

Abstract

Mesoporous silica nanoparticles (MSNs) are considered as promising next-generation nanocarriers for health-related applications. However, their effectiveness mostly relies on their efficient and surface-specific functionalization. In this contribution, we explored different strategies for the rational multistep synthesis of functional MCM-48-type MSNs with selectively created active inner and/or external surfaces. Functional groups were first installed using a combination of (delayed) co-condensation and post-grafting procedures. Both amine [(3-aminopropyl)triethoxysilane (APTS)] and thiol [(3-mercaptopropyl)trimethoxysilane (MPTS)] silanes were used, in various addition sequences. Following this, the different platforms were further functionalized with polyethylene glycol and/or with a pro-chelate ligand used as a magnetic resonance imaging contrast agent (diethylenetriaminepentaacetic acid chelates) and/or loaded with quercetin and/or grafted with an organic dye (rhodamine). The efficiency of the multiple grafting strategies and the effects on the MSN carrier properties are presented. Finally, the colloidal stability of the different systems was evaluated in physiological media, and preliminary tests were performed to verify their drug release capability. The use of MPTS appeared beneficial when compared to APTS in delayed co-condensation procedures to preserve both selective distribution of the functional groups, reactive functionality, and pore ordering. Our results provide in-depth insights into the efficient design of (multi)functional MSNs and especially on the crucial role played by the sequence of step-by-step functionalization methods aiming to produce multipurpose and stable bioplatforms.
Introduction
Mesoporous silica nanoparticles (MSNs) are promising candidates for biomedicine-related applications,1−10 and they are particularly used in the area of targeted and controlled drug delivery,11−16 as well as in medical imaging/diagnosis17−20 or in a combination of both, that is, theranostic systems.21−25 The growing interest for MSNs is directly related to their porosity features. Indeed, these materials are highly porous, with a specific surface area of about 1000–1500 m2 g–1, mesopores of size usually ranging from 2 to 20 nm and pore volume ranging from 0.8 to 1.5 cm3 g–1.26−28 In addition, MSNs are seen as versatile materials because they can be synthesized with well-defined particle size and shape as well as tunable pore network characteristics.29,30 Currently, data regarding the potential adverse effects of MSNs on overall human health on the mid-term and long term are still missing, that is, no clinical trials have been performed yet.31,32 However, amorphous silica is so far considered safe by the Food and Drug Administration and has been extensively used in many of our daily-based products or as excipient in pharmaceutical formulations for many years.33 Owing to their porosity, their relative stability in aqueous media, and degradation in biological media, MSNs became of interest as drug reservoir and bioimaging platforms. A large variety of therapeutic compounds can be accommodated inside their pore network, and many examples using such particles as hosts to improve the administration of simple or more complex therapeutic compounds have been reported.4,34−37 However, in most cases, functionalization of the silica surfaces is required to integrate the desired “biological functions”, for example, enhanced colloidal stability in blood or physiological fluids of interest, increased blood circulation time, hemocompatibility, targeted and/or stimuli-responsive release, imaging capabilities, and so forth.4,38,39 The most common design of “functional MSNs” usually involves the functionalization of two types of “surfaces”, (1) an external surface functionalized by stabilizing groups such as polyethylene glycol (PEG) and/or biotargeting groups such as specific ligands or antibodies40,41 and (2) an internal surface, functionalized or not in order to favor the sequestration and/or release of therapeutic species such as drugs, nutraceuticals, metal complexes, therapeutic peptides, proteins, and so on. From the synthesis point of view, the major issues in such a design are as follows: (1) the difficult distinction between the outer/external and inner/internal pore surfaces of the particles,42 (2) the interference between the different functionalities, (3) the potential pore-blocking and steric hindrance issues resulting from the different grafting and loading protocols,43 and (4) the control of the amount of each guest molecule being grafted/loaded.44 In addition, the multifunctionalization of MSNs should be performed in a minimal number of synthesis steps in order to obtain an easily scalable system and to minimize the costs, with low amounts of harmful solvents and short preparation time. Therefore, while the addition of functionalities is a necessity for the efficient biomedical application of MSNs, preserving as much as possible the porosity features during the early stages of the process appears to be critical and cannot be disregarded.
These issues and the selective surface functionalization of MSNs are challenges that have been faced by researchers over the last recent years, and many strategies have been proposed. The “classical” post-grafting (PG) methods, while being convenient, can result in a nonuniform and poorly controlled silane distribution and usually includes several steps.45−47 Another way to discriminate the different outer/inner surfaces is to use impregnation techniques such as the incipient wetness.39 This method, using an amount of solvent equal to the pore volume of the MSNs, is known to selectively fill the pores by capillarity, resulting in a mostly inner surface functionalization. However, this technique is potentially time-consuming and is mainly used to load inorganics,19,39 even if few examples using organosilanes were reported.48 The last most common way of introducing functionalities is to add the organosilanes during the synthesis of the MSNs.49 This method, known as the co-condensation (CC) or one-pot synthesis, normally leads to a homogenous distribution of the functionalities inside the mesoporous materials. However, control of the spatial distribution is difficult, and some functional groups end up inactive being embedded within the silica framework (the pore walls) with no access to the surface.42,50−53 To overcome these limitations, Kecht et al. developed the delayed CC (DCC) method.54 This strategy is based on the delayed addition of silanes as compared to the main silicon source, to initially allow the formation of the MSNs and then to functionalize the different surfaces. Indeed, if the silanes are added quickly after the main silicon source [e.g., tetraethylorthosilicate (TEOS)], one may obtain a material with functionalities mainly located in the core. On the contrary, if the silanes are introduced after a longer time, the resultant active groups should be mainly grafted on the external surface. This method usually limits the entrapment of functionalities within the pore walls, especially when applied to MSN synthesis with rapid formation kinetics such as nanosized MCM-41 and MCM-48.55 The main advantage of this technique is the possibility to incorporate the desired organosilanes at different spatial locations by simply controlling the addition sequences. For instance, this strategy was successfully applied to functionalize MSNs with polymer moieties56−58 or to produce MSNs exhibiting a pH-responsive gating effect for controlled drug delivery.59 In the vast majority of cases, (3-aminopropyl)triethoxysilane (APTS) is used to first functionalize the MSNs. Indeed, this silane allows functionalizing the surfaces with amine groups which are key active sites for bioconjugation reactions.60−63 Moreover, it is relatively easy to work with APTS, that is, although it should be handled under inert atmosphere, it is not excessively sensitive to humidity and easily accessible. However, the possibility of further reactions of amine-functionalized MSNs was found to be dependent on the functionalization procedure.20,23,24 Co-condensed materials were systematically found to have less accessible amine sites as compared to materials functionalized by other techniques. In the same way, post-functionalization is commonly associated with pore blocking/diffusion issues in the resulting hybrids.64 It is thus proposed to work with other functional silanes such as (3-mercaptopropyl)trimethoxysilane (MPTS). MPTS has been used with ordered mesoporous silica65,66 and like the amine groups, thiols enable also further biofunctionalization and conjugation reactions. Furthermore, among the polymers that were utilized to improve both colloidal stability and the stealth properties of MSNs, PEG is most widely used. It has a high affinity for water and owing to its numerous ether groups, it binds to a large amount of water molecules adopting a “cloud-like” conformation that acts as a water barrier and protects the particle surface.67 Moreover, PEG was shown to improve the blood circulation half-life time and the biocompatibility of the carriers which are both critical parameters in the field of healthcare.68 Nevertheless, the challenges reside in achieving optimal particle modification with given functional groups and placing them in specific locations inside the structure, without altering/losing the well-defined porosity features of the MSNs.
To this aim, we initiated a comprehensive investigation of the modification procedures of MSNs, comparing different strategies, in order to ultimately establish robust and tailor-made functionalization protocols. In this study, MCM-48-type MSNs were chosen as a typical model of mesoporous silica particles. These particles were then modified with APTS and/or MPTS accompanied by PEGylation and/or grafting of the diethylenetriaminepentaacetic acid (DTPA) pro-chelating ligand. Addition sequences are varied and the advantages/limitations of (delayed) CC and PG methods are critically discussed. The materials at different steps of the synthesis were characterized by a variety of methods, including N2 physisorption, transmission electron microscopy (TEM), solid-state nuclear magnetic resonance spectroscopy (ssNMR), low-angle powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), and elemental analysis (EA). In addition, the colloidal behavior of the functionalized MSNs was scrutinized in various pH buffers over a long period of time using dynamic light scattering (DLS). Preliminary drug release tests were also performed with the particles resulting from different protocols. Our results provide in-depth insights into the efficient design of (multi)functional MSNs and especially on the crucial role played by the arrangement of the step-by-step multifunctionalization strategy. Therefore, our findings offer flexible synthesis methods and recipes for tailor-made nanocarriers in a reproducible way, which is expected to be of prime importance regarding the ongoing development of next-generation smart theranostic (nano)platforms.21−25
Results and Discussion
The different functionalization pathways that were investigated in this study are summarized in Scheme 1. Easily reproducible pathways are highlighted in green, while the ones that led to difficulties in reproducibility or where the functionalization yield was quite low are highlighted as black routes. Finally, the ones that did not provide any satisfactory results are highlighted in red. Each of them includes not only different options of functionalization, such as PG, CC, and DCC, but also the grafting/loading of specific species of interest for biorelated applications in order to demonstrate the usefulness of the multifunctionalization method. In all cases, PEGylation was also performed. To obtain meaningful references regarding porosity and pore ordering features, both calcined and chemically extracted mesoporous MCM-48 nanoparticles were first synthesized.69 The characterization of the two samples is presented in Figures S1 and S2. For both materials, TEM images (Figure S1a–d) showed spherical particles of about 130–170 nm. DLS measurements (Figure S1c,f) revealed an average hydrodynamic particle diameter of 192 and 255 nm for calcined and extracted MCM-48 samples, respectively, in agreement with the TEM observations. No sign of major aggregation was found and one can observe the ordering of the pore structure in the TEM images. The cubic Ia3d symmetry of the mesopore network was further evidenced by low-angle XRD measurements, as presented in Figure S1b–e. N2 physisorption analyses (Figure S2) show for the two materials typical type IVb isotherms with a sharp pore condensation/evaporation step, being characteristic of high-quality mesoporous silicas with pores below 4 nm.70 The samples exhibit comparable high specific surface areas of about 1230 m2 g–1 (Table S1). Slightly larger nonlocal density functional theory (NLDFT) pore size and total pore volume were obtained for the extracted material (3.5 nm and 1.0 cm3 g–1) as compared to the calcined one (3.2 nm and 0.8 cm3 g–1) in accordance with the shrinkage occurring during the calcination process.71 All the obtained data are in good agreement with previous reports.15,16,19,69
Scheme 1. Multistep Functionalization Pathways of MSNs.

Amine/Thiol Functionalization
All the amine- and thiol-functionalized samples discussed here were synthesized at least in triplicates to ensure reliable and reproducible results. The C, H, N, and S contents from EA are compiled for all samples in Tables S2 and S3. Amine functionalities were added using a CC or a DCC strategy or via PG. The quantity of amine introduced via PG was varied between 4 and 12 mmol per gram of degassed MSN particles. The TGA data revealed no significant difference in the mass loss of the resulting NH2-MSNs, indicating that 4 mmol of APTS is already in condition of excess of the aminosilane for the grafting procedure (Figure S3). Using more APTS only resulted in unbounded APTS which was washed out during the washing procedures. The PG time dependence on the grafting efficiency was also evaluated as shown in Figure 1. In many reports, classic grafting procedures are carried overnight.72,73 However, in our case, no significant changes in the organic amount being grafted was observed after only half an hour of reaction. This result, being consistent with the relatively fast hydrolysis and condensation kinetics of the APTS in nonneutral medium,74 suggests that grafting procedures could be performed in a faster way and do not require overnight reaction. To compare our data with the literature,75 it was decided to use a PG time of 4 h for the following samples. The nitrogen loading of post-grafted APTS-MSNs after 4 h, obtained by EA, was 1.6 mmol g–1 (Table S3). Typical N2 physisorption isotherms at −196 °C with corresponding NLDFT pore size distribution (PSD) and TGA curves obtained for the calcined MSNs post-grafted with APTS are shown in Figure S4. A satisfactory agreement here between the quadruplicates was obtained with TGA and gas adsorption techniques, so it was decided to average the data for all tabular data presented in this study. For all NH2-MSNs synthesized via CC or DCC, the success of the functionalization was confirmed using solid-state 13C cross polarization (CP) NMR (Figure S5). TGA indicated a mass loss of 12 ± 2% for all samples pointing to a similar amount of grafted amine groups (Table S4). Also, as demonstrated in Figure 2a, APTS functionalization following CC and DCC pathways led to almost identical materials from a physisorption point of view. For both samples, a drastic decrease in the specific surface area (543 m2 g–1, 56% decrease) and the total pore volume (0.3 cm3 g–1, 69% decrease) was observed as compared to the pristine extracted MSNs (Table S1). The porosity features were slightly less affected when both the calcined and extracted MSNs were functionalized with APTS via PG. Indeed, a decrease of 34 and 32% and 54 and 54% for the specific surface area and total pore volume values was noted, respectively. The NLDFT pore size was reduced for all the samples. The variations in the NH2-MSNs obtained using different functionalization techniques may be explained by the effect of the aminosilane on the mesophase during the synthesis and aging processes. Indeed, as shown in Figure 2c, materials derived from the CC pathways, that is, CC and DCC, exhibit poorly resolved low-angle XRD patterns, being typical of samples with no or low pore ordering, whereas the patterns obtained for the post-grafted silicas are still commensurate with the cubic Ia3d symmetry (Figure 2c). Thus, it appears that the presence of the aminosilane destabilizes the synthesis, potentially because of a pH increase and, thus led to the formation of less porous materials without any particular pore organization. Even though it is known that CC often leads to less organized pore networks in the case of 2-D symmetries (e.g., MCM-41),76 this effect is even more pronounced for 3-D-based pore architectures. If ordering of the final particles is a prerequisite, the choice of aminosilanes for early functionalization through one-pot reaction might therefore not be so suitable, especially in the case of MCM-48-type MSNs. Also, even if the pore features and ordering are slightly better preserved for the post-grafted samples, there is still a dramatic decrease of the available pore volume and surface area of the MSNs, most likely originating from pore-blocking/hindering issues related with the grafting method.19 These results are in line with previous work on the accessibility and chemical reactivity of amine sites grafted on ordered mesoporous materials.26−28 In contrast, when MPTS was used during DCC, the resulting thiol-functionalized MSNs retained high porosity. As it can be seen on Figure 2a, a typical type IVb isotherm was obtained, being similar to the one of the pristine MCM-48 silica nanoparticles (NPs). The specific surface area was 1090 m2 g–1, the total pore volume was 0.86 cm3 g–1, and the NLDFT mode pore size was 3.4 nm (Table S1). This represents a reduction of only 11 and 14% for the specific surface area and total pore volume, respectively, as compared to the extracted MSNs. The mode pore size remains almost unaffected and the PSD stayed sharp and narrow (Figure 2b), being a good indication of the absence of any pore blocking. Grafting of MPTS was further confirmed using 13C solid-state NMR (Figure S5) and TGA (Table S4), showing that 12% of MPTS was grafted onto the MSNs. The SH-MSNs synthesized via DCC also showed a perfectly well-ordered pore structure (Figure 2c) with an Ia3d symmetry. Therefore, using MPTS is beneficial in the early functionalization step of the MSNs. Considering that a 10 min delay was applied before adding the MPTS during the DCC process, most of the thiol functionalities should be located mainly toward the external surface of the MSNs. This point is critical because thiol groups will only be used to further attach “external surface functions”, that is, PEG chains for colloidal stability and biocompatibility improvement, using PEG-maleimide for instance. However, “external” thiol groups may not be sufficient to provide a suitable precursor material that can be further poly-grafted with molecules of interest. Therefore, we investigated synthesis options to include both amine and thiol groups using APTS and MPTS, with the aim to preserve as much as possible the porosity features unaltered. These different synthesis pathways are shown in Scheme 1 and can be divided in four groups. In PI to PV (line 2 of Scheme 1), the synthesis design was to use MPTS and APTS together in DCC or to post-graft APTS onto extracted SH-MSNs-DCC. In PVI and PVII, co-condensed NH2-MSNs were further functionalized with MPTS using either DCC or PG, respectively. In PVIII, the material NH2-MSNs-DCC was post-grafted with MPTS. Finally, in PIX to PXI, NH2-MSNs-PG was further post-grafted with MPTS, before or after the rhodamine fluorophore loading (line 2 or 3 of Scheme 1).
Figure 1.
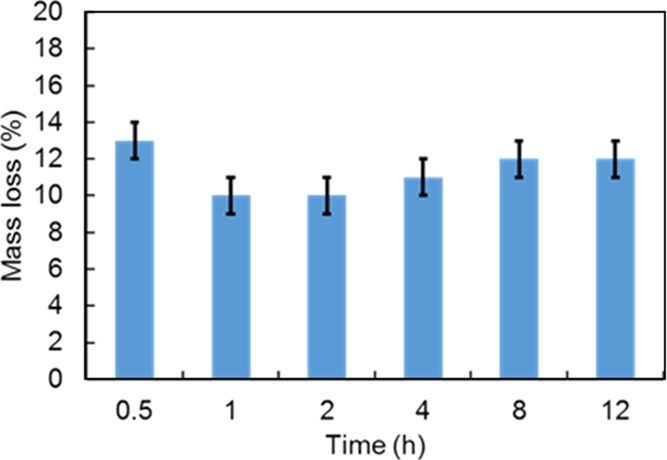
Amount of APTS grafted onto MSNs as a function of the reaction time.
Figure 2.
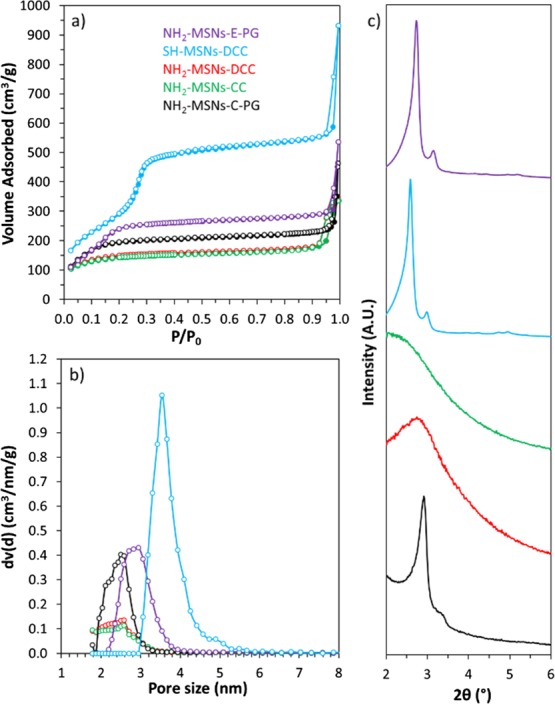
(a) N2 physisorption isotherms at −196 °C, (b) NLDFT PSD, and (c) low-angle XRD patterns of NH2-MSNs with PG performed on calcined particles (black), NH2-MSNs with PG performed on extracted particles (purple), NH2-MSNs-CC (green), NH2-MSNs-DCC (red), and SH-MSNs-DCC (blue).
TEM images of the samples are presented in Figure 3 (for magnified images, see Figure S6). The resulting N2 physisorption isotherms with corresponding NLDFT PSD and low-angle XRD patterns of all the bifunctional MSNs are presented in Figure 4 (full-scale isotherms are found in Figure S7). All the synthesis strategies yielded uniform nanospheres of about 130–190 nm, being similar to pristine MCM-48 MSNs, indicating that the addition of the two organosilanes via CC and/or DCC and/or PG strategies did not interfere much with the final shape and size of the silica particles. In all cases, the presence of both amine and thiol functionalities was also confirmed by 13C CP NMR as shown in Figure S5 and TGA (Table S4). Indeed, in a typical carbon NMR spectrum of MSNs functionalized with MPTS, one can note two resonances at around 9–10 and 27 ppm attributed to the carbons of the silane propyl chain. More specially, the signal at 27 ppm corresponds to the carbons in alpha and beta and at 9–10 ppm to the carbon in gamma of the mercapto group. The absence of resonance lines at 36.5, 39.0, or 55.1 ppm attributed to disulfides, thiosulfonates, or sulfonates77,78 excludes the formation of such species. The presence of APTS triggers the apparition of new signals at 43 and 21 ppm, and these were attributed to carbons in alpha and beta positions of the amine, respectively. The third carbon (in gamma) expected at about 9 ppm overlaps with the MPTS signal at 9–10 ppm. TGA indicated a mass loss of around 14 ± 4% for all bifunctional samples synthesized in CC and/or DCC (PII, PIII, and PVI), pointing to a similar loading of the functional groups. For these three cases, it was not possible to determine which percentage of organics originated from MPTS or APTS. For pathways PIV, PV PVII, PVIII, PIX, and PX, 12 ± 2% of the first function was introduced during the first step (line 1, Scheme 1), whereas only 1–4% were additionally anchored using PG during the second step. EA revealed that the amount of the first functional group was reduced by 0.1 mmol g–1 at the maximum after the second functionalization. For pathways IV and V, the amount of sulfur is 0.4 mmol g–1, and after the PG of the amine function the amount of nitrogen was calculated to be 2.1 mmol g–1, whereas when the amine groups were introduced first (PVII, PVIII, PIX, and PX), the quantity of nitrogen was much lower (1.3 mmol g–1), while the amount of sulfur stays the same (Table S3). Because in these pathways the second active groups were post-grafted, the resulting materials will exhibit a limited number of these latter surface groups. This could potentially decrease the possibilities for subsequent bioconjugation using the second active group (vide infra). This was corroborated by EA which revealed that the nitrogen to sulfur ratio was 1 (PII, PIII and PVI) when introducing both functions at the same time and 6 (PIX and PX) when PG was used. When the amine groups were introduced as a second function via PG (PIV and V), the N/S ratio was 6 because of the high amount of amine functions introduced in the system (Table S2). From the physisorption data, one can easily discriminate two pathways leading to materials with the highest porosity remaining as compared to the pristine MCM-48 MSNs. Amine- and thiol-functionalized materials obtained via DCC using MPTS followed by APTS PG (PIV-2–PV-2, Scheme 1) and via CC using APTS followed by a DCC with MPTS (PVI-2, Scheme 1) still showed specific surface areas of 771 and 811 m2 g–1, total pore volumes of 0.52 and 0.51 cm3 g–1, and NLDFT mode pore sizes of 2.9 and 2.7 nm, respectively (Table S1). Moreover, the PXRD patterns of these two samples (Figure 4c, PVI-2 and PIV-2–PV-2) revealed reasonably well-ordered pore structures, with an Ia3d pore symmetry. A slightly less defined pattern was obtained for amine- and thiol-MSNs resulting from the pathway PVI-2. In this synthesis route, APTS is first added together with TEOS. As discussed above, such a CC method usually led to poorly ordered materials.
Figure 3.

TEM images of amine- and thiol-functionalized MSNs obtained following the different synthesis pathways, (a) PII-2–PIII-2, (b) PIV-2–PV-2, (c) PVI-2, (d) PVII-2, (e) PVIII-2, (f) PIX-2–PX-2, and (g) PXI-3. Higher magnification of the images is available in the Supporting Information (see Figure S6).
Figure 4.

(a) N2 adsorption (plain symbols)–desorption (open symbols) isotherms at −196 °C, (b) corresponding NLDFT PSD, and (c) low-angle powder XRD analyses of MSNs functionalized with both APTS and MPTS using the different synthesis approaches presented in Scheme 1, lines 2 and 3.
However, the subsequent addition of MPTS in the mother liquor to perform an additional DCC step appears to limit or balance the effect of APTS, most probably because of the presence of the acid thiol groups, and it allows to synthesize a final bifunctional material showing some clear signs of cubic Ia3d pore ordering (Figure 4c). Such a strategy is of particular significance as it allows to obtain an ordered bifunctional material with an excellent mesostructure using a one-pot synthesis design. Differently, when APTS is added together with MPTS during DCC (Scheme 1, PII-2–PIII-2), the resulting MSNs showed a noticeably lower mesostructure order with a broader PSD (Figure 4a,b, Table S1) and a less defined diffraction pattern (Figure 4c). Therefore, the sequence of silane addition is of great importance if one wants to design a functional material with high porosity and a specific pore structure. As expected, the use of PG techniques merely to functionalize MSNs with amine and thiol groups did not alter the order of the pore structure (Figure 4c, PIX-2–PX-2). However, the N2 physisorption analyses revealed an even lower specific surface area, pore volume, and pore size as compared to PII-2–PIII-2 (Table S1), in line with the potential pore blocking/hindrance issues related with such PG procedures. Finally, PVII and PVIII synthesis routes, which are based on APTS-MSNs obtained via CC and DCC, respectively, led to the formation of poorly or nonordered bifunctionalized samples with heavily impaired porosity, as shown in Figure 4 and Table S1. These results agree with the “negative” effect of the APTS addition in the mother liquor, as discussed above. In both cases, the amount of subsequent anchored MPTS was very low, about 1% (Table S4) and the amount of sulfur calculated from EA was only half of the amount of nitrogen (Table S3), indicating an ineffective PG procedure.
PEGylation
The PEGylation was performed following the thiol-maleimide “click” chemistry41 using the thiol-functionalized particles and methoxypolyethylene glycol maleimide (see Experimental part and Figure S8). The molecular weight of the polymer was 5 kDa with a hydrodynamic diameter above the pore size of the MCM-48 particles [4.6 nm in phosphate-buffered saline (PBS) 10 mM pH 7.4].79 As mentioned earlier, because a thiol group is needed for this type of PEGylation, all the samples discussed below were functionalized with MPTS (Scheme 1). To optimize the PEGylation protocol, we first investigated the relationship between the amount of PEG-maleimide introduced in the reaction and the resulting amount of grafted PEG, as determined by TGA. The highest grafting efficiency was observed with SH-MSNs-DCC (PI-1, Scheme 1). This is not surprising because these particles exhibit thiol functions which should be mostly located on the outer surface of the material.54 This sample was therefore chosen as the reference material, and as seen in Figure 5, no significant increase in the amount of grafted PEG was observed when more than 1 mass equivalent of PEG-maleimide was used. When 0.5 mass equivalent of PEG was used, an estimated grafting density of 0.6 PEG nm−2 was calculated. This would correspond to a cross-sectional area of 1.75 nm2 for PEG molecules, being in good agreement with the expected size of the PEG-maleimide, that is, 5 kDa,80 and also in line with previous work.29 However, such grafting density implies that only 5–6% of the grafted thiol sites were used for PEGylation, if one considers that all MPTS are available and reactive.
Figure 5.
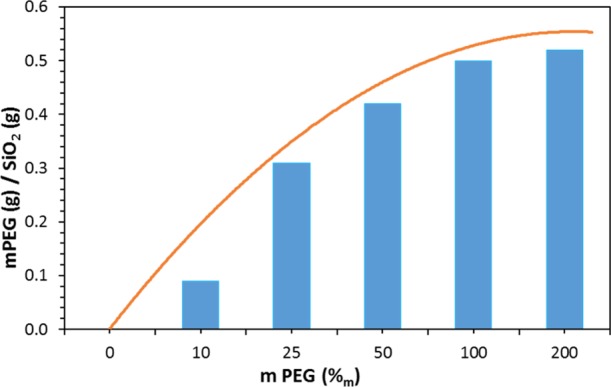
Amount of grafted PEG onto SH-MSNs-DCC as a function of the quantity of introduced PEG during the PEGylation, according to TGA.
The relatively low grafting density difference seen when either 0.5 or 1 mass equivalent of PEG was used (Figure 5) indicates that numerous thiol reactive sites are still available for further functionalization, in both cases. The observed plateau in Figure 5 is thus due to the surface being saturated by PEG or a steric hindrance between the PEG molecules ultimately leading to some reactivity issues. Preliminary visual stability tests showed that at least 1 mass equivalent of PEG was necessary to achieve a proper colloidal stability with no sedimentation. As depicted in Figure 6, PEG-MSNs obtained following the pathway PI are stable in water and Dulbecco’s Modified Eagle’s Medium (DMEM) cell culture medium for an appreciable amount of time. In DMEM, the hydrodynamic diameter varied by 5% only during the first 20 h. This result is significant because satisfactory stability of particles in DMEM is usually the most difficult to obtain, while it is mandatory for most cell incubation studies. In water, PEGylated particles (PI-3) displayed an increase of their hydrodynamic diameter up to 250 nm after 24 h, which then remains stable up to 600 h. This “swelling” was attributed to the large hydration corona provided by the PEG molecules. We thus decided to work with 1 mass equivalent for all other PEGylation of the MSNs. Then, the success of the PEGylation procedure with the different functionalized MSNs was investigated in detail via 13C CP ssNMR. A typical spectrum obtained for MSNs prepared following the pathway PVI is presented in Figure 7. One can immediately note the apparition of an intense peak at around 71 ppm in the PEGylated sample, being related with the repeating unit of the PEG polymer. Despite the use of CP mode, the strong intensity of the peak still indicates the presence of a large amount of PEG moieties, as also supported by TGA. A mass loss of about 10% was associated with the PEG grafting (Table S4, PVI step 3). The signals corresponding to the thiol- and amine-active groups were also detected confirming the stability of both functions during the PEGylation (see Figure 7). 13C CP ssNMR, coupled with TGA, was also of prime importance to study the extent and efficiency of the PEGylation for all the synthesis pathways presented in Scheme 1. Indeed, while CP is not a quantitative technique, there still exists a clear correlation between the amount of grafted PEG and the resulting (relative) intensity of the 13C ssNMR signal at 71 ppm, as shown in Figure 8. One can immediately see the impact on the PEG-related resonance intensity when the grafting strategy is successful with about 20% of PEG moieties added (PI-3, black line), versus a synthesis pathway where the grafting did not work well, that is, few PEG molecules reacted (PV-3, red line). For pathway PI, the intensity of the PEG signal is so dominant that the spectrum had to be magnified in order to observe the other resonances related with the carbons in alpha and beta positions of the thiol groups (inset, Figure 8). These differences in the grafting efficiency can be correlated with the nature, number, and dispersion of the functional groups on the surface of the MSNs. In pathway PI, only thiol functions are present, mostly on the outer part of the particles, thanks to the DCC process. It results in a highly effective grafting. On the contrary, in pathway PV, in parallel to the thiol groups anchored via DCC, amine functionalities were also added via PG, leading to the presence of both types of function on the surface. With this surface configuration, PEG-maleimide species poorly reacted and almost no PEG could be grafted. Nevertheless, as shown in Figure S9, it is still possible to react the surface thiols of this sample with fluorescein maleimide (vide infra). Similar results were obtained with functional MSNs synthesized following pathways PIX and PX where both APTS and MPTS were post-grafted and PVIII where APTS was attached via DCC and MPTS was then post-grafted. In pathways PVII and PVIII, PEGylation seemed to be successful; however, because very few MPTS groups are present on the surface, only 3% was grafted. Thus, the problem seems to originate from the proximity of amine and thiol sites on the surface, ultimately hindering the grafting procedure of large molecules. This hypothesis is consistent with the reproducibility issues faced when trying to graft PEG-maleimide to amine- and thiol-functionalized MSNs synthesized following this pathway (PV). Different attempts led to inconsistent variations, from 1 to 7%, of mass loss attributed to PEG. Successful grafting and better reproducibility were found when PEG-maleimide was reacted with MSNs functionalized with both amine and thiol groups introduced using DCC (PII); however, here also, only 7% of PEG was grafted according to TGA (Table S4). These data thus revealed that the degree of PEG grafting strongly depends on the method used to anchor the amine groups. When they were introduced either via DCC or PG, that is, the reactive NH2 groups are mostly located on the outer region of the particles, the amount of attached PEG was systematically lower as compared to functionalized MSNs where NH2 groups were introduced via a CC method (PVI), as evidenced by TGA and in a qualitative way, by the NMR data (Table S4). Moreover, the position of the PEGylation within the overall functionalization process was found to be of critical importance. Further functionalization of PEG-grafted MSNs with APTS (PI-4, Scheme 1) was ineffective, as TGA revealed no additional mass loss associated with APTS anchoring (Table S4). Furthermore, this sample (PI-4) showed a poor colloidal stability as compared to PEG-MSNs obtained via PI-3. This might be due to a cleavage of the PEG-MSNs bond, that is, reaction of the maleimide cycle with a nucleophile leading to an open carboxylic acid and amide. Therefore, to obtain an effective reaction, it is recommended to PEGylate the mesoporous particles only once all of the primary functional groups, such as −NH2 or −SH or −COOH and so forth, were already added to the MSNs. Ideally, the presence of outer surface active groups that are not necessary for “external” functionalization should be avoided, and more efficient materials, from a colloid point of view, are synthesized when amine functionalities are introduced via a CC process (PVI).
Figure 6.
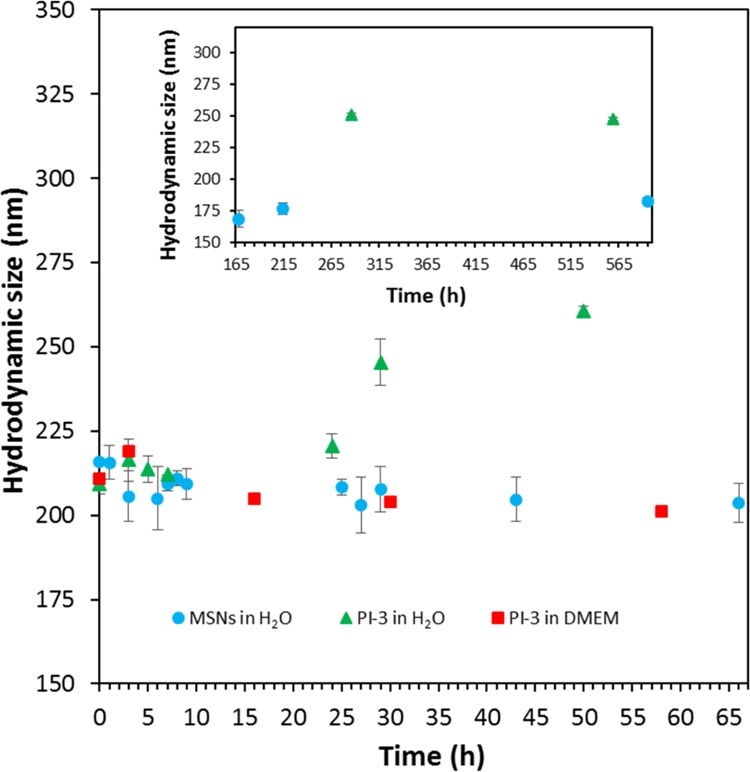
Hydrodynamic diameter of PEG-MSNs (PI-3) measured by DLS in water and DMEM as a function of time.
Figure 7.
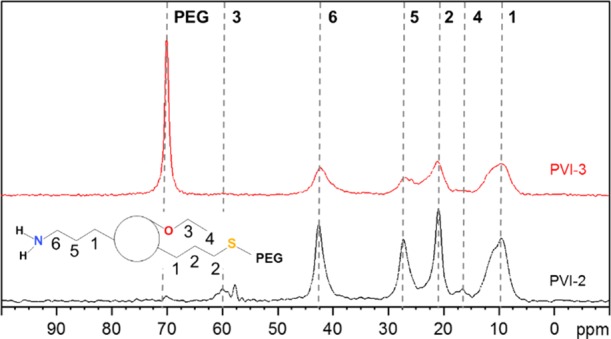
13C CP ssNMR spectra of PEGylated (red line) and non-PEGylated (black line) MSNs obtained following the synthesis pathway PVI of Scheme 1.
Figure 8.
13C CP ssNMR spectra of PEGylated MSNs obtained following the synthesis pathways PI (black line) and PV (red line) of Scheme 1.
The last functionalization concerns the addition of one or more bioactive groups (Scheme 1, steps 3 and 4). In the present report, in order to study in detail the grafting/loading procedure of each active function and its effects on the overall reactivity of the MSNs, we compare three main functionalities separately: (i) drug loading/release, (ii) insertion of a chelate, and (iii) fluorophore grafting, the latter two being for potential use in medical imaging. The goal is to eventually combine these capabilities within a single material to generate a theranostic platform.21−25,29
Drug Adsorption/Release Tests
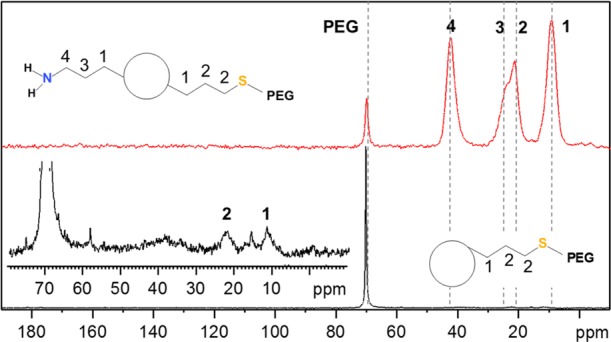
To demonstrate that pores can still be efficiently loaded even if various functionalities were anchored (including bulky PEG), quercetin, a drug with promising antioxidant and tumor necrosis factor alpha inhibiter properties,81−83 was loaded in the MSNs prepared following synthesis pathways PII and PIII. In both cases, the 13C CP ssNMR spectra, presented in Figure 9, and the TGA confirmed the sequestration of quercetin without any degradation, even after extensive washing. About 14 and 18% of quercetin was loaded following PII and PIII, respectively. The main difference between these two pathways is the moment when the PEGylation step was performed in the overall functionalization procedure. In PII, MSNs are first PEGylated and then loaded with quercetin. In PIII, these two steps are inverted. Thus, the amount of loaded drug does not heavily depend on the degree of functionalization because quercetin is a rather small molecule which can reach the pores of the MSNs even if bulky moieties are already grafted on the surface. However, from the 13C ssNMR spectra and the TGA results (Table S4), it is obvious that the pathway PIII is not desirable because PEG moieties are not efficiently grafted. Only 2% of additional mass loss was attributed to PEG. To exclude significant quercetin leaching during the PEGylation, infrared spectroscopy measurements were performed before and after the synthesis (see Figure S10). No change in the absorbance of the peaks related to quercetin was observed. In case of PII, the drug is clearly seen in the spectrum after the drug loading (∼1500 cm–1). Similar to the behavior seen for PI, this result confirms the importance to control wisely the PEGylation timing within the functionalization process for the design of MSN nanocarriers. It appears that PII provides a much more straightforward correlation between the mass loss, and the different synthesis steps and led to a colloidal stable quercetin-loaded PEG-MSNs. N2 physisorption analyses presented in Figure S11a enable to evaluate the effects of each functionalization step on the porosity of the resulting materials prepared according to PII. One can note the progressive decrease in the specific surface area and total pore volume (Table S1), which is consistent with the sequential grafting of PEG and drug loading. The NLDFT mode pore size does not change after PEGylation, confirming the open access to the pores. As expected, no porosity is detected after quercetin loading, in line with the filling of the pores by the drug.
Figure 9.

13C CP solid-state NMR spectra of pure quercetin and quercetin-loaded MSNs synthesized via pathways PII and PIII.
A simple release test was then performed in PBS buffer over a period of 24 h, as presented in Figure 10. The initial concentration of the encapsulated and free quercetin was set to 50 μg mL–1. The release/solubility of quercetin was clearly promoted by the encapsulation into functionalized MSNs, especially during the first 8 h. This result is of particular significance considering the poor solubility of quercetin in PBS buffer at pH = 7.484−86 and its poor stability, that is, about 10 h, in PBS.87 However, no statistical difference was observed between PII and PIII which leads us to the assumption that the PEG does not perturb the release of this drug at pH = 7.4 and merely provides a better colloidal stability to the system.
Figure 10.
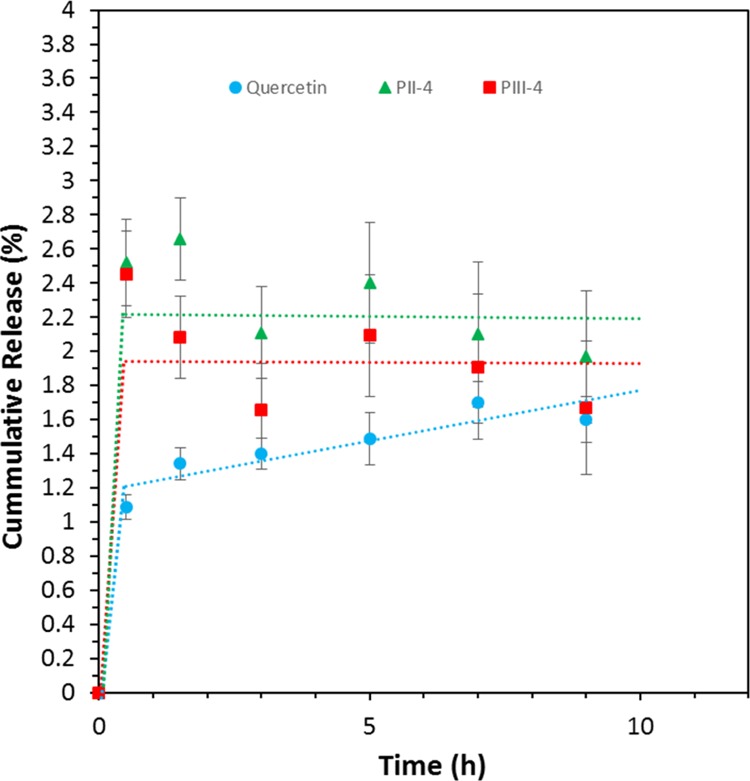
Release of quercetin as a function of time.
Insertion of Gd-Chelates
Grafting of Gd3+ chelates at the surface of the MSNs enables their tracking in the blood system, the organs and/or cells using magnetic resonance imaging (MRI). Paramagnetic Gd-based chelates appear bright in T1-weighted MRI images even at low doses of the chelated substance. Over the last years, several studies reported MSNs labeled with Gd chelates for MRI applications.29,64,88−92 Most of them are based on the grafting of DTPA or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid ligands at the surface of MSNs. Nevertheless, the chemical stability of such metal chelates in vitro and in vivo is still a matter of discussion, the detachment and release of free Gd-complex species being of concern.59 In the same way, proper colloidal stability in the medium of interest is desirable to avoid sedimentation, ultimately resulting in a non-homogeneous distribution of the particles and thus in nonrelevant MRI. Herein, DTPA was first grafted via the amine groups using a reaction scheme described in Figure S12. As shown in Scheme 1, three different pathways were attempted for DTPA anchoring. In PIV and PV, amine functions were post-grafted onto SH-MSNs obtained via DCC. Then, DTPA was reacted with the surface NH2 groups, before (PIV) or after (PV) PEGylation. In both cases, a mass loss of about 5–7% is detected in TGA (for PIV-3 and PV-4, respectively, Table S4) as well as an increase in nitrogen and carbon amount by EA (Tables S2 and S3), confirming the successful grafting of DTPA. It roughly corresponds to a 40% yield with respect to the amount of DTPA introduced. When considering the amount of grafted APTS, it was estimated that about 50–60% of available amine groups reacted (from TGA), leaving some opportunities for further reaction. However, as already discussed above, the amount of PEG grafted in PV-3 is inconsistent from synthesis to synthesis and usually quite low. It results in a material having a variable behavior from a colloidal stability point of view which may be problematic for systematic studies, especially in complex media such as DMEM. Therefore, while the DTPA grafting procedure is effective, this synthesis pathway needs more optimization to be considered as a reliable synthesis route. Unfortunately, PEGylation performed on DTPA-MSNs (PIV-4) was also found to be ineffective (Table S4) and resulted in neglectable amounts of PEG being grafted. In this case, we attributed this behavior to a steric hindrance problem combined with the proximity of available unreacted amine groups with the thiol ones, as mentioned earlier. This pathway was thus not further explored. With regards to the results obtained in PV, it was decided to simplify and optimize the synthesis protocol. This gave rise to the pathway PVI where APTS is no more post-grafted but rather introduced in CC. This choice was made in order to avoid the surface competition between APTS and MPTS which appeared to be an issue. Although this change resulted in a less ordered pore structure, as seen in Figure 4c, because MPTS is still added via DCC, the overall synthesis pathway has the great advantage to provide a material with thiol functions on the external surface and to keep the overall primary functionalization (−NH2 and −SH) in a one-pot synthesis. Furthermore, avoiding the PG with APTS allows to get rid of any extra harmful solvent traces such as toluene. Considering the subsequent PEGylation and DTPA grafting steps, the only solvent being employed is dimethyl sulfoxide (DMSO) which is considered biocompatible and non-toxic for humans.93 Following pathway PVI, it was possible to introduce about 12% w/w of PEG and about 3% of DTPA (Table S4). Success of the two grafting procedures was confirmed by 13C CP ssNMR as shown in Figures 7 and S13. Almost twice the amount of PEG was grafted, as compared to pathway PV, which might be attributed to a more selective functionalization of the thiol active groups. EA showed that the amount of nitrogen for PV-2 was more than three times higher than for PVI-2 (0.5 vs 1.8 mmol g–1), causing problems for the reaction with PEG, while the amount of sulfur was nearly similar. In pathway PVI, there should not be much APTS on the external surface of the MSNs. However, it is estimated that DTPA molecules reacted with only 25–30% of the amine reactive sites. This can be explained considering the fact that not all NH2 groups introduced via CC are accessible. Some of them might be trapped within the pore walls94 making them inactive for any subsequent chemistry. In addition, much less amine sites are present on the outer surface of the MSNs, decreasing the number of easily accessible active sites for large molecules. The last part of the synthesis was to complex Gd3+ as described in the Experimental Section, but will not be discussed in detail here. Zeta potential measurements in nanopure water were in good agreement with the literature29 and showed to be −35 mV before and −24 mV after the Gd chelation. Most importantly, the NLDFT mode pore size of the MSNs does not diminish with the PEGylation and the grafting of the DTPA, confirming that these bulky groups are mostly grafted on the external surface of the mesoporous silica. Compared to previous work,29 the DTPA was here grafted using a significantly less harmful solvent, that is, DMSO instead of toluene, and without the use of a refluxing system, making the procedure noticeably simpler. Even though the amount of PEG (PVI) was higher (12% vs 5%), the NLDFT pore size appeared to be the same as previously reported (2.5 nm). This is a quite remarkable result as it opens the way for further loading of drugs or other substances of biological interest. As it will be discussed in the last part below, the colloidal stability of this sample in various media was also quite remarkable, making this pathway PVI a very potent one for the production of high quality multifunctional MSNs for health-related applications.
Fluorophore Grafting
One way to take advantage of the residual surface reactive groups is to use them to bring a second imaging capability to the particles. In this way, organic fluorophores, rhodamine and fluorescein typically, are particularly interesting as they are already commonly used for cell and organ labeling.95−97 Rhodamine B isothiocyanate was thus first used. In addition to the expected reaction with the free amine groups, rhodamine B isothiocyanate could also react with the thiol functions. Therefore, different pathways of the grafting procedure were performed as presented in Scheme 1 (PX and PXI). The presence of the rhodamine was revealed by the pink coloration of the MSNs, even after extensive washing cycles, and by the UV/vis spectrum in ethanol which showed a peak at 550 nm for both free and grafted rhodamine species (Figure 11). The amount of rhodamine grafted, calculated from TGA, was found to be rather low for PX and PXI (Table S4). However, the particles were nicely colored in both cases, and an identifiable signal from rhodamine was also detected in the 13C CP ssNMR pattern (Figure S14). A low grafting efficiency for fluorophores should not be a major issue because a small amount is usually enough to trigger a good imaging sensitivity.98 Importantly, PX and PXI highlighted again the influence of the addition order of the different functionalities. Indeed, PG of MPTS was heavily hampered by the presence of rhodamine (PXI) and comparable to the effect seen with surface APTS, and the presence of rhodamine and APTS impaired the PEGylation reaction (PX). In contrast, it was possible to add one or two fluorophores on already multifunctionalized materials synthesized according to pathway VI (PVI-4), as shown in Figure S15. Similar to the observations made for PV (Figure S9), rhodamine and/or fluorescein molecules were successfully grafted to the PEG- and Gd-DPTA-containing MSNs using some of the remaining thiol and amine reaction sites. Placing two fluorophores in one single nanocarrier is of interest, especially when the location of the added functional groups is different like it is the case in PVI (CC and DCC).99 Therefore, if one uses proper functionalization procedures, the resulting materials can easily combine dual imaging capabilities while still exhibiting interesting porosity characteristics that can be used for drug loading or other biological functionalities.
Figure 11.
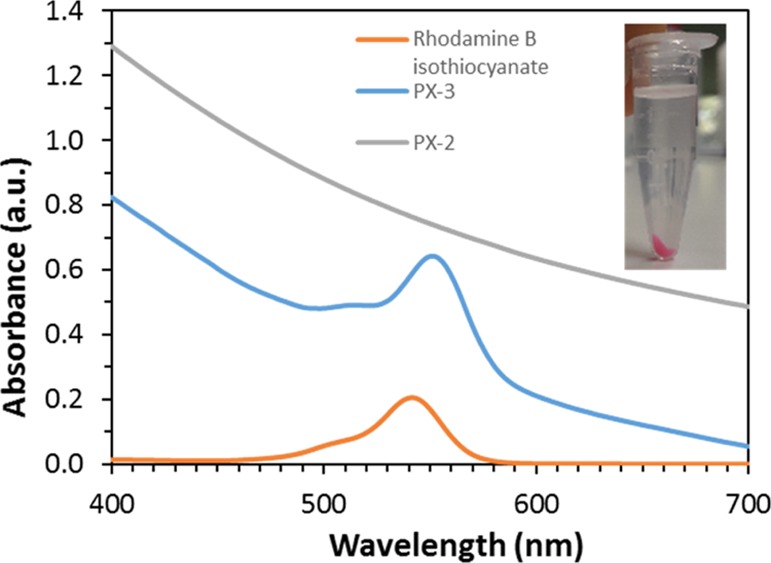
UV/vis spectra of free rhodamine and rhodamine-grafted MSNs synthesized according to PX-3 and PX-2 in EtOH. A photo of the grafted MSNs in EtOH is provided in the inset.
Stability Tests
Finally, colloidal stability was investigated using DLS in different media and at different pH values. For these tests, the most promising synthesis pathway, that is, PVI was selected. As shown in Figure 12, particles obtained after step PVI-4 were suspended in acidic water at pH = 2 and pH = 5. Colloidal stability in biological media of interest, PBS and DMEM in our case, was also studied. The sample was stable in all media and pH values with an average hydrodynamic diameter of about 250 nm, being consistent with the data observed for MSNs synthesized according to PI-3 suspended in water and DMEM (see Figure 6). After 24 h (Figure 12), a maximum standard deviation of 10% was observed for MSNs (PVI-4) suspended in DMEM. Moreover, the particles were still stable in the different media after 5 days, as shown in Figure S16, demonstrating the adequate colloidal behavior of the platform. No visible aggregation/sedimentation was found. The variation of the hydrodynamic diameter after 5 days was rather small in each medium, which indicates that the particles did not undergo a major change within this timeframe. Therefore, this material (PVI-4, where amine and thiol groups were added via CC and DCC, respectively, followed by the sequential PG of PEG, DTPA chelates, and fluorophores) seems to be a great candidate for future in vitro and potentially in vivo studies. It can also be concluded that the PEGylation enhanced dramatically the colloidal behavior of the MSNs, especially if the PEGylation was performed following the proposed “optimized” conditions.
Figure 12.
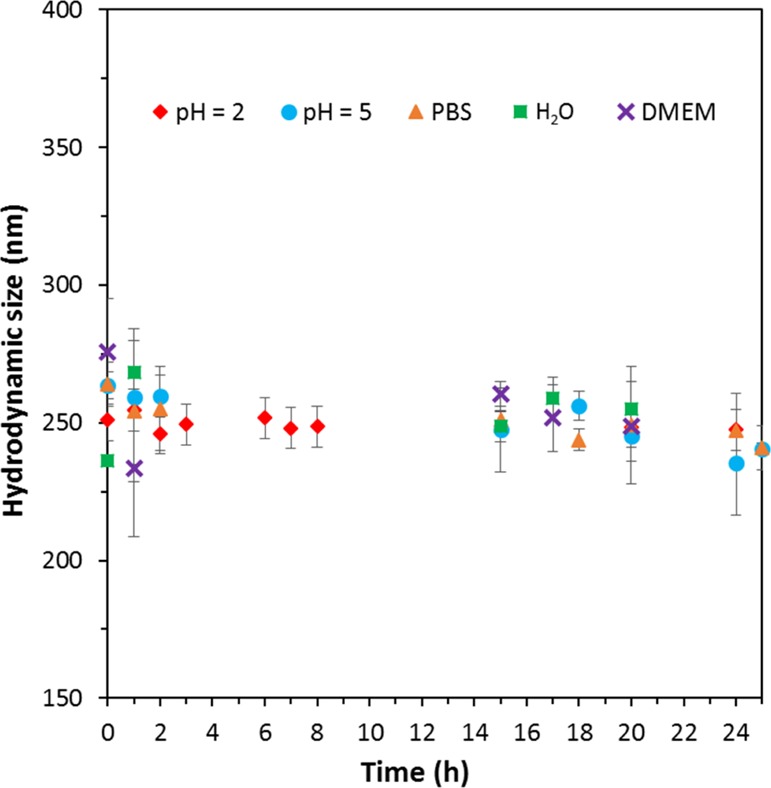
Hydrodynamic diameters of MSNs synthesized according to PVI-4 in different media and pH values over time.
Conclusions
In this work, we demonstrated the importance of the different MSN synthesis and functionalization methods and steps in order to produce the most efficient platform for biorelated applications while preserving the integrity and the features of the mesopore structure. The following conclusions can be formulated:
It appears favorable to work with MPTS instead of APTS, especially for “external surface” functionalization.
If two functionalities are to be included, one should rather choose CC and DCC routes instead of classical PG.
Subsequent functionalization, such as PEGylation, works better if the amount of surface amine functions is limited.
In the same way, PEGylation should always be performed after initial functionalization with the other silanes.
Furthermore, our results showed that unreacted surface groups are still available for further grafting of small molecules, even after dual surface grafting of DTPA and PEG, allowing the synthesis of a potential multi-imaging capability platform.
The next step will be to perform in-depth in vitro and in vivo biological studies with the obtained materials. Furthermore, multi-imaging testing could be implemented, noticeably by combining MRI and fluorescence imaging. Also, because one of the fluorophores could be more distributed on the outer shell of the particles, whereas the other is distributed homogeneously through the entire particle, it could be of great interest to follow, for example, the details in the degradation of the material in different media. Finally, one could also think about extending Scheme 1 by including more functionalities, like halide groups, that may be used for further chemical modifications. However, it has to be kept in mind that the order and the method of functionalization appear to be crucial regarding the final material properties and pore ordering. From a synthesis point of view, the results described here may also be extended and extrapolated to other siliceous particle systems where surface versus core differentiation is mandatory, ultimately allowing an optimal and logical (nano)carrier design.
Materials and Methods
TEOS (98%), n-cetyltrimethylammonium bromide (CTAB, 99%), Pluronic F127 (EO106PO70EO106, BioReagent), diethylenetriaminepentaacetic dianhydride (DTPA dianhydride, 98%), APTS (99%), MPTS (95%), Gd(CH3CO2)3·xH2O (99.9%), PEG maleimide (MW 5000), absolute toluene, PBS, acetic acid, hydrochloric acid (37%), rhodamine B isothiocyanate, fluorescein-5-maleimide, and DMEM (without glucose, l-glutamine, phenol red, sodium pyruvate, and sodium bicarbonate) were obtained from Sigma-Aldrich (Austria) in high purity grade. Absolute ethanol, EtOH (94–96%), DMSO, sodium acetate, and ammonium hydroxide, NH4OH (28–30%), were purchased from Alfa Aesar (Austria).
Mesoporous Silica Nanoparticles
MCM-48 silica nanoparticles were synthesized as reported in the literature.26 Briefly, 0.5 g CTAB (1.37 mmol), 2 g Pluronic F127, and 33.5 g absolute ethanol were stirred at 1000 rpm in a polypropylene (PP) bottle at room temperature. After complete dissolution and at least 30 min, 106.5 mL of a freshly prepared 2.9 wt % NH4OH solution was added at once. The obtained reaction mixture was stirred overnight at 1000 rpm (pH = 11.9). Then, 1.8 g TEOS was added in one shot using a PP beaker. The solution was stirred for 1 more minute at 1000 rpm. After that, it was aged at room temperature in static conditions for 24 h. The resulting white powder was collected by centrifugation, washed with water and ethanol, and left open on a bench for drying overnight at room temperature. Finally, the material was calcined at 550 °C for 5 h (1 °C min–1) or chemically extracted using the following procedure: the obtained particles were filled in a 250 mL round-bottom flask equipped with a Dimroth cooling system, followed by the addition of 150 mL of technical EtOH and 1.5 mL of HCl (37%). The obtained mixture was stirred at 90 °C overnight. Then, the mixture was allowed to cool down to room temperature and centrifuged at 10 000 rpm for 20 min. Then, the particles were washed with water (approx. 200 mL) to remove remaining HCl and with the same amount of EtOH. This procedure was repeated for three times followed by drying the final material overnight in an oven at 45 °C.
MSN Functionalization
Functionalized MSNs were obtained via three different techniques: CC, DCC, and PG.19,26 For CC, the synthesis steps are the same as described above except that the selected organo-silane was mixed with TEOS in a PP beaker and added at once to the synthesis. For DCC, the reaction was carried in a similar way as for the synthesis of MSNs except that after 10 min of aging at room temperature, the synthesis is stirred back at 1000 rpm. Immediately after, the silane was added at once to the solution. The mixture is then stirred for another minute and aged at room temperature for 24 h. For PG, the nanoparticles were first degassed under vacuum overnight at 80 °C if grafting was performed on already functionalized materials or at 150 °C for pristine MSNs. Nanoparticles were then suspended in absolute toluene at 80 °C (for already functionalized material) or 110 °C (for MSNs) under inert atmosphere. After 3 h, the silane was added and the mixture was stirred for 4 h. The amount of added silane was 0.1 molar equivalent as compared to TEOS for CC and DCC, and 4 or 12 mmol g–1 of degassed particles for PG. Washing, template removal, drying, and collection of the functionalized samples were performed exactly as described for the chemically extracted MSNs.
PEGylation
mPEG-maleimide was reacted with the thiol groups of MPTS.100 Briefly, the nanoparticles (0.15 g) were suspended in 10 mL of nanopure water. After stirring at 1000 rpm overnight, 1 mL of PBS (pH 7.4) was added. Then, a mass equivalent amount of mPEG-maleimide (0.15 g) was added to the solution and stirred overnight at room temperature. The resulting product was collected by centrifugation and washed three times with water, followed by drying for at least 12 h at room temperature.
Drug Loading
Quercetin was loaded inside the (functionalized) MSNs using the rotavapor technique.101 Briefly, 20 mg (=20 wt %) quercetin was dissolved in absolute ethanol followed by the addition of 80 mg nanoparticles. The mixture was kept in an ultrasonic bath for at least 1 h. Finally, the ethanol was slowly evaporated using a Buchi rotary evaporator Interface I-100 (pressure difference 50 mbar/step/5 min). The drug solubilization/release test was performed in triplicates in PBS buffer over a period of 24 h. Briefly, 10 mg of particles and 30 mL PBS buffer (=50 μg mL–1) were added in a centrifuge tube and put on a walker at 80 rpm. After a specific period of time, the samples were centrifuged for 5 min at 9000 rpm, and 2 mL of the supernatant was removed. For control purposes, a solubilization test of free quercetin over time was also performed. The drug (1.5 mg) was directly added to 30 mL PBS buffer. The concentration of solubilized drug was then measured by using a UV/vis spectrometer (Onda Spectrophotometer, 800–200 nm), and the measured solution was returned to the PBS buffer. The calibration was performed before each set of measurements using 5 standard solutions (10–1 μg mL–1) and treated in the same way as described above.
DTPA[Gd] Chelate
Approximately 0.15 g nanoparticles was suspended in 4 mL of DMSO followed by the addition of half a mass equivalent of DTPA (0.075 g). After stirring overnight at room temperature, the material was washed with approximately 10 mL of DMSO (twice) and with 10 mL of distilled water (three times). After washing, the material was dried in an oven at 45–50 °C. The obtained material was then added to 15 mL of a 100 mM gadolinium acetate solution in water (1 mL/10 mg particle) and stirred for 1 h to allow the complexation of Gd ions. Finally, the particles were washed two times with approx. 10 mL of an acetate buffer solution (NaOAc/AcOH, 0.1 M, pH = 5) and twice with the same amount of water. The resulting DTPA[Gd] grafted material was dried overnight in an oven at 45 °C.
DLS and Zeta Potential
Stable aqueous suspensions of the material (0.7 mg mL–1) were obtained by performing three cycles of vortex shaking (10 min) and ultrasonic bath treatment (20 min). The supernatant was then analyzed for hydrodynamic diameters and zeta potential values using a Malvern Zetasizer Nano ZS. Measurements were accepted as valid only when satisfactory quality criteria such a polydispersity index and steepness of the correlogram function were fulfilled. For long-term colloidal stability, tests were performed in different media (water, buffer solutions for pH 2, 5, and 8, and PBS buffer). In between tests, the samples were kept in the cuvettes in static conditions, and no further mixing or vortex was used.
Thermogravimetric Analysis
The measurements were performed using a Netsch STA 449-F3 Jupiter analyzer, from room temperature to 800 °C with a heating rate of 10 °C min–1. An air flow of 20 mL min–1 and an additional protective N2 flow of 20 mL min–1 were used. The percentage of grafted moieties was calculated based on the detected mass loss between 150 and 800 °C.
Nitrogen Physisorption Analyses at −196 °C
Nitrogen physisorption analyses at −196 °C were performed with a Quantachrome iQ3 instrument (Anton Parr, Boynton Beach, USA). Prior to the measurements, the samples were outgassed under vacuum overnight at 150 °C for pure silica material and at 80 °C for functionalized particles. The specific surface area (SBET) was obtained using the BET equation in the range of 0.05–0.15 P/P0. The total pore volume was determined at P/P0 = 0.95 according to the Gurvitch rule, and the pore size analysis was calculated using the equilibrium NLDFT model, assuming a cylindrical pore geometry and a silica surface. Calculations were performed with the ASiQwin software v.5.0 provided by Quantachrome.
Transmission Electron Microscopy
The nanoparticles were dispersed in ethanol by using an ultrasonic bath and further deposited on a carbon-coated copper grid. Images were taken with a Philips CM200 transmission electron microscope at an accelerating voltage of 200 keV in bright-field mode. Micrographs were acquired using a Gatan Orius CCD camera with an exposure time of 1 s field.
Solid-State Nuclear Magnetic Resonance Spectroscopy
The spectra were obtained on a Bruker AVANCE NEO 500 wide bore system (Bruker BioSpin, Rheinstetten, Germany) using a 4 mm triple resonance magic angle spinning (MAS) probe. The resonance frequency for 13C NMR was 125.78 MHz, and the MAS rotor spinning was set to 14 kHz. CP was achieved by a ramped contact pulse with a contact time of 2 ms. During acquisition, 1H was high power decoupled using SPINAL with 64 phase permutations. The chemical shifts for 13C are reported in ppm and are referenced external to adamantane by setting the low field signal to 38.48 ppm.
Powder X-ray Diffraction
The low-angle diffractograms of MSNs were recorded on a PANalytical Empyrean diffractometer (Malvern PANanalytical, United Kingdom) in reflection geometry (Bragg–Brentano HD) using Cu Kα1+2 radiation operated at a voltage of 45 kV, a tube current of 40 mA, and with a fixed divergence slit of 0.05 mm. Measurements were performed in continuous mode with a step size 2 theta of 0.013° and a time per step of 300 s. The low-angle XRD pattern of sample PXI-3 was recorded in transmission geometry in continuous mode with a step size 2θ of 0.013°, a time per step of 250 s, and with a fixed divergence slit of 0.76 mm.
Elemental Analysis
EA on C, H, N, and S was done using a Eurovector EA3000 CHNS-O analyzer. The method has been proven to be capable of detecting the organic modifications on various silica materials with the usual accuracy and precision. The uncertainty is below 0.2 wt % for carbon at 20 wt %. The sample amount weighed for an analysis was between 3.5 and 5.0 mg when a sufficient sample amount was available.
Attenuated Total Reflectance Infrared Spectroscopy
Attenuated total reflectance infrared (ATR-IR) spectra were measured with a Bruker Vertex 70 FTIR spectrometer equipped with Specac Golden Gate ATR accessory. Each spectrum was gathered from the acquisition of 72 scans at 4 cm–1 resolution varied from 4000 to 500 cm–1. A new background spectrum gathered from the acquisition of 72 scans at 4 cm–1 resolution was collected before running each sample. All samples were dried at 40 °C before the measurements.
Acknowledgments
The authors acknowledge the funding support of the University of Vienna (Austria). The authors thank Dr. Jean-Luc Bridot (Université Laval) for the fruitful discussions on the different synthesis pathways and Mag. Johannes Theiner (University of Vienna) for performing EA.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02784.
Comparison of calcined and extracted pure MCM-48 MSNs, TGA data of different quantities of aminosilane, reproducibility shown on 4 different batches, 13C CP NMR of functionalized, drug-loaded, DTPA-, and rhodamine-grafted materials, reaction schemes, visual comparison of fluorescein maleimide, long-term stability test, and tables of TGA, EA, and physisorption results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Singh R. K.; Patel K. D.; Leong K. W.; Kim H.-W. Progress in nanotheranostics based on mesoporous silica nanomaterial platforms. ACS Appl. Mater. Interfaces 2017, 9, 10309–10337. 10.1021/acsami.6b16505. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Niu Y.; Li Y.; Gong Y.; Shi H.; Huo Q.; Liu Y.; Xu Q. Stimuli-responsive delivery vehicles based on mesoporous silica nanoparticles: recent advances and challenges. J. Mater. Chem. B 2017, 5, 1339–1352. 10.1039/c6tb03066a. [DOI] [PubMed] [Google Scholar]
- Paris J. L.; Colilla M.; Izquierdo-Barba I.; Manzano M.; Vallet-Regí M. Tuning mesoporous silica dissolution in physiological environments: a review. J. Mater. Sci. 2017, 52, 8761–8771. 10.1007/s10853-017-0787-1. [DOI] [Google Scholar]
- Florek J.; Caillard R.; Kleitz F. Evaluation of mesoporous silica nanoparticles for oral drug delivery - current status and perspective of MSNs drug carriers. Nanoscale 2017, 9, 15252–15277. 10.1039/c7nr05762h. [DOI] [PubMed] [Google Scholar]
- Yu M.; Gu Z.; Ottewell T.; Yu C. Silica-based nanoparticles for therapeutic protein delivery. J. Mater. Chem. B 2017, 5, 3241–3252. 10.1039/c7tb00244k. [DOI] [PubMed] [Google Scholar]
- Albert K.; Huang X.-C.; Hsu H.-Y. Bio-templated silica composites for next-generation biomedical applications. Adv. Colloid Interface Sci. 2017, 249, 272–289. 10.1016/j.cis.2017.04.011. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Yu C. Advances in silica based nanoparticles for targeted cancer therapy. Nanomedicine 2016, 12, 317–332. 10.1016/j.nano.2015.10.018. [DOI] [PubMed] [Google Scholar]
- Möller K.; Bein T. Talented mesoporous silica nanoparticles. Chem. Mater. 2016, 29, 371–388. 10.1021/acs.chemmater.6b03629. [DOI] [Google Scholar]
- Zhang Y.; Hsu B. Y. W.; Ren C.; Li X.; Wang J. Silica-based nanocapsules: synthesis, structure control and biomedical applications. Chem. Soc. Rev. 2015, 44, 315–335. 10.1039/c4cs00199k. [DOI] [PubMed] [Google Scholar]
- Croissant J. G.; Cattoën X.; Wong Chi Man M.; Durand J.-O.; Khashab N. M. Syntheses and applications of periodic mesoporous organosilica nanoparticles. Nanoscale 2015, 7, 20318–20334. 10.1039/c5nr05649g. [DOI] [PubMed] [Google Scholar]
- Butler K. S.; Durfee P. N.; Theron C.; Ashley C. E.; Carnes E. C.; Brinker C. J. Protocells: modular mesoporous silica nanoparticle-supported lipid bilayers for drug delivery. Small 2016, 12, 2173–2185. 10.1002/smll.201502119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekaru H.; Lu J.; Tamanoi F. Development of mesoporous silica-based nanoparticles with controlled release capability for cancer therapy. Adv. Drug Delivery Rev. 2015, 95, 40–49. 10.1016/j.addr.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyo C.; Weiss V.; Bräuchle C.; Bein T. Multifunctional mesoporous silica nanoparticles as a universal platform for drug delivery. Chem. Mater. 2013, 26, 435–451. 10.1021/cm402592t. [DOI] [Google Scholar]
- Wen J.; Yang K.; Liu F.; Li H.; Xu Y.; Sun S. Diverse gatekeepers for mesoporous silica nanoparticle based drug delivery systems. Chem. Soc. Rev. 2017, 46, 6024–6045. 10.1039/c7cs00219j. [DOI] [PubMed] [Google Scholar]
- Guillet-Nicolas R.; Popat A.; Bridot J.-L.; Monteith G.; Qiao S. Z.; Kleitz F. pH-responsive nutraceutical-mesoporous silica nanoconjugates with enhanced colloidal stability. Angew. Chem., Int. Ed. 2013, 52, 2318–2322. 10.1002/anie.201208840. [DOI] [PubMed] [Google Scholar]
- Juère E.; Florek J.; Bouchoucha M.; Jambhrunkar S.; Wong K. Y.; Popat A.; Kleitz F. In Vitro Dissolution, Cellular Membrane Permeability, and Anti-Inflammatory Response of Resveratrol-Encapsulated Mesoporous Silica Nanoparticles. Mol. Pharm. 2017, 14, 4431–4441. 10.1021/acs.molpharmaceut.7b00529. [DOI] [PubMed] [Google Scholar]
- Du X.; Li X.; Xiong L.; Zhang X.; Kleitz F.; Qiao S. Z. Mesoporous silica nanoparticles with organo-bridged silsesquioxane framework as innovative platforms for bioimaging and therapeutic agent delivery. Biomaterials 2016, 91, 90–127. 10.1016/j.biomaterials.2016.03.019. [DOI] [PubMed] [Google Scholar]
- Lim W. Q.; Phua S. Z. F.; Xu H. V.; Sreejith S.; Zhao Y. Recent advances in multifunctional silica-based hybrid nanocarriers for bioimaging and cancer therapy. Nanoscale 2016, 8, 12510–12519. 10.1039/c5nr07853a. [DOI] [PubMed] [Google Scholar]
- Guillet-Nicolas R.; Laprise-Pelletier M.; Nair M. M.; Chevallier P.; Lagueux J.; Gossuin Y.; Laurent S.; Kleitz F.; Fortin M.-A. Manganese-impregnated mesoporous silica nanoparticles for signal enhancement in MRI cell labelling studies. Nanoscale 2013, 5, 11499–11511. 10.1039/c3nr02969g. [DOI] [PubMed] [Google Scholar]
- Kim J.; Kim H. S.; Lee N.; Kim T.; Kim H.; Yu T.; Song I. C.; Moon W. K.; Hyeon T. Multifunctional uniform nanoparticles composed of a magnetite nanocrystal core and a mesoporous silica shell for magnetic resonance and fluorescence imaging and for drug delivery. Angew. Chem., Int. Ed. 2008, 47, 8438–8441. 10.1002/anie.200802469. [DOI] [PubMed] [Google Scholar]
- Niu D.; Li Y.; Shi J. Silica/organosilica cross-linked block copolymer micelles: a versatile theranostic platform. Chem. Soc. Rev. 2017, 46, 569–585. 10.1039/c6cs00495d. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Ma X.; Gao Y.-E.; Hou M.; Xue P.; Li C. M.; Kang Y. Multifunctional silica nanoparticles as a promising theranostic platform for biomedical applications. Mater. Chem. Front. 2017, 1, 1257–1272. 10.1039/c7qm00153c. [DOI] [Google Scholar]
- Borawake D. D.; Pande V. V.; Giri M. A. Mesoporous silica nanoparticles as theranostic platform for smart drug delivery: a review. J. Nanomed. Nanosci. 2017, 5, JNAN-125. 10.29011/JNAN-125. [DOI] [Google Scholar]
- Kempen P. J.; Greasley S.; Parker K. A.; Campbell J. C.; Chang H.-Y.; Jones J. R.; Sinclair R.; Gambhir S. S.; Jokerst J. V. Theranostic mesoporous silica nanoparticles biodegrade after pro-survival drug delivery and ultrasound/magnetic resonance imaging of stem cells. Theranostics 2015, 5, 631–642. 10.7150/thno.11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrogio M. W.; Thomas C. R.; Zhao Y.-L.; Zink J. I.; Stoddart J. F. Mechanized Silica Nanoparticles: A New Frontier in Theranostic Nanomedicine. Acc. Chem. Res. 2011, 44, 903–913. 10.1021/ar200018x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.-W.; Chung P.-W.; Lin V. S.-Y. Facile Synthesis of Monodisperse Spherical MCM-48 Mesoporous Silica Nanoparticles with Controlled Particle Size. Chem. Mater. 2010, 22, 5093–5104. 10.1021/cm1017344. [DOI] [Google Scholar]
- Kim T.-W.; Kleitz F.; Paul B.; Ryoo R. MCM-48-like Large Mesoporous Silicas with Tailored Pore Structure: Facile Synthesis Domain in a Ternary Triblock Copolymer–Butanol–Water System. J. Am. Chem. Soc. 2005, 127, 7601–7610. 10.1021/ja042601m. [DOI] [PubMed] [Google Scholar]
- Kleitz F.; Bérubé F.; Guillet-Nicolas R.; Yang C.-M.; Thommes M. Probing adsorption, pore condensation, and hysteresis behavior of pure fluids in three-dimensional cubic mesoporous KIT-6 silica. J. Phys. Chem. C 2010, 114, 9344–9355. 10.1021/jp909836v. [DOI] [Google Scholar]
- Bouchoucha M.; C.-Gaudreault R.; Fortin M.-A.; Kleitz F. Mesoporous silica nanoparticles: selective surface functionalization for optimal relaxometric and drug loading performances. Adv. Funct. Mater. 2014, 24, 5911–5923. 10.1002/adfm.201400524. [DOI] [Google Scholar]
- Wu S.-H.; Mou C.-Y.; Lin H.-P. Synthesis of mesoporous silica nanoparticles. Chem. Soc. Rev. 2013, 42, 3862–3875. 10.1039/c3cs35405a. [DOI] [PubMed] [Google Scholar]
- Lindén M.Biodistribution and excretion of intravenously injected mesoporous silica nanoparticles: Implications for drug delivery efficiency and safety. The Enzymes; Academic Press, 2018; Vol. 43, pp 155–180. [DOI] [PubMed] [Google Scholar]
- Braun K.; Stürzel C. M.; Biskupek J.; Kaiser U.; Kirchhoff F.; Lindén M. Comparison of different cytotoxicity assays for in vitro evaluation of mesoporous silica nanoparticles. Toxicol. In Vitro 2018, 52, 214–221. 10.1016/j.tiv.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 21CFR172.480, Title 21; CFR Code of Federal Regulations, 2018; Vol. 3.
- Mamaeva V.; Sahlgren C.; Lindén M. Mesoporous silica nanoparticles in medicine-recent advances. Adv. Drug Delivery Rev. 2013, 65, 689–702. 10.1016/j.addr.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Li Y.; Shi J. Hollow-structured mesoporous materials: chemical synthesis, functionalization and applications. Adv. Mater. 2014, 26, 3176–3205. 10.1002/adma.201305319. [DOI] [PubMed] [Google Scholar]
- Croissant J. G.; Fatieiev Y.; Almalik A.; Khashab N. M. Mesoporous silica and organosilica nanoparticles: physical chemistry, biosafety, delivery strategies, and biomedical applications. Adv. Healthcare Mater. 2017, 7, 1700831. 10.1002/adhm.201700831. [DOI] [PubMed] [Google Scholar]
- Croissant J. G.; Fatieiev Y.; Khashab N. M. Degradability and Clearance of Silicon, Organosilica, Silsesquioxane, Silica Mixed Oxide, and Mesoporous Silica Nanoparticles. Adv. Mater. 2017, 29, 1604634. 10.1002/adma.201604634. [DOI] [PubMed] [Google Scholar]
- Liberman A.; Mendez N.; Trogler W. C.; Kummel A. C. Synthesis and surface functionalization of silica nanoparticles for nanomedicine. Surf. Sci. Rep. 2004, 69, 132–158. 10.1016/j.surfrep.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair M. M.; Yen H.; Kleitz F. Nanocast mesoporous mixed metal oxides for catalytic applications. C. R. Chim. 2014, 17, 641–655. 10.1016/j.crci.2014.04.005. [DOI] [Google Scholar]
- Mandal T.; Beck M.; Kirsten N.; Lindén M.; Buske C. Targeting murine leukemic stem cells by antibody functionalized mesoporous silica nanoparticles. Sci. Rep. 2018, 8, 989. 10.1038/s41598-017-18932-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlossbauer A.; Kecht J.; Bein T. Biotin-Avidin as a protease-responsive cap system for controlled guest release from colloidal mesoporous silica. Angew. Chem., Int. Ed. 2009, 48, 3092–3095. 10.1002/anie.200805818. [DOI] [PubMed] [Google Scholar]
- Slowing I.; Trewyn B. G.; Lin V. S.-Y. Effect of surface functionalization of MCM-41-type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J. Am. Chem. Soc. 2006, 128, 14792–14793. 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- Lu A.-H.; Li W.-C.; Kiefer A.; Schmidt W.; Bill E.; Fink G.; Schüth F. Fabrication of magnetically separable mesostructured silica with an open pore system. J. Am. Chem. Soc. 2004, 126, 8616–8617. 10.1021/ja0486521. [DOI] [PubMed] [Google Scholar]
- Narayan R.; Nayak U.; Raichur A.; Garg S. Mesoporous silica nanoparticles: A comprehensive review on synthesis and recent advances. Pharmaceutics 2018, 10, 118. 10.3390/pharmaceutics10030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann F.; Cornelius M.; Morell J.; Fröba M. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chem., Int. Ed. 2006, 45, 3216–3251. 10.1002/anie.200503075. [DOI] [PubMed] [Google Scholar]
- de Juan F.; Ruiz-Hitzky E. Selective functionalization of mesoporous silica. Adv. Mater. 2000, 12, 430–432. . [DOI] [Google Scholar]
- Cheng K.; Landry C. C. Diffusion-Based deprotection in mesoporous materials: A strategy for differential functionalization of porous silica particles. J. Am. Chem. Soc. 2007, 129, 9674–9685. 10.1021/ja070598b. [DOI] [PubMed] [Google Scholar]
- Luan Z.; Fournier J. A.; Wooten J. B.; Miser D. E.; Chang M. J.. Functionalized mesoporous SBA-15 silica molecular sieves with mercaptopropyl groups: Preparation, characterization and application as adsorbents. Studies in Surface Science and Catalysis; Elsevier, 2005; Vol. 156, pp 897–906. [Google Scholar]
- Burkett S. L.; Sims S. D.; Mann S. Synthesis of hybrid inorganic-organic mesoporous silica by co-condensation of siloxane and organosiloxane precursors. Chem. Commun. 1996, 1367–1368. 10.1039/cc9960001367. [DOI] [Google Scholar]
- Radu D. R.; Lai C.-Y.; Huang J.; Shu X.; Lin V. S.-Y. Fine-tuning the degree of organic functionalization of mesoporous silica nanosphere materials via an interfacially designed co-condensation method. Chem. Commun. 2005, 1264–1266. 10.1039/b412618a. [DOI] [PubMed] [Google Scholar]
- Rosenholm J. M.; Lindén M. Wet-chemical analysis of surface concentration of accessible groups on different amino-functionalized mesoporous SBA-15 silicas. Chem. Mater. 2007, 19, 5023–5034. 10.1021/cm071289n. [DOI] [Google Scholar]
- Calvo A.; Joselevich M.; Soler-Illia G. J. A. A.; Williams F. J. Chemical reactivity of amino-functionalized mesoporous silica thin films obtained by co-condensation and post-grafting routes. Microporous Mesoporous Mater. 2009, 121, 67–72. 10.1016/j.micromeso.2009.01.005. [DOI] [Google Scholar]
- Walcarius A.; Delacôte C. Rate of access to the binding sites in organically modified silicates. 3. Effect of structure and density of functional groups in mesoporous solids obtained by the co-condensation route. Chem. Mater. 2003, 15, 4181–4192. 10.1021/cm031089l. [DOI] [Google Scholar]
- Kecht J.; Schlossbauer A.; Bein T. Selective functionalization of the outer and inner surfaces in mesoporous silica nanoparticles. Chem. Mater. 2008, 20, 7207–7214. 10.1021/cm801484r. [DOI] [Google Scholar]
- Blin J. L.; Otjacques C.; Herrier G.; Su B.-L. Kinetic study of MCM-41 synthesis. Int. J. Inorg. Mater. 2001, 3, 75–86. 10.1016/s1466-6049(00)00043-x. [DOI] [Google Scholar]
- Cauda V.; Schlossbauer A.; Bein T. Bio-degradation study of colloidal mesoporous silica nanoparticles: Effect of surface functionalization with organo-silanes and poly(ethylene glycol). Microporous Mesoporous Mater. 2010, 132, 60–71. 10.1016/j.micromeso.2009.11.015. [DOI] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. Poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem., Int. Ed. 2010, 49, 6288–6308. 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Jenkins S. I.; Weinberg D.; al-Shakli A. F.; Fernandes A. R.; Yiu H. H. P.; Telling N. D.; Roach P.; Chari D. M. Stealth nanoparticles evade neural immune cells but also evade major brain cell populations: Implications for PEG-based neurotherapeutics. J. Controlled Release 2016, 224, 136–145. 10.1016/j.jconrel.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Cauda V.; Argyo C.; Bein T. Impact of different PEGylation patterns on the long-term bio-stability of colloidal mesoporous silica nanoparticles. J. Mater. Chem. 2010, 20, 8693–8699. 10.1039/c0jm01390k. [DOI] [Google Scholar]
- Huh S.; Wiench J. W.; Yoo J.-C.; Pruski M.; Lin V. S.-Y. Organic functionalization and morphology control of mesoporous silicas via a co-condensation synthesis method. Chem. Mater. 2003, 15, 4247–4256. 10.1021/cm0210041. [DOI] [Google Scholar]
- Iliade P.; Miletto I.; Coluccia S.; Berlier G. Functionalization of mesoporous MCM-41 with aminopropyl groups by co-condensation and grafting: a physico-chemical characterization. Res. Chem. Intermed. 2011, 38, 785–794. 10.1007/s11164-011-0417-5. [DOI] [Google Scholar]
- Zimmermann J. L.; Nicolaus T.; Neuert G.; Blank K. Thiol-based, site-specific and covalent immobilization of biomolecules for single-molecule experiments. Nat. Protoc. 2010, 5, 975–985. 10.1038/nprot.2010.49. [DOI] [PubMed] [Google Scholar]
- Hermanson G.Bioconjugate Techniques, 3rd ed.; Academic Press, Elsevier, 2013; Chapter 2. [Google Scholar]
- Rath D.; Rana S.; Parida K. M. Organic amine-functionalized silica-based mesoporous materials: an update of syntheses and catalytic applications. RSC Adv. 2014, 4, 57111–57124. 10.1039/c4ra08005j. [DOI] [Google Scholar]
- Möller K.; Kobler J.; Bein T. Colloidal suspensions of mercapto-functionalized nanosized mesoporous silica. J. Mater. Chem. 2007, 17, 624–631. 10.1039/b611931j. [DOI] [Google Scholar]
- Gaslain F. O. M.; Delacôte C.; Walcarius A.; Lebeau B. One-step preparation of thiol-modified mesoporous silica spheres with various functionalization levels and different pore structures. J. Sol-Gel Sci. Technol. 2008, 49, 112–124. 10.1007/s10971-008-1845-6. [DOI] [Google Scholar]
- García K. P.; Zarschler K.; Barbaro L.; Barreto J. A.; O’Malley W.; Spiccia L.; Stephan H.; Graham B. Zwitterionic-Coated Stealth Nanoparticles for Biomedical Applications: Recent Advances in Countering Biomolecular Corona Formation and Uptake by the Mononuclear Phagocyte System. Small 2014, 10, 2516–2529. 10.1002/smll.201303540. [DOI] [PubMed] [Google Scholar]
- He Q.; Zhang J.; Shi J.; Zhu Z.; Zhang L.; Bu W.; Guo L.; Chen Y. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials 2010, 31, 1085–1092. 10.1016/j.biomaterials.2009.10.046. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Liu F.; Nguyen K. T.; Ma X.; Wang X.; Xing B.; Zhao Y. Multifunctional Mesoporous Silica Nanoparticles for Cancer-Targeted and Controlled Drug Delivery. Adv. Funct. Mater. 2012, 22, 5144–5156. 10.1002/adfm.201201316. [DOI] [Google Scholar]
- Thommes M.; Kaneko K.; Neimark A. V.; Olivier J. P.; Rodriguez-Reinoso F.; Rouquerol J.; Sing K. S. W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution. (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. 10.1515/pac-2014-1117. [DOI] [Google Scholar]
- Kleitz F.; Schmidt W.; Schüth F. Calcination behavior of different surfactant-templated mesostructured silica materials. Microporous Mesoporous Mater. 2003, 65, 1–29. 10.1016/s1387-1811(03)00506-7. [DOI] [Google Scholar]
- Huang H. Y.; Yang R. T.; Chinn D.; Munson C. L. Amine-Grafted MCM-48 and Silica Xerogel as Superior Sorbents for Acidic Gas Removal from Natural Gas. Ind. Eng. Chem. Res. 2003, 42, 2427–2433. 10.1021/ie020440u. [DOI] [Google Scholar]
- Bouchoucha M.; Béliveau É.; Kleitz F.; Calon F.; Fortin M.-A. Antibody-conjugated mesoporous silica nanoparticles for brain microvessel endothelial cell targeting. J. Mater. Chem. B 2017, 5, 7721–7735. 10.1039/c7tb01385j. [DOI] [PubMed] [Google Scholar]
- Brochier Salon M.-C.; Belgacem M. N. Hydrolysis-condensation kinetics of different silane coupling agents. Phosphorus, Sulfur Silicon Relat. Elem. 2011, 186, 240–254. 10.1080/10426507.2010.494644. [DOI] [Google Scholar]
- He Y.; Luo L.; Liang S.; Long M.; Xu H. Amino-functionalized mesoporous silica nanoparticles as efficient carriers for anticancer drug delivery. J. Biomater. Appl. 2017, 32, 524–532. 10.1177/0885328217724638. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang N. Gadolinium loaded nanoparticles in theranostic magnetic resonance imaging. Biomaterials 2012, 33, 5363–5375. 10.1016/j.biomaterials.2012.03.084. [DOI] [PubMed] [Google Scholar]
- Díaz I.; Márques-Alvarez C.; Mohino F.; Pérez-Pariente J.; Sastre E. Combined alkyl and sulfonic acid functionalization of MCM-41-type silica: part 2. Esterification of glycerol with fatty acids. J. Catal. 2000, 193, 295–302. 10.1006/jcat.2000.2899. [DOI] [Google Scholar]
- Margolese D.; Melero J. A.; Christiansen S. C.; Chmelka B. F.; Stucky G. D. Direct syntheses of ordered SBA-15 mesoporous silica containing sulfonic acid groups. Chem. Mater. 2000, 12, 2448–2459. 10.1021/cm0010304. [DOI] [Google Scholar]
- Armstrong J. K.; Wenby R. B.; Meiselman H. J.; Fisher T. C. The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys. J. 2004, 87, 4259–4270. 10.1529/biophysj.104.047746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson H. P. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol. Proced. Online 2009, 11, 32–51. 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formica J. V.; Regelson W. Review of the biology of quercetin and related bioflavonoids. Food Chem. Toxicol. 1995, 33, 1061–1080. 10.1016/0278-6915(95)00077-1. [DOI] [PubMed] [Google Scholar]
- Nair M. P.; Mahajan S.; Reynolds J. L.; Aalinkeel R.; Nair H.; Schwartz S. A.; Kandaswami C. The Flavonoid Quercetin Inhibits Proinflammatory Cytokine (Tumor Necrosis Factor Alpha) Gene Expression in Normal Peripheral Blood Mononuclear Cells via Modulation of the NF- System. Clin. Vaccine Immunol. 2006, 13, 319–328. 10.1128/cvi.13.3.319-328.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapino S.; Ugazio E.; Gastaldi L.; Miletto I.; Berlier G.; Zonari D.; Oliaro-Bosso S. Mesoporous silica as topical nanocarriers for quercetin: characterization and in vitro studies. Eur. J. Pharm. Biopharm. 2015, 89, 116–125. 10.1016/j.ejpb.2014.11.022. [DOI] [PubMed] [Google Scholar]
- Cadena P. G.; Pereira M. A.; Cordeiro R. B. S.; Cavalcanti I. M. F.; Barros Neto B.; Pimentel M. d. C. C. B.; Lima Filho J. L.; Silva V. L.; Santos-Magalhães N. S. Nanoencapsulation of quercetin and resveratrol into elastic liposomes. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 309–316. 10.1016/j.bbamem.2012.10.022. [DOI] [PubMed] [Google Scholar]
- Casagrande R.; Georgetti S. R.; Verri W. A. Jr.; Borin M. F.; Lopez R. F. V.; Fonseca M. J. V. In vitro evaluation of quercetin cutaneous absorption from topical formulations and its functional stability by antioxidant activity. Int. J. Pharm. 2007, 328, 183–190. 10.1016/j.ijpharm.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Berlier G.; Gastaldi L.; Ugazio E.; Miletto I.; Iliade P.; Sapino S. Stabilization of quercetin flavonoid in MCM-41 mesoporous silica: positive effect of surface functionalization. J. Colloid Interface Sci. 2013, 393, 109–118. 10.1016/j.jcis.2012.10.073. [DOI] [PubMed] [Google Scholar]
- Cho S. Y.; Kim M. K.; Park K.-s.; Choo H.; Chong Y. Quercetin-POC conjugates: Differential stability and bioactivity profiles between breast cancer (MCF-7) and colorectal carcinoma (HCT116) cell lines. Bioorg. Med. Chem. 2013, 21, 1671–1679. 10.1016/j.bmc.2013.01.057. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Zhang N. Gadolinium loaded nanoparticles in theranostic magnetic resonance imaging. Biomaterials 2012, 33, 5363–5375. 10.1016/j.biomaterials.2012.03.084. [DOI] [PubMed] [Google Scholar]
- Vivero-Escoto J. L.; Taylor-Pashow K. M. L.; Huxford R. C.; Della Rocca J.; Okoruwa C.; An H.; Lin W.; Lin W. Multifunctional mesoporous silica nanospheres with cleavable Gd(III) chelates as MRI contrast agents: synthesis, characterization, target-specificity, and renal clearance. Small 2011, 7, 3519–3528. 10.1002/smll.201100521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet-Nicolas R.; Bridot J.-L.; Seo Y.; Fortin M.-A.; Kleitz F. Enhanced Relaxometric Properties of MRI “Positive” Contrast Agents Confined in Three-Dimensional Cubic Mesoporous Silica Nanoparticles. Adv. Funct. Mater. 2011, 21, 4653–4662. 10.1002/adfm.201101766. [DOI] [Google Scholar]
- Karaman D. Ş.; Sarparanta M. P.; Rosenholm J. M.; Airaksinen A. J. Multimodality imaging of silica and silicon materials in vivo. Adv. Mater. 2018, 30, 1703651. 10.1002/adma.201703651. [DOI] [PubMed] [Google Scholar]
- Laprise-Pelletier M.; Bouchoucha M.; Lagueux J.; Chevallier P.; Lecomte R.; Gossuin Y.; Kleitz F.; Fortin M.-A. Metal chelate grafting at the surface of mesoporous silica nanoparticles (MSNs): physico-chemical and biomedical imaging assessment. J. Mater. Chem. B 2015, 3, 748–758. 10.1039/c4tb01423e. [DOI] [PubMed] [Google Scholar]
- Galvao J.; Davis B.; Tilley M.; Normando E.; Duchen M. R.; Cordeiro M. F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014, 28, 1317–1330. 10.1096/fj.13-235440. [DOI] [PubMed] [Google Scholar]
- Lu J.; Liong M.; Zink J. I.; Tamanoi F. Mesoporous silica nanoparticles as a delivery system for hydrophobic anticancer drugs. Small 2007, 3, 1341–1346. 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- Toseland C. P. Fluorescent labeling and modification of proteins. J. Chem. Biol. 2013, 6, 85–95. 10.1007/s12154-013-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spötl L.; Sarti A.; Dierich M. P.; Möst J. Cell Membrane labeling with fluorescent dyes for the demonstration of cytokine-induced fusion between monocytes and tumor cells. Cytometry 1995, 21, 160–169. 10.1002/cyto.990210208. [DOI] [PubMed] [Google Scholar]
- Fei X.; Gu Y. Progress in modifications and applications of fluorescent dye probe. Prog. Nat. Sci.: Mater. Int. 2009, 19, 1–7. 10.1016/j.pnsc.2008.06.004. [DOI] [Google Scholar]
- Fisher P. Review of using rhodamine B as marker for wildlife studies. Wildl. Soc. Bull. 1999, 27, 318–329. [Google Scholar]
- Kankaanpää P.; Tiitta S.; Bergman L.; Puranen A.-B.; von Haartman E.; Lindén M.; Heino J. Cellular recognition and macropinocytosis-like internalization of nanoparticles targeted to integrin α2β1. Nanoscale 2015, 7, 17889–17901. 10.1039/c5nr06218g. [DOI] [PubMed] [Google Scholar]
- Northrop B. H.; Frayne S. H.; Choudhary U. Thiol-maleimide “click” chemistry: evaluating the influence of solvent, initiator, and thiol on the reaction mechanism, kinetics, and selectivity. Polym. Chem. 2015, 6, 3415–3430. 10.1039/c5py00168d. [DOI] [Google Scholar]
- Limnell T.; Santos H. A.; Mäkilä E.; Heikkilä T.; Salonen J.; Murzin D. Y.; Kumar N.; Laaksonen T.; Peltonen L.; Hirvonen J. Drug Delivery Formulations of Ordered and Nonordered Mesoporous Silica: Comparison of Three Drug Loading Methods. J. Pharm. Sci. 2011, 100, 3294–3306. 10.1002/jps.22577. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.