

Abstract
Craniosynostosis, the premature ossification of cranial sutures, is a developmental disorder of the skull vault, occurring in approximately 1 in 2250 births. The causes are heterogeneous, with a monogenic basis identified in ~25% of patients. Using whole-genome sequencing, we identified a novel, de novo variant in BCL11B, c.7C>A, encoding an R3S substitution (p.R3S), in a male patient with coronal suture synostosis. BCL11B is a transcription factor that interacts directly with the nucleosome remodelling and deacetylation complex (NuRD) and polycomb-related complex 2 (PRC2) through the invariant proteins RBBP4 and RBBP7. The p.R3S substitution occurs within a conserved amino-terminal motif (RRKQxxP) of BCL11B and reduces interaction with both transcriptional complexes. Equilibrium binding studies and molecular dynamics simulations show that the p.R3S substitution disrupts ionic coordination between BCL11B and the RBBP4–MTA1 complex, a subassembly of the NuRD complex, and increases the conformational flexibility of Arg-4, Lys-5 and Gln-6 of BCL11B. These alterations collectively reduce the affinity of BCL11B p.R3S for the RBBP4–MTA1 complex by nearly an order of magnitude. We generated a mouse model of the BCL11B p.R3S substitution using a CRISPR-Cas9-based approach, and we report herein that these mice exhibit craniosynostosis of the coronal suture, as well as other cranial sutures. This finding provides strong evidence that the BCL11B p.R3S substitution is causally associated with craniosynostosis and confirms an important role for BCL11B in the maintenance of cranial suture patency.
Introduction
The flat bones of the mammalian skull and face develop by direct intramembranous ossification of mesenchyme, which is principally derived from mesoderm and neural crest cells (NCCs) (1). Craniofacial sutures are fibrocellular structures that form at the margins of developing bones and limit skull deformation due to both tensile and compressive forces (2). Development and maintenance of sutures is essential and precisely controlled to match skull expansion with the rapid increase in brain size of infants (3).
The major sutures of the skull vault include the metopic and sagittal sutures that separate the paired frontal and parietal bones, respectively, the coronal sutures that form between the parietal and frontal bones and the lambdoid sutures between the parietal and occipital bones. Suture formation, maintenance and subsequent ossification, the latter of which typically begins in the third decade of human life, require the exquisitely choreographed balance of proliferation and differentiation of sutural stem cells and their progeny including bone-forming cells localized at the boundaries of sutures (4).
Craniosynostosis is the most clinically important developmental disorder of the skull vault. Associations of this disorder include restricted skull expansion, increased intracranial pressure and craniofacial dysmorphologies, all of which negatively impact respiration, sensory systems and cognition (5). Failure of the mechanisms that establish the suture or that are responsible for maintaining sutural patency result in craniosynostosis (2).
To date a genetic cause has been identified in approximately a quarter of craniosynostosis cases with important implications for counselling and management (6). Mutations have been detected predominantly in the fibroblast growth factor receptors (FGFR2 and FGFR3), TWIST1 and EFNB1 (7,8). More recently, the use of high-throughput sequencing has implicated mutations in many other genes in the aetiology of craniosynostosis, including TCF12 (9), SMAD6 (10), ERF (11), CDC45 (12) and ZIC1 (13). These findings indicate that the causes of craniosynostosis are heterogeneous and involve many different pathways. In addition, craniosynostosis can be present as a consistent, although rare, feature where mutations have a non-specific impact on osteogenesis, for example affecting osteoblast to osteoclast balance or chromatin modification (8).
Heterozygous mutations in BCL11B have been linked to human pathologies affecting the immune system, intellectual impairment, developmental abnormalities, as well as dermal and craniofacial defects (14,15). BCL11B plays an essential role in the development of the immune (14,16–22), central nervous (23–25) and cutaneous (26) systems and teeth (27,28). Disruption of a single Bcl11b allele in mice results in synostoses within the facial skeleton (29). Bcl11b−/− mice, as well as mice lacking BCL11B in neural crest-derived cells (Bcl11bncc−/−), exhibit abnormally small and misshapen heads, severe midface hypoplasia and malocclusion. Fusion of multiple facial sutures is seen in these mice, including the internasal, naso-premaxillary, interfrontal, premaxillary-maxillary and temporal sutures. Some Bcl11b−/− skulls are characterized by a high degree of porosity, mirroring observations in human patients with increased intracranial pressure (29).
Bcl11b−/−, but not Bcl11bncc−/−, mice exhibit severe coronal suture craniosynostosis, which initiates around embryonic day (E) 16.5—E18.5 (29). BCL11B is expressed in the coronal suture beginning at E14.5 and loss of BCL11B results in ectopic expression of both Runx2 and Fgfr2c within the sutural mesenchyme at E16.5, together with concomitant downregulation of Twist1 expression and ossification of the suture (29). These findings collectively suggest a role for BCL11B in maintaining sutural patency in the mammalian craniofacial skeleton.
Here we describe identification of a patient with craniosynostosis carrying a de novo point mutation in BCL11B and report the generation of a mouse model that recapitulates the human phenotype. The mutation results in a single amino acid substitution, p.R3S, which severely compromises interaction of BCL11B with transcriptional regulatory complexes containing RBBP4 and RBBP7, but not other transcriptional complexes with which BCL11B interacts. Our data demonstrate that the BCL11B p.R3S substitution underlies craniosynostosis in this patient and that interaction of BCL11B with transcriptional regulatory complexes containing RBBP4/7 plays a key role in the ability of BCL11B to suppress the gene expression program underpinning osteogenic differentiation within mammalian cranial sutures.
Results
Clinical description
The index patient was a Caucasian male, delivered by vacuum extraction at 39 weeks of gestation weighing 3390 g. He was the first child of non-consanguineous, unaffected parents. The family history was negative for craniosynostosis. Examined directly after birth, he was noted to have flattening of the forehead and supra-orbital rim on the right side.
Physical examination at 10 months of age revealed a frontal plagiocephalic head shape, mild vertical orbital dystopia, a high philtrum, a short nose and narrow eyebrows (Fig. 1A–C). The skull circumference was 44 cm (−1.81 SD). Skull radiographs and three-dimensional computed tomography (3D-CT) showed synostosis of the right coronal suture and increased fingerprinting (Fig. 1D). Skull radiographs 2 months later showed partial synostosis of the left coronal suture (not shown). A fronto-supraorbital remodelling was performed at the age of 1 year.
Figure 1.
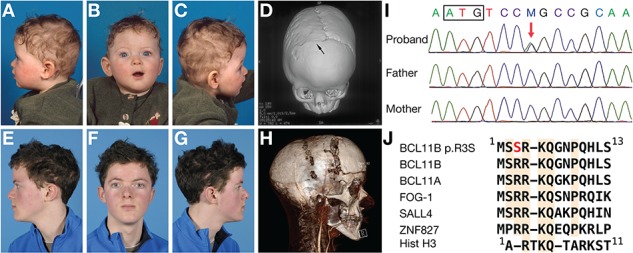
Clinical features. (A–C) Pre-operative facial photographs and 3D-CT scan (D) at the age of 10 months. Right coronal synostosis is indicated by the arrow. (E–G) Post-operative facial photographs at the age of 15 years. (H) 3D-CT and angiogram scan at the age of 12 years just prior to distraction. (I) DNA sequence analysis of proband and parents showing a de novo C>A variant (red arrow) in BCL11B (c.7C>A). The box encloses the start codon. (J) Alignment of amino termini of BCL11B, transcription factors harbouring related sequences and histone H3.
At the age of 5 years, the patient developed impaired vision. Fundoscopy showed papilledema (1.5 dpt right eye and 2 dpt left eye), and a CT scan showed reduced peripheral cerebrospinal fluid spaces, consistent with increased intracranial pressure. This was confirmed by direct measurement (20 mm Hg), and a second fronto-supraorbital advancement was performed. A nonverbal IQ exam, completed at the age of 6 years, indicated a score of 107 (Snijders-Oomen Nonverbal Intelligence Test 2.5-7).
At the age of 11 years, an epileptic insult was observed. Shortly after, vision again declined and fundoscopy showed bilateral papilledema of 2 dpt, while intracranial pressure measurements and imaging (3D-CT and magnetic resonance imaging) were inconclusive. A fronto-orbital advancement and occipital distraction were performed at the age of 12 years. Distraction started after 2 days but was paused after 9 days due to severe headaches. A CT angiogram with 3D-CT reconstruction showed traction on the sagittal sinus and formation of collaterals suboccipitally (Fig. 1H). Distraction was restarted resulting in improvement in vision. Distractors were removed after a period of 4 months. Subsequently, the patient developed normally (Fig. 1E–G) and attended a normal school without additional help up to the age of 16 years.
Genetic analysis
Karyotyping was normal and FGFR1, FGFR2, FGFR3, TWIST1 and ERF were negative for mutations by dideoxy-sequencing. Whole-genome sequencing (WGS) (30) of the parent–child trio was performed, and the data were analysed following a de novo disease model, as described previously (31). Two high-confidence de novo coding variants were identified (Supplementary Material, Table S1); the top ranked of which was in BCL11B (c.7C>A, encoding p.R3S). Sequence analysis confirmed that this was a de novo variant (Fig. 1I). Furthermore, the c.7C>A variant was not present in any database, including the Exome Aggregation Consortium (ExAC) and the Genome Aggregation Database (gnomAD) covering more than 120 000 exomes or genomes from unrelated individuals. The BCL11B variant was thought to likely be causally significant, as Bcl11b is expressed in cranial sutures (29,32), and Bcl11b−/− mice exhibit craniofacial abnormalities, including craniosynostosis (29). Furthermore, the p.R3S substitution occurs within a conserved amino-terminal domain found within a class of NuRD complex interacting transcription factors (Fig. 1J). The second-ranked variant was a de novo missense change in FERMT2. It was considered unlikely to provide a major contribution to the phenotype (Supplementary Material, Table S1). All other coding variants had a low minimal somatic score, and therefore were unlikely to be real. No variants of interest were detected under a recessive or X-linked inheritance hypothesis (data not shown).
To assess the contribution of BCL11B variants to craniosynostosis aetiology more generally, we interrogated existing exome sequencing and WGS data (186 cases) and screened by targeted resequencing a panel of 382 unrelated probands, all without a genetic diagnosis. In addition, we analysed BCL11B exon 1, which encodes the conserved amino-terminal region of the protein, by dideoxy sequencing in a further 115 cases that were not included in the resequencing cohort. Although we identified several rare variants by resequencing (Supplementary Material, Table S2), no plausible causative changes were identified, indicating that mutations of BCL11B leading to craniosynostosis may be rare and/or highly specific.
Effect of p.R3S substitution on interaction of BCL11B with transcriptional regulatory complexes
Co-immunoprecipitation (co-IP) analyses were performed in extracts of transiently transfected HEK293T cells to determine if the p.R3S substitution altered the spectrum of selected transcriptional regulatory complexes with which BCL11B interacts. The mutant protein was well expressed and localized in the nucleus in a manner that was indistinguishable from wild-type protein (Fig. 2A). However, co-IP studies revealed that the amino acid substitution abolished the ability of BCL11B to interact with both the NuRD (compare lanes 4 and 6 of top panel of Fig. 2B; MTA2 and HDAC2 were used as markers of the NuRD complex) and PRC2 complexes (compare lanes 4 and 6 of the middle panel of Fig. 2B; enhancer of zeste 2 [EZH2] was used as a marker of PRC2). The patient-derived substitution did not affect the ability of BCL11B to interact with SIRT1, a class III histone deacetylase (compare lanes 4 and 6 of bottom panel of Fig. 2B), with which BCL11B interacts directly (33). Interaction of BCL11B with PRC2, which to our knowledge has not been reported previously, was hypothesized due to the presence of RBBP4 and RBBP7 in the PRC2 complex and is demonstrated herein.
Figure 2.
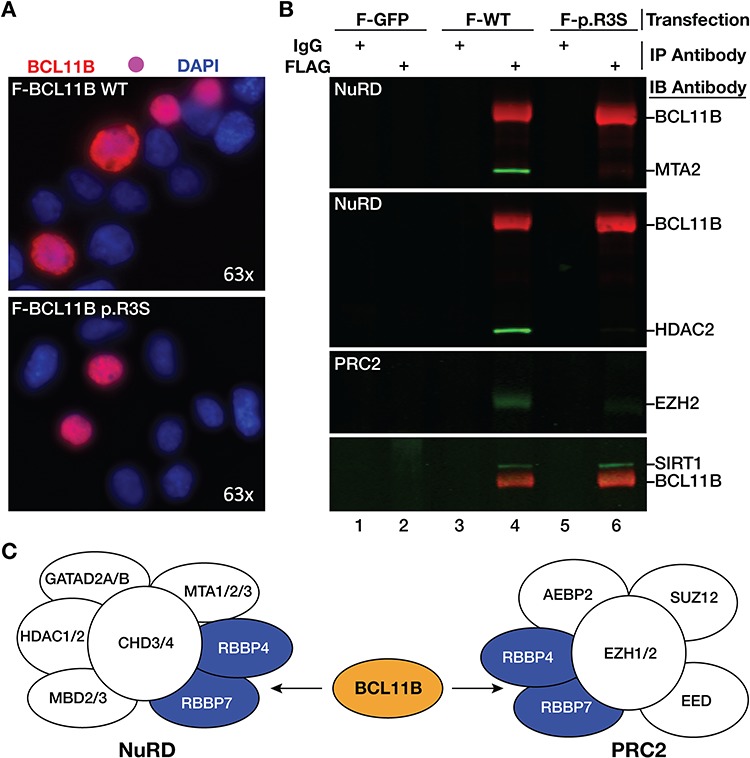
BCL11B p.R3S substitution disrupts interaction with NuRD and PRC2 complexes. (A) Immunocytochemistry of HEK293T cells expressing wild-type BCL11B (top) and BCL11B p.R3S (bottom). Cells were stained with an anti-BCL11B monoclonal antibody and co-stained with DAPI to visualize nuclei. (B) Co-immunoprecipitation studies. BCL11B and BCL11B p.R3S were immunoprecipitated with the anti-FLAG antibody from transiently transfected HEK293T cells, and the corresponding blot was probed with antibodies to BCL11B (red), MTA2 (green), HDAC2 (green), EZH2 (green) and SIRT1 (green). Comparison of lanes 4 (wild-type protein) and 6 (p.R3S) demonstrates that the patient-derived BCL11B mutant does not interact with the NuRD or PRC2 complexes, however, the p.R3S substitution did not alter interaction of the protein with the class III histone deacetylase SIRT1 (33). (C) Schematic outlining BCL11B interaction with the NuRD and PRC2 complexes.
Interaction of BCL11B and BCL11B p.R3S with the RBBP4–MTA1 complex
The amino terminus of the NuRD-associated transcription factor FOG-1 shares a conserved sequence with BCL11B (RRKQxxP, see Fig. 1J). We used the high-resolution structure of FOG-1 peptide bound to RBBP4 (34) to construct a model of BCL11B and BCL11B p.R3S peptides bound to the RBBP4–MTA1 complex (35). The resulting model predicts that BCL11B binds to an electronegative surface on one face of RBBP4, principally via three invariant electropositive residues within the conserved, amino-terminal region of BCL11B. Of these, Arg-4 is bound within an electronegative channel of RBBP4, while Arg-3 and Lys-5 interact with clefts closer to the top surface of RBBP4 (Fig. 3A). Recent reports of the crystal structure of BCL11A and SALL4 in complex with RBBP4 described similar findings, as expected (36,37). The p.R3S substitution removes one of these charged RBBP4-coordinated residues and generates a novel potential site for post-translational modification (PTM; e.g. phosphorylation or O-glycosylation). To evaluate the consequences of this substitution we used fluorescence anisotropy to measure the equilibrium binding affinity of amino-terminal BCL11B peptides to RBBP4–MTA1 complex in vitro. We also conducted molecular dynamics (MD) simulations to gain further insight into this molecular association.
Figure 3.
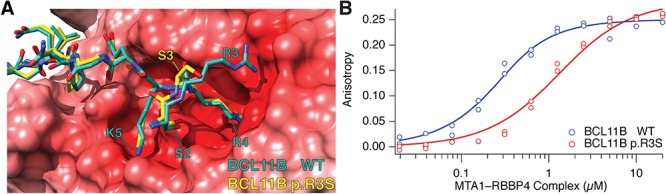
Effect of the p.R3S substitution on interaction of BCL11B with RBBP4–MTA1. (A) Modelled wild-type (green) and p.R3S mutant (yellow) BCL11B peptides (residues 1–15) superimposed and bound to the electrostatic potential surface of the RBBP4–MTA1 complex is shown. The electrostatic potential was calculated with APBS (69) and shown in shades of blue and red to represent positive and negative potential, respectively, and visualized with UCSF Chimera (70). (B) Binding of fluorescein-labelled wild-type and BCL11B p.R3S amino-terminal peptides to RBBP4–MTA1 complex as monitored by fluorescence anisotropy. Steady-state fluorescence anisotropy measurements of each peptide (12.5 nm) are shown with increasing concentration of unlabelled RBBP4–MTA1 complex. Fluorescence anisotropy was calculated using Eq. 1 and fit to Eq. 2. The dissociation constants for wild-type and BCL11B p.R3S peptides were 0.133 ± 0.043 μm and 1.16 ± 0.28 μm, respectively. Data are from replicate independent experiments, and the error estimate is the 95% confidence interval.
Using fluorescein-conjugated wild-type and BCL11B p.R3S peptides, we estimated equilibrium binding affinities from the increase in anisotropy when titrated with unlabelled RBBP4–MTA1 complex. At physiological ionic strength the p.R3S substitution decreased the affinity of the BCL11B amino-terminal peptide by 8.7-fold, from 0.133 ± 0.043 μM, found with the wild-type Arg-3 peptide, to 1.16 ± 0.28 μM for the mutant, Ser-3-containing peptide (Fig. 3B).
We conducted MD simulations to assess the consequence of the p.R3S substitution on potential conformational changes in the amino-terminal tail of BCL11B. The resulting equilibrium structures of wild-type BCL11B peptide (Fig. 4A) were similar to the FOG-1-based model (Fig. 3A) and the BCL11A–RBBP4 structure (36), while the BCL11B p.R3S equilibrium structures revealed a conformational change in the peptide backbone at the site of mutation affecting the coordination of nearby residues (Fig. 4B). The mutant Ser-3 side chain was uncoordinated to RBBP4 and its ψ-bond rotational change oriented both itself and Ser-2 away from the RBBP4 surface. The mutant backbone conformation was indistinguishable from wild type at nearby invariant residues Arg-4, Lys-5 and Gln-6 (Figs 4B and 5A), however, the side chains of these residues favoured alternative positions. While the wild-type BCL11B peptide Arg-4 preferred the axial conformation, deep in the RBBP4 binding site as observed in other amino-terminal motif–RBBP4 structures (34,36–38), the p.R3S mutant peptide preferred an RBBP4 surface conformation coordinated to Asp-198. In the wild-type peptide, Lys-5 was observed in multiple conformations, both coordinated in a binding pocket on the RBBP4 surface and uncoordinated and solvent exposed; but in the p.R3S mutant, the uncoordinated solvent exposed conformations were more frequently observed than with the wild-type peptide (Fig. 5B). The side chain of Gln-6 was also more conformationally flexible in the mutant where it assumed a series of Ser-378-localized and Ser-378-coordinated conformations unique to the p.R3S mutant peptide. Simulation results suggest that the p.R3S substitution induces a peptide backbone conformational change that affects adjacent residues. In one direction, this conformational change exposes to solvent both the known PTM site Ser-2 (39) and the mutant Ser-3. In the other direction, the loss of one of the three conserved ionic interactions affects the coordination of Arg-4 and most prominently Lys-5.
Figure 4.
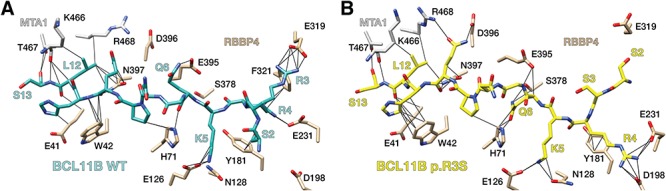
Effect of BCL11B p.R3S substitution on complex bound conformation. RBBP4–MTA1 complex bound structure BCL11B structure of wild-type (A) and p.R3S mutant (B) showing the conformation of representative equilibrium structures from MD simulations.
Figure 5.
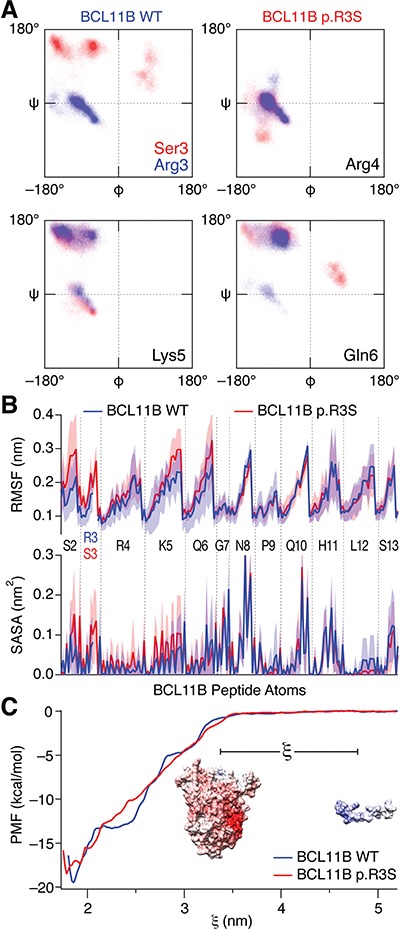
MD analyses of the p.R3S substitution on interaction of BCL11B with RBBP4–MTA1. (A) Ramachandran plots of the p.R3S substitution showing the effects on the site of mutation and adjacent residues. The plots show the distribution of backbone configurations from five independent simulations of wild-type (blue) and p.R3S mutant (red) BCL11B peptides bound to RBBP4–MTA1 complex calculated from MD simulations between 50 and 100 ns. (B) Root-mean-square fluctuation and solvent accessible surface area of the wild-type (blue) and p.R3S mutant (red) BCL11B peptide from MD simulations. The mean of five independent simulations calculated between 50 and 100 ns is shown, and the shaded regions represent the standard deviations. For wild-type Arg-3, only the backbone atoms in common with mutant Ser-3 are shown. (C) PMF from umbrella sampling molecular dynamic simulations. Umbrella sampling simulations for wild-type and BCL11B p.R3S amino-terminal peptides binding to RBBP4–MTA1 complex as a function of reaction coordinate (ξ), which is the centre-of-mass distance between the complex and peptide (illustrated in the inset). The plotted values are the mean of 500 bootstrapped simulations from which the ∆Gbind was −19.5 kcal/mol for the BCL11B peptide and −18.5 kcal/mol for BCL11B p.R3S peptide.
We examined the consequence of the BCL11B p.R3S substitution on the statistical binding free energy by calculating the potential of mean force (PMF) from a series of overlapping 15 ns MD simulations taken along the association–dissociation reaction coordinate of the peptide–complex. The analyses calculated a ∆∆G of the mutation of 1 kcal/mol (Fig. 5C) a value in agreement with fluorescence anisotropy results (Fig. 3B), adding confidence to the MD-based analyses above.
Craniosynostosis in mice harbouring the BCL11B p.R3S substitution
To confirm causality of the BCL11B p.R3S substitution for craniosynostosis and to generate a genetic model of the disorder, we introduced the c.7C>A mutation into the germ line of C57BL/6 mice using CRISPR-Cas9 (clustered, regularly interspaced, short palindromic repeat/CRISPR-associated protein 9) genome editing (40) as outlined in Figure 6 and described in the Reagents and Techniques section.
Figure 6.
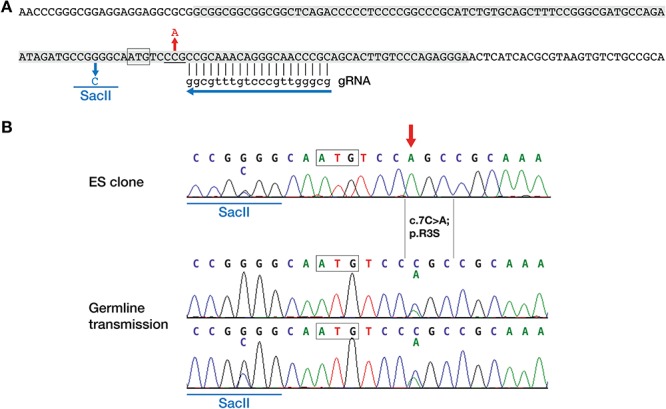
Strategy for generating Bcl11bR3S mice. (A) Mouse Bcl11b genomic sequence around the translation start site (boxed) with CRISPR-Cas9 guide RNA (gRNA) target site and protospacer adjacent motif (underlined) shown. The 127 nucleotides sequence used in the donor oligonucleotide for homology directed repair is shaded. The donor sequence included two mismatches to the genomic sequence to introduce a SacII site for screening purposes (shown in blue) and the c.7C>A; p.R3S mutation (shown in red). (B) DNA sequence from an ES clone with a heterozygous SacII site and homozygous c.7C>A mutation (red arrow, top). Germline transmission of c.7C>A with and without the SacII site (below). The translation start codon is boxed.
Heterozygous, Bcl11bR3S/+ mice were born at Mendelian ratios, survived into adulthood without gross anatomical abnormalities and bred normally. However, examination of calvarial sutures by micro-CT revealed that Bcl11bR3S/+ mice exhibited variable and partial, bilateral osteogenic fusion of the coronal suture that was accompanied by narrowing of the sagittal and lambdoid sutures by ~50% at P0 (compare panels A with B, D with E and G with H of Fig. 7; Supplementary Material, Figs S1 and S2). Other calvarial and facial sutures in the Bcl11bR3S/+ mice were indistinguishable from those of wild-type mice (Fig. 7A–D; Supplementary Material, Fig. S3).
Figure 7.
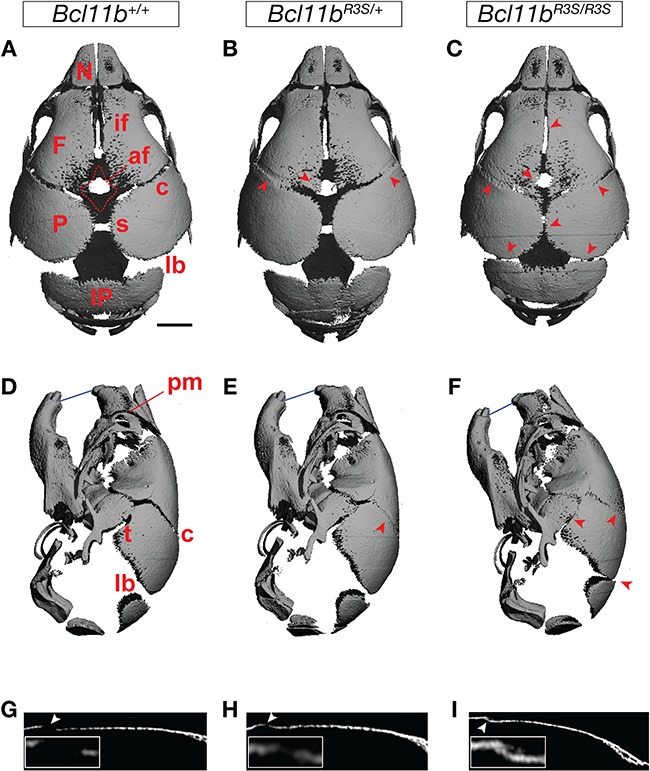
Craniosynostosis in mice harbouring p.R3S substitution. Superior (A–C), lateral (D–F) and sagittal sectional (G–I) views of micro-CT scans of wild-type control, Bcl11bR3S/+ and Bcl11bR3S/R3S in P0 mice skulls. Arrowheads indicate increased mineralization and reduced anterior fontanel (af) and affected coronal (c), interfrontal (if), lambdoid (lb), premaxillary-maxillary (pm), sagittal (s) and temporal (t) sutures. Calvarial interparietal (IP), frontal (F), nasal (N) and parietal (P) bones are indicated in the figure, and the blue line indicates dental occlusion. Scale bar, 1 mm.
Homozygous, mutant mice (Bcl11bR3S/R3S) recapitulated perinatal lethality of Bcl11b−/− mice, due to apparent respiratory insufficiency (26), as well as multi-suture craniosynostosis at P0 involving the coronal (bilateral), interfrontal, sagittal, interparietal and temporal sutures (Fig. 7C, F and I). However, in marked contrast to Bcl11b−/− mice or those lacking BCL11B in cells derived from neural crest (29), Bcl11bR3S/R3S mice did not exhibit fusion of facial sutures (Fig. 7F; Supplementary Material, Fig. S3). The sutural mesenchyme of the facial skeleton is the most sensitive to loss of BCL11B and frequently undergoes premature osteogenic differentiation in Bcl11b+/− heterozygotes (29).
Discussion
This paper describes a novel, de novo mutation of BCL11B in a patient with unilateral coronal suture craniosynostosis. The patient suffered severe sequelae requiring three corrective surgeries before the age of 13 years, suggesting a significant and progressive clinical course. The patient was negative for mutations of known craniosynostosis-related genes, which together with our previous demonstration that Bcl11b−/− mice exhibit craniosynostosis, including of the coronal suture (29), led us to evaluate the molecular consequences of the BCL11B p.R3S substitution and generate a mouse model for this mutation.
BCL11B contains an amino-terminal motif shared among a class of NuRD complex interacting transcription factors and the p.R3S substitution replaces Arg-3 of this highly conserved motif with serine (Fig. 1J). Crystal structures of RBBP4 in complex with the amino-terminal fragments of FOG-1 (34), BCL11A (36) and SALL4 (37) showed the motif residues Arg-3, Arg-4 and Lys-5 coordinated to the acidic core and surface of RBBP4 (Fig. 3A), suggesting that the Arg-3 contributes to the ionic coordination stabilizing its interaction with RBBP4 and closely related RBBP7. In these structures, Arg-3 appeared to be more flexible than the other motif invariant residues. Its electron density was not resolved in the ZNF827–RBBP4 structure (38), and unlike Arg-4 and Lys-5, the side-chain electron densities of Arg-3 fit to four slightly different coordination positions among the three resolved structures. Substitutions at Arg-3 are also more sensitive to context than other motif invariant residues. Site-directed mutagenesis of FOG-1 showed that Arg-4 and Lys-5 were critical for both high-affinity interaction with NuRD complex and transcriptional repressive activity. In contrast, mutation of Arg-3 selectively affected the affinity of FOG-1 for subtypes of NuRD complexes, contributing more to the stability of MTA2–NuRD than to MTA1–NuRD complexes (41). Moreover, mutation of Arg-3 only partly impaired FOG-1-mediated transcriptional repressive activity (41). However, Arg-3, Arg-4 and Lys-5 contribute equally to FOG-2-mediated transcriptional repression, suggesting that the individual residues of these motifs may exhibit a degree of contextual specificity (42). We found the p.R3S substitution in BCL11B eliminated detectable interaction with NuRD complex, also with the RBBP4/7-containing PRC2 complex, without affecting its interaction with SIRT1 (Fig. 2B and C), an interaction previously shown to involve an internal BCL11B domain independent of the amino terminus (33). BCL11B harbours multiple protein–protein interactions domains (33,43–46), and the present results suggest that the effect of the p.R3S substitution may be limited to a subset of interactions that depend on the amino-terminal motif of BCL11B with RBBP4/7-containing transcriptional regulatory complexes.
We examined the interaction of BCL11B and BCL11B p.R3S peptides with a subassembly of the NuRD complex composed of RBBP4 and a large fragment of MTA1. Our in vitro binding data demonstrated that the p.R3S peptide bound to the RBBP4–MTA1 with an affinity that was nearly an order of magnitude less than that of the corresponding wild-type peptide (Fig. 3B). Furthermore, results of MD simulations suggested that the p.R3S substitution additionally induced a conformational change in the BCL11B peptide, increased its disorder, negatively affected binding stability and potentially increased its potential for ectopic PTM (Fig. 5A and B).
Other interactions within the NuRD–BCL11B complex could positively or negatively impact interaction of wild-type BCL11B and/or BCL11B p.R3S proteins with the RBBP4–MTA1 complex. Based on previous structural studies of this subassembly (35) and a report that BCL11B interacts with MTA1 and MTA2 (45), it is conceivable that an MTA protein bound to RBBP4 could contribute to the stability of BCL11B interaction with RBBP4. Indeed, modelling results (Fig. 4) suggest that MTA1 region 466–468 may interact with Leu-12 and Ser-13 of BCL11B to stabilize the interaction of the latter with the holo-NuRD complex in the metazoan nucleus. Unfortunately, the available structural data does not include regions of MTA1/2 that could interact with both RBBP4/7 and the amino-terminal motif containing the substitution site.
The amino termini of BCL11B and related transcription factors share sequence (Fig. 1J) and structural (Fig. 8) homology with the amino-terminal tail of histone H3 when bound to RBBP4/7 factors. One consequence of the BCL11B p.R3S substitution is to expose both the mutant Ser-3 and the adjacent Ser-2 residues to solvent, similar to the position of Thr-3 histone H3 when bound to RBBP4/7 (Fig. 8). In histone H3, Arg-2 and Lys-4 are both methylated and Thr-3 is phosphorylated. With the exception of Ser-2 phosphorylation in transiently transfected cells (39), similar PTMs of the amino tail of BCL11B are unknown, but it is conceivable that the p.R3S substitution renders the amino tail of BCL11B susceptible to other, histone-like PTMs, such as ectopic phosphorylation of the mutant p.R3S site.
Figure 8.
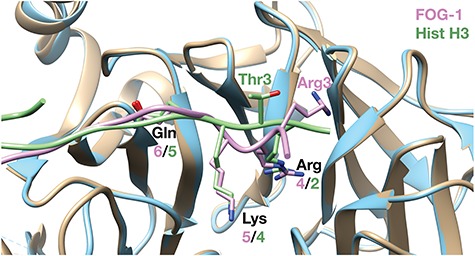
Common structural motif shared with histone H3 tail. The structural alignment of FOG-1 (pink) bound to RBBP4 (blue; PDB ID: 2XU7) (34) and histone H3 (green) bound to RBBP4/7 Drosophila homolog (tan; PDB ID: 2YBA) (71) is shown. BCL11B shares an identical sequence with FOG-1 (structure shown) through Gln-6 (see Fig. 1J).
Disruption of the interaction of BCL11B with RBBP4 and RBBP7 could have profound effects on the transcriptional regulatory activity of BCL11B. BCL11B, a sequence-specific transcription factor, presumably recruits the RBBP4/7-containing complexes to specific genetic loci that are transcriptionally regulated by BCL11B. The reduced affinity of BCL11B p.R3S for RBBP4/7 could greatly compromise BCL11B-dependent recruitment of RBBP4/7-containing transcriptional regulatory complexes to chromatin, resulting in targeted dysregulation of gene expression. Moreover, the sequential nature of recruitment of RBBP4/7-containing transcriptional complexes to target chromatin may serve to amplify the dampened affinity of BCL11B p.R3S for RBBP4/7. For example, BCL11B-mediated repression of a subset of genes may involve sequential NuRD-mediated deacetylation of H3K27ac followed by PRC2-dependent methylation of H3K27, generating a transcriptionally non-permissive environment underpinned by the H3K27me3 modification (47–49). H3K27me3 locks down a transcriptionally repressive state that is physiologically critical in development, cellular differentiation and inactivation of the X chromosome (50–52). As BCL11B-mediated recruitment of both NuRD and PRC2 to this hypothetical locus presumably requires the sequential and binary interaction of BCL11B with RBBP4 (or RBBP7), the p.R3S substitution reported herein could have a multiplicative effect on the transcriptional outcome at this locus. This phenomenon may explain, at least partially, strong ectopic expression—or de-repression—of Runx2 and/or Fgfr2c within the non-osteogenic mesenchyme of the coronal suture in Bcl11b−/− mice at E16.5, followed by ossification of this suture by E18.5 (29).
We generated a mouse model for the BCL11B p.R3S substitution, and this single amino acid change recapitulated coronal suture craniosynostosis in both the heterozygous and homozygous backgrounds. However, craniosynostosis in Bcl11bR3S/+ mice was much less severe and more highly variable than that observed in homozygous Bcl11bR3S/R3S mice. These findings demonstrate that the BCL11B p.R3S substitution is causally related to the craniosynostosis seen in our patient.
Neither Bcl11bR3S/+ nor Bcl11bR3S/R3S mice exhibited synostosis of sutures within the facial skeleton. This finding is surprising because we have previously found that facial sutures are even more sensitive to BCL11B dosage than those of the cranial skeleton (29), suggesting that the transcriptional regulatory complexes that BCL11B may work through to control suture patency in the facial skeleton are different from those required by the transcription factor to regulate cranial suture patency. Consistent with this speculation, our co-immunoprecipitation studies indicated that the p.R3S substitution selectively affected interaction of BCL11B with RBBP4- and RBBP7-containing complexes (NuRD and PRC2), but did not affect interaction with complexes that require other regions of BCL11B for interaction, e.g. the class III HDAC SIRT1 (33). Thus, the Bcl11bR3S/R3S mouse model generated herein may provide a means to dissect the mechanistic basis of BCL11B action in vivo and in several other developmental systems in which the transcription factor plays a critical role.
Recent reports (14,15) have described 11 monoallelic variants at the BCL11B locus that were linked to craniofacial dysmorphisms, neurodevelopmental pathologies, intellectual disabilities, dental anomalies and dermal defects. To date, only one of these, a p.N441K substitution in one of the zinc finger domains of BCL11B, has been investigated mechanistically and is reported to impact binding of BCL11B to known regulatory regions of the genome, alter the specificity of DNA binding by BCL11B and possibly generate a dominant-negative form of the protein (14). Unlike the patients who have multiple developmental anomalies, the patient with craniosynostosis described herein was otherwise healthy and did not present with any extra-craniofacial pathologies (14,15). In fact, the patient is now 19 years old and is continuing to undergo his formal education. Thus, the p.R3S substitution did not have a global effect on BCL11B function in this patient, suggesting that this mutation most likely affected the action of BCL11B primarily in the sutural mesenchyme. It will be of interest to examine the function of BCL11B p.R3S in other developmental systems, such as in the thymocyte, neuronal, skin and tooth lineages.
We hypothesize that the p.R3S substitution results in at least a partial or context-specific loss of BCL11B function, owing to restricted recruitment of NuRD and/or the RBB4/7-containing PRC2 complexes to BCL11B target loci, as described above. However, we cannot rule out a contribution to the phenotype through a gain of BCL11B function, in combination with the loss-of-function effects, in a promoter context-dependent manner. More broadly, mutations that eliminate any of the basic charges on Arg-3, Arg-4 or Lys-5, in BCL11B or in other amino-terminal motif-containing factors, are likely to disrupt interactions with RBBP4/7-containing NuRD and PRC2 complexes. There are two other reported variants affecting the charge on these residues beyond BCL11B p.R3S, namely SALL2 p.R3P and ZNF821 p.R4W (53), but the clinical significance of either substitution is unknown.
In conclusion, we identified a novel mutation in BCL11B associated with coronal suture synostosis, confirmed this association using a newly created mouse model and demonstrated that interaction of BCL11B with the transcriptional machinery was partially disrupted. This mouse model should be useful in dissecting the mechanistic basis for the role of BCL11B in maintenance of sutural patency, as well as in other organ systems in which BCL11B plays a critical developmental role.
Materials and Methods
Whole-genome sequencing
Ethical approval was given for WGS by the board of the Medical Ethical Committee Rotterdam (MEC-2012-140). Informed consent was received from the proband and parents.
WGS was performed on trio DNA by Complete Genomics (Mountain View, CA, USA), as described by Drmanac et al. (30). Data were analysed using CGA Tools version 1.6.0.43. A de novo disease model was tested, using the ‘calldiff script’ (Python script kindly provided by Complete Genomics) as described by Gilissen et al. (31). Prioritization of variants was based on the minimal somatic score and manual curation of the variants (Supplementary Material, Table S1). Confirmation of the BCL11B de novo variant was carried out by dideoxy sequencing. The BCL11B p.R3S variant has been deposited in the Leiden Open Variation Database (https://www.lovd.nl/BCL11B).
Sequence analysis of BCL11B in craniosynostosis
The clinical study was approved by Oxfordshire Research Ethics Committee B (reference C02.143) and Riverside Research Ethics Committee (reference 09/H0706/20). Written informed consent to obtain samples for genetics research was obtained from each child’s parent or guardian. DNA was extracted from whole blood.
BCL11B was resequenced in 382 craniosynostosis probands negative for mutations in known craniosynostosis-related genes. Primer sequences, conditions used and bioinformatics analyses are included in Supplementary Material, Table S3. An additional 115 patients were screened for variants in the first exon of BCL11B by dideoxy sequencing (using standard protocols) or by targeted resequencing (using the primers for BCL11B-Ex1; Supplementary Material, Table S3).
Reagents and techniques
Constructs
The BCL11B construct used herein has been described (54). BCL11B p.R3S was constructed from the parental plasmid using a mutagenic primer and the Q5 Site-Directed Mutagenesis Kit from New England Biolabs (NEB E0554S). The integrity of the entire open-reading frame of BCL11B was verified by dideoxy sequencing.
Generation of mutant mice
Mouse embryonic stem (ES) cells (E14Tg2aIV, 129/Ola) were transfected with pX458 (Addgene 48 138) (40) containing a guide sequence targeting a region near the Bcl11b translation start site (5′-CCGCAAACAGGGCAACCCGC-3′) and an oligonucleotide donor for homology directed repair and introduction of the c.7C>A mutation (5′- gcggcggcggcggctcagaccccctccccggcccgcatctgtgcagctttccgggcgatgccagaatagatgccgGggcaatgtccAgccgcaaacagggcaacccgcagcacttgtcccagaggga-3′; uppercase G indicates site of introduction of a SacII restriction site for screening; the translation start site atg is in boldface; uppercase A denotes the mutation base). Briefly, ES cells were seeded onto gelatin-coated 6-well plates at 3 × 105 cells per well 1 day before transfection with 3.3 μg plasmid, 10 μl of 10 μm donor complexed with 20 μl Fugene HD (Promega) oligonucleotide in a total volume of 100 μl made up with serum free medium. After 24 h, cells were sorted for green fluorescent protein-positive cells using fluorescence-activated cell sorting in a 96-well format. A homozygous c.7C>A ES clone was identified (containing a heterozygous SacII site), and karyotyping was injected into blastocysts (C57BL/6) for chimera generation. Several chimeric mice were produced, and the mutant allele was successfully transmitted through the germline (with and without the SacII site).
Specimen preparation and scanning
Skulls from P0 mice were collected and stored in 70% ethanol prior to scanning. Skulls were scanned using a Scanco μCT40 scanner (Scanco Medical AG, Basserdorf, Switzerland) at 12 μm voxel size, 55 kVp X-ray voltage, 145 μA intensity, with a 200 ms integration time as previously described (29). Stacks of images were also individually analysed in MicroView (version 2.5.0; GE Healthcare Biosciences, London, Ontario, Canada) to extract sagittal sections of the coronal and lambdoid sutures in all genotypes of mice. Mouse work was performed in accordance with UK Home Office regulations under approved project licences and the Oregon State University Institutional Animal Care and Use Committee.
Co-immunoprecipitations
HEK293T cells were transfected with vectors encoding FLAG-BCL11B (54), FLAG-BCL11B p.R3S or Myc-SIRT1 using the calcium phosphate method. Transfected cells were harvested in ice-cold phosphate-buffered saline (PBS) 48 h after transfection. Cell suspensions were centrifuged at 2000g for 5 min, followed by lysis under native conditions in buffer containing 20 mm HEPES, pH 7.4, 250 mm NaCl, 2 mm EDTA, 10% glycerol, 0.5% NP-40, 0.1 mm PMSF, 1 μg/mL pepstatin A, 5 μg/mL leupeptin, 10 μm E64, 25 mm NaF, 5 mm Na2P2O4 and 10 mmN-ethylmaleimide. Cells were incubated on ice for 30 min with vortexing every 5 min, followed by sonication on ice to complete lysis and shear DNA. The lysate was cleared by centrifugation, and BCL11B was immunoprecipitated from native extracts with anti-FLAG antibody (Sigma F4042), as previously described (54). SDS-PAGE immunoblot analyses used rat monoclonal anti-BCL11B (Abcam ab18465, 0.5 μg/ml) and the following rabbit primary antibodies: anti-MTA2 (Bethyl Laboratories A300-395A,1:5000 dilution), anti-HDAC2 (Bethyl A300-705A, 1:5000 dilution), anti-EZH2 (EMD-Millipore 07–689, 1:2000 dilution) and anti-Myc (Cell Signaling 2278, 1:1000 dilution to detect Myc-SIRT1), followed by IRDye680LT-conjugated anti-rat and IRDye800CW-conjugated anti-rabbit secondary antibodies (LI-COR Biosystems, Lincoln, NB, USA) used in combination. Fluorescent signal was recorded on an Odyssey dual-channel near-infrared imager (LI-COR) and analysed with manufacturer-supplied software (Image Studio, version 2.1).
Immunocytochemistry
HEK293T cells were grown on 0.1% gelatin-coated glass coverslips in 24-well plates seeded at 25 000 cells/well for 24 h prior to transfection with vectors encoding FLAG-BCL11B or FLAG-BCL11B p.R3S. Forty-eight hours after transfection, cells were washed twice with PBS, fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized with 0.3% Triton X-100/PBS for 10 min and washed twice with 0.1% Tween/PBS. The fixed and permeabilized cells were then prepared for immunocytochemistry by blocking for 1 h in 10% foetal bovine serum (FBS) in PBS, incubating for 4 h with rat anti-BCL11B monoclonal antibody (Abcam ab18465, 0.25 μg/ml) in 10% FBS/PBS, then incubating for 1.5 h with an anti-rat Cy3-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, 1:500 dilution), followed by a final incubation in 10 μg/ml DAPI in PBS for 10 min. Cells were washed twice with 0.1% Tween/PBS between each immunocytochemistry step, and all incubations were conducted at room temperature. Cells were then washed twice with deionized water, followed by step-wise dehydration with 70% ethanol, 100% ethanol and xylene. Coverslips were then mounted on glass slides with DPX (VWR), dried overnight and stored at 4°C in the dark. Fluoromicrographs were obtained using a Zeiss microscope with appropriate filters and a 63× oil-immersion objective. Colour images were digitally overlaid using Zeiss AxioVs40 software, version 4.8.2.0.
Fluorescence anisotropy
Steady-state fluorescence anisotropy of fluorescein-labelled wild-type BCL11B and BCL11B p.R3S amino-terminal peptides and unlabelled RBBP4–MTA1 complex were recorded in black 96-well plates (Corning) at room temperature using a Victor X5 plate reader (Perkin Elmer). Synthetic BCL11B peptides (residues 1–15) were carboxy-terminal labelled with 5-carboxyfluorescein (5-FAM) and were purchased from Cambridge Research Biochemicals (Billingham, UK), and the complex of full-length RBBP4 and residues 464–546 of MTA1 were prepared as previously described (35). Measurements were conducted in buffer containing 50 mm Tris, pH 7.5, 150 mm NaCl, 0.15 mg/mL BSA and 0.03% Tween-20. Fluorescence was excited at 480 nm and monitored at 535 nm. For each concentration of RBBP4–MTA1, anisotropy, A, was calculated according to
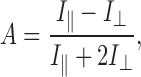 |
[Eq. 1] |
where I‖ is the fluorescence intensity measured in the parallel plane and I⊥ is the fluorescence intensity measured in the perpendicular plane. Anisotropy measurements were fit to the following equation:
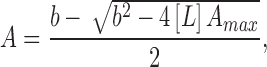 |
[Eq. 2] |
where [L] is the total concentration of RBBP4–MTA1 complex, Amax is the anisotropy of the complex bound fluorescein-labelled peptide, K is the dissociation constant of the peptide and b = Amax + K + [L].
Molecular modelling
MD simulations of the conserved amino-terminal tails of wild-type BCL11B and BCL11B p.R3S bound to the RBBP4–MTA1 complex were based on the high-resolution structures of FOG-1 peptide bound to RBBP4 (PDB ID: 2XU7) (34) and the RBBP4–MTA1 complex using PDB IDs 4PBY (55) and 5FXY (35). Modeller version 9.17 (56,57) and SCWRL4 (58) were used to generate initial simulation models consisting of residues 1–15 of BCL11B and BCL11B p.R3S, 8–411 of RBBP4 and 464–546 of MTA1. MD simulations used the GROMACS software package, version 2016 (59) and were conducted in explicit solvent of 0.15 m NaCl in TIP3P model of water, neutralized with additional sodium ions and parameterized with the CHARMM36m all-atom force field (60), which was chosen to model the inherently disordered BCL11B peptides. The LINCS algorithm (61) was used to constrain all bonds; short-range Coulombic and van der Waals non-bonded interactions were cut off at 1.2 nm, and long-range electrostatics were calculated according to the particle-mesh Ewald algorithm (62). Periodic boundary conditions were applied to all dimensions.
The modelled peptide–complex systems were equilibrated to simulation conditions following steepest descent energy minimization in two steps with positional restraints placed on the protein heavy atoms. First, the temperature was stabilized under a canonical constant volume (NVT) ensemble for 100 ps. Subsequently, pressure was stabilized under isothermal–isobaric ensemble (NPT) for 200 ps. Protein and solvent were coupled to separate temperature baths and maintained with the Nosé–Hoover thermostat (63,64) at 310 K, and pressure was maintained at 1 bar with the Parrinello–Rahman barostat (65). Following equilibration, unrestrained MD simulations were conducted for 100 ns to generate initial configurations for steered molecular dynamics (SMD) simulations (66). During each SMD simulation, the RBBP4 centre of mass was fixed and BCL11B peptides were pulled along the axis orthogonal to the peptide binding site by a force attached to the peptide centre of mass with a harmonic spring constant of 1000 kJ/mol/nm2 at a rate of 0.001 nm/ps for 3.5 ns. Starting configurations for umbrella sampling equilibrium MD were extracted from these SMD trajectories to provide a distribution of overlapping sampling windows along the SMD reaction coordinate. Each extracted configuration was then re-equilibrated with positional restraints placed on protein-heavy atoms under NPT ensemble for 100 ps prior to 15 ns of unrestrained MD simulation. The weighted histogram analysis method (67,68) was used to calculate the one-dimensional PMF as a function of centre-of-mass separation.
Supplementary Material
Acknowledgements
We are grateful to the patient and his parents for participating in this study. Also, we thank Andrew Wilkie for helpful discussions and support throughout this work; Karen Crawford, Sue Butler, Tim Rostron and John Frankland for help with DNA samples, tissue culture and DNA sequencing; Philip Hublitz, Jackie Sharpe and Jackie Sloane-Stanley of the WIMM Genome Engineering and Transgenic Core facilities for support in mouse model generation; Dawn Olson for assisting with the micro-CT data collection; and Drs Ryszard Zielke and Aleksandra Sikora for reagents and advice. We also thank Dr Kateryna Kyrylkova for technical advice throughout this project.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (grant HD088273 to M.L.); Higher Committee for Educational Development, Iraq (to W.H.S.). Modelling studies performed by W.K.V. used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. Molecular graphics were generated using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41-GM103311). JWRS is a Royal Society Wolfson Research Merit Award holder and funded through a Wellcome Trust Senior Investigator Award (WT100237). Work in Oxford was supported by the Wellcome Trust (Investigator Award 102731 to Andrew O.M. Wilkie), the Medical Research Council through the Weatherall Institute of Molecular Medicine Strategic Alliance (G0902418 and MC_UU_12025) and National Institute for Health Research (NIHR) Oxford Biomedical Research Centre Programme (Andrew O.M. Wilkie). The views expressed in this publication are those of the authors and not necessarily those of the National Health Service (NHS), NIHR or the Department of Health.
References
- 1. Jiang X., Iseki S., Maxson R.E., Sucov H.M. and Morriss-Kay G.M. (2002) Tissue origins and interactions in the mammalian skull vault. Dev. Biol., 241, 106–116. [DOI] [PubMed] [Google Scholar]
- 2. Twigg S.R.F. and Wilkie A.O.M. (2015) New insights into craniofacial malformations. Hum. Mol. Genet., 24, R50–R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fitzpatrick D.R. (2013) Filling in the gaps in cranial suture biology. Nat. Genet., 45, 231–232. [DOI] [PubMed] [Google Scholar]
- 4. Ishii M., Sun J., Ting M.-C. and Maxson R.E. (2015) The development of the Calvarial bones and sutures and the pathophysiology of craniosynostosis. Curr. Top. Dev. Biol., 115, 131–156. [DOI] [PubMed] [Google Scholar]
- 5. Johnson D. and Wilkie A.O.M. (2011) Craniosynostosis. Eur. J. Hum. Genet., 19, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilkie A.O.M., Johnson D. and Wall S.A. (2017) Clinical genetics of craniosynostosis. Curr. Opin. Pediatr., 29, 622–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Passos-Bueno M.R., Serti Eacute A.E.A.L., Jehee F.S., Fanganiello R. and Yeh E. (2008) Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front. Oral Biol., 12, 107–143. [DOI] [PubMed] [Google Scholar]
- 8. Twigg S.R.F. and Wilkie A.O.M. (2015) A genetic-pathophysiological framework for craniosynostosis. Am. J. Hum. Genet., 97, 359–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharma V.P., Fenwick A.L., Brockop M.S., McGowan S.J., Goos J.A.C., Hoogeboom A.J.M., Brady A.F., Jeelani N.O., Lynch S.A., Mulliken J.B. et al. (2013) Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat. Genet., 45, 304–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Timberlake A.T., Choi J., Zaidi S., Lu Q., Nelson-Williams C., Brooks E.D., Bilguvar K., Tikhonova I., Mane S., Yang J.F. et al. (2016) Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife, 5, e20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Twigg S.R.F., Vorgia E., McGowan S.J., Peraki I., Fenwick A.L., Sharma V.P., Allegra M., Zaragkoulias A., Sadighi Akha E., Knight S.J.L. et al. (2013) Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat. Genet., 45, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fenwick A.L., Kliszczak M., Cooper F., Murray J., Sanchez-Pulido L., Twigg S.R.F., Goriely A., McGowan S.J., Miller K.A., Taylor I.B. et al. (2016) Mutations in CDC45, encoding an essential component of the pre-initiation complex, cause Meier-Gorlin syndrome and craniosynostosis. Am. J. Hum. Genet., 99, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Twigg S.R.F., Forecki J., Goos J.A.C., Richardson I.C.A., Hoogeboom A.J.M., van den Ouweland A.M.W., Swagemakers S.M.A., Lequin M.H., Van Antwerp D., McGowan S.J. et al. (2015) Gain-of-function mutations in ZIC1 are associated with coronal craniosynostosis and learning disability. Am. J. Hum. Genet., 97, 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Punwani D., Zhang Y., Yu J., Cowan M.J., Rana S., Kwan A., Adhikari A.N., Lizama C.O., Mendelsohn B.A., Fahl S.P. et al. (2016) Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. N. Engl. J. Med., 375, 2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lessel D., Gehbauer C., Bramswig N.C., Schluth-Bolard C., Venkataramanappa S., van Gassen K.L.I., Hempel M., Haack T.B., Baresic A., Genetti C.A. et al. (2018) BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain, 141, 2299–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wakabayashi Y., Watanabe H., Inoue J., Takeda N., Sakata J., Mishima Y., Hitomi J., Yamamoto T., Utsuyama M., Niwa O. et al. (2003) Bcl11b is required for differentiation and survival of αβ T lymphocytes. Nat. Immunol., 4, 533–539. [DOI] [PubMed] [Google Scholar]
- 17. Li L., Leid M. and Rothenberg E.V. (2010) An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science, 329, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Albu D.I., Feng D., Bhattacharya D., Jenkins N.A., Copeland N.G., Liu P. and Avram D. (2007) BCL11B is required for positive selection and survival of double-positive thymocytes. J. Exp. Med., 204, 3003–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kastner P., Chan S., Vogel W.K., Zhang L.-J., Topark-Ngarm A., Golonzhka O., Jost B., Le Gras S., Gross M.K. and Leid M. (2010) Bcl11b represses a mature T-cell gene expression program in immature CD4+ CD8+ thymocytes. Eur. J. Immunol., 40, 2143–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albu D.I., Vanvalkenburgh J., Morin N., Califano D., Jenkins N.A., Copeland N.G., Liu P. and Avram D. (2011) Transcription factor Bcl11b controls selection of invariant natural killer T-cells by regulating glycolipid presentation in double-positive thymocytes. Proc. Natl. Acad. Sci. U. S. A., 108, 6211–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uddin M.N., Zhang Y., Harton J.A., MacNamara K.C. and Avram D. (2014) TNF-α-dependent hematopoiesis following Bcl11b deletion in T cells restricts metastatic melanoma.J. Immunol., 192, 1946–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vanvalkenburgh J., Albu D.I., Bapanpally C., Casanova S., Califano D., Jones D.M., Ignatowicz L., Kawamoto S., Fagarasan S., Jenkins N.A. et al. (2011) Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. J. Exp. Med., 208, 2069–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arlotta P., Molyneaux B.J., Jabaudon D., Yoshida Y. and Macklis J.D. (2008) Ctip2 controls the differentiation of medium spiny neurons and the establishment of the cellular architecture of the striatum. J. Neurosci., 28, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simon R., Baumann L., Fischer J., Seigfried F.A., De Bruyckere E., Liu P., Jenkins N.A., Copeland N.G., Schwegler H. and Britsch S. (2016) Structure-function integrity of the adult hippocampus depends on the transcription factor Bcl11b/Ctip2. Genes Brain Behav., 15, 405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simon R., Brylka H., Schwegler H., Venkataramanappa S., Andratschke J., Wiegreffe C., Liu P., Fuchs E., Jenkins N.A., Copeland N.G. et al. (2012) A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J., 31, 2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Golonzhka O., Liang X., Messaddeq N., Bornert J.-M., Campbell A.L., Metzger D., Chambon P., Ganguli-Indra G., Leid M. and Indra A.K. (2009) Dual role of COUP-TF-interacting protein 2 in epidermal homeostasis and permeability barrier formation. J. Invest. Dermatol., 129, 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Golonzhka O., Metzger D., Bornert J.-M., Bay B.K., Gross M.K., Kioussi C. and Leid M. (2009) Ctip2/Bcl11b controls ameloblast formation during mammalian odontogenesis. Proc. Natl. Acad. Sci. U. S. A., 106, 4278–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kyrylkova K., Kyryachenko S., Biehs B., Klein O., Kioussi C. and Leid M. (2012) BCL11B regulates epithelial proliferation and asymmetric development of the mouse mandibular incisor. PLoS One, 7, e37670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kyrylkova K., Iwaniec U.T., Philbrick K.A. and Leid M. (2016) BCL11B regulates sutural patency in the mouse craniofacial skeleton. Dev. Biol., 415, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Drmanac R., Sparks A.B., Callow M.J., Halpern A.L., Burns N.L., Kermani B.G., Carnevali P., Nazarenko I., Nilsen G.B., Yeung G. et al. (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science, 327, 78–81. [DOI] [PubMed] [Google Scholar]
- 31. Gilissen C., Hehir-Kwa J.Y., Thung D.T., van de Vorst M., van Bon B.W.M., Willemsen M.H., Kwint M., Janssen I.M., Hoischen A., Schenck A. et al. (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature, 511, 344–347. [DOI] [PubMed] [Google Scholar]
- 32. Holmes G., van Bakel H., Zhou X., Losic B. and Jabs E.W. (2015) BCL11B expression in intramembranous osteogenesis during murine craniofacial suture development. Gene Expr. Patterns, 17, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Senawong T., Peterson V.J., Avram D., Shepherd D.M., Frye R.A., Minucci S. and Leid M. (2003) Involvement of the histone deacetylase SIRT1 in chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting protein 2-mediated transcriptional repression. J. Biol. Chem., 278, 43041–43050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lejon S., Thong S.Y., Murthy A., AlQarni S., Murzina N.V., Blobel G.A., Laue E.D. and Mackay J.P. (2011) Insights into association of the NuRD complex with FOG-1 from the crystal structure of an RbAp48·FOG-1 complex. J. Biol. Chem., 286, 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Millard C.J., Varma N., Saleh A., Morris K., Watson P.J., Bottrill A.R., Fairall L., Smith C.J. and Schwabe J.W. (2016) The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife, 5, e13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moody R.R., Lo M.-C., Meagher J.L., Lin C.-C., Stevers N.O., Tinsley S.L., Jung I., Matvekas A., Stuckey J.A. and Sun D. (2018) Probing the interaction between the histone methyltransferase/deacetylase subunit RBBP4/7 and the transcription factor BCL11A in epigenetic complexes. J. Biol. Chem., 293, 2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu B.H., Jobichen C., Chia C.S.B., Chan T.H.M., Tang J.P., Chung T.X.Y., Li J., Poulsen A., Hung A.W., Koh-Stenta X. et al. (2018) Targeting cancer addiction for SALL4 by shifting its transcriptome with a pharmacologic peptide. Proc. Natl. Acad. Sci. U. S. A., 115, E7119–E7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang S.F., Sun A.-A., Shi Y., Li F. and Pickett H.A. (2018) Structural and functional characterization of the RBBP4-ZNF827 interaction and its role in NuRD recruitment to telomeres. Biochem. J., 475, 2667–2679. [DOI] [PubMed] [Google Scholar]
- 39. Dubuissez M., Loison I., Paget S., Vorng H., Ait-Yahia S., Rohr O., Tsicopoulos A. and Leprince D. (2016) Protein kinase C-mediated phosphorylation of BCL11B at serine 2 negatively regulates its interaction with NuRD complexes during CD4+ T-cell activation. Mol. Cell. Biol., 36, 1881–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A. and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc., 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hong W., Nakazawa M., Chen Y.-Y., Kori R., Vakoc C.R., Rakowski C. and Blobel G.A. (2005) FOG-1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA-1. EMBO J., 24, 2367–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin A.C., Roche A.E., Wilk J. and Svensson E.C. (2004) The N termini of friend of GATA (FOG) proteins define a novel transcriptional repression motif and a superfamily of transcriptional repressors. J. Biol. Chem., 279, 55017–55023. [DOI] [PubMed] [Google Scholar]
- 43. Rohr O., Lecestre D., Chasserot-Golaz S., Marban C., Avram D., Aunis D., Leid M. and Schaeffer E. (2003) Recruitment of tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J. Virol., 77, 5415–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marban C., Redel L., Suzanne S., Van Lint C., Lecestre D., Chasserot-Golaz S., Leid M., Aunis D., Schaeffer E. and Rohr O. (2005) COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res., 33, 2318–2331. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Cismasiu V.B., Adamo K., Gecewicz J., Duque J., Lin Q. and Avram D. (2005) BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene, 24, 6753–6764. [DOI] [PubMed] [Google Scholar]
- 46. Topark-Ngarm A., Golonzhka O., Peterson V.J., Barrett B., Martinez B., Crofoot K., Filtz T.M. and Leid M. (2006) CTIP2 associates with the NuRD complex on the promoter of p57KIP2, a newly identified CTIP2 target gene. J. Biol. Chem., 281, 32272–32283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu G. and Wade P.A. (2012) NuRD and pluripotency: a complex balancing act. Cell Stem Cell, 10, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim T.W., Kang B.-H., Jang H., Kwak S., Shin J., Kim H., Lee S.-E., Lee S.-M., Lee J.-H., Kim J.-H. et al. (2015) Ctbp2 modulates NuRD-mediated deacetylation of H3K27 and facilitates PRC2-mediated H3K27me3 in active embryonic stem cell genes during exit from pluripotency. Stem Cells, 33, 2442–2455. [DOI] [PubMed] [Google Scholar]
- 49. Reynolds N., Salmon-Divon M., Dvinge H., Hynes-Allen A., Balasooriya G., Leaford D., Behrens A., Bertone P. and Hendrich B. (2012) NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb repressive complex 2 to direct gene repression. EMBO J., 31, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bracken A.P., Dietrich N., Pasini D., Hansen K.H. and Helin K. (2006) Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev., 20, 1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee T.I., Jenner R.G., Boyer L.A., Guenther M.G., Levine S.S., Kumar R.M., Chevalier B., Johnstone S.E., Cole M.F., Isono K.-I. et al. (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell, 125, 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rougeulle C., Chaumeil J., Sarma K., Allis C.D., Reinberg D., Avner P. and Heard E. (2004) Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X chromosome. Mol. Cell. Biol., 24, 5475–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD, USA) Database of Single Nucleotide Polymorphisms (dbSNP). dbSNP accessions: rs777095208 (SALL2 p.R3P) and rs765940831 (ZNF821 p.R4W), (dbSNP Build ID: 152). Available from: http://www.ncbi.nlm.nih.gov/SNP/
- 54. Zhang L.-J., Vogel W.K., Liu X., Topark-Ngarm A., Arbogast B.L., Maier C.S., Filtz T.M. and Leid M. (2012) Coordinated regulation of transcription factor Bcl11b activity in Thymocytes by the mitogen-activated protein kinase (MAPK) pathways and protein Sumoylation. J. Biol. Chem., 287, 26971–26988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Alqarni S.S.M., Murthy A., Zhang W., Przewloka M.R., Silva A.P.G., Watson A.A., Lejon S., Pei X.Y., Smits A.H., Kloet S.L. et al. (2014) Insight into the architecture of the NuRD complex: structure of the RbAp48-MTA1 subcomplex. J. Biol. Chem., 289, 21844–21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sali A. and Blundell T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol., 234, 779–815. [DOI] [PubMed] [Google Scholar]
- 57. Webb B. and Sali A. (2016) Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci., 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krivov G.G., Shapovalov M.V. and Dunbrack R.L. (2009) Improved prediction of protein side-chain conformations with SCWRL4. Proteins, 77, 778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D. et al. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H. and MacKerell A.D. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hess B. (2008) P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput., 4, 116–122. [DOI] [PubMed] [Google Scholar]
- 62. Darden T., York D. and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys., 98, 10089–10092. [Google Scholar]
- 63. Hoover W.G. (1985) Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A Gen. Phys., 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- 64. Nosé S. (1984) A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys., 81, 511–519. [Google Scholar]
- 65. Parrinello M. and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys., 52, 7182–7190. [Google Scholar]
- 66. Lemkul J.A. and Bevan D.R. (2010) Assessing the stability of Alzheimer's amyloid protofibrils using molecular dynamics. J. Phys. Chem. B, 114, 1652–1660. [DOI] [PubMed] [Google Scholar]
- 67. Hub J.S., de Groot B.L. and van der Spoel D. (2010) g_wham—a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput., 6, 3713–3720. [Google Scholar]
- 68. Kumar S., Rosenberg J.M., Bouzida D., Swendsen R.H. and Kollman P.A. (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem., 13, 1011–1021. [Google Scholar]
- 69. Baker N.A., Sept D., Joseph S., Holst M.J. and McCammon J.A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A., 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. and Ferrin T.E. (2004) UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem., 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 71. Schmitges F.W., Prusty A.B., Faty M., Stützer A., Lingaraju G.M., Aiwazian J., Sack R., Hess D., Li L., Zhou S. et al. (2011) Histone methylation by PRC2 is inhibited by active chromatin marks. Mol. Cell, 42, 330–341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.