

Abstract
Aminopeptidase N/CD13 is expressed by fibroblast-like synoviocytes (FLS) and monocytes (MNs) in inflamed human synovial tissue (ST). This study examined the role of soluble (s)CD13 in angiogenesis, MN migration, phosphorylation of signaling molecules, and induction of arthritis.
The contribution of sCD13 was examined in angiogenesis and MN migration using sCD13, and CD13-depleted RA synovial fluids (SFs). An enzymatically inactive mutant CD13 and intact wild-type (WT) CD13 were used to determine whether its enzymatic activity contributes to the arthritis-related functions. CD13-induced phosphorylation of signaling molecules was determined by Western blotting. The effect of sCD13 on cytokine secretion from RA ST and RA FLS was evaluated. sCD13 was injected into C57Bl/6 mouse knees to assess its arthritogenicity.
sCD13 induced angiogenesis and was a potent chemoattractant for MNs and U937 cells. Inhibitors of Erk1/2, Src, NFκB, Jnk, and PT, a G protein-coupled receptor inhibitor, decreased sCD13-stimulated chemotaxis. CD13-depleted RA SF induced significantly less MN migration than sham-depleted SF, and addition of mutant or WT CD13 to CD13-depleted RA SF equally restored MN migration. sCD13 and recombinant WT or mutant CD13 had similar effects on signaling molecule phosphorylation, indicating that the enzymatic activity of CD13 had no role in these functions. CD13 increased the expression of pro-inflammatory cytokines by RA FLS, and a CD13 neutralizing antibody inhibited cytokine secretion from RA ST organ culture. Mouse knee joints injected with CD13 exhibited increased circumference and pro-inflammatory mediator expression. These data support the concept that sCD13 plays a pivotal role in RA and acute inflammatory arthritis.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease that causes chronic inflammation and destruction of the joints (1). RA fibroblast-like synoviocytes (FLS) and monocytes/macrophages contribute to the joint inflammation by secreting many pro-inflammatory factors, promoting angiogenesis, and contributing to joint damage (1–5). An imbalance in cytokines and chemokines in RA joints leads to the infiltration of the synovium with leukocytes (2, 6). Monocyte (MN) ingress and secretion of pro-inflammatory cytokines amplify the effects of autoimmune responses, resulting in persistent inflammation and progressive destruction of the tissues.
Aminopeptidase N/CD13 is a metalloproteinase which is highly expressed by tumor cells, RA FLS, MNs, endothelial cells (ECs), and mesenchymal stem cells (MSCs) (7, 8). CD13 is a transmembrane protein, that also exists in shed and secreted soluble forms (9). CD13 has been identified in synovial tissue (ST) by immunostaining (10). CD13 is also found in soluble form in serum and synovial fluids (SFs) (11). The concentrations and enzymatic activity of soluble (s)CD13 are significantly higher in RA SFs than in osteoarthritis (OA) SFs or RA sera (12). sCD13 is a potent chemoattractant for T cells and induces cytokine-activated T cell (Tck) migration via G protein-coupled receptors (GPCRs) (12). CD13 plays an important role in MN recruitment into the peritoneum in an acute inflammatory model of peritonitis (7, 13). Cross-linking of membrane bound CD13 induces the phosphorylation of mitogen-activated protein kinases (MAPK) Erk1/2, JNK, and P38 in MNs (7, 14).
Here we determined the role of sCD13 in angiogenesis and MN migration. We assessed whether its enzymatic activity contributes to MN migration. Signaling molecules phosphorylated by sCD13 were examined in RA FLS. Furthermore, we measured sCD13-induced production of pro-inflammatory cytokines in RA ST and FLS, and examined the ability of sCD13 to induce MN ingress to synovium and acute inflammatory arthritis in mice.
Materials and Methods
Cell culture and mice
All procedures involving specimens obtained from human subjects were performed under a protocol approved by the University of Michigan Institutional Review Board. Human dermal microvascular endothelial cells (ECs) (passage 5–8) were maintained in endothelial cell basal medium (EBM) with media supplements and 5% fetal bovine serum (FBS, Lonza, Walkersville, MD). MNs were isolated from the peripheral blood (PB) of healthy volunteers using the MN Isolation Kit II from Miltenyi Biotec. RA FLS were harvested from human STs obtained at arthroplasty or synovectomy from RA joints and propagated as cell lines, which were used at passages 3–8 (15, 16). MN, U937 cells (A human monocytic cell line from ATCC), or RA FLS were maintained in RPMI with 10% FBS. Before stimulation with sCD13, media were switched to reduced serum media, RPMI with 0.1% FBS. Female C57BL/6 wild type (WT) mice (8–10 weeks old) were purchased from the Jax laboratory. All the experiments with mice were performed with approval from the Institutional Animal Care and Use Committee (IACUC).
EC chemotaxis
EC chemotaxis was performed in a modified Boyden chamber (17–20). Test substances included phosphate buffered saline (PBS, negative control), basic fibroblast growth factor (bFGF, 60 nM, positive control), and sCD13 (R&D Systems, Minneapolis, MN).
Matrigel EC tube formation assay
To examine the role of sCD13 in capillary morphogenesis, we performed EC tube formation assay using growth factor reduced Matrigel (GFR, Becton Dickinson, Bedford, MA). sCD13 (500 ng/ml) or vehicle control (PBS for sCD13) were placed directly into the media. The number of tubes formed was quantitated by an observer blinded to the experimental groups. To determine the role of signaling molecules in sCD13-induced EC tube formation, we performed Matrigel EC tube formation using sCD13-induced as a stimulus with or without signaling inhibitors (17–20).
Matrigel plug angiogenesis assay in vivo
To examine the effect of sCD13 in angiogenesis in vivo, we performed mouse Matrigel plug assays by injecting GFR Matrigel (500 μl) subcutaneously (s.c.) into each WT C57BL/6 mouse. After 7 days, the mice were euthanized and the Matrigel plugs were harvested, weighed, and some of them were processed for hemoglobin (Hb) measurement. Hb concentration was determined by comparison to a standard curve in g/dl representing the number of blood vessels in the plugs (17–20). Hb was normalized to plug weight.
MN and U937 cell chemotaxis
MN and U937 chemotaxis was performed in a modified Boyden chamber (17, 21). MN and U937 cells were pre-incubated with chemical signaling inhibitors of Erk1/2 (PD98059, PD), Src (PP2), NFκB (PDTC), Jnk (SP600125, SP), p38 (SB203580, SB), and GPCR (Pertussis toxin, PT) for 30 minutes prior to the assay (9, 12). sCD13 or enzymatically active (WT)/enzymatically inactive (mutant) CD13, purified from the supernatants of CD13-transfected cells as previously described (9, 12), were used as stimuli at 500 ng/ml. This concentration approximates the concentration of sCD13 in RA SF (9, 12). Three high power fields (HPF) (400x) were counted by an observer blinded to the experimental groups. All of the inhibitors were purchased from Calbiochem (La Jolla, CA, USA) and were used at 10μM concentrations except PT and PDTC were used at 100 ng/ml and 200 μM, respectively.
Cyclic adenosine monophosphate (cAMP) ELISA
MNs were placed in 6-well plates and stimulated with or without sCD13 (500 ng/ml) for 5 min, 10 min, 20 min, and 30 min. The levels of cAMP were measured by DetectX® Cyclic AMP (cAMP) Direct Immunoassay kit according to the manufacturer’s instructions (Arbor Assays).
Generation of mutant CD13 and WT CD13
We generated enzymatically inactive mutant CD13 and WT CD13 as previously described, using a CD13 clone (MGC Human ANPEP Sequence-Verified cDNA; GenBank accession no. BC058928 https://www.ncbi.nlm.nih.gov/nuccore/BC058928) that was obtained from Open Biosystems (9, 12).
CD13 depletion from RA SFs and MN chemotaxis assays with CD13 depleted SFs
Rheumatoid factor was removed from RA SF using a kit from Thermo Scientific. CD13 was then immune-depleted from RA SFs and its levels were measured in SFs after immuno-depletion using a CD13 sandwich enzyme linked immunosorbent assay (ELISA) as we have published recently (12). sCD13 was used as a standard to determine CD13 concentration in SFs. MN chemotaxis was performed with CD13-depleted and sham-depleted RA SFs to determine the role of CD13 in MN migration.
To further evaluate the role of non-enzymatic effects of CD13 in MN recruitment, CD13-depleted RA SFs were supplemented with mutant or WT CD13 and MN chemotaxis was performed. We also stimulated MNs with mutant or WT CD13 for 20 minutes and performed Western blots to examine phosphorylation of signaling molecules (17, 21–23).
Western blotting
RA FLS and normal human MNs were incubated with or without chemical signaling inhibitors including PT for 1 hour before stimulation with sCD13 (500ng/ml) for 25 minutes. Cell lysates were subjected to 10% SDS-PAGE and Western blots were performed using antibodies against phospho-Erk1/2, phospho-Src, and phospho-NFĸB (Cell Signaling Technology). The membranes were stripped and reprobed with antibodies against total Erk1/2, Src, NFĸB, and β-actin to assess the equal loading of proteins.
siRNA knockdown
RA FLS were transfected with SignalSilence® p44/42 MAPK (Erk1/2) siRNA or SignalSilence® Control (scrambled) siRNA (Cell Signaling Technology) using TransIT-X2® (Mirus Bio LLC) following the manufacturer’s instructions. RA FLS were transfected for 24–48 hours. Transfected RA FLS were stimulated with sCD13 for 25 minutes and Western blotting was performed to confirm the data obtained with chemical signaling inhibitors.
Determination of sCD13-induced cytokine secretion in FLS cultures by performing quantitative PCR and ELISAs
Total RNA was isolated from RA FLS using the RNAeasy Isolation Kit QIA shredder (Qiagen) according to the manufacturer’s protocol. cDNA was prepared using the cDNA Kit (Life Technologies). Quantitative PCR (qPCR) was performed using Platinum SYBR Green qPCR SuperMix-UDG (Life Technologies) following the manufacturer’s protocol. The primers for human cytokines used were: Monocyte chemoattractant protein-1 (MCP-1/CCL2) forward 5’-ACTCTCGCCTCCAGCATGAA-3’; MCP-1/CCL2 reverse 5’-TTGATTGCATCTGGCTGAGC-3’; interleukin-6 (IL-6) forward 5’-GTAGCCGCCCCACACAGA-3’; IL-6 reverse 5’-CATGTCTCCTTTCTCAGGGCTG-3’; IL-1β forward 5’-AAATACCTGTGGCCTTGGGC-3’; IL-1β reverse 5’-TTTGGGATCTACACTCTCCAGCT-3’ (24). The primers used for β-actin were; forward: 5’-GCTAGGCAGCTCGTAGCTCT-3’ and reverse: 5’-GCCATGTACGTTGCTATCCA-3’ (25). All samples were run in triplicate.
RA FLS were placed onto 6-well plates and stimulated with or without sCD13 (500 ng/ml) for 24 hours and supernatants were collected to perform ELISAs. The cytokine concentrations were measured by the human DuoSet® ELISA (R&D Systems). We used FLS from 6 different RA patients to perform PCR and ELISAs.
Ex vivo culture of RA ST
STs received from the University of Michigan Hospital operating suite were dissected into small pieces of ~1–2 mm3 as described (26). Each piece was incubated in CMRL (1 ml) media containing 10% FBS, 2% L-glutamine, and antibiotics in each well of a 12 well plate. Ex vivo cultures, which contain a representative mixture of the various cell populations present in RA ST in vivo, were incubated for 24 hours, then incubated for an additional 48 hours in fresh medium with or without WM15 (25 μg/ml, a neutralizing antibody against CD13, Biolegend, San Diego, CA) and 1D7, another monoclonal antibody against CD13 (25 μg/ml, University of Michigan Hybridoma Core), or dexamethasone (10 μg/ml) as a positive control for suppression (12). ELISAs were performed on culture supernatants from the 0–24 and 24–72 hour incubation periods to measure secretion of MCP-1/CCL2, IL-6, and IL-8/CXCL8. The ratio of secreted protein at 72 hours to the amount secreted at 24 hours was used to determine fold-change in secretion for each ST piece. We used between 6–12 replicates of ST from 3 different RA patients.
Acute inflammatory arthritis model
To determine whether CD13 could induce acute joint inflammation, C57BL/6 WT mouse knees were injected with sCD13. After anesthetizing mice, sCD13 (5μg in 20μL PBS) was injected into some of the mouse knee joints while PBS (control) was injected into others. Knee joints were measured before (day 0) and 24 hours post injection of PBS or sCD13 (27) by an observer blinded to the experimental groups. Mice were euthanized after 24 hours and knees were harvested. Some of the knees were homogenized for cytokine analysis and others were cryosectioned for histology.
Hematoxylin and Eosin (H&E) and F4/80 staining assay
Some of the mouse knees were decalcified for 2 weeks then embedded in Optimal Cutting Temperature (OCT) compound for cryosectioning. H&E staining was performed to determine inflammation and leukocyte ingress in knees injected with sCD13 or PBS. Some of the mouse knees were cryosectioned without decalcification and immunofluorescence was performed to determine MNs/macrophages, using rat anti-mouse F4/80 (GeneTex) as the primary antibody and Alexa Fluor 594–conjugated goat anti-rat IgG (Life Technologies) (21–23).
Statistics
Results are expressed as mean± standard error of the mean (SEM). Data were analyzed using Student’s t-test. A p-value less than 0.05 was considered significant.
Results
sCD13 induces EC migration and tube formation in vitro
Having previously shown that sCD13 induces migration of activated T cells and FLS (9, 12), we investigated its ability to promote chemotaxis of other cell types essential for RA synovitis. We found that sCD13 induces EC migration in a dose-dependent manner and that maximal sCD13-induced EC migration is significantly higher compared to the bFGF positive control. This increase in EC migration started at 1 ng/ml and was almost 8 fold higher at 1000 ng/ml of sCD13, suggesting that sCD13 is a potent angiogenic factor, at concentrations found in vivo in RA SF (Fig 1A) (12). sCD13 (500 ng/ml) also induced EC tube formation on GFR Matrigel compared to control (p < 0.05) (Fig 1B and C).
Fig. 1). sCD13 induces EC migration, tube formation, and angiogenesis in murine Matrigel plug assays.

A) Human dermal microvascular endothelial cell (EC) chemotaxis was performed in a modified Boyden chamber using sCD13 at various concentrations. sCD13 induces EC migration in a dose-dependent manner. CD13 induced EC migration from 1–1000 ng/ml (p<0.05). Data represent the mean of 3 individual experiments ± SEM. Three high power fields (hpf) (x400) were counted in each replicate well. PBS and bFGF served as negative and positive controls, respectively. B-C) EC tube formation was performed on growth factor reduced (GFR) Matrigel with sCD13. EC tubes formed in response to sCD13 were almost two fold more when compared to PBS, a negative control. Arrows indicate tubes formed in response to sCD13. n = number of replicates in each group. D-E) We examined the role of signaling molecules by performing EC morphogenesis assays using sCD13 as a stimulus in the presence or absence of signaling inhibitors. EC tubes formed by sCD13 were significantly decreased by the inhibitors of NFkB, Jnk, Erk1/2, and GPCR while a p38 inhibitor (SB) did not. F-G) To test the effect of sCD13 in angiogenesis in vivo, Matrigel plug assays using C57BL/6 mice was performed. Each mouse was given a subcutaneous injection of sterile GFR Matrigel (500 μl/injection) containing either sCD13 or PBS, a negative control. Matrigel plugs were harvested after 7 days. Hemoglobin (Hb) was determined. There was more than a 4 fold increase in Hb, an indirect correlate of neovascularization, in the plugs containing sCD13. Results are represented as the mean ± SEM and *p<0.05 was considered significant. On gross appearance, plugs injected with sCD13 had blood vessel formation and appeared red compared to plugs injected with PBS. Arrows indicate redness and new blood vessels formed. n = number of mice per group.
NFkB, Jnk, Erk1/2, and GPCRs are involved in sCD13-mediated EC tube formation
EC tube formation assays on Matrigel were performed to determine the role of signaling molecules in sCD13-mediated EC, morphogenesis. The inhibitors of Jnk (SP), NFkB (PDTC), Erk1/2 (PD), and GPCR (PT), but not p38 (SB), inhibited sCD13-induced EC tube formation (Fig 1D).
sCD13 contributes to angiogenesis in Matrigel plug angiogenesis assays
After finding that sCD13 induces EC migration and tube formation in vitro, we examined sCD13-induced angiogenesis in vivo by performing mouse Matrigel plug assays. On gross appearance, plugs collected from mice injected with sCD13 appeared yellowish-red compared to PBS injected plugs which appeared to be white (Fig 1E). Hemoglobin levels, an indirect measure of angiogenesis, were ~6 fold higher in plugs containing sCD13 (500 ng/ml) compared to the PBS control (Fig 1F).
sCD13 induces MN/U937 migration in vitro
We next determined the role of sCD13 in MN and U937 cell migration in vitro. PBS and N-Formylmethionyl-leucyl-phenylalanine (fMLP) were used as negative and positive controls, respectively. sCD13 induced maximum MN migration at 500ng/ml and this increase in MN migration was significant compared to PBS (Fig 2A). sCD13 also significantly increased U937 cell migration compared to control (Fig 2D and E).
Fig. 2). sCD13 induces MN/U937 migration and various signaling inhibitors including PT reduce sCD13-induced MN and U937 cell migration.

A) sCD13 induced a concentration dependent increase in human MN chemotaxis with a maximum migration at 500 ng/ml. PBS was used as a negative control. Data represent the mean of 4 individual experiments ± SEM and there were 4 replicate wells in each experimental group. Three high-power fields (hpfs) (x400) were counted in each replicate well and results were expressed as cells per 3 hpf. B, C, D, and E) MNs/U937 cells were incubated with signaling inhibitors (10 μM) except PT (100ng/ml) for 30 minutes before performing chemotaxis assay with sCD13. Data represent the mean of ≥10 replicates ± SEM for MNs and the mean of ≥7 replicates ± SEM for U937 cells. PBS served as a negative control. All inhibitors were present during the experiment in addition to the preincubation. hpf=high power field. *p<0.05 was considered significant. We determined the role of GPCRs in MN/U937 cell migration by performing chemotaxis after incubating MNs/U937 cells with PT, a GPCR inhibitor. F) sCD13 increases cAMP in MNs. We detected the functional activity of GPCRs in MNs by measuring the concentration of cAMP at different time points. The levels of cAMP were increased in a time-dependent manner from 5 min to 20 min, and there was an over 50% increase in cAMP at 20 min with group following sCD13 treatment. This indicates that GPCRs are required in CD13-mediated signaling.
Src, Jnk, NFkB, Erk1/2, and GPCRs are involved in sCD13-mediated MN/U937 cell migration
The inhibitors of Src (PP2), NFκB (PDTC), Jnk (SP), and GPCR (PT) significantly reduced sCD13-induced MN migration while inhibitors of p38 (SB) and Erk1/2 (PD) did not (Fig 2B and C). sCD13-induced U937 cell migration was decreased by inhibitors of Src, Erk1/2, NFκB, Jnk, and GPCR, but not by a p38 inhibitor (Fig 2D). There was ~70% decrease in U937 cell migration while normal human MN showed a ~30% decrease in migration with PT, a GPCR inhibitor (Fig 2C and E).
GPCRs are well known to signal via cAMP production (28). To detect the functional activity of GPCRs in MNs, the concentration of cAMP was measured at different time points after sCD13 treatment. The levels of cAMP were increased in a time-dependent manner from 5 min to 20 min, and there was an over 50% increase in the 20 min group following sCD13 treatment (Fig 2F), indicating that GPCRs play a key role in CD13-mediated signaling.
sCD13 increases RA SF-induced MN migration and phosphorylation of signaling molecules independent of its enzymatic activity
In light of our previous finding that the chemotactic effects of sCD13 on T cells are independent of CD13 enzymatic activity (12), we assessed whether the aminopeptidase activity of CD13 contributes to MN migration by using mutant CD13 and WT CD13 (12). WT and mutant CD13 each significantly increased MN chemotaxis which was more than 4 fold compared to control (p≤0.001) (Fig 3A).
Fig. 3). sCD13 contributes to MN chemotaxis and phosphorylation of signaling molecules independent of its enzymatic activity.
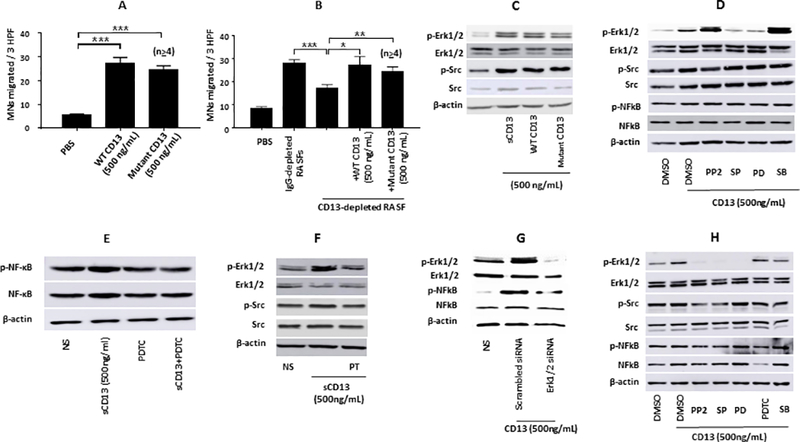
A) To assess whether CD13’s enzymatic activity contributes to MN migration, MN chemotaxis was examined using enzymatically inactive CD13 (mutant CD13) and enzymatically active CD13 (WT CD13) (12). WT and mutant CD13 significantly increased MN chemotaxis more than 4 fold compared to control (*p≤0.001). Data represent the mean of ≥4± SEM for MNs. B) To determine whether sCD13 accounts in part for the MN chemotactic potential of RA SFs, CD13 was depleted from RA SFs using two anti-CD13 antibodies (1D7 and WM15) and mouse IgG was used for sham-depletion. CD13-depleted RA SF induced 38% less MN migration than sham-depleted (*p≤0.001). Addition of mutant or WT CD13 (500 ng/ml of each) to CD13-depleted RA SF restored MN migration to a level similar to the sham-depleted, suggesting that CD13 induces MN migration independent of its enzymatic activity. Data represent mean ± SEM for an n≥4. C) Mutant CD13 and WT CD13 markedly increased the phosphorylation of Erk1/2 and Src in MNs. D-E) sCD13 increased the phosphorylation of Erk1/2, Src, and NFĸB in RA FLS. FLS were incubated with chemical signaling inhibitors for 1 hour before stimulation with sCD13 for 25 minutes. A Jnk inhibitor, SP, and an Erk1/2 (PD) inhibitor reduced the phosphorylation of Erk1/2 and NFkB, indicating that Jnk and Erk1/2 are upstream in sCD13-mediated signaling pathways in RA FLS. PDTC, an inhibitor of NFkB, markedly reduced sCD13-induced phosphorylation of NFkB in RA FLS. F) A GPCR inhibitor, PT, strongly inhibited phosphorylation of Erk1/2 and Src, suggesting that sCD13 mediates its effects via GPCRs. G) We confirmed signaling data by transfecting RA FLS with siRNA against Erk1/2 for 48 hours. RA FLS transfected with Erk1/2 siRNA displayed markedly decreased phosphorylation of Erk1/2 and NFkB compared to control (scrambled) transfected cells. H) sCD13 increased the phosphorylation of Erk1/2, Src, and NFkB in normal human MNs. The inhibitors of Src and Jnk reduced the phosphorylation of Erk1/2 and NFkB, suggesting that Src and Jnk are upstream in sCD13-induced phosphorylation of Erk1/2 and NFkB. MNs were stimulated with sCD13 for 20 minutes after incubating one hour with signaling inhibitors.
To determine whether CD13 accounts for some of the MN chemotactic properties of RA SFs, CD13 was depleted from RA SFs (9, 12). CD13-depleted RA SF induced significantly less MN migration than sham-depleted (p≤0.001). Addition of mutant or WT CD13 to CD13-depleted RA SF restored MN migration to a level similar to the sham-depleted RA SF, suggesting that CD13 contributes to MN ingress in RA in a manner independent of the enzymatic activity of CD13 (Fig 3B).
The effects of mutant versus WT CD13 were also examined in phosphorylation of signaling molecules in MNs. WT and mutant CD13 markedly increased Erk1/2 and Src phosphorylation in MNs, pointing out that CD13 mediates its signaling effects independent of its enzymatic activity (Fig 3C).
sCD13 induces Erk1/2, Src, and NFĸB phosphorylation in RA FLS and normal human MNs
Since sCD13 activates FLS (9), we also analyzed signaling events in FLS that were initiated by sCD13. sCD13 increased the phosphorylation of Erk1/2, Src, and NFκB in RA FLS (Fig 3D). We determined the crosstalk between signaling intermediates in FLS and found that a Jnk inhibitor, SP, reduced the phosphorylation of Erk1/2 and NFkB, indicating that Jnk is upstream in sCD13-mediated signaling pathways in RA FLS (Fig 3D). sCD13-induced NFkB phosphorylation was inhibited by PDTC, an inhibitor of NFkB (Fig 3E). Moreover, we found that sCD13-induced phosphorylation of signaling molecules was strongly inhibited by PT, suggesting that sCD13 mediates its signaling effects via GPCRs (Fig 3F). We confirmed signaling data by transfecting RA FLS with siRNA against Erk1/2 and found that RA FLS transfected with Erk1/2 siRNA displayed decreased sCD13-induced phosphorylation of NFkB as well as Erk1/2 compared to control (scrambled) transfected cells (Fig 3G).
We also evaluated sCD13-stimulated phosphorylation of signaling molecules and cross-talk among these signaling molecules in normal human MNs. sCD13-induced Erk1/2 phosphorylation was decreased by the inhibitors of Src (PP2), Jnk (SP), and Erk1/2 (PD) while inhibitors of NFκB (PDTC) and p38 (SB) did not, suggesting that Src and Jnk are upstream of Erk1/2 in MNs. Similarly, sCD13-mediated NFκB phosphorylation in MNs was inhibited by the inhibitors of Src, Jnk, and Erk1/2 but not by SB, an inhibitor of p38 (Figure 3H).
sCD13 increases inflammatory cytokine expression in RA FLS
FLS are an important source of pro-inflammatory cytokines in RA. To determine the effect of sCD13 on cytokine expression in RA FLS, cells were incubated in serum free RPMI and stimulated for 3 hours to test mRNA expression and 24 hours for protein levels. MCP-1/CCL2 and IL-6 mRNA were significantly induced by sCD13 (Fig 4A and B). ELISAs were performed on conditioned media for MCP-1/CCL2, IL-6, IL-1β, and tumor necrosis factor-α (TNF-α). The levels of MCP-1/CCL2 and IL-6 in RA FLS supernatants were significantly higher after stimulation with sCD13 compared to non-stimulated FLS (Fig 4C and D). No significant changes were found in IL-1β or TNF-α at either the mRNA or protein levels (data not shown).
Fig. 4). sCD13 increases cytokine secretion in RA FLS.

A-D) We also determined the contribution of sCD13 in cytokine secretion at the mRNA and protein levels by stimulating RA FLS with sCD13. The levels of MCP-1/CCL2 and IL-6, but not IL-8/CXCL8 (data not shown), were significantly higher in RA FLS stimulated with sCD13 compared to control at mRNA and protein levels.
Neutralizing antibody against CD13 inhibits secretion of proinflammatory cytokines from RA ST organ cultures
The results shown above, together with our previously published data (9, 12), indicate that sCD13 exerts pro-inflammatory effects on multiple cell lineages that are key constituents of RA synovium. The organ culture system retains the interactions of the various cell types found in RA synovium, including RA FLS, MNs/macrophage, T and B lymphocytes, dendritic cells, ECs, and other cell populations (26). We therefore evaluated the contribution of CD13 in RA ST secretion of inflammatory cytokines by performing ex vivo assays using ST obtained from RA patients cultured with neutralizing antibodies against CD13.
RA ST samples cultured in media containing WM15, an anti-CD13 monoclonal antibody, showed significantly decreased expression of MCP-1/CCL2, IL-6, and IL-8/CXCL8 (Fig. 5A, B, and C). A positive control, dexamethasone, also significantly inhibited expression of these mediators. Another anti-CD13 monoclonal antibody, 1D7, produced no significant change in relative expression of MCP-1/CCL2 or IL-6 and led to a more moderate, while still significant, decrease in IL-8 expression.
Fig. 5). A neutralizing anti-CD13 antibody decreases the production of pro-inflammatory cytokines in ex vivo ST organ cultures.

A-C) Synovial tissue biopsies from RA patients were dissected into small pieces of approximately 2–3 mm in size and each cultured in 1 ml CMRL media with 10% FBS for 24 hours in individual wells of 12-well plates. For blocking studies each piece serves as its own control. Medium harvested from the first 24 hours of culture provides the baseline cytokine production for that piece of tissue. Fresh medium was added with or without anti-CD13 monoclonal antibodies (WM 15 and 1D7, 25ug/ml) and the conditioned media was collected after an additional 48 hours. As a positive control for inhibition of cytokines, some tissue pieces were cultured in dexamethasone (10 μg/ml). ELISAs were performed on baseline and organ cultures supernatants incubated with antibodies and dexamethasone to quantify MCP-1/CCL2, IL-6, and IL-8/CXCL8 expression. The ratio of cytokine at 72 hours/24 hours was used as an index to assess treatment efficacy. Error bars are SEM, and *p<0.05 was considered significant. n ≥ number of replicates in each group.
sCD13 contributes to acute inflammatory arthritis by increasing MN/macrophage infiltrates and increasing the concentration of proinflammatory cytokines MCP-1/CCL2, IL-6, and IL-1β
The organ culture results suggest that CD13 assumes a prominent role in the induction and/or maintenance of synovial inflammation in vivo in RA. We next sought to determine whether sCD13 itself could be directly arthritogenic. Indeed, mouse knees injected with sCD13 exhibited marked swelling and a significant increase in knee circumferences after 24 hours (Fig 6A and B). H&E staining of cryosections of mouse knees injected with sCD13 exhibited increased leukocyte ingress and inflammation compared to PBS injected mouse knee (Figure 6C). F4/80 staining showed a marked increase in the number of MNs/macrophages in mouse knee sections injected with sCD13 compared to controls (Fig 6D). To examine the expression of cytokines in sCD13-injected mouse knees, we performed ELISAs with mouse knee homogenates. MCP-1/CCL2, IL-6, and IL-1β were significantly higher in sCD13-injected knee homogenates compared to PBS (p≤0.01, Fig 6E–G). We did not find an increase in TNF-α in mouse knee homogenates injected with sCD13 (data not shown).
Fig. 6). sCD13 contributes to acute inflammatory arthritis by increasing MN influx, MCP-1/CCL2, IL-6, and IL-1β.

A-B) Mouse knees injected with sCD13 exhibited significant increase in knee circumferences after 24 hours compared to control. C) Mouse knees were cryosectioned after 2 weeks of decalcification. H&E staining exhibits a marked increase in inflammatory cells and disruption of the synovial lining (L) and sublining (SL) in mouse knees injected with sCD13 within 24 hours compared to PBS injected mouse knees. (Magnification 100X). D) sCD13-injected mouse knees show marked increase in F4/80 staining (Red color), a MN/macrophage marker, compared to controls. One of the representative sections from each group is shown. (Magnification 100X). E-G) sCD13-injected mouse knee homogenates showed significantly higher MCP-1/CCL2, IL-6, and IL-1β by ELISAs compared to PBS (*p≤0.01). We did not find an increase in TNF-α in mouse knee homogenates injected with CD13 (Data not shown). n=number of mouse knees in each group. Data represent the mean ± SEM and *p<0.05 was considered significant.
Discussion
Angiogenesis and MN recruitment into the sites of inflammation are critical steps involved in RA pathogenesis. MN ingress into the inflammatory sites is mediated by cytokines and growth factors, such as, TNF-α, MCP-1/CCL2, transforming growth factor-β (TGF-β), CCL5, vascular endothelial growth factor (VEGF), bFGF, IL-1β, and IL-8 (1, 3, 5, 29, 30). Once MNs are recruited into the synovium, they secrete proinflammatory and proangiogenic factors which results in proliferation of the ST and leads to persistence of inflammation in RA. Proangiogenic factors induce angiogenesis, one of the earliest pathological findings in RA. Increased angiogenesis promotes ingress of inflammatory cells at the inflammatory sites (4, 5, 31, 32). In this way, a vicious cycle is formed which results in persistence of the inflammatory response in RA. Targeting angiogenesis is one of the potential alternatives to treat RA, as angiogenesis contributes to pannus development, proliferation, and perpetuation of inflammation. We have previously shown that CD13 is present in soluble form in RA SFs and induces RA FLS proliferation and Tck cell migration in vitro (9, 12). CD13 expression was identified on newly forming capillaries in solid tumors and in RA ST blood vessels, suggesting a contribution of CD13 in angiogenesis (12, 33). CD13 is highly expressed by tumor cells, RA FLS, MNs, ECs, and mesenchymal stem cells(7, 8). It is upregulated on MNs, ECs and FLS in response to lipopolysaccharide (LPS), TGF-β, FBS, bFGF, and VEGF, and IL-17(12, 34–38). Cross-linking of cell surface CD13 with anti-CD13 antibodies induces the phosphorylation of mitogen-activated protein kinases (MAPK) Erk1/2, JNK, and P38 in MNs(7, 14). In the present study, we determined the role of sCD13 in angiogenesis, MN migration, signaling mechanisms in MN migration, phosphorylation of signaling molecules in MN and FLS, and cytokine production in vitro, ex vivo and in vivo.
It has previously been shown that CD13 contributes to migration of various cell types, phagocytosis, and angiogenesis (39–42). There are drawbacks, however, with many of the prior studies of CD13 involving angiogenesis, proliferation, and migration. Most of the above studies were performed with chemical inhibitors against CD13 instead of using sCD13 itself as a stimulus. Data from other groups indicate that the chemical inhibitors, bestatin and actinonin (or occasionally other chemical inhibitors), can lead to apoptosis and could therefore introduce confounding factors (43, 44). We performed all the functional cellular assays using sCD13 or anti-CD13 antibodies rather than using chemical inhibitors against CD13. Our data supports the suggestion that sCD13 is a potent angiogenic factor, as we have found that sCD13 induces EC chemotaxis, tube formation, and angiogenesis in the Matrigel plug angiogenesis assay, a murine model of angiogenesis in vivo (Figures 1A–G).
We also determined the role of signaling molecules in sCD13-mediated EC tube formation and found that the inhibitors of NFκB, Jnk, Erk1/2, and GPCR significantly decreased sCD13-induced tube formation. An inhibitor of p38 (SB) did not reduce EC tube formation, suggesting that p38 is not involved in sCD13-stimulated angiogenesis in vitro (Figures 1D and E). Previous reports have suggested that CD13 contributes to migration of various cell types, phagocytosis, and angiogenesis(39–42, 45). However, most of these studies did not use sCD13 as an agonist, and could not distinguish the roles of cell surface versus cleaved CD13. Moreover, the chemical inhibitors that were used may have other off-target effects(43, 44).
We previously showed that CD13 is involved in the growth and migration of RA FLS, and chemotaxis of Tcks (9, 12, 46). We found here that CD13 also induces MN migration in a dose-dependent manner with maximum effect at 500 ng/ml, the concentration of soluble CD13 present in RA SFs (9, 12, 46). Work ion other systems indicates that CD13 could induce influx of specific subsets of MNs into inflammatory sites. (41, 42). A number of studies have shown that cross-linking of membrane bound CD13 is required for MN ingress (14, 36). In contrast, this is the first report to show that sCD13 is a potent chemotactic factor for MN migration without a requirement for cross-linking of membrane bound CD13.
After finding a role for CD13 in MN migration, we assessed engagement of signaling molecules in this process. Inhibitors of Jnk, Src, NFκB, and GPCR significantly reduced MN migration, suggesting roles for these molecules in sCD13-mediated MN migration. However, inhibitors of Erk1/2 and p38 did not inhibit normal human MN migration in response to sCD13. The finding in this study that sCD13 induces MN migration via GPCRs reinforces our previous data suggesting that sCD13-induced Tck migration is mediated via GPCRs (12). U937 cell migration was inhibited by all the inhibitors of signaling molecules except SB, a p38 inhibitor. The differences between MN and U937 cells may reflect the transformed state of the U937 line.
cAMP is the key mediator of downstream signals initiated by GPCRs via activation of adenylate cyclase. After finding that GPCRs are involved in sCD13-stimulated MN migration, we examined sCD13 as a possible inducer of cAMP in MNs. Our results demonstrate that cAMP was significantly higher in MNs stimulated with sCD13 compared to nonstimulated cells (Fig 2F).
Several studies have identified sCD13 in human sera, tumor effusions, RA synovium, and bronchoalveolar lavage fluids from scleroderma, Sjogren’s syndrome, and myositis patients ((47, 48). CD13 is an ectoenzyme and many studies have found that the enzymatic activity of CD13 contributes to its multiple functions (49–51). We were interested to determine whether sCD13 mediates its effects independent of its enzymatic activity. An enzymatically inactive mutant CD13 was generated and used in a number of functional assays (Fig 3). The results clearly show that sCD13 contributes to MN chemotaxis and increased phosphorylation of signaling molecules independent of its enzymatic activity, as mutant CD13 increased MN migration and phosphorylation of signaling molecules in MNs comparably to WT CD13 (Fig 3B–C).
MNs/macrophages are recruited into STs and secrete pro-inflammatory and proangiogenic cytokines which promote the proliferation of ST cells and more MN infiltration (1, 6). We examined the effect of CD13 in MN migration in RA by performing normal human MN chemotaxis with CD13-depleted and sham-depleted RA SFs. We found CD13-depleted RA SFs resulted in less MN migration compared to sham-depleted RA SFs, pointing out the importance of CD13 in persistence of inflammation in RA (Fig 3B).
Cross-linking of membrane bound CD13 induces the phosphorylation of mitogen-activated protein kinases (MAPK) Erk1/2, JNK, and P38 in MNs (7, 14). We found that sCD13 induced phosphorylation of Erk1/2, Src, and NFκB in RA FLS without a requirement for cross-linking of membrane bound CD13 for phosphorylation of signaling molecules in RA FLS (Fig 3D–E). GPCRs engaged by sCD13 are upstream of Erk1/2, and Src, as an inhibitor of GPCR, PT, inhibited the phosphorylation of these signalling molecules in RA FLS (Fig 3F). We also determined the phosphorylation of signalling molecules in normal human MNs upon stimulation with sCD13 in the presence or absence of chemical signalling inhibitors. sCD13 increased phosphorylation of Erk1/2, Src, and NFκB in MNs. The increased phosphorylation of Erk1/2 was inhibited by the inhibitors of Src and Jnk, suggesting that Src and Jnk are upstream of Erk1/2 in sCD13-induced phosphorylation. sCD13-stimulated phosphorylation of NFκB in MNs was decreased by the inhibitors of Src, Jnk, and NFκB (Fig 3H). We also performed densitometry with all the Western blots as shown in supplementary data (Fig 1 supplementary). All these effects of sCD13 were found without cross-linking of CD13.
Villasenor-Cardoso et. al found that CD13, with or without cross-linking, does not induce IL-6, IL-10, IL-12, and TNFα secretion in human monocyte-derived dendritic cells or macrophages (39). In contrast to the above study, Santos et. al. showed that crosslinking of CD13 plays an important role in proinflammatory cytokine secretion by MNs (14). In this study, we examined proinflammatory cytokine secretion by RA FLS upon stimulation with sCD13. We found that sCD13 has a direct effect on cytokine secretion by RA FLS. Reinhold et. al. found that probestatin and actinonin (two inhibitors of enzymatic activity of CD13) suppress IL-1β, IL-2, and TGF-β production in PBMCs and human T cells, suggesting the involvement of CD13 in cytokine expression (51). Bestatin, another CD13 inhibitor, suppressed IL-6 and IL-8 production in human MNs and induced IL-10 secretion (50). The above studies were performed incubating PBMCs with inhibitors which block the enzymatic activity of CD13 instead of directly stimulating cells with sCD13.
Multiple cell types contribute to the pathologic behavior of RA ST including FLS, MNs/macrophages, B lymphocytes, plasma cells, mast cells, endothelial cells, osteoclasts, and chondrocytes. These various cell types are involved in persistence of inflammatory response in RA by secreting inflammatory mediators and through direct cell-cell interactions mediated by cell surface receptors and their membrane-anchored ligands (2). We performed an RA ST organ ex vivo assay with or without a neutralizing antibody against CD13 to evaluate the secretion of proinflammatory cytokines. In this assay, cell-cell interaction contributes to persistence of inflammation and cytokine secretion in inflammatory conditions such as, RA. WM15, a neutralizing antibody against CD13, significantly inhibited the secretion of key proinflammatory mediators, such as, MCP-1/CCL2, IL-6, and IL-8/CXCL8, which drive the inflammatory response in RA. This suggests that CD13 is a key player involved in cytokine secretion in RA.
sCD13 is present in very high concentrations in human sera, tumor effusions, RA synovium including various autoimmune diseases and is secreted and/or shed by RA FLS, MNs, ECs, tumor cells and mesenchymal stem cells in response to IL-6, TNF-α, IL-1β, LPS, TGF-β, FBS, bFGF, VEGF, and IL-17 (7, 8, 11, 12, 34–36, 38, 47, 48, 50, 51). We and others have demonstrated that sCD13 contributes to the production of proinflammatory cytokines, MN-EC adhesion, migration of cytokine-activated T cells, and MN ingress in a murine model of peritonitis (7, 10, 12–14, 52, 53). These studies point out the importance of CD13 in inflammatory arthritis and potentially other inflammatory or autoimmune diseases. sCD13-driven secretion of cytokines and MN ingress results in initiation and persistence of inflammation. These secreted cytokines and MN ingress cause more sCD13 production by various cell types. In this way, a vicious cycle develops which leads in chronic inflammation, suggesting a pivotal role of sCD13.
We also demonstrated a role for sCD13 in inflammatory arthritis by injecting CD13 into mouse knees and found that sCD13 induced significant inflammation, swelling, and MN recruitment compared to control injected knees (Fig 6A–D). Winnicka et. al. found that CD13 is required for adhesion of MNs to ECs, an essential step for the influx of MNs into inflammatory sites. These authors also found no differences in MN recruitment in thioglycollate-induced peritonitis and collagen antibody-induced arthritis (CAIA) in CD13 knockout compared to wild type mice (54). However, in CAIA, T cells and monocytes may not be critical in the development of arthritis. Our data clearly demonstrate the wide-ranging proinflammatory and arthritogenic properties of sCD13, and imply a pivotal role for sCD13 in RA.
Supplementary Material
Key Points.
Soluble (s)CD13 is a potent angiogenic factor in vitro and in vivo.
sCD13 induces acute inflammatory arthritis in mice and induces MN migration via GPCR.
sCD13 increases cytokine secretion and phosphorylation of signaling molecules.
Grants support
This study was financially supported by NIH 1-R21-AR-072931–01, MICHR (UL1 TR002240), Autoimmunity Centers of Excellence (ACE) from NIH, The Joint Doctoral Program offered by the China Scholarship Council (CSC).
References
- 1.Feldmann M, Maini RN, Bondeson J, Taylor P, Foxwell BM, and Brennan FM. 2001. Cytokine blockade in rheumatoid arthritis. Adv Exp Med Biol 490: 119–127. [DOI] [PubMed] [Google Scholar]
- 2.Fox DA, Gizinski A, Morgan R, and Lundy SK. 2010. Cell-cell interactions in rheumatoid arthritis synovium. Rheum Dis Clin North Am 36: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szekanecz Z, and Koch AE. 2007. Macrophages and their products in rheumatoid arthritis. Curr Opin Rheumatol 19: 289–295. [DOI] [PubMed] [Google Scholar]
- 4.Koch AE 2000. The role of angiogenesis in rheumatoid arthritis: recent developments. Ann Rheum Dis 59 Suppl 1: i65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Folkman J 1995. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1: 27–31. [DOI] [PubMed] [Google Scholar]
- 6.Muller-Ladner U, Pap T, Gay RE, Neidhart M, and Gay S. 2005. Mechanisms of disease: the molecular and cellular basis of joint destruction in rheumatoid arthritis. Nat Clin Pract Rheumatol 1: 102–110. [DOI] [PubMed] [Google Scholar]
- 7.Mina-Osorio P, Winnicka B, O’Conor C, Grant CL, Vogel LK, Rodriguez-Pinto D, Holmes KV, Ortega E, and Shapiro LH. 2008. CD13 is a novel mediator of monocytic/endothelial cell adhesion. J Leukoc Biol 84: 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luan Y, and Xu W. 2007. The structure and main functions of aminopeptidase N. Curr Med Chem 14: 639–647. [DOI] [PubMed] [Google Scholar]
- 9.Morgan RL, Behbahani-Nejad N, Endres J, Amin MA, Lepore NJ, Du Y, Urquhart A, Chung KC, and Fox DA. 2016. Localization, Shedding, Regulation and Function of Aminopeptidase N/CD13 on Fibroblast like Synoviocytes. PloS one 11: e0162008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimizu T, Tani K, Hase K, Ogawa H, Huang L, Shinomiya F, and Sone S. 2002. CD13/aminopeptidase N-induced lymphocyte involvement in inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum 46: 2330–2338. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe Y, Ito K, Iwaki-Egawa S, Yamaguchi R, and Fujimoto Y. 1998. Aminopeptidase N in sera of healthy subjects is a different N-terminal processed derivative from the one obtained from maternal serum. Mol Genet Metab 63: 289–294. [DOI] [PubMed] [Google Scholar]
- 12.Morgan R, Endres J, Behbahani-Nejad N, Phillips K, Ruth JH, Friday SC, Edhayan G, Lanigan T, Urquhart A, Chung KC, and Fox DA. 2015. Expression and function of aminopeptidase N/CD13 produced by fibroblast-like synoviocytes in rheumatoid arthritis: role of CD13 in chemotaxis of cytokine-activated T cells independent of enzymatic activity. Arthritis Rheumatol 67: 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh M, Gerber C, Rahman MM, Vernier KM, Pereira FE, Subramani J, Caromile LA, and Shapiro LH. 2014. Molecular mechanisms regulating CD13-mediated adhesion. Immunology 142: 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santos AN, Langner J, Herrmann M, and Riemann D. 2000. Aminopeptidase N/CD13 is directly linked to signal transduction pathways in monocytes. Cell Immunol 201: 22–32. [DOI] [PubMed] [Google Scholar]
- 15.Amin MA, Mansfield PJ, Pakozdi A, Campbell PL, Ahmed S, Martinez RJ, and Koch AE. 2007. Interleukin-18 induces angiogenic factors in rheumatoid arthritis synovial tissue fibroblasts via distinct signaling pathways. Arthritis Rheum 56: 1787–1797. [DOI] [PubMed] [Google Scholar]
- 16.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, and et al. 1988. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31: 315–324. [DOI] [PubMed] [Google Scholar]
- 17.O’Brien MJ, Shu Q, Stinson WA, Tsou PS, Ruth JH, Isozaki T, Campbell PL, Ohara RA, Koch AE, Fox DA, and Amin MA. 2018. A unique role for galectin-9 in angiogenesis and inflammatory arthritis. Arthritis Res Ther 20: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amin MA, Campbell PL, Ruth JH, Isozaki T, Rabquer BJ, Alex Stinson W, O’Brien M, Edhayan G, Ohara RA, Vargo J, Domino SE, and Koch AE. 2015. A key role for Fut1-regulated angiogenesis and ICAM-1 expression in K/BxN arthritis. Ann Rheum Dis 74: 1459–1466. [DOI] [PubMed] [Google Scholar]
- 19.Amin MA, Volpert OV, Woods JM, Kumar P, Harlow LA, and Koch AE. 2003. Migration inhibitory factor mediates angiogenesis via mitogen-activated protein kinase and phosphatidylinositol kinase. Circ Res 93: 321–329. [DOI] [PubMed] [Google Scholar]
- 20.Tsou PS, Ruth JH, Campbell PL, Isozaki T, Lee S, Marotte H, Domino SE, Koch AE, and Amin MA. 2013. A novel role for inducible Fut2 in angiogenesis. Angiogenesis 16: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amin MA, Ruth JH, Haas CS, Pakozdi A, Mansfield PJ, Haghshenas J, and Koch AE. 2008. H-2g, a glucose analog of blood group H antigen, mediates mononuclear cell recruitment via Src and phosphatidylinositol 3-kinase pathways. Arthritis Rheum 58: 689–695. [DOI] [PubMed] [Google Scholar]
- 22.Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, and Koch AE. 2006. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFkappaB. Blood 107: 2252–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amin MA, Rabquer BJ, Mansfield PJ, Ruth JH, Marotte H, Haas CS, Reamer EN, and Koch AE. 2010. Interleukin 18 induces angiogenesis in vitro and in vivo via Src and Jnk kinases. Ann Rheum Dis 69: 2204–2212. [DOI] [PubMed] [Google Scholar]
- 24.Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, and Vogel SN. 2010. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol 3: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marotte H, Tsou PS, Rabquer BJ, Pinney AJ, Fedorova T, Lalwani N, and Koch AE. 2011. Blocking of interferon regulatory factor 1 reduces tumor necrosis factor alpha-induced interleukin-18 bioactivity in rheumatoid arthritis synovial fibroblasts by induction of interleukin-18 binding protein a: role of the nuclear interferon regulatory factor 1-NF-kappaB-c-jun complex. Arthritis Rheum 63: 3253–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friday SC, and Fox DA. 2016. Phospholipase D enzymes facilitate IL-17- and TNFalpha-induced expression of proinflammatory genes in rheumatoid arthritis synovial fibroblasts (RASF). Immunol Lett 174: 9–18. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida K, Korchynskyi O, Tak PP, Isozaki T, Ruth JH, Campbell PL, Baeten DL, Gerlag DM, Amin MA, and Koch AE. 2014. Citrullination of epithelial neutrophil-activating peptide 78/CXCL5 results in conversion from a non-monocyte-recruiting chemokine to a monocyte-recruiting chemokine. Arthritis Rheum 66: 2716–2727. [DOI] [PubMed] [Google Scholar]
- 28.Tsvetanova NG, and von Zastrow M. 2014. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10: 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Bandt M, Ben Mahdi MH, Ollivier V, Grossin M, Dupuis M, Gaudry M, Bohlen P, Lipson KE, Rice A, Wu Y, Gougerot-Pocidalo MA, and Pasquier C. 2003. Blockade of vascular endothelial growth factor receptor I (VEGF-RI), but not VEGF-RII, suppresses joint destruction in the K/BxN model of rheumatoid arthritis. J Immunol 171: 4853–4859. [DOI] [PubMed] [Google Scholar]
- 30.Clavel G, Valvason C, Yamaoka K, Lemeiter D, Laroche L, Boissier MC, and Bessis N. 2006. Relationship between angiogenesis and inflammation in experimental arthritis. Eur Cytokine Netw 17: 202–210. [PubMed] [Google Scholar]
- 31.Szekanecz Z, Besenyei T, Szentpetery A, and Koch AE. 2010. Angiogenesis and vasculogenesis in rheumatoid arthritis. Curr Opin Rheumatol 22: 299–306. [DOI] [PubMed] [Google Scholar]
- 32.Paleolog EM 1996. Angiogenesis: a critical process in the pathogenesis of RA--a role for VEGF? Br J Rheumatol 35: 917–919. [DOI] [PubMed] [Google Scholar]
- 33.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, and Ruoslahti E. 2000. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res 60: 722–727. [PMC free article] [PubMed] [Google Scholar]
- 34.Kehlen A, Langner J, and Riemann D. 2000. Transforming growth factor-beta increases the expression of aminopeptidase N/CD13 mRNA and protein in monocytes and monocytic cell lines. Adv Exp Med Biol 477: 49–56. [DOI] [PubMed] [Google Scholar]
- 35.Petrovic N, Bhagwat SV, Ratzan WJ, Ostrowski MC, and Shapiro LH. 2003. CD13/APN transcription is induced by RAS/MAPK-mediated phosphorylation of Ets-2 in activated endothelial cells. J Biol Chem 278: 49358–49368. [DOI] [PubMed] [Google Scholar]
- 36.Mina-Osorio P 2008. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med 14: 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mor-Vaknin N, Saha A, Legendre M, Carmona-Rivera C, Amin MA, Rabquer BJ, Gonzales-Hernandez MJ, Jorns J, Mohan S, Yalavarthi S, Pai DA, Angevine K, Almburg SJ, Knight JS, Adams BS, Koch AE, Fox DA, Engelke DR, Kaplan MJ, and Markovitz DM. 2017. DEK-targeting DNA aptamers as therapeutics for inflammatory arthritis. Nat Commun 8: 14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fontijn D, Bosch LJ, Duyndam MC, van Berkel MP, Janmaat ML, and Boven E. 2009. Basic fibroblast growth factor-mediated overexpression of vascular endothelial growth factor in 1F6 human melanoma cells is regulated by activation of PI-3K and p38 MAPK. Cell Oncol 31: 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villasenor-Cardoso MI, Frausto-Del-Rio DA, and Ortega E. 2013. Aminopeptidase N (CD13) is involved in phagocytic processes in human dendritic cells and macrophages. Biomed Res Int 2013: 562984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mina-Osorio P, and Ortega E. 2005. Aminopeptidase N (CD13) functionally interacts with FcgammaRs in human monocytes. J Leukoc Biol 77: 1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahman MM, Ghosh M, Subramani J, Fong GH, Carlson ME, and Shapiro LH. 2014. CD13 regulates anchorage and differentiation of the skeletal muscle satellite stem cell population in ischemic injury. Stem cells 32: 1564–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereira FE, Cronin C, Ghosh M, Zhou SY, Agosto M, Subramani J, Wang R, Shen JB, Schacke W, Liang B, Yang TH, McAulliffe B, Liang BT, and Shapiro LH. 2013. CD13 is essential for inflammatory trafficking and infarct healing following permanent coronary artery occlusion in mice. Cardiovasc Res 100: 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grujic M, Zavasnik-Bergant T, Pejler G, and Renko M. 2005. Actinonin induces apoptosis in U937 leukemia cells. Cancer Lett 223: 211–218. [DOI] [PubMed] [Google Scholar]
- 44.Sekine K, Fujii H, and Abe F. 1999. Induction of apoptosis by bestatin (ubenimex) in human leukemic cell lines. Leukemia 13: 729–734. [DOI] [PubMed] [Google Scholar]
- 45.Riemann D, Kehlen A, and Langner J. 1999. CD13--not just a marker in leukemia typing. Immunol Today 20: 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai A, Ghaffari A, and Ghahary A. 2010. Inhibitory effect of anti-aminopeptidase N/CD13 antibodies on fibroblast migration. Mol Cell Biochem 343: 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Hensbergen Y, Broxterman HJ, Hanemaaijer R, Jorna AS, van Lent NA, Verheul HM, Pinedo HM, and Hoekman K. 2002. Soluble aminopeptidase N/CD13 in malignant and nonmalignant effusions and intratumoral fluid. Clin Cancer Res 8: 3747–3754. [PubMed] [Google Scholar]
- 48.Dan H, Tani K, Hase K, Shimizu T, Tamiya H, Biraa Y, Huang L, Yanagawa H, and Sone S. 2003. CD13/aminopeptidase N in collagen vascular diseases. Rheumatol Int 23: 271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petrovic N, Schacke W, Gahagan JR, O’Conor CA, Winnicka B, Conway RE, Mina-Osorio P, and Shapiro LH. 2007. CD13/APN regulates endothelial invasion and filopodia formation. Blood 110: 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lkhagvaa B, Tani K, Sato K, Toyoda Y, Suzuka C, and Sone S. 2008. Bestatin, an inhibitor for aminopeptidases, modulates the production of cytokines and chemokines by activated monocytes and macrophages. Cytokine 44: 386–391. [DOI] [PubMed] [Google Scholar]
- 51.Reinhold D, Biton A, Goihl A, Pieper S, Lendeckel U, Faust J, Neubert K, Bank U, Tager M, Ansorge S, and Brocke S. 2007. Dual inhibition of dipeptidyl peptidase IV and aminopeptidase N suppresses inflammatory immune responses. Ann N Y Acad Sci 1110: 402–409. [DOI] [PubMed] [Google Scholar]
- 52.Mina-Osorio P, Shapiro LH, and Ortega E. 2006. CD13 in cell adhesion: aminopeptidase N (CD13) mediates homotypic aggregation of monocytic cells. J Leukoc Biol 79: 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramani J, Ghosh M, Rahman MM, Caromile LA, Gerber C, Rezaul K, Han DK, and Shapiro LH. 2013. Tyrosine phosphorylation of CD13 regulates inflammatory cell-cell adhesion and monocyte trafficking. J Immunol 191: 3905–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winnicka B, O’Conor C, Schacke W, Vernier K, Grant CL, Fenteany FH, Pereira FE, Liang B, Kaur A, Zhao R, Montrose DC, Rosenberg DW, Aguila HL, and Shapiro LH. 2010. CD13 is dispensable for normal hematopoiesis and myeloid cell functions in the mouse. J Leukoc Biol 88: 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.