oxidative addition in the bicyclic derivative of cyclopropane (Figure 7b). The hydrogen bond between the hydroxyl group and the chlorine atom makes an acute angle between the two planes that distorts the geometry from its normal one. However, under dimeric conditions, such distortion in geometry of the transition structure has not been observed in 1DTS1 (Figure 7c) as the hydroxyl group forms a hydrogen bond with the chlorine ligand of the second metal atom leaving the chlorine ligand associated to the first metal atom engaged in the oxidative addition process. The conformational change of the hydroxyl group converts 1DI2 to 1DI3 through the transition structure 1DTS2 (activation barrier 5.62 kcal mol–1). Cleavage of the C1–Pt bond in 1DI3 (C1–Pt = 2.35 Å, Figure 8) occurs through the transition-state 1DTS3 (C1–Pt = 2.47 Å, Figure 8) to generate 1DI4 (activation barrier 1.05 kcal mol–1) by increasing the C1–Pt bond distance from 2.35 Å (1DI3) to 2.98 Å (1DI4).
Figure 7.

(a) Ideal geometry of the transition state for oxidative addition of cyclopropane to the metal center. (b) Distorted geometry of the oxidative addition to the bicyclic substrate when the hydroxyl group is present at the ring junction under monomeric catalytic conditions and (c) undistorted geometry of the oxidative addition to the bicyclic substrate when the hydroxyl group is present at the ring junction under dimeric catalytic conditions.
Figure 8.
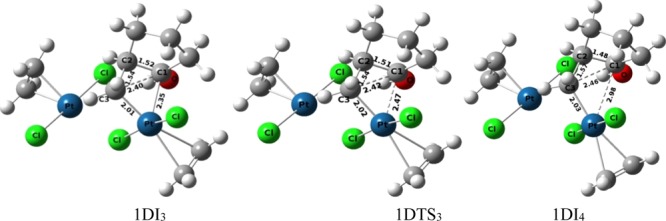
Ball and Stick models of the transition state and intermediate for the cleavage of the C1–Pt bond under the catalytic condition of the dimeric form of Pt-salt.
The final step of protodemetallation occurs from the intermediate 1DI4 through the transition structure 1DTS4 (activation barrier 11.12 kcal mol–1) resulting in the final product 1DI5 (methyl-cyclopentane and Zeise’s dimer). Both the attachment of proton and the detachment of the metal center in 1DTS4 (O–H = 1.36 Å, H–C3 = 1.48 Å, Figure 9) occur from the same face of the C3 atom (Figure 9). We have envisioned another possible way of protodeplatination such that the incoming proton and the eliminating Pt-metal should orient in the opposite face of the C3 atom. Such study results the divergent pathway-1Da that starts by a conformational reorganization of the cyclopentane ring through the transition structure 1DTS4a (activation barrier 1.43 kcal mol–1), resulting in the intermediate 1DI5a. Another conformation change of 1DI5a results in 1DI6a, which is necessary for the final SN2 type mechanism for protodeplatination. Final protodeplatination occurs through transition-state 1DTS6a (O–H = 1.34 Å, H–C3 = 1.36 Å, Figure 9) with an activation barrier of 16.08 kcal mol–1. However, this SN2 type protodemetallation makes the global activation barrier of pathway-1Da, 22.61 kcal mol–1, which is larger than the global activation barrier of pathway-1D (19.58 kcal mol–1). This double activation process of pathway-1D is also more favorable than the previously discussed pathway-1M as revealed from Table 1, thus suggesting the involvement of two Pt metals in catalyzing the reaction.
Figure 9.
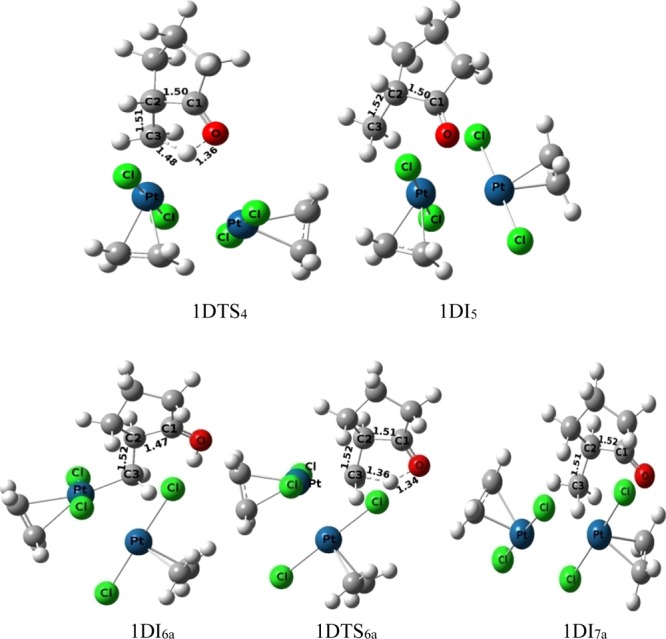
Ball and Stick models of the transition state and intermediate for the protodeplatination step of pathway-1D (upper) and pathway-1Da (lower) under the catalytic condition of the dimeric form of Pt-salt.
Table 1. Comparison of Gibbs Free Energy Barriers of the Several Segments of Different Pathways and Global Activation Energy (kcal mol–1).
| relative
energy required (kcal mol–1) (ΔG#) |
|||||
|---|---|---|---|---|---|
| pathway no. | oxidative addition of the metal atom | cleavage of the platinacyclobutane ring | protode-platination | global activation barrier | relative energy of the transition state that leads to the final product (ΔG) |
| Rearrangement of 1-Hydroxybicyclo[3.1.0]hexane | |||||
| path-1Ma (Figure 2) | 18.99 | 3.10 | 26.41 | 35.95 (34.56)a | 35.95 |
| path-1M (Figure 2) | 18.99 | 3.10 | 13.62 | 26.09 (25.77)a | 23.16 |
| path-1Da (Figure 2) | 1.69 | 1.05 | 16.08 | 22.61 (22.51)a | 22.61 |
| path-1D (Figure 2) | 1.69 | 1.05 | 11.12 | 19.58 (23.37)a | 17.65 |
| Rearrangement of 1-Siloxy-bicyclo[3.1.0]hexane | |||||
| path-2M (Figure 10) | 18.85 | 3.81 | 11.83 | 28.71 (28.95)a | 28.71 |
| path-2D (Figure 10) | 18.61 | 3.09 | 12.05 | 25.71 (25.10)a | 24.69 |
Calculation of the free energies was done using M06 functional.
Study on the Mechanism of Rearrangement of 1-Siloxy-bicyclo[3.1.0]hexane
The mechanism of the rearrangement of the silyl derivative of the bicyclic compound may also be considered as the composite of three separate steps. The first two steps, that is the oxidative addition of the metal atom and the cleavage of the platinacyclobutane ring are similar to that of the rearrangement of 1-hydroxy-bicyclo[3.1.0]hexane. However, the last step, that is the concerted 1,2-shift of hydrogen and the demetallation of this rearrangement differs from the step of protodemetallation of the previous path (Scheme 3). The catalysis under the monometallic and dimetallic condition of this reaction was studied, and photoelectron spectroscopy (PES) of both of them is presented in Figure 10. The mechanistic pathway under the single activation of the Pt-metal is designated here as pathway-2M (violet color in Figure 10), whereas the catalysis under double activation is designated as pathway-2D (orange color in Figure 10).
Figure 10.

PES along with the thermodynamic parameters and structures of the stationary points of pathway-2M (violet color) and pathway-2D (orange color) under the catalytic condition of the monomeric and dimeric form of Pt-salt. Energy values not enclosed in brackets are calculated using M06-2X functional. Values indicated in the round bracket are calculated using M06 functional.
Catalytic Activity of Monometallic Complex
The initial oxidative addition of pathway-2M occurs through the transition-state 2MTS1 (activation barrier 18.85 kcal mol–1) from the intermediate 2MI1, resulting in platinacyclobutane intermediate 2MI2. It has been revealed from the mechanistic pathway that this step is endergonic in nature. The distance between the oxygen atom of the siloxy group (−OSiH3) and coordinated Pt-metal is 2.19 Å in 2MI1 (Figure 11), and the C1–C3 bond of the cyclopropane moiety increases from 1.48 Å in 2MI1 to 2.32 Å in 2MI2 (through C1–C3 bond distance 1.96 Å in 2MTS1) during the insertion of the Pt-metal in the cyclopropane moiety.
Figure 11.
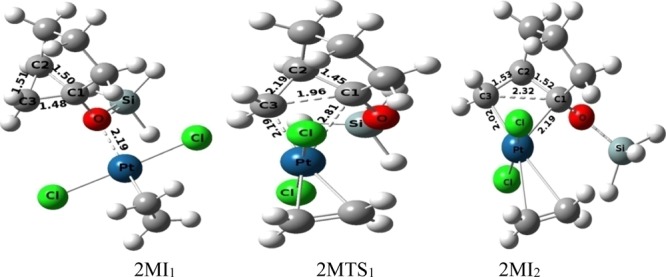
Ball and stick models of the stationary points during the formation of the platinacyclobutane intermediate (of the substrate 1-siloxy-bicyclo[3.1.0]hexane) under the catalytic condition of the monomeric form of Pt-salt.
In the following step of C1–Pt bond cleavage, the carbon atom attached to the siloxy group (−OSiH3) decreases the electron density around the oxygen atom and facilitates the breaking of the C1–Pt bond of the four-membered platinacyclobutane intermediate (2MI2) to generate η1-complex 2MI3 (through transition-state 2MTS2 with an activation barrier of 3.81 kcal mol–1). This heterolytic cleavage of the C1–Pt bond gives the zwitter ionic intermediate, 2MI3, the formation of which is followed by a conformational rearrangement of the siloxy group for the formation of intermediate 2MI4 (transition structure, 2MTS3 with an activation barrier of 5.68 kcal mol–1). There is another conformational change of the cyclopentane ring of intermediates 2MI4 through transition structure 2MTS4 (activation barrier 4.43 kcal mol–1) to generate intermediate 2MI5. 1,2-hydrogen shift from C2 to C1(Figure 12) of intermediate 2MI5 followed by a reductive elimination generates the final product 2MI6 (transition structure 2MTS5 with an activation barrier of 11.83 kcal mol–1). The global activation barrier of pathway-2M under the catalytic condition of the monomeric form of Pt-metal is 28.71 kcal mol–1.
Figure 12.
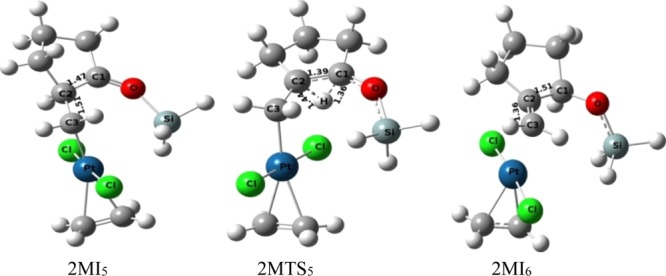
Ball and stick models of the stationary points of the protodeplatination step (of the substrate 1-siloxy-bicyclo[3.1.0]hexane) under the catalytic condition of the monomeric form of Pt-salt.
Catalytic Activity of Dimetallic Complex
Pathway-2D (Figure 10), which consists of three major steps, also follows the mechanism similar to that of pathway-2M under the catalysis of Zeise’s salt. In the first step of the mechanism, oxidative addition reaction takes place to form the platinacyclobutane intermediate (2DI2) from the initial substrate complex, 2DI1, by crossing an activation barrier of 18.61 kcal mol–1(transition structure 2DTS1 in which C1–Pt = 2.95 Å, C3–Pt = 2.18 Å, and C1–C3 = 1.89 Å, Figure 13). The next step is the heterolytic cleavage of the C1–Pt bond of the intermediate 2DI2 to generate the intermediate 2DI3 through transition structure 2DTS2 (C1–Pt distance 2.55 Å, Figure 13) with an activation barrier of 3.09 kcal mol–1. For initiating the proton shift and demetallation process, two necessary successive conformational changes have been occurred through the transition structures 2DTS3 and 2DTS4 and the associated intermediate 2DI4 in between them. The first conformational change is responsible for altering the orientation of the −OSiH3 group from the syn- to antiposition (transition structure 2DTS3 with an activation barrier of 5.08 kcal mol–1) for the formation of intermediate 2DI4. The next one flips the envelope structure of the cyclopentane ring from one conformation to other generating the intermediate 2DI5 (through transition-state 2DTS4, with an activation barrier of 1.44 kcal mol–1). The last mandatory step consists of two nuclear movements in which the 1,2-hydrogen shift occurs from C2 to C1 and the detachment of the catalytic metal center from C3 to generate intermediate 2DI6. The activation barrier associated to the last step is 12.05 kcal mol–1 through the transition structure 2DTS5. The global activation barrier of pathway-2D is 25.71 kcal mol–1. However, the overall comparison between pathway-2M and pathway-2D shows that the global activation barrier of the latter pathway (25.71 kcal mol–1) is more favorable than the former one (28.71 kcal mol–1, Table 1).
Figure 13.
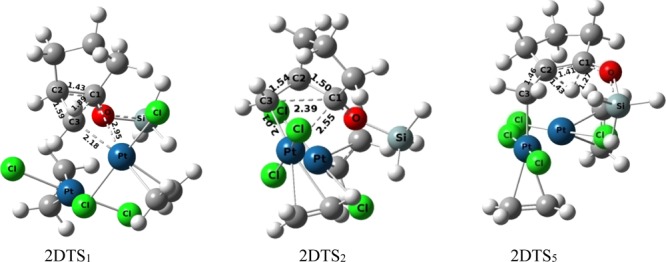
Ball and stick models of the stationary points of the substrate (1-siloxy-bicyclo[3.1.0]hexane) under the catalytic condition of the dimeric form of Pt-salt.
From the previous discussion, it is clear that one of the two Pt atoms of Zeise’s dimer is actively involved in the catalytic process. It participates in oxidative addition with the cyclopropane ring, consequently involved in the bond-making and bond-breaking process throughout the course of the rearrangement reaction. The second Pt provides some secondary support by coordination with various substituents having the donor atom present in the substrate. To study the role of other transition metals of the same group, we replaced second Pt by Pd and Ni in Zeise’s dimer and recalculated the global activation barrier of the most favorable pathway. The calculated global activation barrier suggests that heteronuclear Zeise’s dimer bearing one Pt-atom replaced by Pd or Ni may generate processes which have a comparable catalytic activity such as homonuclear Zeise’s dimer.
Conclusions
In summary, our DFT study reveals that the formation of monocyclic derivatives from the substrate 1-hydroxy-bicyclo[3.1.0]hexane and 1-siloxy-bicyclo[3.1.0]hexane takes place in three major steps under catalytic conditions of monomeric and dimeric forms of Pt-salt. The first step undergoes the oxidative addition process for the formation of the platinacyclobutane intermediate which is followed by the cleavage of the Pt–C bond to open up platinacyclobutane as a second step. The final step is protodeplatination to generate the end products. Comparison of the energy barriers associated to different pathways reveals that double activation by the two Pt metal atoms leads to the energetically more favorable mechanism to form the products. The energy barriers associated to the pathway-1D and 2D (19.58 and 25.71 kcal mol–1) are quite reasonable to explain the room temperature conditions required for the rearrangement. Outcome of our study on the catalytic activity of heteronuclear Zeise’s dimer where one of the Pt atoms is replaced by Pd or Ni may provide importantly to the synthetic chemist to develop a new catalytic system with multimetallic complexes.
Computational Methods
All calculations were carried out with the Gaussian 09 computational program package.12 Geometry optimization of all species13 was performed using M06-2X functional14 in the DFT method. The 6-31G(d,p) basis set15 was employed for all nonmetal atoms and the LANL2DZ basis set16 was employed for the Pt atom. The computational method has been found reliable to study this ring-opening reaction and rearrangement reactions.17 Frequency calculations at the same level of theory were performed to obtain the gas phase free energies and to confirm each stationary point to be either a minimum (no imaginary frequency) or a transition structure (only one imaginary frequency). Intrinsic reaction coordinate calculations were carried out to confirm the connection of each transition state to its corresponding reactants and products.18 Single-point self-consistent reaction field calculations based on the polarizable continuum model were used for examining the solvation effect in gas-phase optimized structures.19 To obtain more accurate energies of the stationary points, single-point calculations were performed using the LANL2TZ basis set and effective core potential (ECP) with f polarization function for the metal center.20 Diethylether for 1-hydroxy-bicyclo[3.1.0]hexane and dichloromethane for 1-siloxy-bicyclo[3.1.0]hexane were used as the solvent. The natural bond order (NBO) analysis was carried out using the NBO program, where the basis set is 6-31G(d,p) for nonmetal atoms and LANL2DZ (ECP) for metals are employed.21 All energies, reported in the Results and Discussion section, are relative free energies (ΔG#) at 298.15 K. The global activation barrier was recalculated by optimizing the relevant stationary points using M06 functional.22
Acknowledgments
We are thankful to the UGC, New Delhi, India, for providing financial assistance. We are also grateful to the DST, New Delhi, India, for providing financial assistance to one of our author (K.C.) in the form of the research project (SR/WOS-A/CS-153/2016). R.S. thanks DST-INSPIRE (IF160987), New Delhi, India, for fellowship. We are also thankful to Visva-Bharati for providing us the necessary infrastructural facility to perform the research work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02344.
PES of all pathways, Cartesian coordinates, absolute energies, frequencies, and detailed thermodynamic parameters of all computed stationary points along with the structure in the ball and stick model (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Bishop K. C. III Transition metal catalyzed rearrangements of small ring organic molecules. Chem. Rev. 1976, 76, 461–486. 10.1021/cr60302a003. [DOI] [Google Scholar]; b Crabtree R. H. The organometallic chemistry of alkanes. Chem. Rev. 1985, 85, 245–269. 10.1021/cr00068a002. [DOI] [Google Scholar]; c Soriano E.; Ballesteros P.; Marco-Contelles J. A Theoretical Investigation on the Mechanism of the PtCl2-Mediated Cycloisomerization of Heteroatom-Tethered 1,6-Enynes. J. Org. Chem. 2004, 69, 8018–8023. 10.1021/jo048828h. [DOI] [PubMed] [Google Scholar]; d Soriano E.; Marco-Contelles J. Theoretical Analysis of the High Versatility in PtCl2-Mediated Cycloisomerization of Enynes on a Common Mechanistic Basis. J. Org. Chem. 2005, 70, 9345–9353. 10.1021/jo0514265. [DOI] [PubMed] [Google Scholar]; e Ye L.; Chen Q.; Zhang J.; Michelet V. PtCl2-Catalyzed Cycloisomerization of 1,6-Enynes for the Synthesis of Substituted Bicyclo[3.1.0]hexanes. J. Org. Chem. 2009, 74, 9550–9553. 10.1021/jo902083r. [DOI] [PubMed] [Google Scholar]; f Nevado C.; Ferrer C.; Echavarren A. M. New Annulations via Platinum-Catalyzed Enyne Cyclization and Cyclopropane Cleavage. Org. Lett. 2004, 6, 3191–3194. 10.1021/ol0486573. [DOI] [PubMed] [Google Scholar]
- a Hamilton J. G.; Palke W. E. Bonding in cyclopropane. J. Am. Chem. Soc. 1993, 115, 4159–4164. 10.1021/ja00063a038. [DOI] [Google Scholar]; b Bertolini T. M. Visualizing Bent Bonds in Cyclopropane. J. Chem. Educ. 2004, 81, 818. 10.1021/ed081p818. [DOI] [Google Scholar]; c Fleming I.Molecular Orbitals and Organic Chemical reactions. Reference; John Wiley and Sons Ltd.: Chichester, 2010; pp 1–67. [Google Scholar]
- a Hidai M.; Orisaku M.; Uchida Y. Rhodium Catalyzed Carbonylation Reactions Of Cyclopropanes. Chem. Lett. 1980, 9, 753–754. 10.1246/cl.1980.753. [DOI] [Google Scholar]; b Gassman P. G.; Bonser S. M. The transition metal complex promoted isomerization of trans-bicyclo[4.1.0]hept-3-ene to cis-bicyclo[4.1.0]hept-3-ene. Tetrahedron Lett. 1983, 24, 3431–3434. 10.1016/s0040-4039(00)86005-0. [DOI] [Google Scholar]; c Campbell H.; Jennings P. W. New evidence in the iridium(I)-catalyzed reaction of endo-tricyclo[3.2.1.02,4]oct-6-ene. Organometallics 1983, 2, 1460–1461. 10.1021/om50004a038. [DOI] [Google Scholar]; d Parsons E. J.; Jennings P. W. Platinacyclobutane chemistry: cis-disubstituted platinacyclobutane complex from bicyclo[4.1.0]heptane. Organometallics 1988, 7, 1435–1437. 10.1021/om00096a032. [DOI] [Google Scholar]; e Hoberg J. O.; Jennings P. W. Carbon-carbon and carbon-oxygen bond formation from the reaction of platinum(II) with bicyclo[4.1.0]hept-2-ene and related derivatives. Organometallics 1992, 11, 3452–3456. 10.1021/om00046a053. [DOI] [Google Scholar]
- a Lautens M.; Ren Y.; Delanghe P.; Chiu P.; Ma S.; Colucci J. 1994 Merck Frosst Award Lecture New strategies for the stereoselective synthesis of natural and unnatural products via organometallic reagents and catalysts1. Can. J. Chem. 1995, 73, 1251–1257. 10.1139/v95-153. [DOI] [Google Scholar]; b Donaldson W. A.; Stepuszek D. J. Reactivity of (3-chloro-2-methylenecycloalkyl)palladium chloride dimers: a palladium-mediated ring homologation—functionalization approach to 4-aryltropones related to colchicine. J. Org. Chem. 1992, 57, 1309–1313. 10.1021/jo00030a049. [DOI] [Google Scholar]; c Saigo K.; Shimada S.; Shibasaki T.; Hasegawa M. Lewis Acid-Mediated Reaction of 2,2-Dialkoxycyclopropanecarboxylic Esters with Ketene Silyl Acetals. Synthesis of Cyclopentenones. Chem. Lett. 1990, 19, 1093–1096. 10.1246/cl.1990.1093. [DOI] [Google Scholar]; d Stewart F. F.; Neilsen W. D.; Ekeland R. E.; Larsen R. D.; Jennings P. W. Synthesis of 1,3-divinylcyclopentane derivatives from platina(IV)cyclobutane complexes. Organometallics 1993, 12, 4585–4591. 10.1021/om00035a050. [DOI] [Google Scholar]
- a Jennings P. W.; Johnson L. L. Metallacyclobutane Complexes of the Group Eight Transition Metals: Synthesis, Characterizations, and Chemistry. Chem. Rev. 1994, 94, 2241–2290. 10.1021/cr00032a003. [DOI] [Google Scholar]
- a Parsons E. J.; Jennings P. W. Platinacyclobutane chemistry: platinacyclobutanes from bicyclo[X.1.0]hydrocarbons. J. Am. Chem. Soc. 1987, 109, 3973–3977. 10.1021/ja00247a023. [DOI] [Google Scholar]; b Parsons E. J.; Jennings P. W. Platinacyclobutane chemistry: cis-disubstituted platinacyclobutane complex from bicyclo[4.1.0]heptane. Organometallics 1988, 7, 1435–1437. 10.1021/om00096a032. [DOI] [Google Scholar]
- Malapit C. A.; Chitale S. M.; Thakur M. S.; Taboada R.; Howell A. R. Pt-Catalyzed Rearrangement of Oxaspirohexanes to 3-Methylenetetrahydrofurans: Scope and Mechanism. J. Org. Chem. 2015, 80, 5196–5209. 10.1021/acs.joc.5b00604. [DOI] [PubMed] [Google Scholar]
- Hoberg J. O.; Jennings P. W. Platinum(II)-Catalyzed Isomerization of Alkoxycyclopropanes to Alkylated Ketones. Organometallics 1996, 15, 3902–3904. 10.1021/om960157t. [DOI] [Google Scholar]
- Ikura K.; Ryu I.; Kambe N.; Sonoda N. Room temperature isomerization of siloxycyclopropanes to silyl ethers of 2-methylenealkanols catalyzed by Zeise’s dimer. J. Am. Chem. Soc. 1992, 114, 1520–1521. 10.1021/ja00030a080. [DOI] [Google Scholar]
- Beyer J.; Skaanderup P. R.; Madsen R. Platinum-Catalyzed Ring Opening of 1,2-Cyclopropanated Sugars withO-Nucleophiles. Convenient Synthesis of 2-C-Branched Carbohydrates. J. Am. Chem. Soc. 2000, 122, 9575–9583. 10.1021/ja001558+. [DOI] [Google Scholar]
- Mondal S.; Chatterjee A.; Saha R.; Ghosh A.; Chakrabarty K.; Das G. K. Study on the mechanism of isomerization of oxaspirohexane catalyzed by Zeise’s Dimer. Mol. Catal. 2018, 452, 247–259. 10.1016/j.mcat.2018.04.003. [DOI] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, Revision C.01; Gaussian Inc: Wallingford CT, 2010. (Full reference is given in Supporting Information).
- a Camiletti G. G.; Machado S. F.; Jorge F. E. Gaussian basis set of double zeta quality for atoms K through Kr: Application in DFT calculations of molecular properties. J. Comput. Chem. 2008, 29, 2434–2444. 10.1002/jcc.20996. [DOI] [PubMed] [Google Scholar]; b Kang R.; Chen H.; Shaik S.; Yao J. Assessment of Theoretical Methods for Complexes of Gold(I) and Gold(III) with Unsaturated Aliphatic Hydrocarbon: Which Density Functional Should We Choose?. J. Chem. Theory Comput. 2011, 7, 4002–4011. 10.1021/ct200656p. [DOI] [PubMed] [Google Scholar]; c Basak A.; Chakrabarty K.; Ghosh A.; Das G. K. Mechanism of the Gold(III)-Catalyzed Isomerization of Substituted Allenes to Conjugated Dienes: A DFT Study. J. Org. Chem. 2013, 78, 9715–9724. 10.1021/jo401400x. [DOI] [PubMed] [Google Scholar]; d Ghosh A.; Basak A.; Chakrabarty K.; Mondal S.; Chatterjee A.; Das G. K. Au-Catalyzed Hexannulation and Pt-Catalyzed Pentannulation of Propargylic Ester Bearing a 2-Alkynyl-phenyl Substituent: A Comparative DFT Study. ACS Omega 2018, 3, 1159–1169. 10.1021/acsomega.7b01889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Latouche C.; Skouteris D.; Palazzetti F.; Barone V. TD-DFT Benchmark on Inorganic Pt(II) and Ir(III) Complexes. J. Chem. Theory Comput. 2015, 11, 3281–3289. 10.1021/acs.jctc.5b00257. [DOI] [PubMed] [Google Scholar]; b Zhao Y.; Truhlar D. G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]; c Walker M.; Harvey A. J. A.; Sen A.; Dessent C. E. H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A 2013, 117, 12590–12600. 10.1021/jp408166m. [DOI] [PubMed] [Google Scholar]
- a Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]; b Hehre W. J.; Ditchfield R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]; c Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/bf00533485. [DOI] [Google Scholar]; d Hariharan P. C.; Pople J. A. Accuracy of AHnequilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 1974, 27, 209–214. 10.1080/00268977400100171. [DOI] [Google Scholar]; e Dill J. D.; Pople J. A. Self-consistent molecular orbital methods. XV. Extended Gaussian-type basis sets for lithium, beryllium, and boron. J. Chem. Phys. 1975, 62, 2921–2923. 10.1063/1.430801. [DOI] [Google Scholar]; f Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. 10.1063/1.444267. [DOI] [Google Scholar]; g Mitin A. V.; Baker J.; Pulay P. An improved 6–31G* basis set for first row transition metals. J. Chem. Phys. 2003, 118, 7775–7782. 10.1063/1.1563619. [DOI] [Google Scholar]; h Rassolov V. A.; Pople J. A.; Ratner M. A.; Windus T. L. 6-31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223–1229. 10.1063/1.476673. [DOI] [Google Scholar]
- a Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]; b Wadt W. R.; Hay P. J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. 10.1063/1.448800. [DOI] [Google Scholar]; c Hay P. J.; Wadt W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. 10.1063/1.448975. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. 10.1021/ct800246v. [DOI] [PubMed] [Google Scholar]
- a Fukui K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. 10.1021/j100717a029. [DOI] [Google Scholar]; b Fukui K. The path of chemical reactions - the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. 10.1021/ar00072a001. [DOI] [Google Scholar]; c Gonzalez C.; Schlegel H. B. Reaction path following in Mass-Weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. 10.1021/j100377a021. [DOI] [Google Scholar]
- Tomasi J.; Persico M. Molecular Interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. 10.1021/cr00031a013. [DOI] [Google Scholar]
- a Roy L. E.; Hay P. J.; Martin R. L. Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. 10.1021/ct8000409. [DOI] [PubMed] [Google Scholar]; b Ehlers A. W.; Böhme M.; Dapprich S.; Gobbi A.; Höllwarth A.; Jonas V.; Köhler K. F.; Stegmann R.; Veldkamp A.; Frenking G. A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc—Cu, Y—Ag and La—Au. Chem. Phys. Lett. 1993, 208, 111–114. 10.1016/0009-2614(93)80086-5. [DOI] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Catalyzation of Alkynes: Revisiting Baldwin’s Rules for Ring Closure. Chem. Rev. 2011, 111, 6513–6556. 10.1021/cr200164y. [DOI] [PubMed] [Google Scholar]
- Jacquemin D.; Perpète E. A.; Ciofini I.; Adamo C.; Valero R.; Zhao Y.; Truhlar D. G. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. J. Chem. Theory Comput. 2010, 6, 2071–2085. 10.1021/ct100119e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.