

Abstract

Tailoring the polymer melt rheology and the chain relaxation dynamics permits easy handling of polymer processing and enables broader range of applications. Novel strategy to control the polymer melt rheology and the chain relaxation dynamics was devised. A simple process for molecular structural change in a polyamide (nylon 6) to easily generate a long-chain branching in a controllable manner without forming a network structure led to unusually large enhancements in the relaxation dynamics. The zero shear viscosity of the polyamide has increased more than 200 folds of linear chains viscosity, whereas the molar mass change was ca. 1.6 times. Storage modulus and the loss modulus at low frequency increased more than 104 and 103 times to those of neat polyamide without forming a network structure. The rheological properties of the polymer (nylon 6) melts can be finely tailored by this simple process to cover a broad range of applications.
Introduction
Most polymers synthesized by step polymerization are known to have low molar mass compared to other polymers synthesized by free radical polymerization. Due to the low molar mass, their melts generally exhibit quite distinctive rheological properties from the other polymers; they have a wide Newtonian viscosity plateau over a wide range of shear rates and the melt viscosity is quite low.1,2 The consequence of this property is a lack of processibility, particularly in extrusion process such as a blow molding process, due to a low shear viscosity at low shear rates. Moreover, their weak strain hardening gives rise to difficulties in processing operations in which elongational properties dominate.2 The issue is then how to control the rheological properties of those polymers to easily meet the requirement for various processing conditions. One possible method to cure the characteristic rheological properties is to change the molar mass and structure of the molecules in a controllable manner because the chain relaxation dynamics and rheological properties of polymer melts are profoundly influenced by the molar mass, polymer molecule’s structure, and ensuing molecular entanglements.
Entangled polymer melts can display a wide range of rheological behaviors depending on the molecular interactions. Sufficiently entangled polymer melts show a strong viscosity (η) increase with their molar mass (M), η ∼ M3, compared to unentangled polymers (η ∼ M).3 Reptation theory with contour length fluctuations and constraint-release effects predicts that the shear viscosity of entangled linear polymer melts scales with the molecular weight as M3.4.3 On the other hand, long-chain branching polymers, prepared by reacting chain-end functional groups with a multifunctionalized molecules, generally exhibit properties that differ significantly from those of linear polymers, even with a low degree of branches.4,5 Dynamics of branched polymers can be described by a tube theory, but they cannot reptate easily within the relaxation times of normal linear polymers because of their central branch point.6 Instead, they renew their configurations and thus relax stresses by contour length fluctuations of the arms. In this case, the total stress after a step strain is proportional to the length of the tube still unvisited by a chain end.7 Theoretical calculation of full relaxation spectrum by Pearson and Helfand showed that the relaxation of branched chain molecules like a star polymer or a H-polymer can be exponentially slower than those of linear molecules due to their long arms.8 However, general star or H-polymer molecules of long chain length have some problems in processing due to their extremely slow relaxation and chain dynamics.5,8,9
In this study, we tried to solve those rheological problems of polymers synthesized by the step polymerization method using a simple method, i.e., to increase the molar mass by a linking agent that can connect two chains in a linear matter first to double its molar mass and then later generate 3- or 4-armed polymers in a controllable manner. Here, we adopt a simple reactive extrusion to change the structure of a polyamide (nylon 6) with a linking agent. The chain relaxation dynamics were varied over a few orders of magnitude through the control of the linking agent concentration. This is ascribed to the tailoring of long-chain branching without a network structure (or a gel) generation in the melt. The relaxation dynamics and the flow properties of a polymer melt could undergo unusually large enhancements in a controllable magnitude depending on the amount of the linking agent by the 3- or 4-armed star polymer molecules of which branching emanates from the connected part of the molecules.
Results and Discussion
Early intention of a diepoxy (4,4′-di(2,3-epoxypropyloxy)phenyl benzoate, abbreviated DEPPB) addition was to enhance the molar mass of nylon 6 (Scheme 1a) by connecting two molecules through the additive molecule because the molar mass increase would increase more chain entanglements. The chemical structure of the reaction (a) product, an extended polyamide 6, in Scheme 1 could be verified by the heteronuclear single quantum coherence (HSQC) spectra (Figure 1). 1H NMR spectra of neat KN171 and sample #6 containing DEPPB of which the ratio between amine and epoxy groups was 1:2 were nearly identical. However, the weak signal of aromatic protons of the extender (DEPPB) appeared at 6.8–8.2 ppm in a spectrum of extended polyamide 6. The HSQC spectra of an extended polyamide 6 showed that all aromatic protons were correlated with aromatic carbons and their signal was attributed to CH (Figure 1a). Furthermore, chemical shift of E carbon (67.9 ppm) due to the reaction between epoxide and diamine could be confirmed (Figure 1b), though the reaction (b) of Scheme 1 could not be identified because the signal of the tertiary amine was overlapped with that of other amines.
Scheme 1. Reaction of the Linking Agent (DEPPB) with Nylon 6 Molecules.

(a) The reactive group molar ratio is 1:1 (linking of two nylon 6 molecules). (b) The reactive group molar ratio is greater than 1:1 (3- or 4-armed starlike polymer formation).
Figure 1.

(a) HQSC spectra of an extended polyamide 6, which showed that all aromatic protons were correlated with aromatic carbons and their signal was attributed to the CH. (b) Confirmation of the chemical shift of E carbon (67.9 ppm) due to the reaction between epoxide and amine end group of nylon 6.
Figure 2 shows the melt viscosities for differently modified nylon 6 as a function of frequency. There is a broad Newtonian plateau of low viscosity for neat nylon 6 melt at the frequencies below 100 rad/s, above which it exhibits a weak shear thinning. Modified nylon 6 reveals a different trend as the linking agent was added. The shear thinning region widens and the absolute value of the melt viscosity, η, at low frequencies increases significantly. When the specimen had more molar amount of epoxy group than the amine group of nylon 6 (over 1:1 ratio), the melt viscosities increased substantially higher than that of neat nylon 6. Surprisingly, there is no apparent Newtonian plateau for these samples, but only a strong shear-thinning behavior is observed throughout the whole frequency range. To the best of our knowledge, this is the first observation that neat nylon 6 melt without any other fillers shows only shear thinning behavior without the appearance of the Newtonian plateau in the frequency range from 0.1 to 500 rad/s. The shear thinning behavior of the modified nylon 6 melt in Figure 2 could be well described by the simple Carreau equation with Cox–Merz rule, η(γ)/η0 ≈ (1 + (γτn)2)(n−1)/2, where η0 is the zero shear rate viscosity, γ is the shear rate, and τn is the characteristic time.11 The zero shear rate viscosity obtained from the Carreau model (Table 1) increased with the added DEPPB concentration. After reaching the peak of 1.09 × 105 Pa s for DEPPB(2), it decreased to 9.07 × 104 Pa s for DEPPB(2.25). The melt viscosity at the peak was more than 200 times larger than that of the pure nylon 6 melt.
Figure 2.
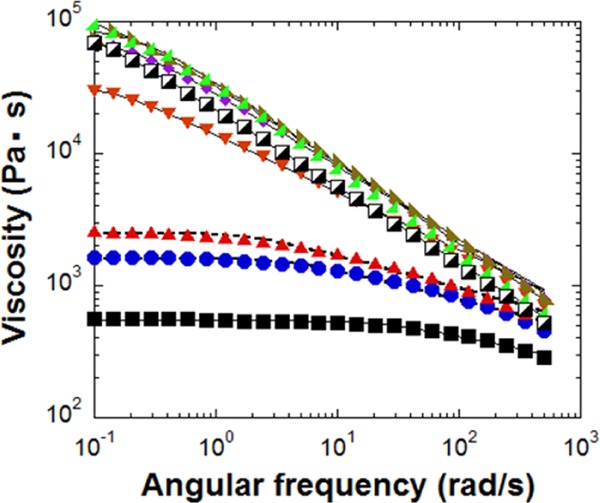
Dynamic melt viscosities of neat nylon 6 (■) and modified nylon 6 (approximate ratio between amine and epoxy groups: (●) 1:0.2, (▰) 1:0.4, (▼) 1:0.6, (⧫) 1:1, (◪) 1:1.25, (Black Lower Right Triangle) 1:2, and (Black Lower Left Triangle) 1:2.25 measured at 250 °C).
Table 1. Viscosity Molar Mass Calculated from the Mark–Houwink Equation13 a.
| molar ratio between the epoxy group of the linking agent and the amine group of nylon 6 | viscosity molar mass (g/mol) after the reaction | zero shear rate viscosity (Pa s) from the Carreau equation |
|---|---|---|
| 0.0 (nylon 6) | 3.24 × 104 | 540 |
| 0.2 | 3.38 × 104 | 1600 |
| 0.4 | 3.76 × 104 | 2500 |
| 0.6 | 3.97 × 104 | 3.33 × 104 |
| 1. | 4.54 × 104 | 7.24 × 104 |
| 1.25 | 4.91 × 104 | 8..85 ×104 |
| 2. | 4.97 × 104 | 10.9 × 104 |
| 2.25 | 4.18 × 104 | 9.07 × 104 |
Standard deviation is less than ±2%.
The intrinsic viscosity was measured to calculate the molar mass using the Mark–Houwink equation (Table 1).12 Like the melt viscosity, the viscosity average molar mass reached the maximum when the molar ratio of the reactive functional groups was 2 (DEPPB(2)), beyond which it decreased. It should be noted that the viscosity average molar mass of DEPPB(2) was ca 1.6 times of pure nylon 6. For the linear polymer chains, this slight molar mass change (1.6 times) cannot enhance the melt viscosity more than 5 times of pure nylon 6, assuming them to follow the 3.4 power-law (η ∼ M3.4).6 Hence, remarkable increase in the melt viscosity of DEPPB(2) with a strong shear thinning without the Newtonian plateau cannot be reconciled with the increase in the linear molar mass by the linking agent.
Huge increase in the melt viscosity may hint the occurrence of the cross-linking reaction that DEPPB molecules trigger to form a gel-like structure. However, the melt viscosity of other samples containing more DEPPB than DEPPB(2) exhibited reductions in the melt viscosity (Figure 2). Hence, it cannot be reconciled within the frame work of cross-linking reactions because more DEPPB addition definitely incurs more cross-linking reaction, which results in the higher melt viscosity. More importantly, three dimensionally cross-linked polymers are incapable of macroscopic viscous flow because, at moderate level, the cross-links prevent the network chains from flowing past one another.12 When the first network molecule is formed in a nonlinear polymer networks, it encompasses the whole reactant mixture, which instantly becomes immobilized: this corresponds to the gel point.4,7 However, the present reacted extrudates can be reprocessed many times without showing any signs of solidification in extrudates (Figure 3). The extrudates look like normal nylon 6 pellets. If cross-linking reaction has happened, the extrudates should be in the form of a powder or display anomalous topology. Under the observation of optical microscope, all specimen films became transparent above the melt temperature of nylon 6 (ca. 240 °C). Another point excluding the possibility of gelation is that the starting monomers should have functionality of three or higher to form the complex network structure.12,16 To proceed the cross-linking reaction, the reacting polymers should have diamine groups, but the nylon 6 molecules have monoamine group at the chain end and the diepoxides have the functionality of two. The other chain end group of nylon 6 is the initiator of ε-caprolactam polymerization. Thus, they cannot cause any cross-linking reaction at all.18 Also, the light-scattering analysis of the polymer solution did not detect any insoluble particles in the formic acid. The consequence of all these observations is that the cross-linking reaction or gelation could not be the reason for the strong shear thinning and unexpected high melt viscosity at low shear rates.
Figure 3.

Photographs of extruded strands after pelletization.
Similar drastic changes can be observed in the dynamic moduli (storage modulus (G′) and loss modulus (G″)) in Figure 4. The storage modulus is in phase with the deformation, whereas the loss modulus is out of phase with the deformation.4 In non-cross-linked linear polymer melts, G′ is expected to be proportional to ω2 and G″ to ω at low frequency, showing the linear viscoelastic (LVE) behavior.7 The flow of nylon 6 melt follows this behavior, but modified nylon 6 melts exhibit large deviations from the linear viscoelastic behavior. The power-law dependence of G′ and G″ on the frequency becomes monotonically weaker with the addition of the linking agent. The significance of this behavior is that the dynamics steadily changes from the LVE behavior to nonlinear viscoelastic behavior. The G′ value of DEPPB(2) at low frequency shows an increase of almost 4 order (104 times) higher than that of nylon 6, whereas G″ value of the same sample exhibits ca. 3 order (103 times) increase. After DEPPB (2), G′ and G″ values decreased similarly to the melt viscosity change. The interaction frequency (ωc) of the G′ and the G″ provides the chains’ relaxation time, τ = 1/ωc.7 The relaxation time of the modified melts was the longest for DEPPB(2) (≈ca. 15 s). Hence, the molecules relax most slowly for the sample that displays the maximum zero-shear rate viscosity.
Figure 4.

(a) Storage modulus and (b) loss modulus of neat nylon 6 and modified nylon 6. (c) Comparison of storage modulus and loss modulus of nylon 6 and two modified nylons (DEPPB(1) and DEPPB(2)). (d) The reduced van Gurp–Palmen (rvGP) plot of nylon 6 and modified nylon 6. The rheological properties were measured at 250 °C.
Less than 1.6 times molar mass change for DEPPB(2) cannot describe the whole rheological property changes (Table 1 and Figures 2 and 4). Possible topological structure like a gel was also excluded due to the downturn of the viscosity and the relaxation time with more addition of DEPPB after DEPPB(2). The other possible and plausible topological structure suitable for the explanation of all the rheological behaviors is the branching structure.9,13 DEPPB can react with the amine end groups of two nylon 6 molecules through the chain-linking reaction (Scheme 1a). Connecting two nylon 6 molecules into one doubles the molar mass which can generate more entanglements between chains and induce higher viscosity because of slow relaxation. But, it turns out that further reaction of the secondary amine after the primary amine reaction can be proceeded to produce 3- or 4-armed (H-type) star polymers. The chemical reaction between the epoxy group of DEPPB and primary amine group of nylon 6 is plausibly proceeded first as in Scheme 1a. The most commonly observed reaction mechanism between an epoxy group and an amine group is autocatalytic, where the epoxy ring opens to form a hydroxyl group, which is able to catalyze further the amine-epoxy reactions.10 After the first reaction, the secondary amine can further react with other epoxy groups to form a three- or four-armed (H-shape) chains (reaction (b) of Scheme 1). Due to the steric hindrance and low reactivity, amide groups of nylon 6 chains cannot undergo branch-forming reaction as actively as the secondary amine group of the linking agent, which generates three- or four-armed (H-shape) star polymers (reaction (b) of Scheme 1). If 4-armed (H-shape) chains are mainly produced in the extruder, the reacting functional group ratio (epoxy to amine) is simply calculated as 1.5 for four-armed (H) polymers. The data show the highest melt viscosity for DEPPB (2), which is ascribable to some dangling DEPPB moiety rather than connecting two chains and forming branches.
The branched chains have longer relaxation times to show complex thermorheological behavior in which time−temperature superposition fails.9,13 One way of distinguishing the complex thermorheological behavior from the linear chain’s behavior is to plot the phase angle (δ) of the loss tangent (tan δ = G″/G′) versus the absolute value of the complex modulus, |G*(ω)|, called the van Gurp–Palmen (vGP) plot.14 The van Gurp–Palmen plot was found to be quite useful to characterize different architecture and topology such as polydispersity of linear polymers, long chain branching (LCB), gelation, and filler interaction in nanocomposites.15−17 To sketch the data of different samples directly in one figure regardless of their chemical constitution or chemical topology, so-called reduced van Gurp–Palmen (rvGP) plot was adopted to plot the phase angle (δ) versus the reduced absolute value of the complex modulus, |G*(ω)|/GN0, where GN0 is the plateau modulus. Here, the plateau modulus, GN0, was determined by the crossover modulus-based method (GN0 = Gc′ = Gc″ at angular frequency of ω = ωc).20 Keunings et al. compared several methods to determine the plateau modulus, GN0.20 According to their analysis, because G″ sometimes has no maximum nor a minimum when the MWD is very broad and/or the MW very low, especially if the polymer is semicrystalline, other methods are not applicable to determine the plateau modulus. Though the crossover modulus-based method underestimates a little bit for polyolefin polymers, it is useful for the semicrystalline polycondensates and ring-opening polymers, e.g., poly (caprolactam) (Ny 6), poly(hexamethylene adipamide) (Ny 66).20 The x-axis in the rvGP plot is in logarithmic scale, which reduces the curve movement in the horizontal direction. Thus, the qualitative nature of the rvGP plot is not remarkably affected. The rvGP plot rather than vGP plot was used in this study because it turned out to be quite useful to distinguish the long-chain branching topology.17−19
Polymers with a linear topology (linear molecules) have a unique course of a curve in the rvGP plot like those presented in Figure 4d. It is apparent from the rvGP plot and the data in Table 1 that nylon 6 mixtures with DEPPB up to 0.4 functional group molar ratio exhibit similar rheological behavior (simple fluids of linear polymers), although the curves shifted downward because of increased molar mass and molecular weight distribution.15−17 Other samples containing more DEPPB molecules than DEPPB(1) show a totally different behavior; the angle decreased rapidly and then became almost flat. This is rheologically complex fluid behavior by the long-chain branching or gelation.4,16 Rapid decrease is due to the enhancement of elasticity. The gelation effect is excluded again because, if this is due to the gelation (cross-linking), the angle |δ| should go down further.17 Instead, there appears a plateau-like flat region with the increase of modulus, which indicates that the melts are strongly viscoelastic but not dominantly elastic like a gel.17−19 δ vs |G*(ω)|/GN0 does exhibit significant differences with the modifier (DEPPB) content, reflecting the influence of the branches on the relaxation spectrum. The more DEPPB is added, the stronger is the deviation from the curve of the linear chains. This indicates the generation of more branched molecules. However, excessive modifier forms dangling chains rather than connecting two nylon 6 chains or forming branched molecules, whose relaxation spectrum decreases due to the less branched molecules.
Moving from high to low modulus values, the phase angle δ in the rvGP plot rises to reach the limiting value at the plateau modulus, GN0.17 This characteristic curvature is found for semicrystalline linear polymers.18 For amorphous polymers like polystyrene or poly(methyl methacrylate), the phase angle δ drops, passes a minimum, rises again, moves through an inflection point, and fully approaches its limiting value of 90° while moving from high to low modulus values.17 For DEPPB(1.25) sample, the δ(|G*(ω)|/GN0) curve differs significantly from that of the linear samples and is the same for all other samples containing more DEPPB than DEPPB(1.25). Their shapes cannot be superposed to make a master curve. In the case of thermorheologically complex fluids, G′(ω) as well as G″(ω) cannot be superimposed, as not all relaxation times have the same temperature dependence, whereas thermorheologically simple fluids can form a master curve due to the same temperature dependence of all the relaxation times.4,7 As the DEPPB amount increases, the δ value decreases rapidly with increasing |G*(ω)|/GN0 to reach a flat region. This behavior is different from that of a gel in the rvGP plot. The angle δ of a gel increases, passes through the maximum, and then decreases.19 The relaxation behavior of long-chain branching (LCB) molecules are known to be similar to that of a gel network because LCBs are entangled in the melt state, which is similar to that of a physical gel.21 However, rvGP plot differentiate the LCB behavior from that of a gel. This feature agrees with the other branched polymer’s behavior; δ curves for DEPPB(1.25) to DEPPB(2) samples are shifted downward in comparison to the curve for linear samples, demonstrating the influence of such chain branching.
In polymer systems of long-chain branches, the relaxation times are significantly increased due to the chain entanglements of the branches.7 The melt viscosity of a branched polymer grows exponentially with the number of entanglements per arm (η0 ∼ (Ma/Me)3/2 exp(νMa/Me), where Ma is the arm’s molecular weight, Me is the entanglement molecular weight, and ν is the constant13) and is independent of the number of arms, f (3 or 4 gave the same results9). The tube model gives ν value of 15:8, but the dynamic dilution of effective entanglement gives a smaller value by a factor of 3, which was in good agreement with the reported data.13 Using the zero shear rate viscosity from the Carreau model and the viscosity average molar mass gives ν value of 0.32 ± 0.03 for the samples showing high shear thinning behavior (DEPPB(1.25), DEPPB(2), and DEPPB(2.25) in Figure 2 and Figure 4). This is smaller than the experimental data of Fetters et al.9 (ν was 0.47), possibly due to incomplete branching structure, wide polydispersity of nylon 6 and the use of the viscosity average molar mass rather than the weight average molar mass.
The chain relaxation is also reflected in the glass transition temperature (Tg). Addition of the linking agent into the neat nylon 6 melts results in the increase in Tg due to the restriction of chain relaxation by chain branching.7Figure 5a shows the glass transition temperature of modified nylon 6 samples having a higher Tg than that of neat nylon 6. DEPPB(2.25) shows a downward shift, indicating that it is not because of the gelation (or cross-linking) of the polymers.16 The topology variation does not hamper chain ordering in the cooling process (crystallization) because branches do not take part in the same folding sheet of nylon 6, or they belong to other folding sheet.22 In a system that undergoes significant structural topology change in the crystal lattice, the melt dynamics can display a complex rheological behavior. However, X-ray diffraction (XRD) measurement in Figure 5b reveals that there is no effect of chain branching on the crystal lattice. The pair of peaks at 2θ = 20.5 and 24° are distinctive features of the α-plane in nylon 6.22 They are the reflections from the (020) and (002) planes.22,23 The degree of crystallinity determined using the X-ray peaks was ca 39.5% for neat nylon 6 and 39 (±0.5%) for other modified ones.
Figure 5.

(a) Variation in the glass transition temperature with the linking agent (line is a guide for eyes). Data are average values of three independent measurements. (b) X-ray diffraction patterns (approximate ratio between reacted amine and epoxy groups: (black line) nylon 6, (green line) 1:0.4, (blue line) 1:1, and (red line) 1:2.5). They show almost the same crystallinity of ca. 39.5% for neat nylon 6 and 39 (+0.5%) for other modified ones.
Conclusions
The chain relaxation dynamics variation over a few orders of magnitude could be easily controlled by the tailoring of chains topology in the melt through the addition of proper amount of the linking agent. A simple process for molecular structural change to easily generate a long chain branching in a controllable manner without forming a network structure was devised on the basis of the secondary amine reaction. The relaxation dynamics and the rheological properties of a polymer melt can undergo unusually large enhancements by the probable generation of 3- or 4-armed (H) star polymer molecules, whose branching emanates from the linking molecules. The zero shear viscosity increased more than 200 times the linear chains viscosity. Storage modulus and the loss modulus at low frequency increased more than 104 and 103 times that of that of neat polyamides without forming a network structure. These findings overcome the rheological barrier of polyamide melts to open up new possibilities for wider applications.
Experimental Section
As a linking agent, diepoxy (4,4′-di(2,3-epoxypropyloxy)phenyl benzoate, shortly DEPPB) was synthesized with the following chemical structure.

DEPPB was synthesized through the following procedure. (1) (4,4′-Dihydroxyphenyl benzoate) (DHPB) was synthesized first. 4-Hydroxybenzoic acid (60 g, 0.43 mol) and NaOH (38 g, 0.96 mol) were added to 900 mL of water and the mixture stirred for 10 min at 5 °C. Ethylchloroformate (54 g, 0.5 mol) was added dropwise into the solution and then stirred for 10 min. A 2N HCl solution (500 mL) was poured into the solution and the resulting white precipitate was filtered and washed with water several times. The solid product was recrystallized from acetone and white needle-like crystals were obtained. These crystals and a few drops of N,N-dimethylformamide were poured into thionyl chloride and boiled for 1 h. The excess thionyl chloride was evaporated under vacuum at 10–2 mmHg. The product was dissolved in 300 mL of CH2Cl2 and the solution was added dropwise to a tetrahydrofuran solution containing hydroquinone (55 g, 0.5 mol) and pyridine (40 mL, 0.5 mol). After reaction for 12 h at room temperature, the solution was precipitated in 2N NaOH solution. After filtration, the precipitate was washed with excess water and then dissolved in ethanol. A 2N NaOH solution and acetic acid were added successively to the ethanol solution. After stirring for 2 h, ethanol was evaporated and the product washed with water and then recrystallized from methylcellulose acetate. The yield was 68% (67 g). (2) A mixture of DHPB (33 g, 0.2 mol), 3-bromopropene (26 mL, 0.30 mol), and K2CO3 (70 g, 0.5 mol) was added to 400 mL of acetone and then boiled for 24 h. The solid was filtered and acetone was evaporated for 24 h. The remaining solid was washed successively with 5% Na2CO3 solution, excess water, and 200 mL of cold ethanol. After drying, a white powder was obtained, which was recrystallized from acetonitrile/isopropanol (1:1). The yield was 74% (46 g). (3) Diallyl monomer (31 g, 0.1 mol) from step 2 was oxidized with 3-chloroperoxybenzoic acid and the product recrystallized from acetonitrille/isopropanol (1:1). The yield was 75% (25 g). The melting temperature (Tm) of the product was found to be 119 °C. 1H NMR (CDCl3): 2.79 (2H, dd, CH2 of epoxy), 2.92 (2H, dd, CH2 of epoxy), 3.39 (2H,m, CH of epoxy), 3.94 (CH2 of glycidyl), 4.27 (CH2 of glycidyl), 7.02 (4H, d, aromatic), 8.15 (2H,d, aromatic). IR (KBR pellet): 921 cm–1 (oxirane), 1256 cm–1 (−C–O−), 1728 cm–1 (carbonyl).
Nylon 6 was a Kolon product (KN171, Korea). The weight average molar mass was found to be 8.5 × 104 g/mol with a polydispersity index of 3.5. This molar mass was much higher than Me of nylon 6 (2233 g mol–1).4 Nylon 6 pellets and DEPPB powder were dried in a vacuum oven at 100 and 80 °C, respectively, for 24 h. The DEPPB powder was then premixed in a container with dried nylon 6 pellets at a predetermined weight ratio. Finally, the mixture was blended in a twin screw extruder (PRISM) at 280 °C. The extrudates were pelletized to be used for further characterization. All the extrudates showed the same shape (cylindrical rod) as the nylon 6 extrudates, as shown later, which means no structural variation such as gellation (or cross-linking) happened. If gellation or cross-linking occurred, the extrudates come out as a brittle powder.
Fourier transform infrared spectra were obtained using a Bruker 200 spectrometer (IF 66) with an average of 200 scans at a resolution of 4 cm–1. The NMR samples were prepared by dissolving the pellets in formic acid-d2 with heating. Formic acid was used as both solvent and internal standard (1H, 13C). 1H and 13C heteronuclear single quantum coherence (HSQC) NMR spectra were collected on an 850 MHz Bruker AVANCE HD III spectrometer at the NCIRF at Seoul National University. The XRD patterns for the prepared specimens were recorded on a Rigaku D/max 3C diffractometer using a filtered Cu Kα radiation (λ = 0.15406 nm). The diffractometer was operated at 40 kV and 40 mA. The XRD data were collected from 10 to 35° (2θ) in a fixed time mode with a step interval of 0.02° (2θ).
Differential scanning calorimetry (DSC, Mettler DSC 30) measurements were performed under nitrogen atmosphere. The rheological properties were measured at 250 °C with a UDS200 (Physica, Germany) rheometer on which 25 mm diameter cone and plate were mounted. The frequency range was set at 0.1–500 rad/s, and the applied strain was 5%. Before the measurement, the samples were prepared with a compression molder at 280 °C. The measurements were carried out under a nitrogen atmosphere. The viscosity molar mass was calculated by the Mark–Houwink equation of [η] = k[M]a with k = 0.0226 and a = 0.82, where [η] is the intrinsic viscosity, which was measured using an Ubelhode viscometer at 25 °C.13 Formic acid (85%) was used as a solvent.
Acknowledgments
This work was supported by NRF (BK21Plus MSE SNU) and MIKE (RIAM No. 10037237, Fundamental R&D Program for Core Technology of Materials).
The authors declare no competing financial interest.
References
- Laun H. M. Das viskoelastische verhalten von polyamid-6-schmelzen. Rheol. Acta 1979, 18, 478–491. 10.1007/BF01736954. [DOI] [Google Scholar]
- Dijkstra D. J. Guidelines for rheological characterization of polyamide melts. Pure Appl. Chem. 2009, 81, 339–349. 10.1351/PAC-REP-08-07-22. [DOI] [Google Scholar]
- de Gennes P. G.Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, 1980. [Google Scholar]
- Dealy J. M.; Larson R. G.. Structure and Rheology of Molten Polymers; Hanser: Munich, 2006. [Google Scholar]
- Read D. J.; Auhl D.; Das C.; den Doelder J.; Kapnistos M.; Vittorias I.; McLeish T. C. B. Linking models of polymerization and dynamics to predict branched polymer structure and flow. Science 2011, 333, 1871–1874. 10.1126/science.1207060. [DOI] [PubMed] [Google Scholar]
- Doi M.; Edwards S. F.. The Rheology of Polymer Dynamics; Oxford University Press: Oxford, 1986. [Google Scholar]
- Rubinstein M.; Colby R.. Polymer Physics; Oxford University Press: Oxford, 2003. [Google Scholar]
- Pearson D. S.; Helfand E. Viscoelastic properties of star-shaped polymers. Macromolecules 1984, 17, 888–895. 10.1021/ma00134a060. [DOI] [Google Scholar]
- Fetters L. J.; Kiss A. D.; Pearson D. S.; Quack G. F.; Vitus F. J. Rheological behavior of star-shaped polymers. Macromolecules 1993, 26, 647–654. 10.1021/ma00056a015. [DOI] [Google Scholar]
- Wang X.; Gillham J. K. Studies on the preparation and properties of conductive polymers. IV. Novel method to prepare metallized plastics from metal chelates of polyamides-imides. J. Appl. Polym. Sci. 1991, 43, 2267–2277. 10.1002/app.1991.070431216. [DOI] [Google Scholar]
- Carreau P. J.; De Kee D. C. R.; Chhabra R. P.. Rheology of Polymeric Systems; Hanser: Munich, 1997. [Google Scholar]
- Young R. J.; Lovell P. A.. Introduction to Polymers, 3th ed.; CRC Press: Boca Raton, 2011. [Google Scholar]
- Ball R. C.; McLeish T. C. B. Dynamic dilution and the viscosity of star-polymer melts. Macromolecules 1989, 22, 1911–1913. 10.1021/ma00194a066. [DOI] [Google Scholar]
- van Gurp M.; Palmen J. Time-temperature superposition for polymeric blends. J. Rheol. Bull. 1998, 64, 5–8. [Google Scholar]
- Stange J.; Wächter S.; Münstedt H.; Kasper H. Linear rheological properties of the semifluorinated copolymer tetrafluoroethylene-hexafluoropropylene-vinylidenfluoride (THV) with controlled amounts of long-chain branching. Macromolecules 2007, 40, 2409–2416. 10.1021/ma0626867. [DOI] [Google Scholar]
- García-Franco C. A.; Lohse D. J.; Robertson C. G.; Georjon O. Relative quantification of long chain branching in essentially linear polyethylenes. Eur. Polym. J. 2008, 44, 376–391. 10.1016/j.eurpolymj.2007.10.030. [DOI] [Google Scholar]
- Trinkle S.; Walter P.; Friedrich C. Van Gurp-Palmen Plot II—classification of long chain branched polymers by their topology. Rheol. Acta 2002, 41, 103–113. 10.1007/s003970200010. [DOI] [Google Scholar]
- Dordinejad A. K.; Jafari S. H. A qualitative assessment of long chain branching content in LLDPE, LDPE and their blends via thermorheological analysis. J. Appl. Polym. Sci. 2013, 130, 3240–3250. 10.1002/app.39560. [DOI] [Google Scholar]
- Schlatter G.; Fleury G.; Muller R. Fourier transform rheology of branched polyethylene: Experiments and models for assessing the macromolecular architecture. Macromolecules 2005, 38, 6492–6503. 10.1021/ma0505530. [DOI] [Google Scholar]
- Liu C.; He J.; van Ruymbeke E.; Keunings R.; Bailly C. Evaluation of different methods for the determination of the plateau modulus and the entanglement molecular weight. Polymer 2006, 47, 4461–4479. 10.1016/j.polymer.2006.04.054. [DOI] [Google Scholar]
- Kjøniksen A.-L.; Nystrom B. Effects of polymer concentration and cross-linking density on rheology of chemically cross-linked poly(vinyl alcohol) near the gelation threshold. Macromolecules 1996, 29, 5215–5222. 10.1021/ma960094q. [DOI] [Google Scholar]
- Li Y.; Goddard W. A. III Nylon 6 crystal structures, folds and lamellae from theory. Macromolecules 2002, 35, 8440–8455. 10.1021/ma020815n. [DOI] [Google Scholar]
- Holmes D. R.; Bunn C. W.; Smith D. J. The crystal structure of polycaprolactams: Nylon 6. J. Polym. Sci. 1955, 17, 159–177. 10.1002/pol.1955.120178401. [DOI] [Google Scholar]