

Abstract
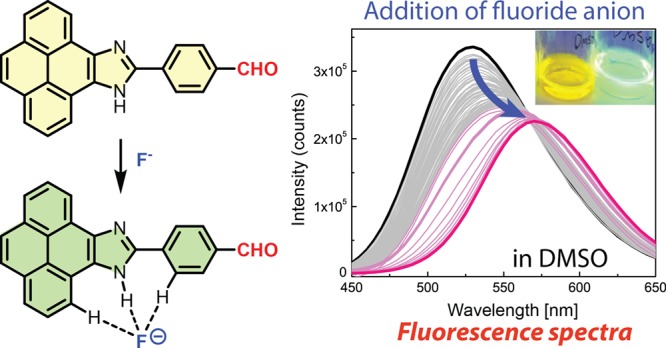
Two structural isomers of (9H-pyreno[4,5-d]imidazol-10-yl)-benzaldehyde, with para and meta substitution patterns, were synthesized by condensation of 4,5-pyrenedione with terephthalaldehyde and isophthalaldehyde, respectively. These new pyrenoimidazole derivatives were characterized by single-crystal X-ray crystallography, UV–vis absorption spectroscopy, fluorescence spectroscopy, and cyclic voltammetry to elucidate their structural, solid-state packing, and electronic properties. Interactions of these compounds with fluoride anions in polar organic solvents (acetone and dimethyl sulfoxide) were investigated by NMR, UV–vis, and fluorescence techniques in conjunction with density functional theory calculations. UV–vis analysis showed that the binding of the two pyrenoimidazolyl benzaldehydes with fluoride anions resulted in significant colorimetric responses, while fluorescence studies showed that the para-pyrenoimidazolyl benzaldehyde behaved as an intramolecular charge transfer fluorescent probe, exhibiting ratiometric sensing performance to efficiently detect and quantify fluoride anions at the sub-millimolar level.
Introduction
The design and synthesis of functional organic π-conjugated molecules are of considerable importance to the field of nanoscale electronics and optoelectronics1−6 as research in this area continuously adds new molecular building blocks to the synthetic toolbox and accelerates the development of materials and devices with improved performance or unprecedented function. Pyrene is a small polycyclic aromatic hydrocarbon with a planar, rigid π-conjugated molecular backbone and hence exhibits fascinating photophysical, electrochemical, and supramolecular properties.7 For decades, pyrene has been widely used as an active fluorophore in materials science. Currently, the number of pyrene-based molecules is growing rapidly owing to the great efforts made by synthetic chemists.7−9
9H-Pyreno[4,5-d]imidazole (PI) is a heterocycle-fused pyrene system that can be readily prepared by the condensation reaction between 4,5-pyrenedione and a suitable aldehyde in the presence of ammonium acetate.10,11 The extended π-framework and the ability of imidazole to form complexes with various metal ions and acids have rendered PI an active component in the preparation of functional molecules, ranging from ligands for metal ions,12 fluorescence emitters,13−17 dye-sensitized solar cells,18−20 and electrocatalysts.11 The imidazole unit in the PI system is important because it provides effective binding sites for the capture of anions and/or metal cations.21−24 In particular, hydrogen bonding can occur between the imidazolyl N–H proton and various anions. Indeed, a number of PI-based sensors have relied upon these interactions. For example, Baitalik and co-workers prepared Ru(II) and Os(II) complexes containing a PI ligand and demonstrated their colorimetric and fluorescence sensing function toward a range of anions.25,26 Fluorescence quenching was observed when these complexes were mixed with some basic anions, such as F–, CN–, and CH3COO–, due to deprotonation of the imidazolyl moiety. Acidic anions (e.g., H2PO4–), on the other hand, interacted with the pyrenoimidazolyl unit through hydrogen bonding to induce fluorescence enhancement. In 2015, a pyrene-bisimidazole system was reported by the Baitalik group and it showed significant absorption and emission responses to CN– and Cu2+ ions.27 Such properties allowed this compound to act as a ratiometric optical sensor for CN– in aqueous media and a molecular memory device. Most recently, Karthik et al. reported the synthesis of a series of substituted 10-aryl-PIs and investigated their photophysical properties and reactivity toward Ru(II)-catalyzed oxidative annulation.16
Synthetically, aryl-PIs (particularly the phenyl-PIs) can be easily and economically prepared owing to the abundant availability of benzaldehyde precursors from commercial sources. They should therefore constitute a versatile platform for the development of novel chromophores/fluorophores with diverse functionalities and activities. In the literature, however, the family of phenyl-PIs is still underdeveloped and there is a lack of demonstration of their applicability in molecule-based optoelectronic devices, such as colorimetric and fluorescence chemosensors. Awareness of this background thus motivated us to design and characterize a new class of 10-phenyl-PIs in which the phenyl group is functionalized with an electron-withdrawing group (EWG). Generally speaking, the imidazolyl group is electron-accepting in nature and can provide an acidic N–H proton to interact with various anions through hydrogen bonds.28−33 The hydrogen bonding interaction, in theory, polarizes the imidazolyl N–H bond to alter the electron density on the imidazolyl ring. In a phenyl-PI system such as that illustrated in Figure 1, intramolecular charge transfer (ICT) can conceivably occur through π-electron delocalization.34,35 Such an effect might be expected to significantly modulate the electronic and photophysical properties, particularly the fluorescence behavior. So far, there has been no report of PI-based chemosensors that operate with an ICT mechanism. In the design of chemosensors, the ICT mechanism has often been used to achieve ratiometric fluorescence sensors, which detect analytes (e.g., anions) at two or more significant emission wavelengths. For practical application, ratiometric fluorescence measurements are advantageous over the detection methods that rely on only single wavelength emission in terms of accuracy, reproducibility, and detection range.36−41 In this work, we have investigated the fundamental properties of benzaldehyde-substituted PI systems in terms of their structural, crystallographic, and electronic properties. Furthermore, the interactions of these new PI systems with fluoride anion were examined to demonstrate the possibility of tuning this type of fluorophores to give ratiometric fluorescence sensing function for fluoride anions.
Figure 1.

Proposed ICT occurring upon hydrogen bonding interactions between an anion (A–) and 10-phenyl-PI bearing an EWG.
Results and Discussion
Synthesis of Pyrenoimidazolyl-Benzaldehyde Isomers 4 and 5
In this work, two pyrenoimidazolyl-benzaldehyde (PI-BAL) derivatives (4 and 5 in Scheme 1) were designed and synthesized. In the structures of the two compounds, an electron-withdrawing formyl group is attached to the phenyl group at the para and meta positions, respectively. The two structural isomers were expected to help understand their photophysical properties in relation to substitution patterns, particularly the effects of resonance (para) versus induction (meta). The synthesis of compounds 4 and 5 was conducted through a direct condensation reaction between the now readily available 4,5-pyrenedione (1)42 and terephthalaldehyde (2) or isophthalaldehyde (3), in the presence of ammonium acetate and glacial acetic acid. In the reactions, the molar ratio of 4,5-pyrenedione (1) versus 2 or 3 was set as 1:3, so that only one of the formyl groups in 2 or 3 underwent condensation and the other remained intact. The condensation reactions proceeded smoothly upon heating at 110 °C for 1–3 h. Compounds 4 and 5 were obtained in reasonably good yields (50–60%) after silica flash column chromatographic separation.
Scheme 1. Synthesis of PI-BALs 4 and 5.

Solid-State Structural Properties
The obtained PI-BALs 4 and 5 were characterized initially by infrared (IR), mass spectrometry (MS), 1H and 13C NMR analyses. Detailed spectroscopic data are included in the Supporting Information. Recrystallization of compound 4 from acetone/hexanes (3:7, v/v) resulted in the formation of crystals in different forms; fast recrystallization afforded thin orange needles, whereas slow crystallization yielded larger translucent single crystals with a brownish color. A crystal of 4 was subjected to single-crystal X-ray crystallographic analysis to determine its solid-state structure.
In the crystal structure of 4, the phenyl ring does not assume the expected co-planar orientation relative to the PI moiety, even though co-planarity is favored by maximal π-conjugation. Instead, the phenyl group is rotated by 21.6° with respect to the PI group. The slightly twisted conformation can be ascribed to crystal packing forces, as density functional theory (DFT) calculations show that the minimum-energy structure of compound 4 in the gas phase or solvents is fully planar when intermolecular interactions are not taken into consideration (vide infra). One interesting feature observed in the solid-state packing of compound 4 is that hydrogen-bonded dimers are formed through the interactions of the imidazolyl N–H protons (donor) and the C=O oxygens (acceptor) with an intermolecular O···H distance of 2.27 Å and an N–H···O angle at 169.5° as shown in Figure 2A. In the solid state, the hydrogen-bonded dimers are closely packed together through π-stacking as revealed by Figure 2B, and such a tight packing motif leaves no space for solvent molecules to co-crystalize. Overall, intermolecular hydrogen bonding and π-stacking are the two major factors dictating the solid-state structure of compound 4.
Figure 2.

(A) Oak ridge thermal ellipsoid plot (ORTEP; 50% probability) of the hydrogen-bonded dimer of PI-BAL 4 (CCDC 1851836). (B) Solid-state packing diagram of 4. The parallelogram highlights a hydrogen bonded dimer viewed from its side. (C) ORTEP plot (50% probability) of PI-BAL 5 co-crystallized with dimethyl sulfoxide (DMSO; CCDC 1851835). (D) Solid-state packing diagram of 5.
PI-BAL 5 was recrystallized from a mixed solvent of hexanes/DMSO (9:1, v/v) to give dark-brown single crystals. X-ray structural analysis showed that, in contrast to 4, the molecule of 5 adopts a fully planar conformation when packed in the solid state (see Figure 2C). Also of note is that two molecules of DMSO are present with compound 5 in the unit cell, whereby multiple hydrogen bonds among them can be identified. In particular, the imidazolyl N–H proton forms a significant hydrogen bond with the oxygen atom of one DMSO molecule at an O···H distance of 2.12 Å, while another DMSO molecule interacts with the first DMSO molecule through S–O and CH3 interactions (Figure 2C). The crystal structure of 5 also reveals that the phenyl and pyrenyl protons, which are in close proximity to the imidazolyl N–H group, form hydrogen bonds with the first DMSO molecule. The participation of DMSO molecules in the crystallization of 5 thus leads to a solid-state packing motif dramatically different than that of its para-isomer 4. As shown in Figure 2D, π-stacking occurs but there are no hydrogen bonding interactions among the molecules of 5. The association of DMSO molecules with 5 through hydrogen bonds makes the crystal packing less tight than the case of 4.
Solvent Effects on UV–Vis Absorption and Fluorescence Properties
The UV–vis absorption and fluorescence emission properties of PI-BALs 4 and 5 were studied in a range of organic solvents to assess the solvent effects. Figure 3 illustrates the UV–vis and fluorescence spectra measured in three organic solvents (toluene, DMSO, and methanol), which are highlighted here to represent nonpolar, dipolar aprotic, and dipolar protic solvents, respectively. For compound 4, the UV–vis absorption spectrum features two long-wavelength bands at 394 and 410 nm in toluene (Figure 3A). A similar pattern can be seen in the spectrum measured in benzene. When the solvents are switched to methylene chloride and chloroform, which are also nonpolar in nature, only one absorption band appears in this spectral region (see Figure S7 in the Supporting Information for details). In view of the hydrogen-bonded dimer observed in the crystal structure of 4, the spectral variations are rationalized as follows: in aromatic solvents (benzene and toluene), compound 4 forms a hydrogen-bonded dimer, in which the hydrogen bonds polarize the imidazolyl N–H and the C=O groups to enhance the “push-and-pull” effect, resulting in the π → π* transition bands being bathochromically shifted. UV–vis analysis of 4 in toluene at various concentrations (ca. 10–5 to 10–3 M) shows that the relative intensity of the peak at 410 nm decreases with decreasing concentration of 4 (see Figure S11A in the Supporting Information). When the toluene solution of 4 was titrated with methanol, the two long-wavelength bands gradually merged into one absorption peak (Figure S11B in the Supporting Information). On the basis of these experimental observations, the peak at 394 nm in toluene is attributed to the free molecule of 4, while the peak at 410 nm is assigned to the hydrogen-bonded dimer of 4.
Figure 3.

(A) Normalized UV–vis absorption and (B) fluorescence emission spectra of 4 measured in various solvents. (C) Normalized UV–vis absorption and (D) fluorescence emission spectra of 5 measured in various solvents.
In polar solvents, the long-wavelength absorption band of 4 shows only a small degree of bathochromic shift relative to those measured in nonpolar solvents. This trend generally agrees with the DFT computational analysis, which shows that in the solution phase compound 4 becomes more delocalized when the solvent polarity increases. Time-dependent DFT (TD-DFT) calculations reveal that the lowest-energy absorption band of 4 is mainly due to the highest occupied molecular orbital (HOMO) → lowest unoccupied molecular orbital (LUMO) transition, and it does not vary very significantly in different solvents (see Table S4 in the Supporting Information), which is in agreement with the experimentally observed weak solvent effects in the UV–vis analysis. Similarly, the absorption profiles of 5 show only slight variations when changing the solvent polarity from nonpolar to polar (Figure 3C). However, in methanol the UV–vis absorption bands show notable hypsochromic shift relative to the others. It can be rationalized that in a dipolar protic solvent such as methanol the imidazolyl C=N group, which is a hydrogen bond acceptor, can form hydrogen bond(s) with the solvent molecule(s). Such interactions would be expected to cause the hypsochromic shift of UV–vis peaks in methanol. Overall, the UV–vis absorption analysis indicates that the π → π* transitions in both compounds 4 and 5 are more significantly influenced by the protic nature of solvent than solvent polarity.
The fluorescence spectra of 4 and 5 exhibit a high degree of dependence on both solvent polarity and hydrogen bonding interactions. As can be seen in Figure 3B, the maximum emission wavelength of 4 in toluene appears at 464 nm and it is bathochromically shifted to 529 nm in DMSO, which is a hydrogen bond acceptor. In methanol, which acts as both hydrogen bond donor and acceptor, the emission of 4 is almost completely quenched. For compound 5, the maximum emission wavelength is shifted from 445 nm (in toluene) to 542 nm (in DMSO), but in methanol it is blue-shifted to 403 nm (see Figure 3D). The drastic solvent effects observed in the fluorescence spectra of 4 and 5 suggest that the excited states of these compounds undergo diverse radiative and nonradiative deactivation pathways which are sensitive to solvent–solute interactions; especially, the mechanism of imidazole-based excited-state proton transfer43−46 could play an important role in the photo-excitation/deactivation processes.
Electrochemical Redox Properties
The electrochemical redox activities of PI-BALs 4 and 5 were examined by cyclic voltammetry (CV). Scanned in the positive potential window, there were no significant redox peaks detected for the two compounds, indicating that they cannot be oxidized easily. In the negative potential window, compounds 4 and 5 exhibit very similar redox features in their CV profiles (Figure 4). In acetone, two redox couples were detected in the cyclic voltammograms of both compounds, while in DMSO the two compounds were found to undergo three distinctive redox processes in the negative potential window. The three steps of electron transfers are tentatively assigned to the injection of electrons to the benzaldehyde, imidazolyl, and pyrenyl groups respectively. The positions of the formyl group (para and meta) appear to have the same impact on the overall electrochemical redox properties of the PI-BAL system.
Figure 4.

Cyclic voltammograms of PI-BAL 4 measured in (A) acetone and (B) DMSO, and PI-BAL 5 measured in (C) acetone and (D) DMSO. Experimental conditions: electrolyte: Bu4NBF4 (0.1 M), working electrode: glassy carbon, counter electrode: Pt wire, reference electrode: Ag/AgCl, scan rate: 100 mV/s.
Interactions with the Fluoride Anion
As mentioned previously, the acidic imidazolyl N–H group in the PI system can participate in hydrogen bonding interactions with various hydrogen bond acceptors (e.g., anions).28−33 The binding in theory would modulate the electronic properties of the PI system, leading to significant changes in electronic absorption and emission behavior. In a sense, this could serve as the mechanism for efficient colorimetric and fluorescence sensing of certain anions. To investigate these properties, PI-BALs 4 and 5 were subjected to titration with tetrabutylammonium fluoride (TBAF) as a fluoride anion source in acetone and/or DMSO, and the titration processes were monitored by 1H NMR, UV–vis absorption, and fluorescence spectroscopic analyses, respectively.
Figure 5 shows the results of 1H NMR titration of compound 4 with TBAF in acetone-d6. Before titration, the eight pyrenyl protons of 4 appear as discrete signals without degeneracy in the 1H NMR spectrum, suggesting that the imidazolyl proton (N–H) does not undergo very rapid exchange processes (e.g., tautomerization) in acetone. Upon addition of TBAF at ca. 0.2 molar equivalent, the 1H NMR spectral pattern changes immediately to feature twofold symmetry and degeneracy for the pyrenyl protons; for instance, the pyrenyl protons at the 4,5-positions (labeled as g and g′ in Figure 5) can be now seen as a singlet in the spectra. Such an observation suggests that the interactions of fluoride anion with 4 considerably accelerate the tautomerization of the imidazolyl moiety, rendering the PI unit twofold symmetric in the 1H NMR spectrum. DFT calculations show that fluoride anion can form a hydrogen bond with the imidazolyl N–H group (vide infra), which weakens the N–H bond and facilitates more rapid tautomerization. It is also noted that the N–H signal at 13.04 ppm disappears after ca. 0.1 molar equivalent of TBAF is added, due to rapid proton exchange processes, most likely tautomerization. As the titration continues, the aldehyde proton (labeled as a in Figure 5) shows a notable upfield shift, from 10.14 to 9.97 ppm, and the titration reaches an equilibrium after the addition of more than 2 molar equivalents of TBAF. The results suggest that PI-BAL 4 interacts with fluoride anions through two steps as outlined in Figure 5. In the first step, a 1:1 complex is formed through hydrogen bonding interactions, while in the second step compound 4 is deprotonated along with the formation of [HF2–] ion.37 During each of the two steps, the imidazolyl group experiences increased electron density which can be delocalized to the para-formyl group through the resonance effect, causing the aldehyde proton to significantly shift toward upfield. Some of the aryl protons of 4 also show significant upfield or downfield shifts during the titration, and these shifts can be reasonably correlated with the fluoride anion binding and subsequent deprotonation of the imidazolyl N–H group of 4.
Figure 5.

1H NMR (300 MHz) spectra monitoring the titration of PI-BAL 4 with TBAF in acetone-d6.
The electronic absorption and emission properties of compound 4 in response to the interactions with fluoride anions were examined by UV–vis absorption and fluorescence spectroscopic analyses. Figure 6A shows the UV–vis titration results of 4 with TBAF in acetone. Upon addition of TBAF from 0 to ca. 3 molar equivalents, the long-wavelength absorption region (ca. 400–520 nm) shows a steady increase in intensity and the absorption band at 355 nm decreases significantly. Two isosbestic points at 382 and 399 nm are clearly observable, and this stage of spectral changes can be attributed to the formation of a 1:1 complex between 4 and fluoride anion through hydrogen bonding interactions. After titration with more than 3 molar equivalents of TBAF, the UV–vis spectra exhibit another stage of changes in which a long-wavelength absorption band at 448 nm grows prominently, while the short-wavelength absorption bands in the region of 350 nm to 400 nm continuously decrease. TD-DFT calculations suggest that the strongly increasing new band is consistent with the HOMO → LUMO transition of deprotonated 4. Accordingly, the second stage of spectral changes is linked to the deprotonation of compound 4, through which an anionic product [4–] is formed. This species is stabilized by the resonance effect exerted by the electron-withdrawing formyl group at the para position of the phenyl ring.
Figure 6.

Titration of PI-BAL 4 with TBAF monitored by UV–vis absorption spectroscopy in (A) acetone and (B) DMSO, fluorescence spectroscopy in (C) acetone and (D) DMSO.
In DMSO, the UV–vis absorption of 4 responds to TBAF titration in a different way than in acetone. As shown in Figure 6B, there is only one stage of spectral changes observed during the whole process of titration, with three isosbestic points clearly observable at 409, 330, and 311 nm. Compared to the titration results obtained in acetone, the different spectral responses observed in DMSO suggest that in DMSO the second step (i.e., deprotonation) occurs more readily than the first hydrogen bond forming step.
Subsequent to the UV–vis titration experiments, the fluorescence spectral changes of compound 4 upon titration with TBAF in acetone or DMSO were determined. In acetone, the maximum emission peak of 4 shows a dramatic redshift from 506 to 557 nm during the titration of up to more than 30 molar equivalents of TBAF, with the intensity of emission peak attenuated by approximately 50% (see Figure 6C). In DMSO, similar fluorescence spectral changes can be observed (6D). As discussed above, the interactions of 4 with fluoride anions result in deprotonation of the imidazolyl N–H group to form a delocalized anion. The resonance effect between the imidazolyl anion and the electron-withdrawing formyl group at the para positions should facilitate access to an emissive CT state during the photo-excitation processes, leading to the bathochromic shift of its emission band. In this way, compound 4 behaves as an ICT fluorophore and could therefore function as an effective ratiometric fluorescence probe for the detection and quantification of fluoride anion in solution. To evaluate such sensory function, correlations of the ratio of fluorescence intensities at two different wavelengths with the concentration of TBAF were made. Herein, the two fluorescence intensities were determined at the maximum emission wavelengths before the addition of TBAF and after the saturation of titration was reached. As can be seen from Figure 7A, in acetone the ratio of fluorescence intensities at 507 and 561 nm versus the concentration of TBAF shows two linear relationships. In the sub-millimolar range, the correlation features satisfactory sensitivity for quantification of fluoride anion. In DMSO (Figure 7B), the correlations appear to be more complex, with three linear relationships identified. The linearity in the low-concentration range (<0.3 mM) shows the best sensitivity in terms of sensor performance, but the detection range is a bit narrower than that in acetone.
Figure 7.

Plots of the ratio of fluorescence intensities of PI-BAL 4 at two different wavelengths against the concentration of TBAF. (A) Fluorescence intensities at 507 and 561 nm (F507/F561) in acetone, (B) fluorescence intensities at 530 and 585 nm (F530/F585) in DMSO. Solid lines are the linear least squares fitting for data points in selected ranges and associated R2 values are indicated.
The interactions of compound 5 with TBAF in the solution phase were also studied. Figure 8 shows the 1H NMR titration spectra obtained in acetone-d6. During the titration, the aldehyde proton shows an upfield shift from 10.21 to 10.14 ppm. The magnitude of this shift (0.07 ppm) is significantly smaller than that observed in the titration of 4 with TBAF (0.17 ppm). This can be explained by the meta relationship between the formyl group and the PI unit, which precludes a direct resonance interaction between the negatively charged N atom and the formyl group. It is also interesting to note that the chemical shifts of the other aryl proton signals all change only slightly during the addition of the first equivalent of TBAF. This observation suggests that the formation of a 1:1 complex between 5 and fluoride anions does not polarize the imidazolyl N–H bond as much as in the case of 4. In principle, the imidazolyl N–H of 5 is less acidic than that of 4 due to the lack of a direct resonance interaction with the formyl group. As such, 5 is predicted to form a weaker hydrogen bond with the fluoride ion than its para-isomer 4. This point is corroborated by DFT computational analysis (vide infra).
Figure 8.

1H NMR (300 MHz) spectra monitoring the titration of PI-BAL 5 with TBAF in acetone-d6.
The results of the UVvis titration of PI-BAL 5 with TBAF in acetone and DMSO are shown in Figure 9A,B. As in the case of its para-isomer 4, the UV–vis absorption of 5 shows a two-stage response to the fluoride anion in acetone. Upon titration from 0 to ca. 3.9 molar equivalents of TBAF, the spectra show an increase in absorption intensity in the long-wavelength region (390–480 nm) and a slight decrease in intensity in the short-wavelength region. There are three isosbestic points observed at 386, 382, and 368 nm in the first stage of spectral change. In the second stage, a long-wavelength band at 409 nm grows pronouncedly and it can be assigned to the π → π* transition of the deprotonated 5 upon interactions with the second equivalent of the fluoride anion. In DMSO, the UV–vis absorption spectra only exhibit one-stage responses to the titration with TBAF, with five isosbestic points observed at 388, 384, 375, 337, and 307 nm. In contrast to the fluorescence titration results of 4, meta-pyrenoimidazolyl benzaldehyde 5 responds to the fluoride ion in acetone and DMSO with only fluorescence attenuation (see Figure 9C,D). Comparing the different fluorescence behavior of the two structural isomers, 4 and 5, in response to fluoride anion titration, it is evident that the resonance effect plays a crucial and indispensable role in inducing ICT emission for the PI system. It is also worth noting that the fluorescence responses of 4 to other halide anions (Cl–, Br–, and I–) were observed to be insignificant particularly in DMSO (see Figures 10, S23 and S24 in the Supporting Information). These results demonstrate that compound 4 can indeed serve as a selective fluorescence probe to detect fluoride among other halide anions. Further understanding of the detailed structure–photophysical property relationship as well as the effect of solvent will be of great value for the rational design and tuning of highly efficient and sensitive fluorescence sensors based on the PI fluorophore.
Figure 9.

Titration of PI-BAL 5 with TBAF monitored by UV–vis absorption spectroscopy in (A) acetone and (B) DMSO, fluorescence spectroscopy in (C) acetone and (D) DMSO.
Figure 10.

Photographic image of the solutions of compound 4 (86.0 μM) and its 1:1 mixtures with various halide sources (Bu4NF, Bu4NCl, Bu4NBr, and Bu4NI) under UV light irradiation.
Theoretical Modeling Studies
DFT computational analysis was carried out on PI-BALs 4 and 5 to gain deeper insight into their structural and electronic properties. Figure 11 illustrates the optimized ground-state structures of compounds 4 and 5 in the gas phase, with their frontier molecular orbitals (FMOs) displayed as well. It can be seen that the major differences between the structural isomers lie in their unoccupied (antibonding) orbitals (e.g., LUMO and LUMO + 1), while the occupied (bonding) orbitals (HOMO and HOMO – 1) of the two compounds appear to be very similar in shape and spatial distribution. Owing to the resonance effect, the LUMO of para-isomer 4 is more delocalized than that of meta-isomer 5. For the LUMO + 1 of meta-isomer 5, however, the orbital exhibits more extended spatial distribution than that of para-isomer 4. Also of note is that para-isomer 4 has a smaller HOMO–LUMO gap (3.04 eV) than meta-isomer 5 (3.23 eV) because of the resonance effect. The different FMO energies and distributions thus account for their different electronic transition properties in the ground and excited states.
Figure 11.
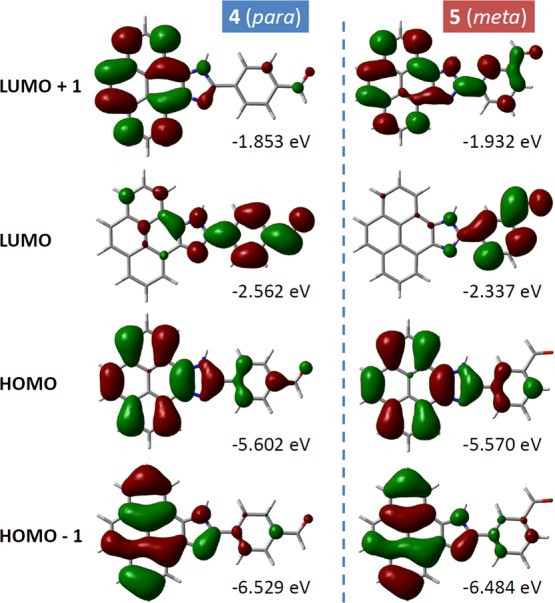
Contour plots (isovalue = 0.02 e/Å3) and eigenvalues of FMOs for PI-BALs 4 and 5 calculated at the B3LYP/6–31++G(d,p) level of theory in the gas phase.
To better understand the interactions (particularly, hydrogen bonding) between PI-BALs and the fluoride anion, the 1:1 complexes of [4···F–] and [5···F–] were modeled by DFT calculations. Herein, the computational studies took both the gas and solution phases into consideration, with three representative solvents (benzene, acetone, and DMSO). As shown in Figure 12A, the fluoride anion binds with compound 4 through three hydrogen bonding interactions in the gas phase. The primary binding force comes from the imidazolyl N–H1···F– interaction, while the pyrenyl proton H1 and phenyl proton H3 also contribute hydrogen bonds to bind with the fluoride anion in a way similar to the [DMSO···5] interactions observed in the crystal structure of 5 (Figure 2C). In the gas phase, H1···F– shows a bond distance at 1.04 Å and N–H1 distance at 1.42 Å, indicating a significant degree of deprotonation at the imidazolyl N–H position upon binding with fluoride ion. The nature of the bonding interactions between 4 and F– was also assessed by quantum theory of atoms in molecules (QTAIM) analysis.47 Through the analysis, three bond critical points (BCPs) were identified between F– anion and each of the three protons labeled as H1, H2, and H3 in Figure 12. The imidazolyl H1···F– interaction shows a weak covalent bond character, with ρ(BCP) = 0.240 au and ∇2ρ(BCP) = −0.879. The two BCPs between F– and pyrenyl H2/phenyl H3 show similar ρ(BCP) values (ca. 0.011–0.013 au), which are in the range of 0.002–0.040 au for a hydrogen bond.48,49 In the solution phase, however, the H1···F– bond elongates and the imidazolyl N–H1 bond shortens with increasing solvent polarity, suggesting that the degree of N–H deprotonation is attenuated in polar organic media. QTAIM analysis also reveals that the H1···F– interaction is weakened to exhibit a hydrogen bond character in polar organic solvents (see Table S10 in the Supporting Information for details).
Figure 12.

Optimized geometries of the 1:1 complexes of (A) [4···F–] and (B) [5···F–] in the gas phase at the B3LYP/6-31++G(d,p) level of theory, and variation of hydrogen bond distances in different solvents calculated by the polarizable continuum model (PCM) solvent model.
For the optimized structures of [5···F–] in the gas phase and various solvents (see Figure 12B), the N–H1 and H1···F– bonds are shorter than those of [4···F–]. These results suggest that the N–H1···F– interaction in [5···F–] is relatively weak. Again, this can be attributed to the meta relationship between the formyl group and the imidalzolyl unit in 5, which rules out a direct resonance interaction between them. To quantitatively assess the thermodynamic properties of the fluoride anion interactions with 4 and 5, the Gibbs free energy changes (ΔG°) for the 1:1 complexation reactions between the fluoride anion with 4 and 5 and subsequent deprotonation reactions in the gas phase and various solvents were computed. Scheme 2 lists the detailed thermodynamic data, which show that the two steps of fluoride interactions are both exergonic and increasing the solvent polarity reduces the thermodynamic driving force significantly.
Scheme 2. Gibbs Free Energy Changes for the Interactions of 4 and 5 with the Fluoride Ion in the Gas Phase and Various Solvents.

Calculated at the B3LYP/6-31++G(d,p) with the PCM used for solvents.
Conclusions
In conclusion, we have synthesized and studied the structural and electronic properties of two structural isomers of PI-BALs. Hydrogen bonding interactions have been found to play an important role in the solid-state structures of these compounds. Of particular note is that para-isomer 4 tends to form hydrogen-bonded dimers while meta-isomer can cocrystalize with hydrogen bond acceptors, such as DMSO. Significant solvents effects were observed in the UV–vis absorption and fluorescence emission properties of compounds 4 and 5. Cyclic voltammetric studies show that both compounds 4 and 5 have similar redox activities in polar solvents (acetone and DMSO). In the presence of fluoride anions, the two compounds undergo similar two-step reactions in polar solvents: (i) forming a hydrogen-bonded 1:1 complex and (ii) deprotonation of the imidazolyl N–H group. The UV–vis absorption spectra of the two compounds in response to fluoride anion titration are similar, showing colorimetric responses as a result of fluoride-induced deprotonation of the PI chromophore. The fluorescence responses of 4 and 5 to fluoride anion titration, however, are dramatically different. For para-isomer 4, the fluorescence is retained to some extent but the maximum emission band is bathochromically shifted. Such properties enable compound 4 to act as a fluorescence ratiometric sensor for detection of fluoride anion in solution. The interactions of fluoride anions with meta-isomer 5, on the other hand, result in fluorescence attenuation. Comparison of the fluorescence properties of the two pyrenoimidazolyl benzaldehyde isomers underscores the key role of the resonance effect in inducing the ICT mechanism. Our current studies lay a fundamental foundation for further efforts to design and develop new functional ICT fluorophores based on the PI motif.
Experimental Section
Reagents and solvents were purchased from commercial sources and used without purification unless otherwise noted. 1H and 13C NMR spectra were recorded on a Bruker 300 MHz ADVANCE III spectrometer. IR spectra were recorded using a Bruker Alfa spectrometer. High-resolution APPI–TOF MS analyses were performed on a GCT premier Micromass Technologies instrument. UV–visible absorption spectra were recorded using a Cary 6000i spectrophotometer. Fluorescence spectra were measured on a Photon Technology International (PTI) QuantaMaster spectrofluorometer. CV analyses were performed on a standard three-electrode setup (glassy carbon working electrode, Ag/AgCl reference electrode, and Pt wire counter electrode) controlled by a BASi Epsilon potentiostat. Single-crystal X-ray diffraction analysis was conducted on a Bruker D8/APEX II CCD diffractometer equipped with a Cu Kα (1.54178 Å) microfocus source. Crystal structures were solved and refined with the SHELXT software package. DFT and TD-DFT computational studies were carried out using the hybrid B3LYP functional50,51 with the 6-31++G(d,p) basis set as implemented in the Gaussian 09 software package.52 Optimized ground-state geometries were confirmed by frequency calculations to show the presence of no imaginary frequencies. Solvent effects were calculated using the PCM53 implemented in Gaussian 09. Visualization of molecular structures and orbitals were done using CYLview54 and GaussView 5.55 QTAIM analysis was carried out using the Multiwfn program.56
Synthesis of Compound 4
A mixture of 4,5-pyrenedione (1) (0.200 g, 0.861 mmol), terephthalaldehyde (2) (0.350 g, 2.58 mmol), ammonium acetate (1.33 g, 17.2 mmol), and glacial acetic acid (99.7%, 7 mL) was heated at 110 °C for 50 min. The solution was slowly cooled to room temperature and the resulting precipitate was collected by vacuum filtration and sequentially washed with glacial acetic acid, saturated aqueous NaHCO3 solution, and water. The crude product was purified by silica flash column chromatography using acetone/hexanes (3:7, v/v) as eluent to afford pure compound 4 as an orange solid (0.178 g, 0.516 mmol, 60%). mp > 280 °C (decomp.); FTIR (neat): 3361, 3052, 2849, 1662, 1602, 1569, 1432, 1162, 821, 713 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 14.00 (s, 1H), 10.12 (s, 1H), 8.86 (d, J = 8.5 Hz, 2H), 8.60 (d, J = 8.3 Hz, 2H), 8.34–8.19 (m, 8H) ppm; 13C NMR (75 MHz, DMSO-d6): δ 192.6, 148.0, 138.0, 136.0, 135.4, 131.6, 131.4, 130.2, 128.9, 127.9, 127.4, 126.5, 126.3, 126.0, 124.7, 124.4, 122.0, 121.5, 119.2, 119.1 ppm; HRMS (APPI–TOF, positive mode) m/z: calcd for C24H15N2O [M + H]+, 347.1184; found 347.1172.
Synthesis of Compound 5
A mixture of 4,5-pyrenedione (1) (0.200 g, 0.861 mmol), isophthalaldehyde (3) (0.350 g, 2.58 mmol), ammonium acetate (1.33 g, 17.2 mmol), and glacial acetic acid (99.7%, 7 mL) was heated at 110 °C for 3 h. The solution was slowly cooled to room temperature and the resulting precipitate was collected by vacuum filtration and sequentially washed with glacial acetic acid, saturated aqueous solution of NaHCO3, and water. The crude product was purified by silica flash column chromatography using acetone/hexanes (1:9, v/v) as eluent to afford pure compound 5 as a dark brown solid (0.149 g, 0.430 mmol, 50%). mp > 272 °C (decomp.); FTIR (neat): 3341, 3033, 2916, 2815, 2732, 1685, 1604, 1446, 1309, 1280, 1181, 977, 908, 831, 722, 674 cm–1; 1H NMR (300 MHz, DMSO-d6): δ 13.96 (s, 1H), 10.21 (s, 1H), 8.93–8.80 (m, 3H), 8.73–8.69 (m, 1H), 8.30–8.13 (m, 6H), 8.09–8.05 (m, 1H), 7.88 (t, J = 7.7 Hz, 1H); 13C NMR (75 MHz, DMSO-d6): δ 193.1, 161.9, 148.2, 137.6, 136.9, 131.8, 131.6, 131.4, 131.2, 130.7, 130.0, 128.5, 127.9, 127.5, 126.4, 126.3, 126.2, 126.0, 124.4, 124.3, 121.90, 121.86, 121.6, 119.0 ppm; HRMS (APPI–TOF, positive mode) m/z: calcd for C24H15N2O [M + H]+, 347.1184; found 347.1173.
Acknowledgments
The authors thank NSERC, CFI, and Memorial University of Newfoundland for funding support. Compute Canada and Ace-net are acknowledged for support in the computational studies. Drs. Robert MacDonald and Michael Ferguson at the University of Alberta are acknowledged for conducting the X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02482.
Details of spectroscopic characterization data and X-ray crystallographic data of compounds 4 and 5; detailed results of UV-vis and fluorescence spectral analysis; results of DFT and TD-DFT calculations; and interactions of compound 4 with other halide anions (PDF)
Crystallographic information file, CCDC 1851836 (CIF)
Crystallographic information file, CCDC 1851835 (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cicoira F.; Santato C.. Organic Electronics: Emerging Concepts and Technologies; Wiley-VCH, 2013. [Google Scholar]
- Sun S.-S.; Dalton L. R.. Introduction to Organic Electronic and Optoelectronic Materials and Devices; CRC Press, 2016. [Google Scholar]
- Facchetti A. π-Conjugated Polymers for Organic Electronics and Photovoltaic Cell Applications. Chem. Mater. 2011, 23, 733–758. 10.1021/cm102419z. [DOI] [Google Scholar]
- Wang C.; Dong H.; Hu W.; Liu Y.; Zhu D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. 10.1021/cr100380z. [DOI] [PubMed] [Google Scholar]
- Ostroverkhova O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. 10.1021/acs.chemrev.6b00127. [DOI] [PubMed] [Google Scholar]
- Li Y.Organic Optoelectronic Materials; Springer, 2016. [Google Scholar]
- Figueira-Duarte T. M.; Müllen K. Pyrene-Based Materials for Organic Electronics. Chem. Rev. 2011, 111, 7260–7314. 10.1021/cr100428a. [DOI] [PubMed] [Google Scholar]
- Mateo-Alonso A. Pyrene-Fused Pyrazaacenes: From Small Molecules to Nanoribbons. Chem. Soc. Rev. 2014, 43, 6311–6324. 10.1039/c4cs00119b. [DOI] [PubMed] [Google Scholar]
- Ghasemabadi P. G.; Yao T.; Bodwell G. J. Cyclophanes Containing Large Polycyclic Aromatic Hydrocarbons. Chem. Soc. Rev. 2015, 44, 6494–6518. 10.1039/c5cs00274e. [DOI] [PubMed] [Google Scholar]
- Valdés H.; Poyatos M.; Peris E. A Pyrene-Based N-Heterocyclic Carbene: Study of Its Coordination Chemistry and Stereoelectronic Properties. Organometallics 2013, 33, 394–401. 10.1021/om401134w. [DOI] [Google Scholar]
- Therrien J. A.; Wolf M. O.; Patrick B. O. Polyannulated Bis(N-heterocyclic carbene)palladium Pincer Complexes for Electrocatalytic CO2 Reduction. Inorg. Chem. 2015, 54, 11721–11732. 10.1021/acs.inorgchem.5b01698. [DOI] [PubMed] [Google Scholar]
- Liu B.; Lystrom L.; Kilina S.; Sun W. Tuning the Ground State and Excited State Properties of Monocationic Iridium(III) Complexes by Varying the Site of Benzannulation on Diimine Ligand. Inorg. Chem. 2017, 56, 5361–5370. 10.1021/acs.inorgchem.7b00467. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Thomas K. R. J.; Lin C.-C.; Jou J.-H. Pyrenoimidazole-Based Deep-Blue-Emitting Materials: Optical, Electrochemical, and Electroluminescent Characteristics. Chem.—Asian J. 2013, 8, 2111–2124. 10.1002/asia.201300271. [DOI] [PubMed] [Google Scholar]
- Peng Y.-X.; Dai Y.; Wang N.; Huang W. Symmetrical Fluorescent Oligothiophene and Benzene Centered Bispyrenoimidazole Derivatives with Double n-Dodecyl Chains Showing High Thermal Stability. Tetrahedron Lett. 2014, 55, 5984–5987. 10.1016/j.tetlet.2014.09.003. [DOI] [Google Scholar]
- Jadhav T.; Dhokale B.; Mobin S. M.; Misra R. Aggregation Induced Emission and Mechanochromism in Pyrenoimidazoles. J. Mater. Chem. C 2015, 3, 9981–9988. 10.1039/c5tc02181b. [DOI] [Google Scholar]
- Karthik S.; Ajantha J.; Nagaraja C. M.; Easwaramoorthi S.; Gandhi T. Synthesis and photophysics of extended π-conjugated systems of substituted 10-aryl-pyrenoimidazoles. Org. Biomol. Chem. 2016, 14, 10255–10266. 10.1039/c6ob01760f. [DOI] [PubMed] [Google Scholar]
- Shan T.; Liu Y.; Tang X.; Bai Q.; Gao Y.; Gao Z.; Li J.; Deng J.; Yang B.; Lu P.; et al. Highly Efficient Deep Blue Organic Light-Emitting Diodes Based on Imidazole: Significantly Enhanced Performance by Effective Energy Transfer with Negligible Efficiency Roll-off. ACS Appl. Mater. Interfaces 2016, 8, 28771–28779. 10.1021/acsami.6b10004. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Thomas K. R. J.; Lee C.-P.; Ho K.-C. Novel Pyrenoimidazole-Based Organic Dyes for Dye-Sensitized Solar Cells. Org. Lett. 2011, 13, 2622–2625. 10.1021/ol2006874. [DOI] [PubMed] [Google Scholar]
- Chang J.; Lee C.-P.; Kumar D.; Chen P.-W.; Lin L.-Y.; Thomas K. R. J.; Ho K.-C. Co-Sensitization Promoted Light Harvesting for Organic Dye-Sensitized Solar Cells Using Unsymmetrical Squaraine Dye and Novel Pyrenoimidazole-Based Dye. J. Power Sources 2013, 240, 779–785. 10.1016/j.jpowsour.2013.04.075. [DOI] [Google Scholar]
- Kumar D.; Thomas K. R. J.; Lee C.-P.; Ho K.-C. Triarylamine-Free Pyrenoimidazole-Containing Organic Dyes with Different π-Linkers for Dye-Sensitized Solar Cells. Asian J. Org. Chem. 2013, 4, 164–172. 10.1002/ajoc.201402214. [DOI] [Google Scholar]
- Zapata F.; Caballero A.; Espinosa A.; Tárraga A.; Molina P. Cation Coordination Induced Modulation of the Anion Sensing Properties of a Ferrocene–Imidazophenanthroline Dyad: Multichannel Recognition from Phosphate-Related to Chloride Anions. J. Org. Chem. 2008, 73, 4034–4044. 10.1021/jo800296c. [DOI] [PubMed] [Google Scholar]
- Saha D.; Das S.; Maity D.; Dutta S.; Baitalik S. Synthesis, Structural Characterization, and Photophysical, Electrochemical, Intercomponent Energy-Transfer, and Anion-Sensing Studies of Imidazole 4,5-Bis(benzimidazole)-Bridged Os(II)Os(II) and Ru(II)Os(II) Bipyridine Complexes. Inorg. Chem. 2010, 50, 46–61. 10.1021/ic100905u. [DOI] [PubMed] [Google Scholar]
- Bhaumik C.; Maity D.; Das S.; Baitalik S. Anion Sensing Studies of Luminescent Bis-tridentate Ruthenium(II) and Osmium(II) Complexes Based on Terpyridyl-imidazole Ligand Through Different Channels. Polyhedron 2013, 52, 890–899. 10.1016/j.poly.2012.07.025. [DOI] [Google Scholar]
- Das S.; Karmakar S.; Mardanya S.; Baitalik S. Synthesis, structural characterization, and multichannel anion and cation sensing studies of a bifunctional Ru(ii) polypyridyl-imidazole based receptor. Dalton Trans. 2014, 43, 3767–3782. 10.1039/c3dt52424h. [DOI] [PubMed] [Google Scholar]
- Maity D.; Bhaumik C.; Mondal D.; Baitalik S. Ru(II) and Os(II) Complexes Based on Terpyridyl-imidazole Ligand Rigidly Linked to Pyrene: Synthesis, Structure, Photophysics, Electrochemistry, and Anion-Sensing Studies. Inorg. Chem. 2013, 52, 13941–13955. 10.1021/ic401582m. [DOI] [PubMed] [Google Scholar]
- Mardanya S.; Karmakar S.; Maity D.; Baitalik S. Ruthenium(II) and Osmium(II) Mixed Chelates Based on Pyrenyl-Pyridylimidazole and 2,2′-Bipyridine Ligands as Efficient DNA Intercalators and Anion Sensors. Inorg. Chem. 2014, 54, 513–526. 10.1021/ic502271k. [DOI] [PubMed] [Google Scholar]
- Mardanya S.; Karmakar S.; Mondal D.; Baitalik S. An imidazolyl-pyreno-imidazole conjugate as a cyanide sensor and a set-reset memorized sequential logic device. Dalton Trans. 2015, 44, 15994–16012. 10.1039/c5dt01317h. [DOI] [PubMed] [Google Scholar]
- Causey C. P.; Allen W. E. Anion Binding by Fluorescent Biimidazole Diamides. J. Org. Chem. 2002, 67, 5963–5968. 10.1021/jo020098v. [DOI] [PubMed] [Google Scholar]
- Kang J.; Kim H. S.; Jang D. O. Fluorescent Anion Chemosensors Using 2-Aminobenzimidazole Receptors. Tetrahedron Lett. 2005, 46, 6079–6082. 10.1016/j.tetlet.2005.07.002. [DOI] [Google Scholar]
- Zhang M.; Li M.; Li F.; Cheng Y.; Zhang J.; Yi T.; Huang C. A novel Y-type, Two-Photon Active Fluorophore: Synthesis and Application in Ratiometric Fluorescent Sensor for Fluoride Anion. Dyes Pigm. 2008, 77, 408–414. 10.1016/j.dyepig.2007.07.003. [DOI] [Google Scholar]
- Mo H.-J.; Shen Y.; Ye B.-H. Selective Recognition of Cyanide Anion via Formation of Multipoint NH and Phenyl CH Hydrogen Bonding with Acyclic Ruthenium Bipyridine Imidazole Receptors in Water. Inorg. Chem. 2012, 51, 7174–7184. 10.1021/ic300217v. [DOI] [PubMed] [Google Scholar]
- Molina P.; Tárraga A.; Otón F. Imidazole Derivatives: A Comprehensive Survey of Their Recognition Properties. Org. Biomol. Chem. 2012, 10, 1711–1724. 10.1039/c2ob06808g. [DOI] [PubMed] [Google Scholar]
- Esteves C. I. C.; Raposo M. M. M.; Costa S. P. G. New 2,4,5-Triarylimidazoles Based on a Phenylalanine Core: Synthesis, Photophysical Characterization and Evaluation as Fluorimetric Chemosensors for Ion Recognition. Dyes Pigm. 2016, 134, 258–268. 10.1016/j.dyepig.2016.07.020. [DOI] [Google Scholar]
- Lin W.; Long L.; Yuan L.; Cao Z.; Chen B.; Tan W. A Ratiometric Fluorescent Probe for Cysteine and Homocysteine Displaying a Large Emission Shift. Org. Lett. 2008, 10, 5577–5580. 10.1021/ol802436j. [DOI] [PubMed] [Google Scholar]
- Long L.; Zhou L.; Wang L.; Meng S.; Gong A.; Zhang C. A ratiometric Fluorescent Probe for Iron(III) and Its Application for Detection of Iron(III) in Human Blood Serum. Anal. Chim. Acta 2014, 812, 145–151. 10.1016/j.aca.2013.12.024. [DOI] [PubMed] [Google Scholar]
- Kubo Y.; Yamamoto M.; Ikeda M.; Takeuchi M.; Shinkai S.; Yamaguchi S.; Tamao K. A Colorimetric and Ratiometric Fluorescent Chemosensor with Three Emission Changes: Fluoride Ion Sensing by a Triarylborane–Porphyrin Conjugate. Angew. Chem., Int. Ed. 2003, 42, 2036–2040. 10.1002/anie.200250788. [DOI] [PubMed] [Google Scholar]
- Peng X.; Wu Y.; Fan J.; Tian M.; Han K. Colorimetric and Ratiometric Fluorescence Sensing of Fluoride: Tuning Selectivity in Proton Transfer. J. Org. Chem. 2005, 70, 10524–10531. 10.1021/jo051766q. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Xiao Y.; Qian X.; Cui J.; Cui D. Ratiometric and Selective Fluorescent Sensor for CuIIBased on Internal Charge Transfer (ICT). Org. Lett. 2005, 7, 889–892. 10.1021/ol0473445. [DOI] [PubMed] [Google Scholar]
- Liu B.; Tian H. A Ratiometric Fluorescent Chemosensor for Fluoride Ions Based on a Proton Transfer Signaling Mechanism. J. Mater. Chem. 2005, 15, 2681–2686. 10.1039/b501234a. [DOI] [Google Scholar]
- Zhang J. F.; Lim C. S.; Bhuniya S.; Cho B. R.; Kim J. S. A Highly Selective Colorimetric and Ratiometric Two-Photon Fluorescent Probe for Fluoride Ion Detection. Org. Lett. 2011, 13, 1190–1193. 10.1021/ol200072e. [DOI] [PubMed] [Google Scholar]
- Chen S.; Hong Y.; Liu Y.; Liu J.; Leung C. W. T.; Li M.; Kwok R. T. K.; Zhao E.; Lam J. W. Y.; Yu Y.; et al. Full-Range Intracellular pH Sensing by an Aggregation-induced Emission-active Two-Channel Ratiometric Fluorogen. J. Am. Chem. Soc. 2013, 135, 4926–4929. 10.1021/ja400337p. [DOI] [PubMed] [Google Scholar]
- Walsh J. C.; Williams K.-L. M.; Lungerich D.; Bodwell G. J. Synthesis of Pyrene-4,5-dione on a 15 g Scale. Eur. J. Org. Chem. 2016, 2016, 5933–5936. 10.1002/ejoc.201601198. [DOI] [Google Scholar]
- Bräuer M.; Mosquera M.; Pérez-Lustres J. L.; Rodríguez-Prieto F. Ground-State Tautomerism and Excited-State Proton-Transfer Processes in 4,5-Dimethyl-2-(2’-hydroxyphenyl)imidazole in Solution: Fluorescence Spectroscopy and Quantum Mechanical Calculations. J. Phys. Chem. A 1998, 102, 10736–10745. 10.1021/jp983291v. [DOI] [Google Scholar]
- Park S.; Kwon O.-H.; Kim S.; Park S.; Choi M.-G.; Cha M.; Park S. Y.; Jang D.-J. Imidazole-Based Excited-State Intramolecular Proton-Transfer Materials: Synthesis and Amplified Spontaneous Emission From a Large Single Crystal. J. Am. Chem. Soc. 2005, 127, 10070–10074. 10.1021/ja0508727. [DOI] [PubMed] [Google Scholar]
- Park S.; Kwon O.-H.; Lee Y.-S.; Jang D.-J.; Park S. Y. Imidazole-Based Excited-State Intramolecular Proton-Transfer (ESIPT) Materials: Observation of Thermally Activated Delayed Fluorescence (TDF). J. Phys. Chem. A 2007, 111, 9649–9653. 10.1021/jp072212p. [DOI] [PubMed] [Google Scholar]
- Li G.-Y.; Zhao G.-J.; Liu Y.-H.; Han K.-L.; He G.-Z. TD-DFT Study on the Sensing Mechanism of a Fluorescent Chemosensor for Fluoride: Excited-State Proton Transfer. J. Comput. Chem. 2010, 31, 1759–1765. 10.1002/jcc.21466. [DOI] [PubMed] [Google Scholar]
- Bader R. F.Atoms in Molecules; Oxford University Press: New York, USA, 1994. [Google Scholar]
- Koch U.; Popelier P. L. A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. 10.1021/j100024a016. [DOI] [Google Scholar]
- Grabowski S. J. Hydrogen bonding strength-measures based on geometric and topological parameters. J. Phys. Org. Chem. 2004, 17, 18–31. 10.1002/poc.685. [DOI] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.. et al. Gaussian09, Revision E.01; Gaussian Inc.: Wallingford CT, 2009.
- Miertuš S.; Scrocco E.; Tomasi J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of AB Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. 10.1016/0301-0104(81)85090-2. [DOI] [Google Scholar]
- Legault C. Y.CYLview, 1.0b; Université de Sherbrooke, 2009. http://www.cylview.org.
- Dennington R.; Keith T.; Millam J.. GaussView Version 5; Semichem Inc.: Shawnee Mission KS: 2009.
- Lu T.; Chen F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.